

Abstract
The AAA+ ATPase p97 regulates ubiquitin-dependent protein homeostasis and has been pursued as a cancer drug target. The ATP-competitive inhibitor CB-5083 and allosteric inhibitor NMS-873 are the most advanced p97 inhibitors described to date. Previous studies have reported that their cytotoxicity can be readily overcome and involves single p97 mutations in the linker between the D1 and D2 ATPase domains and within D2. We report here that the proline 472 to leucine (P472L) mutation, in the D1–D2 linker and identified in CB-5083–resistant cells, desensitizes p97 to both inhibitor classes. This mutation does not disrupt the distinct D2-binding sites of the inhibitors. Instead, P472L changes ATPase domain communication within the p97 hexamer. P472L enhances cooperative D2 ATP binding and hydrolysis. This mechanism alters the function of the D1–D2 linker in the control of D2 activity involving the ATP-bound state of D1. Although increased D2 activity is sufficient to desensitize the P472L mutant to NMS-873, the mutant's desensitization to CB-5083 also requires D1 ATPase domain function. Our study highlights the remarkable adaptability of p97 ATPase domain communication that enables escape from mechanistically distinct classes of cytotoxic p97 inhibitors.
Keywords: ubiquitin, proteasome, ATPases associated with diverse cellular activities (AAA), protein degradation, protein homeostasis, protein quality control, protein unfolding
Introduction
p97 (also known as valosin-containing protein and Cdc48 in yeast) is an essential AAA+ ATPase that functions as a protein segregase (1–3). The enzyme uses mechanical force generated by ATP-dependent conformational changes to extract ubiquitin-modified proteins from membranes, interacting proteins, and DNA. This function is critical for protein quality control to clear misfolded proteins by the proteasome (4). The over-reliance of cancers on this mechanism has led to the development of p97 inhibitors as candidate therapeutic agents (5, 6).
p97 functions as a hexamer where each subunit contains an N domain and two ATPase domains, D1 and D2, that are separated by short intervening linkers (7, 8). Each ATPase domain assembles into ring-like structures in the hexamer with the D1 ring stacked on top of the D2 ring surrounding a central pore. Structural and biochemical studies have identified distinct roles for D1 and D2 in substrate processing. ATP binding to D1 re-orients the N domain upward to enable the binding of accessory proteins that deliver ubiquitinated substrates (9–12). D2 activity processes the substrate as it transits through the central pore (13). D1 ATP hydrolysis and partial ubiquitin removal by the N domain–bound deubiquitinating enzyme promote substrate release (13, 14).
Coordinated communication between the ATPase domains of p97 is necessary for its mechanism. Intra-subunit control of D1 and D2 activity is mediated by conformational changes involving the D1–D2 linker (9). Mutation of Walker A or B motifs found in both D1 and D2 that disrupt ATP binding or impair hydrolysis shows that ATP binding to one domain stimulates the other domain's ATPase activity by increasing its affinity for ATP over ADP (15–18). D2 contributes most of the overall ATPase activity of p97 and, unlike D1, requires allosteric D2–D2 interactions for cooperative ATP hydrolysis (17, 19–22). This cooperativity involves arginine finger residues Arg-635 and Arg-638 from an adjacent D2 domain to catalyze γ-phosphate hydrolysis (15, 19–21, 23).
p97 mutations cause genetic disorders known as multisystem proteinopathy type-1 (MSP-1)2 (24–26). These progressively degenerative diseases manifest in muscle, bone, and/or nervous system and are histologically identified by accumulated ubiquitin-modified proteins in afflicted tissues (26). The dominant, single p97 mutations found in MSP-1 patients are localized within the N and D1 region (25). Many MSP-1 mutants have increased D2 ATPase activity, decreased D1 ADP affinity, and aberrant N domain movement (11, 12, 27–34). A recent biochemical study found that an MSP-1 mutant unfolds a model ubiquitinated substrate faster than WT p97 (14). These findings suggest improperly coordinated ATPase activities of p97 may underlie MSP-1 pathogenesis.
p97 inhibitors developed as candidate cancer therapeutics provide new tools to understand the biological function of the enzyme. CB-5083 is an ATP-competitive and D2-selective inhibitor (35, 36). NMS-873 exclusively targets D2 activity but relies on an allosteric mechanism requiring D2 with ATP bound to inhibit cooperative D2 ATP hydrolysis (37). We and others have found that prolonged exposure of cancer cell lines to cytotoxic concentrations of these inhibitors results in the emergence of resistant cells (35, 38, 39). The resistant cells harbor single amino acid mutations in D2 and the D1–D2 linker of p97 and are distinct from those found in MSP-1 patients (Fig. 1A). How the p97 mutants overcome the mechanism of action of CB-5083 or NMS-873 is unclear.
Figure 1.

P472L mutation increases p97 ATPase activity and desensitizes the enzyme to ATP-competitive and allosteric inhibitors. A, p97 mutations found in NMS-873- or CB-5083–resistant HCT116 cells are localized to the D1–D2 linker and D2 and are distinct from disease-causing MSP-1 mutations. N615V and N616F are mutations shown to disrupt NMS-873 analog binding from cross-linking studies (37). Residues implicated in NMS-873 (pink) and CB-5083 (cyan) resistance are shown (PDB code 5FTL) with inhibitor-binding sites indicated. Additional mutations found in HCT116 cells resistant to CB-5083 and NMS-873 during this study are in bold. B, ATP titration experiments were performed with p97 and the P472L mutant to obtain steady-state kinetic parameters. Resulting Michaelis-Menten constants (KM), turnover numbers (kcat), and catalytic efficiencies (kcat/KM) are shown in Table 1. C, sensitivities of the ATPase activities of p97 and the P472L mutant to CB-5083 or NMS-873 were evaluated in inhibitor titration experiments. IC50 values obtained from dose-response curves are shown in Table 1. Data points represent the mean (n = 4) with standard deviation (S.D.) error.
In this study, we biochemically analyzed p97 mutants identified from CB-5083–resistant and NMS-873–resistant cells. We found that the CB-5083–resistant mutant harboring the proline 472 to leucine (P472L) mutation in the D1–D2 linker is also desensitized to NMS-873 but without disrupting the D2-binding sites of either inhibitor. The P472L mutation alleviates intra-subunit control of D2 activity by enhancing this domain's cooperative ATP binding and hydrolysis. These findings suggest adapted ATPase domain communication circumvents the mechanisms of action of both ATP-competitive and allosteric p97 inhibitors to provide the enzyme's essential function.
Results
P472L mutation increases p97 ATPase activity and desensitizes the enzyme to its inhibitors
We expressed and purified p97-harboring mutations identified from CB-5083–resistant (P472L, Q473P, G481A, N660K, or T688A) and NMS-873–resistant (A530T, R567H, or L639F) HCT116 cells (Fig. 1A) (35, 39). Mutants previously shown to disrupt the NMS-873 analog binding in vitro (K615V or N616F (37)), MSP-1 mutants harboring R95G or R155H (12, 26, 33, 40), and WT p97 were also included in the panel. Native gel analyses demonstrated that all mutants had essentially the same mobility as p97, with a predominant species consistent with the hexameric state of the enzyme (Fig. S1A).
We measured the ATPase activities of these proteins using colorimetric phosphate detection. After empirically determining enzyme concentrations that yielded linear rates of ATP hydrolysis, we performed ATP titration experiments to obtain steady-state kinetic parameters (Fig. S1B and Table 1). Like MSP-1 mutants (11, 12, 28, 32, 33), most mutants from CB-5083- and NMS-873–resistant cells had increased catalytic efficiencies (kcat/KM) except the N660K and T688A mutants. The P472L mutant, from CB-5083–resistant cells, had the highest activity measured due to an increased kcat and decreased KM values (Fig. 1B and Table 1).
Table 1.
Kinetic properties of p97 mutants and IC50 values of CB-5083 and NMS-873
Values shown are mean ± S.E. (n = 4).
| KM | kcat | kcat/KM | CB-5083 | NMS-873 | |
|---|---|---|---|---|---|
| μm | min−1 | μm−1 min−1 × 10−3 | nm | nm | |
| WT | 162 ± 22 | 13.4 ± 0.6 | 83 ± 12 | 8.4 ± 0.1 | 14 ± 1 |
| MSP-1 mutants | |||||
| R95G | 88 ± 12 | 13 ± 0.5 | 147 ± 21 | 5.5 ± 0.1 | 10 ± 1 |
| R155H | 100 ± 13 | 21.5 ± 0.9 | 215 ± 29 | 3.3 ± 0.1 | 10 ± 1 |
| NMS-873 mutants | |||||
| A530T | 91 ± 13 | 52 ± 2 | 572 ± 87 | 7.7 ± 0.1 | 46 ± 4 |
| R567H | 143 ± 17 | 21.6 ± 0.8 | 151 ± 18 | 8.1 ± 0.2 | 80 ± 8 |
| K615V | 173 ± 21 | 30 ± 1 | 173 ± 22 | 4.3 ± 0.4 | 97 ± 8 |
| N616F | 93 ± 11 | 15 ± 1 | 161 ± 19 | 14 ± 2 | >4000a |
| L639F | 123 ± 13 | 42 ± 1 | 341 ± 38 | 5.9 ± 0.1 | 140 ± 16 |
| CB-5083 mutants | |||||
| P472L | 51 ± 7 | 55 ± 2 | 1084 ± 147 | 105 ± 10 | 569 ± 54 |
| Q473P | 95 ± 12 | 43 ± 2 | 446 ± 59 | 11.4 ± 0.8 | 14 ± 1 |
| V474A | 110 ± 17 | 52 ± 3 | 471 ± 77 | 34 ± 3 | 25 ± 3 |
| G481A | 280 ± 47 | 34 ± 2 | 123 ± 22 | 33 ± 3 | 47 ± 5 |
| N660K | 105 ± 11 | 8.4 ± 0.3 | 80 ± 9 | 202 ± 19 | 8 ± 0.8 |
| T688A | 94 ± 10 | 1.23 ± 0.04 | 13 ± 1 | 248 ± 31 | 36 ± 3 |
a Activity inhibition was not observed over the concentration range tested.
We next evaluated the biochemical sensitivities of the enzymes to CB-5083 and NMS-873 in inhibitor titration experiments (Fig. S1C). Mutants identified from cells resistant to either CB-5083 or NMS-873 demonstrated biochemical desensitization to their corresponding inhibitor except the Q473P mutant from CB-5083–resistant cells (Table 1). The mutants from NMS-873–resistant cells all remained sensitive to CB-5083. However, several of the mutants isolated from CB-5083–resistant cells showed cross-resistance to NMS-873 with the P472L mutant most robustly desensitized (Fig. 1C). Thus, the P472L mutation causes increased p97 ATPase activity and markedly desensitizes the enzyme to both ATP-competitive and allosteric p97 inhibitors.
P472L mutant is sufficient to desensitize cells to p97 inhibitors
We sought to determine whether the desensitization of the P472L mutant to CB-5083 and NMS-873 in biochemical experiments is also observed in cells. We used homology-directed repair with CRISPR to introduce a cytosine to thymine missense mutation to encode P472L in the p97 gene of HCT116 cells. Genomic DNA samples isolated from populations of transfected and then CB-5083–selected cells were sequenced to verify incorporation of the missense mutation and a silent mutation that prevents targeted Cas9 cleavage (Fig. 2A).
Figure 2.

p97 P472L mutant cells are resistant to ATP-competitive and allosteric p97 inhibitors. A, P472L mutation was introduced into the p97 gene using homology-directed repair with CRISPR in HCT116 cells. Sequencing chromatograms from genomic DNA samples show incorporation of the missense C/T mutation to alter the Pro-472 codon and a silent A/G mutation that disrupts the targeted PAM in transfected and CB-5083–selected cell populations. B, P472L-edited HCT116 cell populations, control HCT116 cells, and NMS-873–resistant HCT116 cells harboring the A530T p97 mutation were tested in cell viability experiments. Inhibitors (2-fold serial dilutions from 5 μm CB-5083 or 20 μm NMS-873; 3-fold dilutions from 200 nm bortezomib) were added for 72 h (CB-5083 and NMS-873) or 48 h (bortezomib) prior to measuring ATP by luminescent detection. Resulting measurements were normalized to the maximum luminescence signal set at 100%. Data points represent the mean (n = 3) with S.D. IC50 values with standard error (S.E.) from treatments that resulted in valid dose-response curves are indicated (ND, not determined). C, HCT116 and p97 P472L mutant cells were stably transfected with the p97 and proteasome substrate Ub–G76V–GFP and seeded to 96-well plates prior to incubating with CB-5083 (DMSO, 0.123, 0.37, and 1.1 μm) and NMS-873 (DMSO, 0.37, 1.1, and 3.3 μm) for 8 h. Integrated intensities (individual data points, mean, and, S.D. error) are shown. D, protein extracts from HCT116 and P472L cells treated with vehicle (DMSO) or inhibitors (0.3 or 2.5 μm, CB-5083; 2 or 5 μm, NMS-873) for 6 h were analyzed by Western blotting for ubiquitin, p97, ATF4, and CHOP.
The resulting cells were tested in cell viability dose-response experiments with CB-5083, NMS-873, and the proteasome inhibitor bortezomib using luminescent ATP detection. The parental HCT116 cells were used as a positive control, and NMS-873–resistant cells harboring the A530T p97 mutation were also included because they remain sensitive to CB-5083 (39). Although all the cells responded similarly to bortezomib based on resulting IC50 values, the P472L cells treated with either CB-5083 or NMS-873 and the A530T cells treated with NMS-873 did not (Fig. 2B). Thus, the P472L mutant overcomes the cytotoxicity of both classes of p97 inhibitors.
To measure cellular p97 activity, we generated HCT116 and P472L mutant cells that stably express the p97- and proteasome-dependent reporter ubiquitin–G76V–GFP (Ub–G76V–GFP). This constitutively ubiquitinated reporter is unfolded by p97 prior to its degradation by the proteasome (41, 42). p97 inhibition results in increased GFP fluorescence as a direct readout of perturbed ubiquitinated substrate unfolding (41). Vehicle control (DMSO)-treated HCT116 and P472L mutant cells had similar steady-state levels of GFP fluorescence to indicate that the P472L mutant cells did not have increased substrate turnover (Fig. 2C). HCT116 cells demonstrated concentration-dependent reporter accumulation with CB-5083 and NMS-873. Consistent with the P472L mutant supporting p97 activity in the presence of both inhibitors, GFP fluorescence was largely unchanged with increasing concentrations of either inhibitor.
We performed Western blotting experiments to examine ubiquitin–protein conjugates, ATF4 and CHOP, that accumulate in response to p97 inhibition (35, 37). DMSO-treated P472L mutant cells showed unaffected ubiquitin–protein conjugates (Fig. 2D). CB-5083- and NMS-873–treated HCT116 cells and CB-5083–treated NMS-873–resistant A530T mutant cells had concentration-dependent increases in ubiquitin–protein conjugates ATF4 and CHOP. The P472L mutant cells did not appreciably accumulate these proteins in response to either inhibitor. These data indicate the P472L mutant does not overtly alter ubiquitin-dependent protein homeostasis and is sufficient to desensitize cells to both classes of p97 inhibitors.
P472L mutation does not disrupt the binding sites of p97 inhibitors
On-target mutations can disrupt inhibitor-binding sites to cause resistance to selective cytotoxic molecules (43). Because Pro-472 is in the D1–D2 linker and makes direct contact with an α-helix in D2 (see Fig. 1A), the leucine mutation could cause structural rearrangements in D2 that disrupt both CB-5083- and NMS-873–binding sites. To test this possibility, we performed biophysical experiments to compare the binding affinities of CB-5083 or NMS-873 to the P472L mutant and p97.
We used isothermal titration calorimetry (ITC) to measure the binding of CB-5083 to the D2 ATP-binding pockets of p97 and the P472L mutant. In experiments titrating CB-5083 on p97 and the P472L mutant (Fig. 3A), we obtained similar dissociation constants (KD values) for CB-5083. Thus, the P472L mutant maintains an intact CB-5083–binding site.
Figure 3.

P472L mutation does not disrupt the binding sites of p97 inhibitors. A, ITC was used to measure the binding of CB-5083 to p97 and the P472L mutant. Top panels are raw data, and lower panels show isotherms with fitted curves and representative KD values. B, effect of increasing NMS-873 concentrations on TR-FRET generated by p97 and the P472L mutant in the presence of BODIPY-FL–ATP, and a terbium-labeled anti-His tag antibody was measured (n = 4, S.D.). Apparent KD values extrapolated from NMS-873–dependent increases in TR-FRET are shown. C, fluorescence polarization experiments were performed (n = 2, S.D.) to measure the enhanced binding of EDA–ADP–ATTO-495 to p97 and the P472L mutant with increasing NMS-873 concentrations. Apparent KD values with S.E. are shown.
To measure allosteric NMS-873 binding (37), we used two different approaches. p97 with bound BODIPY-FL–ATP and a terbium-labeled antibody that binds the N-terminal hexahistidine tag of recombinant p97 generates TR-FRET (39). Increasing concentrations of NMS-873 cause increased TR-FRET to indicate that the inhibitor enhances ATP binding to p97. This enables measurement of the apparent dissociation constant (KD) of NMS-873 to ATP-bound p97. Increasing concentrations of NMS-873 added to the ATP-bound P472L mutant caused increasing TR-FRET (Fig. 3B). The resulting apparent dissociation constant was only modestly increased compared with that obtained for ATP-bound p97. We also measured NMS-873 binding to p97 with bound EDA–ADP–ATTO-495 by fluorescence polarization (Fig. 3C). Previous work demonstrated that ADP binding to p97 as measured by anisotropy is increased by NMS-873 in a concentration-dependent manner (37). We obtained similar dissociation constants for NMS-873 with respect to ADP-bound p97 and the P472L mutant. Moreover, NMS-873 had higher affinity to both proteins in the presence of bound ATP over ADP, reflecting its allosteric mechanism of action (37).
P472L mutation increases D2 ATPase activity and alters D1–D2 communication
Because P472L does not disrupt the binding sites of CB-5083 or NMS-873, the increased ATPase activity of the mutant could cause its desensitization to the inhibitors. To evaluate this, we first sought to determine whether the mutant's increased activity is attributable to the D1 and/or D2 ATPase domains. We expressed and purified p97 and the P472L mutant with Walker A or Walker B motif mutations in D1 or D2 (Fig. 4A) (8, 16, 17, 44). A Walker A mutation (K251A in D1 and K524A in D2) disrupts ATP binding to the mutated domain to eliminate intra-subunit communication through the D1–D2 linker necessary for controlling the ATPase activity of the other domain. A Walker B mutation (E305Q in D1 and E578Q in D2) allows ATP binding to the mutated domain but impairs its hydrolysis. This type of mutation supports intra-subunit communication whereby ATP bound to the mutated domain stimulates the activity of the other domain through the bidirectional function of the D1–D2 linker (9, 16).
Figure 4.

P472L mutation increases D2 ATPase activity and alters its intra-subunit control by D1. A, schematic shows the location of the P472L mutation relative to Walker A and Walker B mutations in D1 and D2. B, ATP titration experiments (n = 4, S.D. error) were performed to obtain steady-state kinetic parameters of p97 and the P472L mutant proteins containing the indicated Walker motif mutations. The sensitivities of the ATPase activities of p97 and the P472L mutant with Walker motif mutations in D1 or D2 were evaluated in the presence of increasing concentrations of CB-5083 (C) and NMS-873 (D) (n = 4, S.D. error). Resulting kinetic properties and IC50 values are summarized in Table 2.
After determining enzyme concentrations that resulted in linear rates of ATP hydrolysis using colorimetric phosphate detection, ATP titration experiments were performed to obtain steady-state kinetic parameters of the enzymes (Fig. 4B and Table 2). The Walker motif mutations in D2 resulted in similar D1 catalytic efficiencies for the P472L mutant and p97 where both responded to the ATP-bound status of D2. Although these Walker motif mutations in D1 of p97 demonstrated ATP-dependent stimulation of D2 activity, D2 of the P472L mutant maintained elevated activity that modestly increased with ATP-bound D1.
Table 2.
Kinetic properties and IC50 values of CB-5083 and NMS-873 for p97 and the P472L mutant with D1 or D2 Walker motif mutations
Values shown are mean ± S.E. (n = 4).
| KM | kcat | kcat/KM | CB-5083 | NMS-873 | |
|---|---|---|---|---|---|
| μm | min−1 | μm−1 min−1 × 10−3 | nm | nm | |
| WT | 91 ± 25 | 11.4 ± 0.9 | 125 ± 35 | 5 ± 0.5 | 14 ± 1 |
| D1 K251A | 43 ± 5 | 0.27 ± 0.01 | 6.2 ± 0.7 | 43 ± 6 | 32 ± 4 |
| D1 E305Q | 66 ± 8 | 12.0 ± 0.4 | 187 ± 23 | 10 ± 1 | 16 ± 2 |
| D2 K524A | 115 ± 14 | 0.25 ± 0.01 | 2.2 ± 0.3 | NDa | ND |
| D2 E578Q | 37 ± 4 | 2.37 ± 0.07 | 64 ± 7 | 20 ± 5 | 513 ± 101 |
| P472L | 48 ± 6 | 49 ± 2 | 1009 ± 119 | 138 ± 16 | 569 ± 54 |
| D1 K251A | 41 ± 5 | 19 ± 0.6 | 460 ± 55 | 29 ± 4 | 1837 ± 389 |
| D1 E305Q | 38 ± 5 | 29 ± 1 | 752 ± 109 | 48 ± 6 | 4299 ± 1433 |
| D2 K524A | 331 ± 43 | 0.68 ± 0.04 | 2.0 ± 0.3 | ND | ND |
| D2 E578Q | 22 ± 4 | 1.64 ± 0.06 | 73 ± 12 | 53 ± 10 | >10,000b |
a ND means not determined.
b Activity inhibition was not observed over the concentration range tested.
We next evaluated whether the resistance of the P472L mutant to CB-5083 and NMS-873 relies exclusively on its increased D2 activity or whether altered intra-subunit communication between D1 and D2 is also involved. We measured ATPase activity inhibition of the P472L mutant and p97 proteins containing individual Walker motif mutations in D1 or D2 with increasing concentrations of CB-5083 or NMS-873. The enzymes with the Walker A mutation in D2 were not evaluated because both inhibitors require an intact D2 ATP-binding pocket to bind p97 (16, 35, 37).
Consistent with previous studies (35), impaired D2 ATP hydrolysis for p97 caused decreased sensitivity to CB-5083 with the corresponding Walker B mutation in D1 having a negligible decrease (Fig. 4C and Table 2). However, p97 with impaired ATP binding to D1 demonstrated the most robust desensitization. These data suggest the sensitivities of these p97 proteins to CB-5083 are related to their ATPase activities. Decreased p97 activity through impaired intra-subunit communication is associated with decreased inhibitor sensitivity. By contrast, the P472L proteins with the individual Walker motif mutations all had increased sensitivities to CB-5083 that did not appear related to their ATPase activities. The Walker B ATP hydrolysis mutation in D1 or D2 resulted in similar CB-5083 sensitivities for the P472L mutant proteins, and the D1 ATP-binding defective protein had the largest increase.
In experiments evaluating NMS-873, we found, as expected from previous studies, that disrupting D2 ATP hydrolysis desensitizes p97 to the molecule with D1 Walker motif mutations causing negligible changes (Fig. 4D, Table 2) (16). For the P472L mutant, each individual Walker motif mutation resulted in additional desensitization of the proteins. Like p97, disrupted D2 ATP hydrolysis for the P472L mutant caused the most robust desensitization with inhibition not observed over the concentration range tested. However, unlike what was seen for p97, impairment of ATP binding or hydrolysis by D1 decreased the NMS-873 sensitivities of the P472L mutant proteins. Collectively, these data indicate NMS-873 resistance of the P472L mutant relies on its increased D2 activity with CB-5083 resistance also involving altered D1–D2 communication.
P472L mutation enhances cooperative ATP binding and hydrolysis by D2
We sought to determine the mechanism of increased D2 ATPase activity and its altered control by D1. Our data indicate that the P472L mutant maintains the CB-5083–binding site in the D2 ATP-binding pocket (see Fig. 3A). Molecular docking studies suggest CB-5083 binds within the adenosine region and an adjacent hydrophobic stretch, while bypassing the triphosphate-binding region (36). Within this latter region, arginine finger residues Arg-635 and Arg-638 of the neighboring D2 domain are functionally relevant because they mediate cooperative γ-phosphate hydrolysis of bound ATP (Fig. 5A) (19, 23). Because these residues are important for cooperative ATP hydrolysis by D2 and do not participate in CB-5083 binding, we sought to examine their contributions to the increased activity of the P472L mutant. Previous studies demonstrated that R635A or R638A mutations support p97 hexamer assembly and nucleotide binding but impair ATPase activity (19, 23).
Figure 5.

P472L mutation enhances cooperative D2 ATP binding and hydrolysis. A, p97 schematic (top) and D2 catalytic pocket model (bottom, PDB code FTM) show arginine finger residues Arg-635 and Arg-638 from an adjacent D2 domain involved in cooperative D2 ATP hydrolysis. The affinities of ADP (B) or ATP (C) to the D2 domains of the indicated p97 or P472L mutant proteins were measured by fluorescence polarization (n = 2, S.D. error). D, kinetic properties of p97 and the P472L mutant proteins in the presence or absence of R635A or R638A were obtained from ATP titration experiments (n = 4, S.D. error). E, sensitivities of p97 and the P472L mutant proteins to CB-5083 and NMS-873 were tested in inhibitor titration experiments (n = 4, S.D. error). Dissociation constants for ATP and ADP binding to D2, enzyme kinetic properties, and IC50 values are summarized in Table 3.
We first measured the binding of ADP and ATP to D2 of p97 and the P472L mutant in the presence or absence of the arginine finger mutations R635A or R638A. These experiments used purified proteins that contain only an intact D2 ATP-binding pocket (Walker A mutation in D1 and Walker B mutation in D2). Increasing concentrations of the proteins were mixed with fluorescent ADP or ATP to obtain KD values by anisotropy. The D2 domains of p97 and the P472L mutant had similar ADP affinities, and R635A or R638A caused modest changes (Fig. 5B and Table 3). However, D2 of the P472L mutant had markedly higher ATP affinity than p97 (Fig. 5C and Table 3). The arginine finger mutations caused modestly reduced ATP affinities for p97 but more robustly decreased the ATP affinity of the P472L mutant. These data indicate that the P472L mutation changes D2 to increase its affinity for ATP over ADP. This change involves alterations to allosteric D2–D2 interactions where adjacent arginine finger residues promote increased D2 ATP affinity.
Table 3.
D2 nucleotide binding, kinetic properties, and IC50 values of CB-5083 and NMS-873 for p97 and the P472L mutant with R635A or R638A arginine finger mutations
Values shown are mean ± S.E. (n = 2, ATP and ADP binding; n = 4, activity).
| KD, ADP | KD, ATP | KM | kcat | kcat/KM | CB-5083 | NMS-873 | |
|---|---|---|---|---|---|---|---|
| μm | μm | μm | min−1 | (μm−1 min−1 × 10−3) | nm | nm | |
| WT | 0.6 ± 0.07 | 1.8 ± 0.2 | 77 ± 9 | 8.6 ± 0.3 | 112 ± 14 | 8.1 ± 0.8 | 12 ± 1 |
| R635A | 0.8 ± 0.1 | 4.6 ± 0.6 | 88 ± 11 | 0.29 ± 0.01 | 3.2 ± 0.4 | >8000a | >8000a |
| R638A | 1.2 ± 0.1 | 3.5 ± 0.4 | 48 ± 4 | 0.39 ± 0.01 | 8.1 ± 0.7 | >8000a | >8000a |
| P472L | 1.1 ± 0.1 | 0.4 ± 0.06 | 40 ± 5 | 37 ± 1 | 924 ± 114 | 118 ± 17 | 674 ± 58 |
| R635A | 0.9 ± 0.1 | 2.6 ± 0.2 | 29 ± 5 | 0.94 ± 0.04 | 32 ± 5 | 31 ± 4 | 72 ± 12 |
| R638A | 1.4 ± 0.1 | 4.0 ± 0.4 | 36 ± 3 | 1.14 ± 0.02 | 31 ± 3 | 79 ± 18 | 254 ± 29 |
a Activity inhibition was not observed over the concentration range tested.
We expressed and purified the P472L mutant and p97 containing these single D2 arginine finger mutations to measure their ATPase activities. After empirically determining enzyme concentrations that resulted in linear rates of ATP hydrolysis, we performed ATP titration experiments to obtain steady-state kinetic parameters of the enzymes (Fig. 5D and Table 3). As expected (19, 23), either arginine finger mutation in p97 resulted in severely impaired catalytic efficiencies through decreased substrate turnover. Although the P472L mutant proteins also demonstrated impairment, they maintained higher catalytic efficiencies and substrate turnover than the corresponding p97 proteins. These observations indicate that the P472L mutation does not exclusively increase ATP affinity to D2. It also enhances ATP hydrolysis by the enzyme whereby the presence of a single intact arginine finger residue provides higher ATPase activity than that for p97.
We next tested whether the arginine finger mutations alter the sensitivities of the P472L mutant and p97 to CB-5083 or NMS-873 (Fig. 5E and Table 3). The arginine finger mutations in p97 completely desensitized these proteins to inhibition. Thus, impaired p97 D2 activity prevents productive inhibition by the molecules. For the P472L mutant, however, the residual D2 activity sensitized the enzymes to both CB-5083 and NMS-873 with R635A resulting in the most prominent changes.
Discussion
In this study, we have provided evidence that adapted ATPase domain communication desensitizes the AAA+ ATPase p97 to its current small molecule inhibitors. Through a biochemical analysis of p97 mutants identified in cells resistant to ATP-competitive or allosteric inhibitors, we determined that the P472L mutation, localized within the linker between its D1 and D2 ATPase domains, desensitizes the enzyme to both inhibitor classes. The underlying mechanism does not rely on disrupting the distinct D2-binding sites of the inhibitors. Our data suggest P472L alleviates intra-subunit control of D2 by D1 through enhanced cooperative D2 activity (Fig. 6). These changes circumvent the distinct mechanisms of action of CB-5083 and NMS-873 necessary for productive p97 inhibition and support the enzyme's essential function.
Figure 6.
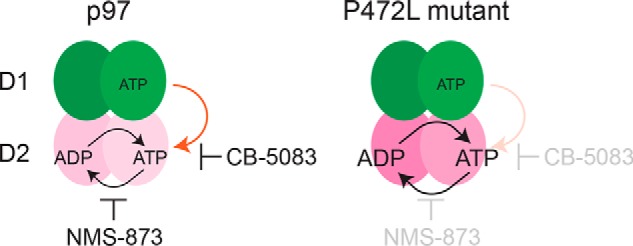
Proposed desensitization mechanism of p97 to its ATP-competitive and allosteric inhibitors. NMS-873 allosterically inhibits p97 activity by blocking cooperative D2 ATP hydrolysis. CB-5083 functions as an ATP-competitive and D2-selective inhibitor. Our data suggest that intra-subunit control of D2 activity through ATP-bound D1 is critical for p97 sensitivity to this molecule (orange arrow). The P472L mutant, identified from CB-5083–resistant cells, is desensitized to both inhibitors. This mechanism involves enhanced cooperative ATP binding and hydrolysis by D2 that alter its intra-subunit control by ATP-bound D1.
P472L alters the bidirectional role of the D1–D2 linker in intra-subunit communication. For p97, ATP binding to either D1 or D2 controls the other domain's ATPase activity (16). This mechanism involves conformational changes mediated by the D1–D2 linker to increase ATP affinity over ADP (9, 15). The P472L mutant has similar D1 activity to p97 that is stimulated by ATP binding to D2, indicating that this function of the linker is unchanged. By contrast, the elevated D2 activity of the mutant is markedly less responsive to ATP-bound D1. This altered dependence on intra-subunit communication to control D2 can be attributed to changes in the D2 ATP-binding pocket. In the absence of ATP binding to D1, D2 of the P472L mutant has higher affinity for ATP than for ADP. This is in marked contrast to WT p97 where ATP-bound D1 is necessary for this to occur.
P472L increases D2 activity by altering allosteric interactions in the D2 ring required for cooperative ATP hydrolysis by the domain. We have identified changes to the D2 ATP-binding pocket that do not disrupt ATP-competitive CB-5083 binding. Our data indicate P472L alters the triphosphate-binding region where arginine finger residues of an adjacent D2 domain promote increased ATP affinity. Although mutation of either D2 arginine finger Arg-635 or Arg-638 to alanine results in severely impaired p97 ATPase activity, the corresponding P472L mutant proteins remain more active even with similar D2 ATP affinities. This observation suggests the P472L mutation changes the orientations of D2 arginine finger residues to enhance ATP hydrolysis.
NMS-873 allosterically inhibits p97 by blocking cooperative D2 ATP hydrolysis (37). This mechanism is largely independent of intra-subunit communication because Walker motif mutations that disrupt ATP binding or hydrolysis by D1 cause negligible changes in the sensitivity of p97 to the inhibitor (16). These mutations in the context of P472L result in further enzyme desensitization, suggesting that intra-subunit communication decreases the mutant's resistance to NMS-873. By contrast, D2 arginine finger mutations R635A or R638A sensitize the P472L mutant to the inhibitor. Based on these observations, we propose that increased cooperative D2 activity is sufficient to desensitize p97 to NMS-873.
CB-5083 functions as a D2-selective and ATP-competitive p97 inhibitor (35, 36). Our data demonstrate, however, that disrupted ATP binding to D1 desensitizes p97 to the compound. This unexpected finding suggests intra-subunit communication of the ATP-bound status of D1 to D2 is critical for CB-5083 to optimally inhibit p97. We propose that the P472L mutant is desensitized to CB-5083 by overcoming this mechanism through increased cooperative D2 ATP binding and hydrolysis. This model is supported by our findings that impaired D2 activity and disrupted intra-subunit communication both sensitize the mutant to CB-5083.
Collectively, our study establishes that adapted ATPase domain communication can desensitize p97 to its current ATP-competitive and allosteric inhibitors. The underlying mechanism reveals unanticipated plasticity of the enzyme to provide its essential biological function. Unlike MSP-1 disease-causing mutants that also have elevated D2 ATPase activities (11, 12, 28, 32–34), the P472L mutant does not appear to generally impact ubiquitin-dependent protein homeostasis in cells. Considering that p97 function involves distinct roles attributed to D1 and D2 ATPases (13), we propose that the mutant has sufficiently controlled D1 activity for substrate recruitment and/or release to prevent aberrant processing through its increased D2 activity.
Experimental procedures
p97 inhibitors
NMS-873 was purchased from XcessBio and Sigma. CB-5083 was purchased from Selleckchem. Dry powders were stored at room temperature in a humidity-controlled desiccator with 10 mm liquid stocks prepared in 100% DMSO and stored at −80 °C. Compound identity and purity were assessed by LC-MS.
Recombinant DNA, protein expression, and purification
Baculovirus expression and purification of p97 were as described (39, 45). The p97 reporter Ub–G76V–GFP was generated as described (41, 42). Site-directed mutagenesis used QuikChange (Agilent). Because of low expression in insect cells, D2 arginine finger mutants and the K251A mutants were expressed from pET11b using Rosetta 2(DE3) bacteria (Novagen). Cultures were grown to an A600 of 0.8 and induced with 0.8 mm isopropyl 1-thio-β-d-galactopyranoside for 4 h at 30 °C, and proteins were purified by nickel-nitrilotriacetic acid chromatography as for baculovirus-expressed p97. Protein concentrations were determined by Coomassie-stained SDS-polyacrylamide gel band intensity with BSA as a standard using a Licor Odyssey scanner and Image Studio software.
Cell culture, gene editing, and inhibitor profiling
HCT116 cells were purchased from ATCC, tested for mycoplasma, and authenticated before and at several points throughout the study. Sf9 and Hi5 insect cells were purchased from ThermoFisher Scientific. HCT116 cell lines resistant to CB-5083 or NMS-873 were isolated and characterized as described and used 0.5 μm CB-5083 or 2 μm NMS-873 (39). Single amino acid p97 mutations identified during this study are indicated in bold in Fig. 1A. To generate engineered P472L cell lines, HCT116 cells were co-transfected with a Cas9-expressing plasmid (PX458, Addgene plasmid no. 48138) containing an sgRNA corresponding to bases 35,060,894 to 35,060,875 of the p97 gene exon 12 on chromosome 9 and a linear repair template corresponding to 35,060,974 to 35,060,787 with mutations at nucleotide 35,060,873 (G to A; disrupts PAM) and 35,060,868 (C to T; mutates Pro-472 codon to Leu). Exact sequences of the sgRNA and repair template DNA will be provided upon request. For cell viability experiments evaluating CB-5083, NMS-873, and bortezomib (Fig. 2B), transfected and CB-5083–selected (1.5 μm, 72 h) cell populations were used. Sequence-verified P472L mutant cell lines isolated by limited dilution cloning without CB-5083 selection were used for Ub–G76V–GFP and Western blotting experiments.
Cells (HCT116, NMS-873–resistant p97 A530T (39), and engineered p97 P472L) were seeded to 96-well plates (5000 cells per well). After 24 h, 2-fold serial dilutions of CB-5083 (maximum 5 μm), NMS-873 (maximum 20 μm), and bortezomib (maximum 200 nm) were added, followed by an additional 48-h (bortezomib) or 72-h (CB-5083 and NMS-873) incubation. Cell viability was measured using Cell Titer-Glo (Promega). Resulting data were analyzed and fit (where appropriate) to a 4-parameter variable slope inhibition equation using GraphPad Prism 7.
For p97 substrate reporter experiments, HCT116 and P472L cells were transfected with a plasmid encoding Ub–G76V–GFP and selected using 600 μg/ml geneticin. After seeding to 96-well plates (10,000 cells per well), CB-5083 or NMS-873 was added for 8 h prior to imaging and analysis on a Celigo cytometer to obtain integrated fluorescence intensities. Western blotting experiments used 6-h CB-5083 or NMS-873 treatments (39). Image acquisition was on a Licor Odyssey scanner.
p97 ATPase activity and inhibitor profiling
ATPase activity assays were as described previously (39) and performed in 384-well format using colorimetric phosphate detection (PiColorLock Gold, Novus) with a FlexStation 3 microplate reader (Molecular Devices). Reaction velocities at 200 μm ATP were initially measured at different enzyme concentrations to identify linear rates after 2 h at room temperature. ATP titration experiments were performed (1 mm maximum, 2-fold serial dilutions, and eight concentrations) with KM and kcat values calculated using the Michaelis-Menten equation. Enzyme concentrations were as follows: 22 nm WT; 20 nm R95G; 12 nm R155H; 12.5 nm K615V; 15 nm N616F; 5 nm P472L; 6.25 nm Q473P; 6 nm V474A; 12 nm G481A; 30 nm N660K; 200 nm T688A; 3 nm A530T; 14 nm R567H; 8 nm L639F; 540 nm K251A; 21 nm E305Q; 1040 nm K524A; 95 nm E578Q; 10 nm K251A/P472L; 8 nm E305Q/P472L; 500 nm P472L/K524A; 115 nm P472L/E578Q; 1500 nm R635A; 700 nm R638A; 240 nm P472L/R635A; and 230 nm P472L/R638A.
Inhibitor profiling experiments were performed with 200 μm ATP, and compounds were serially diluted from the indicated maximum. Relative rates were fit to a 4-parameter inhibition dose-response equation using GraphPad Prism 7 to obtain IC50 values with standard error.
Ligand binding
ITC measurements were performed on a MicroCal iTC200 using 25 mm HEPES, pH 7.6, 1 mm MgCl2, 100 mm NaCl, and 0.5 mm tris(2-carboxyethyl)phosphine at 23 °C with 32 μm p97 or P472L mutant. Each titration used 19 injections of 2 μl of 300 μm CB-5083. Resulting data were fit using Origin software (Malvern) with a one-site binding model to obtain KD values.
TR-FRET assays were performed as described and used 36 nm p97 or P472L mutant proteins, 1 nm terbium-labeled anti-His antibody, and 50 nm BODIPY-FL–ATP with 2-fold serial dilutions of NMS-873 to 8 μm (39). After 1 h at room temperature, signals were recorded on an Envision microplate reader with excitation at 385 nm and emission at 485 and 520 nm. The emission ratio (520:485) data were fit to a hyperbolic equation in GraphPad Prism 7 to obtain apparent KD (EC50) values with respect to NMS-873.
Fluorescence polarization assays used the same buffer as ITC experiments supplemented with 0.05% Tween 20 and performed at room temperature. These experiments used p97 or P472L mutant proteins where the D1 ATPase has the Walker A mutation (K251A) and D2 has the Walker B mutation (E578Q) to attribute nucleotide binding to D2 without ATP hydrolysis. The 2-fold serial dilutions of p97 proteins (maximum 40 μm, purified from bacteria) were mixed with 20 nm BODIPY-FL–ATP (ThermoFisher Scientific) or 10 nm EDA–ADP–ATTO-495 (Jena Bioscience) in black low volume 384-well plates. After 30 min, fluorescence anisotropy was recorded on an Envision microplate reader (PerkinElmer Life Sciences) with excitation at 485 nm and emission at 595 nm. Background fluorescence (proteins without added fluorophores) was measured and subtracted from parallel and perpendicular intensities prior to anisotropy calculations. Data were fit in GraphPad Prism 7 using a ligand-binding quadratic equation to obtain KD values.
Author contributions
Y. W., J. I. T., and M. D. P. conceptualization; Y. W., J. I. T., G. A. B., A. A. B., and M. D. P. resources; Y. W., J. I. T., G. A. B., A. A. B., and M. D. P. investigation; Y. W., J. I. T., G. A. B., and A. A. B. methodology; Y. W., J. I. T., G. A. B., A. A. B., and M. D. P. writing-review and editing; M. D. P. supervision; M. D. P. funding acquisition; M. D. P. writing-original draft.
Supplementary Material
Acknowledgments
We thank Eduard Sergienko and Anthony Pinkerton for technical advice and assistance, Dieter Wolf for invaluable input on the manuscript, and Dorit Hanein, Niels Volkmann, Robert Liddington, and Guy Salvesen for helpful discussions. PX458 was a gift from Feng Zhang (Addgene plasmid no. 48138).
This work was supported in part by a Padres Pedal the Cause Postdoctoral Fellowship (to Y. W.), National Institutes of Health Grants R01CA180150 and R01CA185300 (to M. D. P.), and Sanford Burnham Prebys NCI Cancer Center Support Grant P30 CA030199 from the National Institutes of Health. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Fig. S1.
- MSP-1
- multisystem proteinopathy type-1
- ITC
- isothermal titration calorimetry
- PDB
- Protein Data Bank
- PAM
- protospacer adjacent motif
- TR-FRET
- time-resolved fluorescence energy transfer.
References
- 1. Stach L., and Freemont P. S. (2017) The AAA+ ATPase p97, a cellular multitool. Biochem. J. 474, 2953–2976 10.1042/BCJ20160783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. van den Boom J., and Meyer H. (2018) Vcp/p97-mediated unfolding as a principle in protein homeostasis and signaling. Mol. Cell 69, 182–194 10.1016/j.molcel.2017.10.028 [DOI] [PubMed] [Google Scholar]
- 3. Ye Y., Tang W. K., Zhang T., and Xia D. (2017) A mighty “protein extractor” of the cell: structure and function of the p97/cdc48 ATPase. Front. Mol. Biosci. 4, 39 10.3389/fmolb.2017.00039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Meyer H., Bug M., and Bremer S. (2012) Emerging functions of the vcp/p97 AAA-ATPase in the ubiquitin system. Nat. Cell Biol. 14, 117–123 10.1038/ncb2407 [DOI] [PubMed] [Google Scholar]
- 5. Chapman E., Maksim N., de la Cruz F., and La Clair J. J. (2015) Inhibitors of the AAA+ chaperone p97. Molecules 20, 3027–3049 10.3390/molecules20023027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Deshaies R. J. (2014) Proteotoxic crisis, the ubiquitin-proteasome system, and cancer therapy. BMC Biol. 12, 94 10.1186/s12915-014-0094-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. DeLaBarre B., and Brunger A. T. (2003) Complete structure of p97/valosin-containing protein reveals communication between nucleotide domains. Nat. Struct. Biol. 10, 856–863 10.1038/nsb972 [DOI] [PubMed] [Google Scholar]
- 8. Huyton T., Pye V. E., Briggs L. C., Flynn T. C., Beuron F., Kondo H., Ma J., Zhang X., and Freemont P. S. (2003) The crystal structure of murine p97/vcp at 3.6a. J. Struct. Biol. 144, 337–348 10.1016/j.jsb.2003.10.007 [DOI] [PubMed] [Google Scholar]
- 9. Banerjee S., Bartesaghi A., Merk A., Rao P., Bulfer S. L., Yan Y., Green N., Mroczkowski B., Neitz R. J., Wipf P., Falconieri V., Deshaies R. J., Milne J. L., Huryn D., Arkin M., and Subramaniam S. (2016) 2.3 A resolution cryo-EM structure of human p97 and mechanism of allosteric inhibition. Science 351, 871–875 10.1126/science.aad7974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bodnar N. O., Kim K. H., Ji Z., Wales T. E., Svetlov V., Nudler E., Engen J. R., Walz T., and Rapoport T. A. (2018) Structure of the cdc48 ATPase with its ubiquitin-binding cofactor ufd1-npl4. Nat. Struct. Mol. Biol. 25, 616–622 10.1038/s41594-018-0085-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Schuetz A. K., and Kay L. E. (2016) A dynamic molecular basis for malfunction in disease mutants of p97/vcp. Elife 5, e20143 10.7554/eLife.20143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tang W. K., Li D., Li C. C., Esser L., Dai R., Guo L., and Xia D. (2010) A novel ATP-dependent conformation in p97 n-d1 fragment revealed by crystal structures of disease-related mutants. EMBO J. 29, 2217–2229 10.1038/emboj.2010.104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bodnar N. O., and Rapoport T. A. (2017) Molecular mechanism of substrate processing by the cdc48 ATPase complex. Cell 169, 722–735.e9 10.1016/j.cell.2017.04.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Blythe E. E., Olson K. C., Chau V., and Deshaies R. J. (2017) Ubiquitin- and ATP-dependent unfoldase activity of p97/vcp*nploc4*ufd1l is enhanced by a mutation that causes multisystem proteinopathy. Proc. Natl. Acad. Sci. U.S.A. 114, E4380–E4388 10.1073/pnas.1706205114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Briggs L. C., Baldwin G. S., Miyata N., Kondo H., Zhang X., and Freemont P. S. (2008) Analysis of nucleotide binding to p97 reveals the properties of a tandem aaa hexameric ATPase. J. Biol. Chem. 283, 13745–13752 10.1074/jbc.M709632200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chou T. F., Bulfer S. L., Weihl C. C., Li K., Lis L. G., Walters M. A., Schoenen F. J., Lin H. J., Deshaies R. J., and Arkin M. R. (2014) Specific inhibition of p97/vcp ATPase and kinetic analysis demonstrate interaction between d1 and d2 ATPase domains. J. Mol. Biol. 426, 2886–2899 10.1016/j.jmb.2014.05.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Song C., Wang Q., and Li C. C. (2003) Atpase activity of p97-valosin-containing protein (vcp). d2 mediates the major enzyme activity, and d1 contributes to the heat-induced activity. J. Biol. Chem. 278, 3648–3655 10.1074/jbc.M208422200 [DOI] [PubMed] [Google Scholar]
- 18. Ye Y., Meyer H. H., and Rapoport T. A. (2003) Function of the p97-ufd1-npl4 complex in retrotranslocation from the ER to the cytosol: dual recognition of nonubiquitinated polypeptide segments and polyubiquitin chains. J. Cell Biol. 162, 71–84 10.1083/jcb.200302169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hänzelmann P., and Schindelin H. (2016) Structural basis of ATP hydrolysis and intersubunit signaling in the AAA+ ATPase p97. Structure 24, 127–139 10.1016/j.str.2015.10.026 [DOI] [PubMed] [Google Scholar]
- 20. Wang Q., Song C., and Li C. C. (2003) Hexamerization of p97-vcp is promoted by ATP binding to the d1 domain and required for ATPase and biological activities. Biochem. Biophys. Res. Commun. 300, 253–260 10.1016/S0006-291X(02)02840-1 [DOI] [PubMed] [Google Scholar]
- 21. Wang Q., Song C., Yang X., and Li C. C. (2003) D1 ring is stable and nucleotide-independent, whereas d2 ring undergoes major conformational changes during the ATPase cycle of p97-vcp. J. Biol. Chem. 278, 32784–32793 10.1074/jbc.M303869200 [DOI] [PubMed] [Google Scholar]
- 22. Wendler P., Ciniawsky S., Kock M., and Kube S. (2012) Structure and function of the AAA+ nucleotide binding pocket. Biochim. Biophys. Acta 1823, 2–14 10.1016/j.bbamcr.2011.06.014 [DOI] [PubMed] [Google Scholar]
- 23. Wang Q., Song C., Irizarry L., Dai R., Zhang X., and Li C. C. (2005) Multifunctional roles of the conserved arg residues in the second region of homology of p97/valosin-containing protein. J. Biol. Chem. 280, 40515–40523 10.1074/jbc.M509636200 [DOI] [PubMed] [Google Scholar]
- 24. Meyer H., and Weihl C. C. (2014) The vcp/p97 system at a glance: connecting cellular function to disease pathogenesis. J. Cell Sci. 127, 3877–3883 10.1242/jcs.093831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tang W. K., and Xia D. (2016) Mutations in the human AAA(+) chaperone p97 and related diseases. Front. Mol. Biosci. 3, 79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Watts G. D., Wymer J., Kovach M. J., Mehta S. G., Mumm S., Darvish D., Pestronk A., Whyte M. P., and Kimonis V. E. (2004) Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nat. Genet. 36, 377–381 10.1038/ng1332 [DOI] [PubMed] [Google Scholar]
- 27. Bulfer S. L., Chou T. F., and Arkin M. R. (2016) p97 disease mutations modulate nucleotide-induced conformation to alter protein-protein interactions. ACS Chem. Biol. 11, 2112–2116 10.1021/acschembio.6b00350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Halawani D., LeBlanc A. C., Rouiller I., Michnick S. W., Servant M. J., and Latterich M. (2009) Hereditary inclusion body myopathy-linked p97/vcp mutations in the nh2 domain and the d1 ring modulate p97/vcp ATPase activity and d2 ring conformation. Mol. Cell. Biol. 29, 4484–4494 10.1128/MCB.00252-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ju J. S., Miller S. E., Hanson P. I., and Weihl C. C. (2008) Impaired protein aggregate handling and clearance underlie the pathogenesis of p97/vcp-associated disease. J. Biol. Chem. 283, 30289–30299 10.1074/jbc.M805517200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Manno A., Noguchi M., Fukushi J., Motohashi Y., and Kakizuka A. (2010) Enhanced ATPase activities as a primary defect of mutant valosin-containing proteins that cause inclusion body myopathy associated with paget disease of bone and frontotemporal dementia. Genes Cells 15, 911–922 [DOI] [PubMed] [Google Scholar]
- 31. Niwa H., Ewens C. A., Tsang C., Yeung H. O., Zhang X., and Freemont P. S. (2012) The role of the n-domain in the ATPase activity of the mammalian AAA ATPase p97/vcp. J. Biol. Chem. 287, 8561–8570 10.1074/jbc.M111.302778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tang W. K., and Xia D. (2013) Altered intersubunit communication is the molecular basis for functional defects of pathogenic p97 mutants. J. Biol. Chem. 288, 36624–36635 10.1074/jbc.M113.488924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Weihl C. C., Dalal S., Pestronk A., and Hanson P. I. (2006) Inclusion body myopathy-associated mutations in p97/vcp impair endoplasmic reticulum-associated degradation. Hum. Mol. Genet. 15, 189–199 10.1093/hmg/ddi426 [DOI] [PubMed] [Google Scholar]
- 34. Zhang X., Gui L., Zhang X., Bulfer S. L., Sanghez V., Wong D. E., Lee Y., Lehmann L., Lee J. S., Shih P. Y., Lin H. J., Iacovino M., Weihl C. C., Arkin M. R., Wang Y., and Chou T. F. (2015) Altered cofactor regulation with disease-associated p97/vcp mutations. Proc. Natl. Acad. Sci. U.S.A. 112, E1705–E1714 10.1073/pnas.1418820112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Anderson D. J., Le Moigne R., Djakovic S., Kumar B., Rice J., Wong S., Wang J., Yao B., Valle E., Kiss von Soly S., Madriaga A., Soriano F., Menon M. K., Wu Z. Y., Kampmann M., et al. (2015) Targeting the AAA ATPase p97 as an approach to treat cancer through disruption of protein homeostasis. Cancer Cell 28, 653–665 10.1016/j.ccell.2015.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhou H. J., Wang J., Yao B., Wong S., Djakovic S., Kumar B., Rice J., Valle E., Soriano F., Menon M. K., Madriaga A., Kiss von Soly S., Kumar A., Parlati F., Yakes F. M., et al. (2015) Discovery of a first-in-class, potent, selective, and orally bioavailable inhibitor of the p97 AAA ATPase (cb-5083). J. Med. Chem. 58, 9480–9497 10.1021/acs.jmedchem.5b01346 [DOI] [PubMed] [Google Scholar]
- 37. Magnaghi P., D'Alessio R., Valsasina B., Avanzi N., Rizzi S., Asa D., Gasparri F., Cozzi L., Cucchi U., Orrenius C., Polucci P., Ballinari D., Perrera C., Leone A., Cervi G., et al. (2013) Covalent and allosteric inhibitors of the ATPase vcp/p97 induce cancer cell death. Nat. Chem. Biol. 9, 548–556 10.1038/nchembio.1313 [DOI] [PubMed] [Google Scholar]
- 38. Bastola P., Wang F., Schaich M. A., Gan T., Freudenthal B. D., Chou T. F., and Chien J. (2017) Specific mutations in the d1-d2 linker region of vcp/p97 enhance ATPase activity and confer resistance to vcp inhibitors. Cell Death Discov. 3, 17065 10.1038/cddiscovery.2017.65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Her N. G., Toth J. I., Ma C. T., Wei Y., Motamedchaboki K., Sergienko E., and Petroski M. D. (2016) p97 composition changes caused by allosteric inhibition are suppressed by an on-target mechanism that increases the enzyme's ATPase activity. Cell Chem. Biol. 23, 517–528 10.1016/j.chembiol.2016.03.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ritz D., Vuk M., Kirchner P., Bug M., Schütz S., Hayer A., Bremer S., Lusk C., Baloh R. H., Lee H., Glatter T., Gstaiger M., Aebersold R., Weihl C. C., and Meyer H. (2011) Endolysosomal sorting of ubiquitylated caveolin-1 is regulated by vcp and ubxd1 and impaired by vcp disease mutations. Nat Cell Biol. 13, 1116–1123 10.1038/ncb2301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Chou T. F., and Deshaies R. J. (2011) Quantitative cell-based protein degradation assays to identify and classify drugs that target the ubiquitin-proteasome system. J. Biol. Chem. 286, 16546–16554 10.1074/jbc.M110.215319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Dantuma N. P., Lindsten K., Glas R., Jellne M., and Masucci M. G. (2000) Short-lived green fluorescent proteins for quantifying ubiquitin/proteasome-dependent proteolysis in living cells. Nat. Biotechnol. 18, 538–543 10.1038/75406 [DOI] [PubMed] [Google Scholar]
- 43. Holohan C., Van Schaeybroeck S., Longley D. B., and Johnston P. G. (2013) Cancer drug resistance: an evolving paradigm. Nat. Rev. Cancer 13, 714–726 10.1038/nrc3599 [DOI] [PubMed] [Google Scholar]
- 44. Walker J. E., Saraste M., Runswick M. J., and Gay N. J. (1982) Distantly related sequences in the α- and β-subunits of atp synthase, myosin, kinases and other ATP-requiring enzymes and a common nucleotide binding fold. EMBO J. 1, 945–951 10.1002/j.1460-2075.1982.tb01276.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Locke M., Toth J. I., and Petroski M. D. (2014) Lys11- and Lys48-linked ubiquitin chains interact with p97 during endoplasmic-reticulum-associated degradation. Biochem. J. 459, 205–216 10.1042/BJ20120662 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.