

 This work is licensed under a
This work is licensed under a Summary
IgG4-related hypophysitis is an important diagnostic consideration in patients with a pituitary mass or pituitary dysfunction and can initially present with headaches, visual field deficits and/or endocrine dysfunction. Isolated IgG4-related pituitary disease is rare, with most cases of IgG4-related disease involving additional organ systems. We report the case of a teenage female patient with isolated IgG4-related hypophysitis, diagnosed after initially presenting with headaches. Our patient had no presenting endocrinologic abnormalities. She was treated with surgical resection, prednisolone and rituximab with no further progression of disease and sustained normal endocrine function. This case, the youngest described patient with isolated IgG4-related hypophysitis and uniquely lacking endocrinologic abnormalities, adds to the limited reports of isolated pituitary disease. The use of rituximab for isolated pituitary disease has never been described. While IgG4-related hypophysitis has been increasingly recognized, substantial evidence concerning the appropriate treatment and follow-up of these patients is largely lacking.
Learning points:
IgG4-related hypophysitis most often occurs in the setting of additional organ involvement but can be an isolated finding. This diagnosis should therefore be considered in a patient presenting with pituitary abnormalities.
Most patients with IgG4-related hypophysitis will have abnormal pituitary function, but normal functioning does not exclude this diagnosis.
Corticosteroids have been the mainstay of therapy for IgG4-related disease, with other immunosuppressive regimens being reserved for refractory cases. Further research is needed to understand the effectiveness of corticosteroid-sparing regimens and whether there is utility in using these agents as first-line therapies.
Background
Broadly, IgG4-related disease is a fibroinflammatory autoimmune condition characterized by IgG4-positive plasma cell infiltration of various organs (1). Because IgG4-related disease has been described in virtually every organ system, some refer to it as analogous to sarcoidosis: a systemic disease affecting variable organs but with the same pathologic features (1). IgG4-related hypophysitis, a specific manifestation of IgG4-related disease, was described as early as 2004, though formal diagnostic criteria lagged until 2011 (2, 3). Since then, it has been increasingly recognized in the diagnostic considerations of hypophysitis.
In 2015, the largest case series of 125 patients with IgG4-related disease was published (4). This series had no patients with pituitary involvement; however, a case series published in 2012 described 2 of 25 patients (8%) with pituitary involvement (5). Other case series follow suit with either no or very few reports of pituitary disease (6). Isolated IgG4-related hypophysitis is even more rarely described, with about a dozen cases identified in the literature (2, 3, 6, 7, 8, 9). Whether isolated or with other organ involvement, there is only one reported case of IgG4-related pituitary disease with normal pituitary function (9).
The following case report of a teenage patient with isolated IgG4-related hypophysitis adds to the small number of such reports and is the youngest documented case. Her normal pituitary function also distinguishes her case. Lastly, rituximab as a primary therapy for isolated pituitary disease has never been described in the literature.
Case presentation
A previously healthy 14-year-old female was evaluated in urgent care due to persistent headaches. Brain MRI was obtained and showed a pituitary lesion. Findings were most consistent with a cystic pituitary adenoma, and neurosurgical referral was made. Under the direction of the neurosurgery team, follow-up MRI within a week of her initial presentation showed a decrease in the size of the lesion (Fig. 1A) The continued working diagnosis was a pituitary adenoma. Consultation with endocrinology was completed. Menarche occurred at age 12 years and menstrual periods were normal. There was no history of galactorrhea. Laboratory evaluations to rule out pituitary abnormalities associated with the mass were unremarkable with the exception of a slightly elevated prolactin (Table 1).
Figure 1.

Brain MRI. Post contrast sagittal and coronal images of pituitary gland, aligned vertically by date. (A) At presentation to our facility. (B) Just prior to surgery, approximately 2 months after initial presentation. (C) Four months post resection. (D) Nine months post resection. Description of images: (A) Hypointense cystic lesion in posterior aspect of anterior pituitary with no fluid–fluid level. Pituitary stalk with mild nodular enhancement and mild displacement. Not seen in this image: The pituitary stalk is mildly displaced to the left. 0.4 × 1.4 × 0.5 cm (AP by transverse by craniocaudal). (B) Increase in size of hypointense mass lesion within the anterior pituitary with a large central non contrast enhancing cystic portion measuring approximately 1.2 × 2.1 × 1.7 cm (AP by transverse by craniocaudal). (C) Resection of cystic lesion with residual cystic component in posterior aspect of the anterior pituitary measuring 0.5 × 1.5 × 0.4 cm (AP by transverse by craniocaudal). (D) Asymmetrical residual pituitary tissue. 3 mm hypointense focus in posterior aspect of the anterior pituitary.
Table 1.
Initial laboratory investigations.
| Parameter (normal values) | Results |
|---|---|
| Sodium (133–143 mmol/L) | 134 |
| Potassium (3.4–5.3 mmol/L) | 3.9 |
| Chloride (96–110 mmol/L) | 103 |
| Bicarbonate (20–32 mmol/L) | 24 |
| Creatinine (0.39–0.73 mg/dL) | 0.72 |
| Glucose (70–99 mg/dL) | 90 |
| Calcium (9.1–10.3 mg/dL) | 9.5 |
| White blood cells (4–11 × 10−9/L) | 8.5 |
| Hemoglobin (11.7–15.7 g/dL) | 14.7 |
| Hematocrit (35–47%) | 42.9 |
| Platelets (150–450 × 10−9/L) | 235 |
| CRP (0–8 mg/L) | <2.9 |
| ESR (0–15 mm/h) | 8 |
| TSH (0.4–4 mU/L) | 2.26 |
| Free T4 (0.76–1.46 ng/dL) | 1.02 |
| Lutropin (0.5–31.2 IU/L) | 5.1 |
| FSH (0.9–12.4 IU/L) | 4.8 |
| Estradiol ultrasensitive (15–350 pg/mL) | 118 |
| Prolactin (3–27 µg/L) | 36 |
| IGF binding protein 3 (3.5–9.7 µg/mL) | 5.7 |
| IGF 1 (219–483 ng/mL) | 429 |
| Adrenal corticotropin (10–47 pg/mL) | 23 |
| Baseline cortisol (4–22 µg/dL) | 27.2 |
| Cortisol 1 h post stimulation testing | 40.6 |
Initial laboratory investigations revealed no concerning abnormalities.
Approximately 2 months after her initial presentation, repeat brain MRI, performed due to worsening headaches, revealed an enlarging mass with characteristics of hemorrhage into the cystic lesion (Fig. 1B). Surgical intervention was pursued, ultimately being performed about 4 months from her initial presentation. There were no new signs of pituitary dysfunction. Prior to resection, she was taking as needed analgesics for headache, and her physical examination was normal. Breast and pubic hair development were Tanner stage 5. Ophthalmologic evaluation demonstrated no visual field deficits. There was no family history of autoimmunity or endocrinologic disorders.
Investigation
Pre-surgical laboratory investigations were reassuring, with no evidence of pituitary dysfunction except for a very mild elevation of her prolactin (Table 1), likely related to stalk compression by the pituitary mass. At the time of surgical decompression, purulent fluid exuded from the core of the mass, atypical for an adenoma. This prompted consideration of additional etiologies, with the differential including a ruptured Rathke’s cleft cyst and primary inflammatory processes such as lymphocytic hypophysitis. Histopathologic review of the resected specimen was consistent with IgG4-related hypophysitis (Fig. 2), with greater than 40 IgG4-positive cells per 400× microscopic field. There were no cyst wall remnants to suggest Rathke’s cleft cyst, and inflammatory changes were consistent with fibrotic adenohypophysitis, typical for IgG4-related disease. Post-surgical labs were notable for fluctuating hyponatremia and hypernatremia due to diabetes insipidus and related desmopressin therapy.
Figure 2.
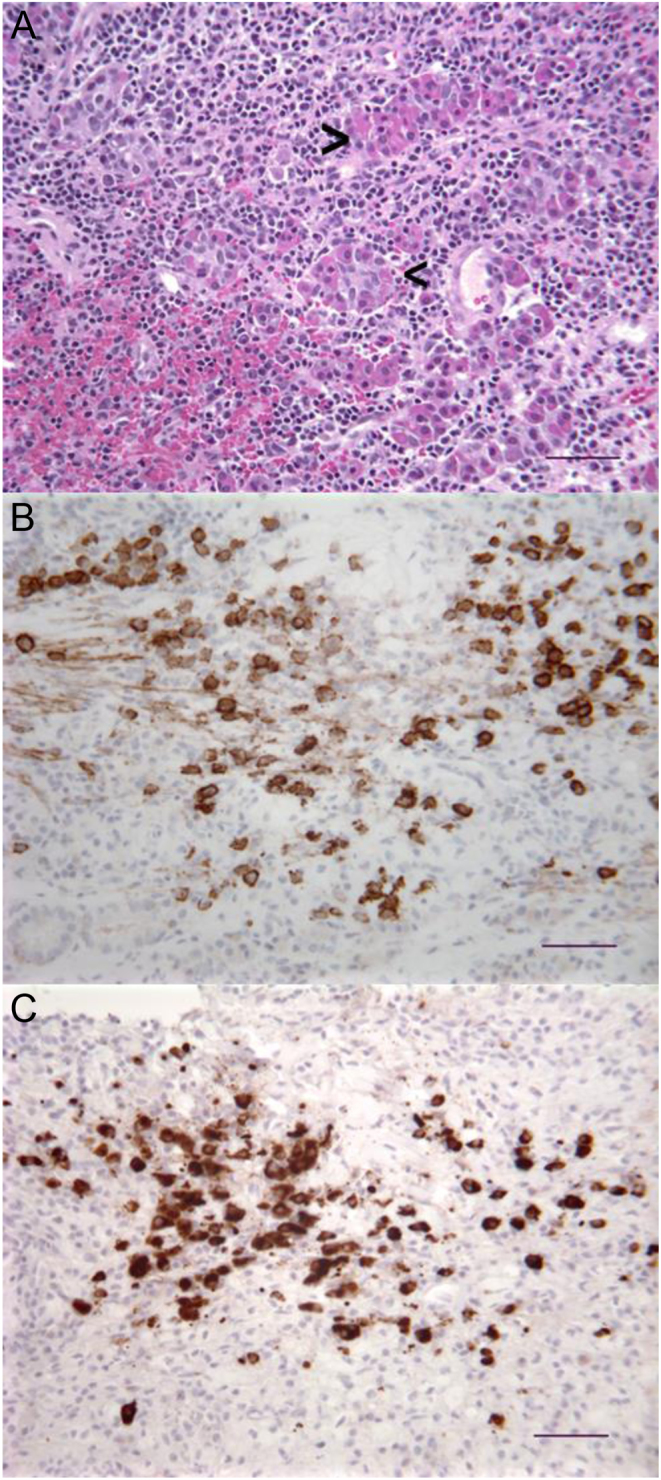
Biopsy pathology. (A) Hematoxylin-and-eosin-stained section with lymphoplasmacytic infiltrates intercalated among acinar elements (arrowheads) of the adenohypophysis. (B) Immunohistochemical stain for CD138, a plasma cell marker, showing large numbers of plasma cells within the infiltrate. Diaminobenezidine reaction product. (C) Immunohistochemical stain for IgG4 in the same area as (B), showing that the majority of plasmacytes are immunoreactive for IgG4 (>40 IgG4 positive cells in the high power field with greater than 40% of the CD138 labeled plasma cells being IgG4 positive). Diaminobenezidine reaction product. Scale bars = 50 µm. Photomicrographs were taken with a Leitz Diaplan microscope and Spot Insight digital camera.
To investigate the possibility of an underlying autoimmune disease or other IgG4-related organ involvement, additional workup was undertaken during the post-operative period (Table 2). These were performed after pathologic findings suggested IgG4-related disease, before the initiation of medical treatment. Importantly, pre-treatment serum IgG4 levels were normal, as has been described in other cases of IgG4-related disease (1), including cases of IgG4 hypophysitis (2, 3). Whole-body 2-[18F]-fluoro-2-deoxy-d-glucose–positron emission tomography/computed tomography (FDG-PET/CT) scan, completed to look for additional foci of disease, was reassuring. There was no increased signal in the pituitary. Ultimately, with paucity of findings to suggest another autoimmune/inflammatory etiology and in the absence of other foci of disease, isolated IgG4-related hypophysitis was the final diagnosis.
Table 2.
Further laboratory investigations following diagnosis of IgG4-related hypophysitis.
| Parameter (normal values) | Result |
|---|---|
| Total protein (6.8–8.8 g/dL) | 5.2 |
| Albumin (3.4–5 g/dL) | 2.8 |
| Alkaline phosphatase (70–230 U/L) | 59 |
| ALT (0–50 U/L) | 15 |
| AST (0–35 U/L) | 10 |
| Total bilirubin (0.2–1.3 mg/dL) | 0.2 |
| Total IgG (695–1620 mg/dL) | 628 |
| IgG1 (300–856 mg/dL) | 294 |
| IgG2 (64–495 mg/dL) | 182 |
| IgG3 (24–192 mg/dL) | 31 |
| IgG4 (11–157 mg/dL) | 61 |
| IgM (60–265 mg/dL) | 50 |
| IgA (70–380 mg/dL) | 84 |
| IgE (0–150 KIU/L) | 1587 |
| Antinuclear antibody (<1) | <1 |
| RNP antibody IgG (0–0.9 AI) | <0.2 |
| Scleroderma antibody (0–0.9 AI) | <0.2 |
| Smith ENA antibody (0–0.9 AI) | <0.2 |
| SSA (Ro) ENA antibody (0–0.9 AI) | <0.2 |
| SSB (La) ENA antibody (0–0.9 AI) | <0.2 |
| ds-DNA (0–29 IU/mL) | <15 |
| Total complement (60–144 CAE Units) | 73 |
| Complement C3 (76–169 mg/dL) | 57 |
| Complement C4 (15–50 mg/dL) | 10 |
| Thyroid peroxidase antibody (<35 IU/mL) | <10 |
| Thyroglobulin antibody (<40 IU/mL) | <20 |
| Tissue transglutaminase IgA | Negative |
| Urine protein/creatinine ratio (0–0.2 g/g) | 0.45 |
| Blood urine (negative) | Trace |
Additional laboratory investigations, prompted by the diagnosis of IgG4-related hypophysitis, were performed to assess for any underlying autoimmune disease and/or IgG4-related involvement of other organs. These were drawn in the post-op period, prior to initiation of treatment, and are notable for a low total protein and albumin, low total serum IgG but normal IgG4, hypocomplementemia (low C3/C4) and mild hematuria and proteinuria. There are no values to suggest an underlying autoimmune disease or other IgG4-related disease. Hematuria and proteinuria were attributed to her urinary catheter and/or acute post-operative status and resolved. Total IgG normalized after 6 months. C3 normalized within a year. C4 remained slightly low, the significance of which was unclear since no other disease foci were detected and no clear residual pituitary disease was identified. Pituitary cell antibodies were not investigated.
Treatment
After completion of additional workup and an ultimate diagnosis of IgG4-related hypophysitis, the patient was started on oral prednisolone (1 mg/kg/day, which was 40 mg), continued at a stable dose for three-and-a-half weeks, and then tapered off by 6 months from the start date. Within a month of diagnosis, even before response to prednisolone could be fully assessed, rituximab was administered as a first-line corticosteroid-sparing agent at a dose of 1000 mg IV for two doses, given 3 weeks apart due to planning logistics. Post-operative diabetes insipidus was treated with desmopressin, which was discontinued within a month of surgical decompression, and serum sodium normalized.
Outcome and follow-up
Pituitary function remained normal for 20 months of post-resection follow-up. Repeat brain MRIs at four (Fig. 1C), nine (Fig. 1D) and 20 months post-resection showed significant reduction in the pituitary mass. Post-resection images demonstrated a small residual cystic area in the posterior sella and a small amount of enhancing tissue in the right and left lateral sella. It was unclear whether these findings were due to post-surgical changes or small foci of residual disease. These areas remained stable without any interval change so residual disease was felt to be less likely.
Clinically, headaches continued post-operatively, and neurologic evaluation raised concern for daily tension headaches. Headaches did not worsen with the prednisolone taper, and their persistence was therefore felt to be unrelated to the pituitary concerns. This also called into question the relationship between the initial headaches and the pituitary mass. Despite advice to the contrary, this patient was lost to follow-up.
Discussion
Patients with IgG4-related hypophysitis most often present with pituitary dysfunction, many involving both the posterior and anterior pituitary (6, 7, 8). Because abnormalities may be subclinical, thorough pituitary function evaluation should be undertaken whenever IgG4-related pituitary disease is suspected. In the absence of pituitary dysfunction, patients may present with visual field deficits and/or headaches. The only previously described patient lacking pituitary insufficiency presented with bitemporal hemianopsia and an elevated serum IgG4 level (9). Our patient is therefore unique in the literature, being the youngest described patient with isolated hypophysitis and lacking endocrinologic abnormalities.
The workup of IgG4-related disease includes laboratory and imaging studies to identify other organ involvement. Diagnostic criteria for IgG4-related hypophysitis include pathological features, though diagnosis can be made without this (2). IgG4 levels are sometimes elevated but can be normal (1). There is some literature to suggest a correlation between serum IgG4 concentration and the number of organs affected (4), though this is debatable (1). There has been increasing interest in imaging studies, including PET. It is important to note the limits of PET, however, which identifies areas of active inflammation but would not necessarily identify older areas, where fibrosis rather than active inflammation is playing a role. In our patient’s case, the pituitary did not have an increased signal on PET, but this does not exclude involvement. Her abnormal MRI, surgical findings and pathology are consistent with IgG4-related disease despite the PET not showing increased uptake post-operatively.
Of the documented cases of IgG4-related hypophysitis, corticosteroids are the mainstay of therapy, regardless of other organ involvement or isolated pituitary disease (2, 6, 7, 8). While other immunosuppressive medications (e.g azathioprine, methotrexate, mycophenolate) have been used in cases of corticosteroid-resistant IgG4-related disease, data regarding the effectiveness of these are certainly not strong, and current recommendations are based largely on expert opinion (10). More recently, rituximab has shown favorable response rates in IgG4-related disease (11, 12). To date, no clear descriptions of rituximab use in the setting of pituitary disease, particularly isolated pituitary disease, are published. Additionally, it is generally reserved for relapsed disease, after failure of other agents. Overall, the literature remains unclear regarding which therapy option(s) are best, as well as the timing and duration.
In our patient, several factors prompted the decision to administer rituximab before primary failure of corticosteroids and over other corticosteroid-sparing agents. Two possible treatment approaches were discussed with the family. The first was the standard of care, systemic corticosteroid monotherapy. Review of cases using this approach shows that corticosteroid courses are long, many months to years, with a more prolonged maintenance course often being favored due to reported relapses with tapering or withdraw (6). The other discussed approach was early introduction of a corticosteroid-sparing agent, which we felt could possibly obviate the need for a more prolonged corticosteroid course. This was based on review of literature at the time, which suggested that while glucocorticoids are the only proven treatment to induce remission, other therapies show promise for remission maintenance (12).
Ultimately, while definitive evidence for an early corticosteroid-sparing agent was lacking, the family felt strongly about pursuing this option. Understanding the adverse effects of prolonged corticosteroids, they firmly wanted to minimize exposure. We agreed that this was reasonable and that the potential risks of adding a corticosteroid-sparing agent might be lower than the risks of prolonged corticosteroids.
The decision to administer a corticosteroid-sparing agent early was also based on relapse data for IgG4-related disease, which appears to occur in at least a third to half of patients after primary corticosteroid treatment, often after discontinuation (4, 12). Exact data on relapse rates and time to relapse varies substantially, but the literature is clear that relapses pose a major challenge and often require prolonged corticosteroids (11, 12). Because most patients with IgG4-related pituitary dysfunction require lifelong endocrine replacement therapy, experts have questioned whether the development of pituitary dysfunction suggests significant irreversible pathologic damage (13). It has been suggested that the sooner an immunosuppressive therapy is started, the greater the likelihood that pituitary function will be maintained (13). With the family being averse to prolonged corticosteroids, we felt that early initiation of a corticosteroid-sparing agent could possibly reduce relapse risk.
With a lack of evidence regarding the superiority of one corticosteroid-sparing agent over another, we felt the safety profile of rituximab was favorable. Adverse effects in cases of IgG4-related disease treated with rituximab have mainly included infection-related complications (11, 12). In our otherwise healthy patient, we felt this risk was low. Rituximab use also aligned with the family’s preference to avoid frequent administration of medications, particularly medications with day-to-day side effects. Finally, while disease relapse is described even after the use of rituximab (11, 12), there is no evidence to suggest relapse rates are greater than standard of care.
Monitoring treatment response can be challenging. When serum IgG4 levels are elevated, normalization can indicate treatment response, though there are cases of remission despite ongoing elevation of serum IgG4 levels (1). In the case of hypophysitis, there are descriptions of serum IgG4 normalization ranging from rapid (e.g. days) to more delayed (e.g. months) (3). In our patient, her pre-treatment serum IgG4 level was normal, rendering it an ineffective measure.
It is yet unknown whether early, aggressive therapy prevents evolution of disease and its spreading to additional organs. There is no way of predicting which patients will have a relapse, either in the same organ or in new organs. Thus, regular follow-up is necessary. Monitoring relies on a combination of history, physical examination, laboratory assessments and perhaps follow-up imaging. There are unfortunately no clinical guidelines regarding the frequency of follow-up, nor the precise evaluations that should be done to follow this disease over time. Our patient was unfortunately lost to follow-up 20 months from her initial presentation.
In conclusion, our case of a teenage female with isolated IgG4-related hypophysitis and an absence of hypopituitarism is distinctive, as is her treatment with rituximab and sustained remission after 20 months. Our therapy decisions were based on review of the available literature, family preferences and the recognition that evolution of pituitary dysfunction could be devastating and lifelong. Compared to other corticosteroid-sparing agents, the relative safety profile of rituximab was felt to be favorable. There is scarce guidance for the treatment of IgG4-related disease, particularly for pituitary disease, and this case highlights the need for further research to advance our understanding of the most effective treatments.
Declaration of interest
Dr Miller is a consultant for Abbvie, Ascendis, Ferring, Genentech, Novo Nordisk, Pfizer, Sandoz, Sanofi and Tolmar and has received research support from Alexion, Ascendis, Endo Pharmaceuticals, Genentech, Novo Nordisk, Opko, Sandoz, Shire, Tolmar, Ultragenyx and Versartis. The other authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported..
Funding
This research did not receive any specific grant from any funding agency in the public, commercial or not-for-profit sector.
Patient consent
We previously received a generic consent from the patient’s parent to publish this case. Due to a lack of ongoing clinical follow-up, informed consent using the specific form provided by this journal was not obtained from the patient's parents. The case has been anonymised as much as possible. We have provided a copy of our generic consent, which was deemed acceptable.
Author contribution statement
D R B is the corresponding author and was primarily responsible for manuscript preparation. D R B, P M H and B S M were all involved in the clinical care and decision making for the presented patient and all assisted in reviewing the literature. H B C was the pathologist involved in this case and contributed the article’s pathology images/legends in addition to reviewing the pathology content of the main text. P M H, B S M and H B C were responsible for manuscript revisions. All authors have read and approved the final version.
References
- 1.Stone JH, Zen Y, Deshpande V. IgG4-related disease. New England Journal of Medicine 2012. 366 539–551. ( 10.1056/NEJMra1104650) [DOI] [PubMed] [Google Scholar]
- 2.Leporati P, Landek-Salgado MA, Lupi I, Chiovato L, Caturegli P. IgG4-related hypophysitis: a new addition to the hypophysitis spectrum. Journal of Clinical Endocrinology and Metabolism 2011. 96 1971–1980. ( 10.1210/jc.2010-2970) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yuen KCJ, Moloney KJ, Mercado JU, Rostad S, McCullough BJ, Litvack ZN, Delashaw JB, Mayberg MR. A case series of atypical features of patients with biopsy-proven isolated IgG4-related hypophysitis and normal serum IgG4 levels. Pituitary 2018. 21 238–246. ( 10.1007/s11102-017-0852-4) [DOI] [PubMed] [Google Scholar]
- 4.Wallace ZS, Deshpande V, Mattoo H, Mahajan VS, Kulikova M, Pillai S, Stone JH. IgG4-related disease: clinical and laboratory features in one hundred twenty-five patients. Arthritis and Rheumatology 2015. 67 2466–2475. ( 10.1002/art.39205) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ebbo M, Daniel L, Pavic M, Seve P, Hamidou M, Andres E, Burtey S, Chiche L, Serratrice J, Longy-Boursier M, et al IgG4-related systemic disease: features and treatment response in a French cohort: results of a multicenter registry. Medicine 2012. 91 49–56. ( 10.1097/MD.0b013e3182433d77) [DOI] [PubMed] [Google Scholar]
- 6.Iseda I, Hida K, Tone A, Tenta M, Shibata Y, Matsuo K, Yamadori I, Hashimoto K. Prednisolone markedly reduced serum IgG4 levels along with the improvement of pituitary mass and anterior pituitary function in a patient with IgG4-related infundibulo-hypophysitis. Endocrine Journal 2014. 61 195–203. ( 10.1507/endocrj.EJ13-0407) [DOI] [PubMed] [Google Scholar]
- 7.Bando H, Iguchi G, Fukuoka H, Taniguchi M, Yamamoto M, Matsumoto R, Suda K, Nishizawa H, Takahashi M, Kohmura E, et al The prevalence of IgG4-related hypophysitis in 170 consecutive patients with hypopituitarism and/or central diabetes insipidus and review of the literature. European Journal of Endocrinology 2013. 170 161–172. ( 10.1530/EJE-13-0642) [DOI] [PubMed] [Google Scholar]
- 8.Sosa GA, Bell S, Christiansen SB, Pietrani M, Glerean M, Loto M, Lovazzano S, Carrizo A, Ajler P, Fainstein Day P. Histologically confirmed isolated IgG4-related hypophysitis: two case reports in young women. Endocrinology, Diabetes and Metabolism Case Reports 2014. 2014 140062 ( 10.1530/EDM-14-0062) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hattori Y, Tahara S, Ishii Y, Kitamura T, Inomoto C, Osamura RY, Teramoto A, Morita A. A case of IgG4-related hypophysitis without pituitary insufficiency. Journal of Clinical Endocrinology and Metabolism 2013. 98 1808–1811. ( 10.1210/jc.2013-1088) [DOI] [PubMed] [Google Scholar]
- 10.Khosroshahi A, Wallace ZS, Crowe JL, Akamizu T, Azumi A, Carruthers MN, Chari ST, Della-Torre E, Frulloni L, Goto H, et al International Consensus Guidance Statement on the management and treatment of IgG4-related disease. Arthritis and Rheumatology 2015. 67 1688–1699. ( 10.1002/art.39132) [DOI] [PubMed] [Google Scholar]
- 11.Khosroshahi A, Carruthers MN, Deshpande V, Unizony S, Bloch DB, Stone JH. Rituximab for the treatment of IgG4-related disease: lessons from 10 consecutive patients. Medicine 2012. 91 57–66. ( 10.1097/MD.0b013e3182431ef6) [DOI] [PubMed] [Google Scholar]
- 12.Carruthers MN, Topazian MD, Khosroshahi A, Witzig TE, Wallace ZS, Hart PA, Deshpande V, Smyrk TC, Chari S, Stone JH. Rituximab for IgG4-related disease: a prospective, open-label trial. Annals of the Rheumatic Diseases 2015. 74 1171–1177. ( 10.1136/annrheumdis-2014-206605) [DOI] [PubMed] [Google Scholar]
- 13.Caturegli P, Iwama S. From Japan with love: another tessera in the hypophysitis mosaic . Journal of Clinical Endocrinology and Metabolism 2013. 98 1865–1868. ( 10.1210/jc.2013-1912) [DOI] [PubMed] [Google Scholar]