

Abstract
Conophylline is a Vinca alkaloid from leaves of the tropical plant Ervatamia microphylla and has been shown to mimic the effect of the growth and differentiation factor activin A on pancreatic progenitor cells. However, activin A stimulates fibrosis of pancreatic stellate cells, whereas conophylline inhibits it, suggesting that this compound may serve as an antifibrotic drug. Here we investigated the effects of conophylline on human foreskin fibroblasts, especially focusing on extracellular matrix (ECM) proteins. A gene microarray analysis revealed that conophylline remarkably suppressed expression of the gene for hyaluronan synthase 2 (HAS2) and of its antisense RNA, whereas the expression of collagen genes was unaffected. Of note, immunostaining experiments revealed that conophylline substantially inhibits incorporation of versican and collagens into the ECM in cells treated with transforming growth factor β (TGFβ), which promotes collagen synthesis, but not in cells not treated with TGFβ. Moreover, a protein biosynthesis assay disclosed that conophylline decreases collagen biosynthesis, concomitant with a decrease in total protein biosynthesis, indicating that conophylline-mediated inhibition of fibrosis is not specific to collagen synthesis. Conophylline affected neither TGFβ-induced nuclear translocation of SMAD family member 2/3 (SMAD2/3) nor phosphorylation of SMAD2. However, conophylline substantially inhibited phosphorylation of extracellular signal–regulated kinase 1/2 (ERK1/2), suggesting that conophylline inhibits HAS2 expression via TGFβ-mediated activation of the ERK1/2 pathway. Taken together, our results indicate that conophylline may be a useful inhibitor of ECM formation in fibrosis.
Keywords: extracellular matrix, fibroblast, transforming growth factor β (TGFβ), hyaluronan, collagen, fibrosis, anti-fibrotic, conophylline, stellate cell, Vinca alkaloid
Introduction
Fibrosis is the formation of excess fibrous connective tissue in an organ or tissue in a reparative or reactive process (1). There, stromal fibroblasts become activated and trans-differentiated into myofibroblasts, and these cells synthesize high levels of the extracellular matrix (ECM)2 molecules, including collagens. In response to injury, including wound healing, fibrosis occurs as scarring (2). When fibrosis occurs in organs such as the lungs and liver, excessive connective tissue in the stroma obliterates the architecture and interferes with their function, leading to serious diseases. In addition, some chemical agents used for disease treatment, such as bleomycin, are known to cause fibrosis as a side effect (3). Therefore, understanding the mechanisms of fibrosis and its regulation have been general and important issues in the field of clinical medicine.
Conophylline (CNP) is a Vinca alkaloid extracted from leaves of the tropical plant Ervatamia microphylla (4). This compound was initially found to mimic the effect of activin A on the differentiation of pancreatic progenitor cells (5). It induces differentiation of pancreatic progenitor cells into insulin-producing β-cells and converts cultured ductal cells to β-cells in vitro (5) and in vivo (6). Interestingly, although activin A up-regulates the expression of α-smooth muscle actin (αSMA) and collagens of pancreatic stellate cells toward pancreatic fibrosis (7), CNP suppresses their expression (5). CNP inhibits progression of nonalcoholic steatohepatitis by inhibiting fibrosis (8). These results suggest that CNP may serve as an anti-fibrosis drug.
Here we investigated the effects of CNP on the behavior of human foreskin fibroblasts (NB1RGB). Our microarray analysis revealed that CNP remarkably suppressed hyaluronan synthase 2 (HAS2) expression, leading to a decrease in hyaluronan (HA). CNP inhibited collagen biosynthesis by a decrease in total protein synthesis. Further analysis suggested that CNP inhibits the TGFβ-mediated pathway, especially to ERK1/2, but not the Smad2/3 pathway.
Results
Initially, we treated growing and confluent NB1RGB fibroblasts with different concentrations of CNP and examined its cytotoxicity (Fig. 1). In a growing phase, CNP at a concentration of 0.1 μg/ml and higher decreased the cell number, which became apparent as early as day 2 after treatment (Fig. 1B). On day 5, the cells treated with 0.1 μg/ml CNP decreased to ∼40% that of nontreated cells (Fig. 1C). In contrast, treatment with 0.025 μg/ml CNP in the growing phase showed only 20% inhibition of cell proliferation on day5 (Fig. 1C). When cells were plated at confluence and cultured further, the MTT levels increased slightly, indicating that cells proliferated slightly and piled up. When CNP was added to confluent NB1RGB cells, the cell number decreased at 0.025 μg/ml and higher concentrations, which became apparent as early as day 2 (Fig. 1B). At 0.1 μg/ml CNP, the decrease became obvious on day 5 (Fig. 1C). As observed under a microscope, cells were viable and attached on the dishes. In addition, both a trypan blue exclusion test and alamarBlue® assay showed no clear cytotoxicity (Fig. S3). These results indicate that CNP inhibits cell proliferation without cytotoxicity, at least up to 0.1 μg/ml. Therefore, we treated cells with CNP at 0.025 or 0.1 μg/ml in the following experiments.
Figure 1.
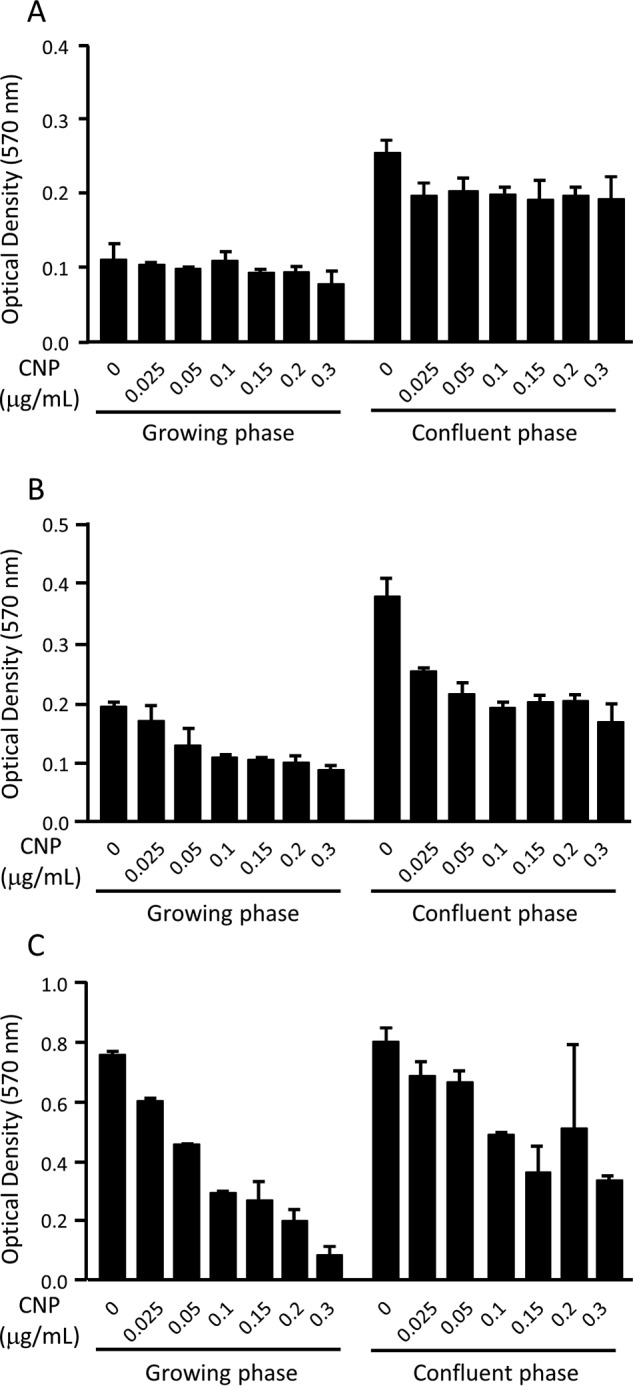
Effects of CNP on the viability of NB1RGB cells. A–C, cells in growth phase (left) and at confluence (right) were treated with CNP at the indicated concentrations for 24 h (A), 48 h (B), and 120 h (C), and viability was assessed by MTT. The experiment was performed twice with essentially the same results.
To detect specific molecules whose expression is affected by CNP treatment, we performed a microarray analysis. When 27,958 genes in total were analyzed, 39 genes were up-regulated more than 5-fold, and 53 genes were down-regulated less than 0.2-fold after 6 h of CNP treatment in the presence or absence of TGFβ (Fig. 2A). Panther classification analysis (PCA) showed that 22% of CNP-suppressed genes encoded proteins related to the heterotrimeric G protein signaling pathway (the Gαq- and Gα0-mediated pathways, e.g. BDKRB1, RASGRP1, SSTR1, and BDKRB2), and 11% of them encode proteins related to the inflammation signaling pathway (e.g. IL6 and NFATC4) (Fig. 2B). In addition, PCA by cellular component showed that CNP down-regulates expression of membrane-related genes, including HAS1 and HAS2 (Fig. 2C).
Figure 2.

Gene expression analysis. NB1RGB cells were cultured for 16 h and treated with CNP or not. After 1 h, cells were treated with TGFβ(Tβ) or not and cultured for an additional 6 h. Next, total RNA from CNP-treated and untreated cells was extracted and subjected to cDNA microarray analysis using a Human Gene Expression 4 × 44K microarray chip (Agilent Technologies). A, heatmap of up-regulated genes (39 genes; -fold change, >5.0) and down-regulated genes (53 genes, -fold change <0.2) after CNP treatment in the presence or absence of TGFβ. The heatmap was constructed using normalized values for each sample and Treeview software. The corresponding gene names are annotated on the right. B–D, gene ontology analyses using the Panther classification system. The up-regulated and/or down-regulated genes were classified using PANTHER gene list analysis (www.pantherdb.org/). (Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party–hosted site.) Shown are a pie chart indicating the percentages of genes classified into each molecular pathway (B), cellular component (C), and molecular function (D). E, differential gene expression between CNP-treated cells and untreated cells. Raw fluorescence values obtained by scanning were utilized for comparison of gene expression of HAS2 and GAPDH. *, p < 0.01; **, p < 0.001; significant difference.
Furthermore, PCA by molecular function showed that CNP down-regulates the expression of genes encoding catalytic activity (23%; e.g. DGK1, HAS1, HAS2, and NOX4), receptor activity (23%; BDKRB1, IL7R, IL21R, and SSTR1), and binding activity (39%; ADAMTS4, CXCL12, and NFATC4) (Fig. 2D). Accordingly, the raw fluorescence intensities of both HAS1 and HAS2, which encode hyaluronan-synthesizing enzymes, significantly decreased after CNP treatment compared with no treatment (Fig. 2E). These results suggest that CNP down-regulates the expression of HAS genes in NB1RGB fibroblasts.
To validate the microarray results, we performed qRT-PCR. The levels of HAS2 expression decreased dramatically 3 h after treatment with CNP at either 0.025 or 0.1 μg/ml (Fig. 3A). As HAS2 expression is regulated by its antisense RNA (HAS2 AS), we also examined its expression levels and found that it decreases substantially (Fig. 3B). As CNP was initially found to mimic the effect of activin A, we examined whether activin A inhibits HAS2 expression. Interestingly, activin A showed little effect on HAS2 expression, suggesting specific inhibition by CNP (Fig. 3C).
Figure 3.

Expression of HAS2 and HAS2AS, hyaluronan secretion and coat formation, and versican localization. A and B, expression levels, assessed by qRT-PCR, of both HAS2 (A) and HAS2AS (B) of cells treated with CNP for the indicated times (n = 3, mean ± S.D.; **, p < 0.05). C, expression levels of HAS2 in cells treated with activin A (n = 3, mean ± S.D.). D, NB1RGB cells, treated with CNP and TGFβ as indicated, under a particle exclusion assay. E and F, HA levels in cells/the ECM (E) and conditioned medium (F) measured by ELISA (n = 3, mean ± S.D.; **, p < 0.05). G, immunostaining patterns of versican in cultured fibroblasts treated with CNP and TGFβ as indicated. H, quantification of versican by measurement of the pixels of the immunostaining pictures (mean ± S.D. of 10 fields; **, p < 0.05). The experiments were performed twice (C, E, and F), three times (A, B, G, and H), or four times (D) with essentially the same results. Representative results are shown.
By erythrocyte exclusion assay, a pericellular HA coat was observed in both TGFβ-treated and nontreated cells (Fig. 3D, left panels, arrowheads). CNP treatment at a concentration of 0.025 or 0.1 μg/ml abrogated the pericellular HA coat in both TGFβ-treated and nontreated cells (Fig. 3D, center and right panels). When HA amounts in the cell lysate/ECM and conditioned medium after 3 days of culture were measured using ELISA, significant decreases of HA levels in the conditioned medium and a trend (not statistically significant; p = 0.3, n = 6) of decrease in HA deposition, correlated with CNP concentrations, were observed in both TGFβ-treated and nontreated samples (Fig. 3, E and F). Interestingly, TGFβ slightly increased HA deposition during 3 days of culture (Fig. 3F).
Formation of a pericellular HA coat requires HA-binding molecules, including versican, TSG-6, pentraxin-3, and inter-α-trypsin inhibitor (ITI) (9–11). As seen by immunofluorescence staining, versican expression was substantially up-regulated by TGFβ treatment. In both treated and nontreated samples, versican was localized along with fibers and did not accumulate in the pericellular coat (Fig. 3G). Although CNP had little effect on versican deposition of cell culture without TGFβ treatment, it substantially inhibited its deposition with TGFβ treatment (Fig. 3, G and H). Our microarray data revealed little effect of CNP on expression of TSG6 and pentraxin-3, although TGFβ substantially up-regulated expression of TSG6 and down-regulated that of pentraxin 3 (Fig. S1).
We further mined the transcriptomics data for the effects of CNP on glycolytic pathways genes, including those for production of UDP sugar precursors. No genes related to HA metabolism, except for HAS1 and HAS2, showed more than 3-fold changes of expression (Table S1). Taken together, the decrease in HA secretion in CNP-treated cells is mainly due to a remarkable reduction in HAS expression.
As CNP has been reported to inhibit fibrosis of pancreatic tissue (5), we investigated the effects of CNP on collagen secretion and its incorporation into the ECM. As seen by immunofluorescence staining, CNP treatment substantially diminished type I collagen incorporation into the ECM (Fig. 4A, top panels). TGFβ treatment increased collagen incorporation into the ECM. Under this condition, CNP treatment dramatically decreased its incorporation (Fig. 4A, bottom panels). Quantification of the staining intensity confirmed the decreased levels, with statistical significance, of collagen incorporation observed under the microscope (Fig. 4B). We also investigated expression of type III collagen, known to be involved in fibrosis. Immunofluorescence staining displayed essentially the same patterns, with lower staining levels than type I collagen (Fig. 4, C and D). The specificity of immunostaining patterns for collagens was confirmed by immunostaining using nonimmune IgG in place of primary antibodies (Fig. 4E). Immunoblot analysis confirmed that CNP substantially inhibited type I collagen incorporation (Fig. 4F). By SircolTM assay, CNP at both 0.025 and 0.1 μg/ml did not affect collagen levels in the cell lysate and the ECM of cell culture without TGFβ treatment. When co-treated with TGFβ, CNP inhibited them (Fig. 4G). Taken together, these results clearly indicate that CNP suppresses the incorporation of collagens into the ECM, which is more remarkable when cells are treated with TGFβ.
Figure 4.

Incorporation into the ECM and biosynthesis of collagen by NB1RGB cells treated with CNP and TGFβ. A, immunofluorescent staining for type I collagen. Cells were cultured for 72 h in the presence of CNP and TGFβ as indicated, and then immunostaining was performed. B, quantification of collagen by measurement of the pixels of the immunostaining pictures (mean ± S.D. of 10 fields; **, p < 0.05). C, immunofluorescence staining for type III collagen. Cells were cultured and treated as above, and then immunostaining was performed. D, quantification of collagen by measurement of the pixels of the immunostaining pictures (mean ± S.D. of 10 fields; **, p < 0.05). E, negative control of immunostaining, where nonimmune rabbit IgG was used in place of primary antibody. The picture was taken with a longer exposure. F, Western blotting for type I collagen. Cells were treated with CNP and TGFβ as above, and cells/ECM were collected and applied to Western blotting. The bands of α1 and α2 chains of type I collagen are indicated by arrows. The numbers below indicate the level of band density, with a nontreated sample as 100. G, quantification of collagen by SilcolTM assay (n = 3, mean ± S.D.; **, p < 0.05). H, biosynthesis of total proteins and collagens. Cells were treated with CNP and/or TGFβ and then labeled with [3H]proline for 24 h. Radioactivity incorporated in the cells was measured. Total protein levels (left panel, n = 3, mean ± S.D.; **, p < 0.05) and collagen synthesis levels (percent) of total proteins (right panel) are shown. The experiments were performed twice (G), three times (C, D, F, and H), or five times (A, B, and E) with essentially the same results. Representative results are shown.
Next we labeled fibroblasts with [3H]proline and examined the effects of CNP on biosynthesis of collagens and total proteins. The levels of total protein synthesis decreased by treatment with CNP at concentrations of both 0.025 and 0.1 μg/ml by ∼40%. TGFβ stimulation up-regulated protein biosynthesis by 50%, and under such conditions, CNP treatment inhibited total protein biosynthesis in a manner similar to that of unstimulated cells, i.e. by ∼35% and 50% at 0.025 and 0.1 μg/ml, respectively (Fig. 4H, left panel). The ratio of collagen per total protein in nonstimulated cells was ∼2% in both CNP-treated and nontreated cells. That in TGFβ-stimulated cells was ∼6% and was unaffected by CNP treatment (Fig. 4H, right panel). These results suggest that CNP inhibits total protein biosynthesis and that its inhibition is not specific to collagen. We further mined the transcriptomics data for the effects of CNP on collagen gene expression, which confirmed that CNP has no specific effects on collagen expression (Table S2).
The fact that CNP inhibited collagen incorporation more strongly when the cells were treated with TGFβ (Fig. 4) suggests that CNP inhibits TGFβ-mediated pathways. TGFβ facilitates differentiation of fibroblasts toward myofibroblasts, which actively synthesize collagens. Therefore, we examined whether CNP affects the TGFβ-mediated pathway and differentiation. When double-stained for total Smad2/3 and αSMA, Smad2/3 was mainly localized in the cytoplasm in all TGFβ-nontreated cells. When treated with TGFβ, Smad2/3 was translocated to the nuclei in all samples regardless of CNP treatment. Without TGFβ treatment, CNP did not affect the number of αSMA-positive cells. When treated with TGFβ, the number of αSMA-positive cells increased at 24 h and decreased with CNP treatment (Fig. 5, A and B). These results indicate that CNP inhibits TGFβ-mediated differentiation of fibroblasts toward myofibroblasts.
Figure 5.

Differentiation toward myofibroblasts and Smad2/3 nuclear translocation. A, cells were cultured for 24 h in the presence of CNP and TGFβ as indicated. Immunostaining for Smad2/3 (red) and αSMA (green) was performed, and nuclei were stained with 4′,6-diamidino-2-phenylindole (blue). Note that Smad2/3 nuclear translocation by TGFβ is not affected by CNP treatment. B, the ratio of αSMA-positive cells per total cells (mean ± S.D. of 10 fields; **, p < 0.05). The immunofluorescence staining was performed five times with essentially the same results. Representative pictures are shown.
The results shown in Fig. 5 suggested that CNP has little effect on Smad2/3 signal transduction. When analyzed by Western blotting, TGFβ treatment substantially increased phophoSmad2, and CNP at both 0.025 and 0.1 μg/ml had little effect on phosphorylation of Smad2 (Fig. 6A, first panel and graph), confirming the results of Smad2/3 nuclear translocation (Fig. 5A). Next we investigated other signaling pathways mediated by TGFβ. CNP inhibited phosphorylation of ERK1/2 at both 0.025 and 0.1 μg/ml. TGFβ treatment substantially inhibited it, and combined treatment with CNP further inhibited phosphorylation in a dose-dependent manner (Fig. 6A, second panel and graph). CNP inhibited phosphorylation of p38 MAPK in a dose-dependent manner. TGFβ substantially inhibited it; however, combined treatment with CNP did not affect it (Fig. 6A, third panel and graph).
Figure 6.

Signal transduction pathways affected by CNP. A, immunoblot panels for Smad2/3, ERK1/2, p38 MAPK, and Akt together with their phosphorylation forms. The lines in the pAkt and tAkt panels indicate a splice where the same image was reordered so that the data are presented in the same order as the other panels in this figure. The right panels are quantification data of their band density. Each experiment was performed five times with essentially the same results. Statistical analysis was performed using the results of all experiments. B, suppression of RAS-ERK signaling by CNP. The GSEA was conducted using GSEA software v2.2.4 and the Molecular Signatures Database (Broad Institute). All raw data were formatted and applied to all Gene Ontology (GO) gene sets (C5). A representative GSEA enrichment plot and corresponding heatmap image of the indicated gene set are shown for CNP-treated and untreated cells, respectively. Genes contributing to the enrichment are shown in rows, and the samples are shown in columns on the heatmap. Expression is shown as a gradient from high (red) to low (blue). FDR, false discovery rate; NES, normalized enrichment score.
To investigate whether the RAS–ERK1/2 pathway was suppressed by CNP treatment, we analyzed the microarray data using the GSEA software version 2.2.4 and the Molecular Signatures Database (Broad Institute). The representative GSEA enrichment plot and corresponding heatmap image of the indicated gene set for CNP-treated and untreated cells confirmed suppression of the RAS–ERK1/2 pathway by CNP treatment (Fig. 6B).
To examine whether the PI3K/AKT pathway is involved in CNP effects, we analyzed the transcriptomics data and found that CNP slightly inhibits the expression of AKT2 and AKT3 but not AKT1 (Table S3). When the expression of four representative genes downstream of the PI3K/AKT pathway was examined, we observed no remarkable changes with CNP treatment (Fig. S2). Western blot analysis demonstrated little effect on the phosphorylation of Akt by CNP (Fig. 6A, fourth panel and graph).
Discussion
In this study, we investigated the effects of CNP, a Vinca alkaloid that mimics activin A function, on fibroblast behavior and formation of the ECM. The interesting features of this study were as follows: CNP dramatically inhibits HAS 1, HAS2, and HAS2 AS expression, resulting in a decrease in HA secretion; CNP inhibits cell proliferation and biosynthesis of collagens at the same ratio with total protein biosynthesis; CNP inhibits versican incorporation into the ECM; CNP inhibits TGFβ-mediated differentiation of fibroblasts toward myofibroblasts; and although CNP does not affect Smad2/3 signaling, it inhibits ERK1/2 activation, which may be responsible for CNP action. Taken together, these findings compellingly indicate inhibitory effects of CNP on ECM formation by fibroblasts.
Our microarray analysis revealed marked decreases in HAS1 and HAS2 expression with CNP treatment among ECM-related genes. In addition, HAS2AS was substantially reduced by the treatment, which was validated by qRT-PCR. The measurement of HA in cell lysate and the ECM and an erythrocyte exclusion assay confirmed its decreased levels by CNP treatment. HAS2 transcription is regulated by a variety of signal transduction pathways involving many molecules (12–14). Among them, both EGF family factors and G protein–coupled receptors (GPCRs) are known to be major molecules that regulate HAS2 expression. EGFs increase HAS2 expression (13). Early growth response-1 (EGR-1) induces CD44v6, which then sustains ERK signaling, up-regulating HAS2 expression (15). GPCRs act via protein kinase A, and its downstream CREB1 binds to its response elements in the HAS2 promoter, inducing HAS2 transcription (16–19). We have shown that CNP inhibits ERK1/2 phosphorylation. Our microarray data using the GSEA software program confirmed suppression of RAS-ERK1/2 pathway by CNP. The G protein–coupled receptor (GPCRs)/protein kinase A/CREB1 cascade up-regulates HAS2 transcription (19). Our gene ontology analysis of microarray data shows that CNP profoundly inhibits G protein–mediated pathways (Fig. 2B). Therefore, it is likely that CNP inhibits both EGF-mediated and GPCR-mediated pathways, leading to down-regulation of HAS2 transcription. Although the PI3K/AKT pathway has been reported to up-regulate HAS2 expression (14), our microarray analysis did not suggest involvement of this pathway (Fig. S2 and Table S3). HAS2AS1 forms a duplex with HAS2 mRNA and stabilizes the HAS2 transcript (20). In addition, HAS2AS1 binds to O-GlcNAcylated p65 and induces HAS2 transcription (12, 21). Reduced HAS2AS levels with CNP treatment may prompt inhibition of HAS2 transcription (22–25).
Incorporation of HA into the ECM involves HA-binding ECM molecules such as versican. We have shown that TGFβ substantially increases versican expression and that CNP inhibits its incorporation into the ECM without affecting its transcription. Although versican deposition decreased to ∼60% with CNP treatment, the HA amount in the cells/ECM and the conditioned medium decreased only slightly. This is consistent with our previous results showing that mouse embryonic fibroblasts with ∼20% expression levels of versican exhibit ∼85% levels of HA in the ECM (26). These observations suggest that versican is not a prerequisite for HA deposition. Interestingly, these mouse embryonic fibroblasts exhibit sustained HA-mediated signaling and attain premature senescence. Similarly, CNP treatment may alter HA-mediated signaling. Pericellular HA coat formation involves HA-binding molecules, including TGS6, pentraxin 3 (11), heavy chains of ITI (10), and versican (9). Our microarray analysis revealed that CNP has little influence on the expression of TSG6 and pentraxin 3. Therefore, these molecules are unlikely to affect the HA coat sizes of CNP-treated fibroblasts. Recently, it has been shown that versican G1 fragments but not full-length versican form aggregates with HA and heavy chains of ITI in the pericellular matrix (27), which agrees with our observation that intact versican is not accumulated in pericellular matrix.
Although CNP has been reported to inhibit fibrosis of the pancreas (7), our microarray analysis showed no significant differences in mRNA levels of collagens. Our biosynthesis assay revealed that TGFβ up-regulates biosynthesis of total proteins, including collagens, and that CNP inhibits protein biosynthesis both in the presence and absence of TGFβ (Fig. 5A). Although TGFβ treatment increased the ratio of collagens per total proteins, CNP had little effect on the ratio, which agrees with our microarray results.
By both immunostaining and SircolTM assay, the inhibition of collagen deposition by CNP was statistically significant only when treated with TGFβ, although there was a tendency of inhibition in the absence of TGFβ. In contrast, when treated with TGFβ to facilitate collagen synthesis, CNP substantially inhibited its deposition, suggesting that the inhibitory activity of CNP on collagen levels is mainly exerted via the TGFβ-mediated pathway. CNP altered the collagen fiber structure from diffuse fine fibers to broader woven fibers with more spaces among fibers. A previous study showed similar collagen fiber patterns by inhibiting HA deposition using 4-methylumbelliferone, HA oligosaccharide, and hyaluronidase (28). Formation of woven collagen fibers with CNP treatment may be due to a decrease in HA deposition.
We demonstrated that CNP did not affect the Smad2/3 signal transduction pathway but did affect both the ERK1/2 and p38/MAPK pathways. CNP inhibited ERK1/2 phosphorylation in both the presence and absence of TGFβ, whereas CNP inhibited p38 phosphorylation only in its absence. These inhibition patterns suggest that the inhibitory effect of CNP occurs via the ERK1/2 signaling pathways. As ERK1/2 up-regulates cell proliferation (29) and HAS2 gene expression by activating its downstream CREB, the inhibitory effects of CNP are likely due to down-regulation of the ERK1/2 pathway. Cross-talk between TGFβ signaling and ERK1/2 signaling has been reported (30, 31). EGF treatment has been shown to inhibit basal and TGFβ-induced expression of type I collagen and αSMA (31), contrary to our observations. Two signaling pathways may interact differently, depending on the expression of other molecules, cell types, and culture conditions. Previously, CNP has been reported to inhibit cell proliferation and collagen secretion via the p38 MAPK pathway in rat pancreatic stellate cells (4, 7, 32) and mainly via c-Jun N-terminal kinase and partly via both the ERK1/2 and p38 pathways in Lx-2 hepatic stellate cells (33). The effects of CNP may be mediated by different MAPK pathways in various types of fibroblastic cells.
Our results indicate that CNP affects MAPK pathways but not the Smad2/3 pathway, which facilitates COL1A2 transcription. This supports our observation that CNP did not inhibit collagen biosynthesis in a specific manner. CNP was first identified as a compound that shares the receptor with activin A, but interestingly, it inhibits fibrosis, whereas activin A facilitates it (5). Although activin A binds to its receptor and induces Smad2/3 signal transduction similar to TGFβ (34–37), CNP had little effect on the Smad2/3 pathway. The pathway-specific inhibitory effects of CNP eliminate its competitive inhibition as a ligand with activin A and TGFβ. It may modify the receptor complex or inhibit the function of intervening molecules between TGFβ receptors and MAPK.
In this study, we have demonstrated that CNP inhibits fibroblast proliferation and ECM formation, decreasing incorporation of HA, versican, and collagens, without cytotoxicity. CNP may be used as an inhibitor of ECM formation under pathological conditions, where excess ECM formation is involved in several pathological processes, such as liver and lung fibrosis and keloid and hypertrophic scarring. CNP may be a good tool to control these pathological conditions.
Experimental procedures
Cell culture
Human foreskin fibroblasts (NB1RGB) were provided by the RIKEN BRC through the National Bio-Resource Project of the MEXT Japan and were grown in DMEM containing 10% fetal bovine serum (FBS) at 37 °C in a 5% CO2 incubator. Fibroblasts up to three passages were used for all experiments.
MTT assay, alamarBlue® assay, and trypan blue exclusion test
NB1RGB cells were plated in 96 wells at growth and confluent phases and cultured for 48 h. The medium was replaced with fresh medium, CNP (final concentrations of 0.025, 0.1, 0.15, 0.1, 0.2, and 0.3 μg/ml) was added, and the cells were cultured for 24, 48, and 120 h. Then MTT (Sigma-Aldrich) dye solution was added to each well and incubated for 4 h, and absorption at 570 nm was determined with an automatic ELISA plate reader (Multiskan; Thermo Electron, Vantaa, Finland) (38). NB1RGB cells at 60% confluence were treated with CNP for days as indicated. The alamarBlue® assay (Invitrogen) was performed according to the manufacturer's instructions. For the trypan blue exclusion test, after staining with 5 mg/ml trypan blue for 1 min, both stained and unstained cells were counted under a microscope.
Microarray analysis
Cells were plated at 80% confluence and cultured for 16 h. CNP at each concentration (0.025 and 0.1 μg/ml) was added, and the cells were incubated for 1 h. TGFβ (hereafter a final concentration of 10 ng/ml) was then added, and cells were cultured for 6 h. After washing with PBS, cells and the extracellular matrix were collected and subjected to a microarray analysis. We performed a comprehensive gene expression analysis and compared the results among the TGFβ + and −, and CNP + and − groups using a Human Gene Expression 4 × 44K microarray chip (G4845A, Agilent Technologies, Santa Clara, CA), which can examine 27,958 genes. For analysis of gene expression profiling, total RNA was isolated using the RNeasy Mini Kit (Qiagen) according to the manufacturer's instructions. The quality of the isolated RNA was ascertained using a NanoDrop 1000 spectrophotometer (Thermo Fisher Scientific). The cDNA microarray analysis was performed according to the manufacturer's instructions (Agilent Technologies). In brief, cDNA synthesis and cRNA labeling with cyanine 3 (Cy3) dye were performed using the Agilent Low Input Quick Amp Labeling Kit (Agilent Technologies).
The Cy3-labeled cRNA was purified, fragmented, and hybridized on a Human Gene Expression 4 × 44 K microarray chip containing 43,377 oligonucleotide probes with the Gene Expression Hybridization Kit (Agilent Technologies). After incubation at 60 °C for 17 h, the microarray slide was washed and scanned using a DNA microarray scanner (Agilent Technologies).
The scanned data were quantified using Feature Extraction software version 11.0.1.1 (Agilent Technologies). The signal intensities were then normalized as described previously (39). Gene expression changes were detected as follows. The difference of the normalization value = (averaged normalization value of CNP-treated cells (0.025 μm and 0.1 μm) − untreated cells); an up-regulated gene was determined by a difference from the normalization value of more than 0.55 (-fold change, >5.0) and a down-regulated gene by a difference from the normalization value of less than 0.55 (-fold change, <-0.2). The raw and normalized microarray data have been submitted to the GEO database at NCBI under accession number GSE104813. Data mining was performed using a pathway database, REACTOME (https://reactome.org/what-is-reactome).3
Quantitative real-time RT-PCR
Cells were plated in 35-mm dishes (cell density, 1 × 105 cells/dish) and incubated for 24 h at 37 °C. Then CNP was added at the aforementioned concentrations. In other experiments, activin A (R&D Systems) at a concentration of 10 ng/ml was added. After 0, 3, 6, and 12 h, cells were washed with PBS and collected, and total RNA was extracted with the RNeasy Mini KitTM (Qiagen), and with 2 μg of total RNA, cDNA was synthesized using the SuperScript VILOTM cDNA Synthesis Kit (Qiagen). qRT-PCR was performed using StepOnePlusTM Real-Time PCR System (Applied Biosystems) and Power SYBR PCR Master Mix (Life Technologies) according to the manufacturer's protocol. The oligonucleotides used for PCR were as follows: HAS forward, GTCATGTACACAGCCTTCAGAGC; HAS2 reverse, ACAGATGAGGCTGGGTCAAGCA; HAS2AS forward, TCGAATAAACTGGAAATGATGC; HAS2AS reverse, GATGTCAAAACCTGAAAGGGAT; GAPDH forward, TGCACCACCAACTGCTTAGC; GAPDH reverse, GGCATGGACTGTGGTCATGAG. The levels of PCR products were determined with StepOnePlusTM equipment (Life Technologies) and analyzed using the accessory software. The relative abundance of transcripts was normalized to the GAPDH mRNA level.
Erythrocyte exclusion assay
Assessment of the pericellular HA coat was performed as described previously (40). Cells were plated in 35-mm dishes (cell density, 3 × 104 cells/dish), cultured for 2 days, and treated with CNP and/or TGFβ for an additional 2 days. Then conditioned medium was removed, and glutaraldehyde-fixed rabbit erythrocytes were added. After 15 min, the cells were observed by phase-contrast microscopy.
Quantification of HA levels
Cells were plated in 35-mm dishes at 60% confluence (∼cell density, 1 × 105 cells/dish) and cultured for 24 h. After washing twice with PBS, the medium was replaced with DMEM without FBS, and cells were incubated for 16 h. Then CNP and TGFβ were added in the same manner as above, and the cells were cultured for 3 days. The conditioned medium was collected. After washing with PBS twice, cells together with the extracellular matrix were recovered with CytoBuster ProteinTM. Hyaluronan was quantified using an HA assay kit (Cosmo Bio Co. Ltd.).
Immunocytochemistry
Cells were plated on a coverglass in a 6-well plate at 60% confluence and cultured overnight. After replacing the medium, CNP at the concentration described above was added or not, and cells were cultured for 1 h. Then TGFβ was added or not, and cells were cultured for 3 days. After removal of the medium, the cells were fixed with 4% paraformaldehyde for 20 min, rinsed with PBS for 5 min three times, and blocked with 10% BSA for 30 min at room temperature. After washing with PBS for 5 min three times, the cells were treated with mouse anti-human versican (1:500, 2B1, Seikagaku), rabbit anti-human type I collagen (1:200, LSL, LB-1196), or rabbit anti-human type III collagen (1:200, LSL, LB-1393) overnight at 4 °C. After washing, they were treated with Alexa 594–conjugated goat anti-mouse or goat anti-rabbit antibody for 1 h at room temperature. After washing with PBS for 5 min three times, the nuclei were stained with 4′,6-diamidino-2-phenylindole (Wako Chemicals) for 5 min. After washing briefly, the coverslip was flipped over and mounted onto a slide glass. The cells were then observed under a fluorescent microscope (Keyence, BZ-9000).
SircolTM assay
Cells were plated in 35-mm dishes at 60% confluence (∼cell density, 1 × 105 cells/dish) and cultured for 24 h. Then the medium was replaced with fresh medium, CNP and TGFβ were added in the same manner as above, and the cells were cultured for 3 days. After washing with PBS twice, cells together with the extracellular matrix were recovered, and their collagen concentration was measured using the SircolTM assay kit (BioSite) according to the manufacturer's protocol.
Analysis of collagen biosynthesis
Analysis of collagen biosynthesis was carried out as described previously (41). Cells were plated at 80% confluence in 35-mm dishes (cell density, 1.3 × 105 cells/dish) and cultured for 16 h until confluence. The medium was replaced with 1 ml of DMEM containing 10% FBS and CNP at each concentration (0, 0.025, and 0.1 μg/ml) and 0.5 mm β-aminopropionitrile (Tokyo Kasei). 200 μm L-ascorbyl phosphate magnesium phosphate n-hydrate (Wako Pure Chemicals) was added and incubated for 1 h. Then TGFβ (final concentration of 10 ng/ml) was added, and cells were cultured. In the control dishes, cells were cultured in the absence of TGFβ. After 2 h, cells were labeled with 10 Ci [3H]proline for 24 h. Then both cells and conditioned medium were collected and sonicated. 200 μl of 0.5% BSA and 50 l of 0.2% gelatin were added to a 400-μl suspension, and the sample was boiled for 10 min. After cooling down on ice, the sample was dialyzed against 5 mm acetate and lyophilized.
The lyophilized sample was dissolved in 950 μl of NaOH, sonicated, and neutralized with 2 m Tris-HCl (pH 7.5). The sample was then aliquoted into 100 μl. One sample was treated with 400 μg/ml collagenase (Advance Biofactures Corp., Form III) at 37 °C for 4 h. Another sample was left untreated by addition of collagenase solvent. Then 250 μl of 0.5% tannic acid–10% trichloroacetate was added. The sample was placed on ice for 15 min and centrifuged for 5 min, and the supernatant was processed for liquid scintillation counting (collagens (COL)).
500 μl of 0.5% SDS and 5 mm DTT were added to the precipitate, and the sample was sonicated, boiled for 5 min, and processed to liquid scintillation counting (non-collagenous proteins (NCP)). Collagen biosynthesis (percent) was calculated with the formula 100 × COL/COL + 5.4 × NCP.
Western blotting
Cells were plated at 60% confluence (cell density, 1 × 105 cells/dish) for 24 h, the medium was replaced with DMEM without FBS, and cells were incubated for 16 h. CNP was added or not, and after 1 h, TGFβ was added or not. After 30 min, cells were collected as samples. After measurement of protein amounts using the MicroBCA kit (Thermo Fisher Scientific), samples with the same protein amount were subjected to SDS-PAGE and then transferred to a polyvinylidene difluoride membrane. The membrane was blocked with 5% skim milk in 50 mm Tris-HCl (pH 7.5) and 150 mm NaCl containing 0.05% Tween 20 (TBST) and treated with the indicated antibodies diluted in CanGetSignalTM Solution I overnight at 4 °C. After washing with TBST, the membrane was incubated with a goat anti-rabbit polyclonal antibody conjugated to peroxidase (Cappel, 1:1000) diluted in CanGetSignalTM Solution II for 1 h. After washing with TBST, the bands were detected with amplified chemiluminescence (ECL) using LAS4000 (GE Healthcare). The band density was measured using ImageJ (National Institutes of Health). The antibodies used were as follows: rabbit monoclonal antibodies against total Smad2/3 (1:1000, CST), phosphoSmad2 (1:1000, CST), total ERK1/2, phosphoERK1/2 (1:1000, CST), total p38 (1:2000, CST), phospho-p38 (1:2000, CST), and rabbit polyclonal antibodies against phosphoAkt1 (1:1000, CST) and total Akt (1:1000, CST).
For Western blotting of type I collagen, cells were plated in 35-mm dishes at 90% confluence (cell density, 1.5 × 105 cells/dish) and cultured for 16 h. Then cells were treated with or without CNP and TGFβ as above and cultured for 72 h. After washing with PBS twice, cells together with the extracellular matrix were collected and applied to Western blot analysis as above. The primary antibody used was rabbit anti-human type I collagen (1:1000, LSL, LB-1196).
Statistical analyses
Data are presented as the means ± S.D. Statistical analyses were performed with post hoc tests (Bonferroni) using ANOVA. Probability values of 0.05 were considered statistically significant.
Author contributions
T. T., A. O., S. K., K. Y., D. V., A. P., K. U., and H. W. conceptualization; T. T., A. O., S. K., D. V., A. P., S. H., K. U., and H. W. data curation; T. T., A. O., S. K., and S. H. formal analysis; T. T., A. O., S. K., D. V., A. P., S. H., K. U., and H. W. investigation; T. T., A. O., Y. H., D. V., A. P., S. H., K. U., and H. W. methodology; T. T., A. O., S. K., and H. W. writing-original draft; A. O., S. K., K. U., and H. W. validation; K. M. and H. W. resources; K. M., K. Y., Y. H., A. P., K. U., and H. W. supervision; K. M., K. Y., Y. H., D. V., and H. W. project administration; K. Y. and H. W. funding acquisition; H. W. writing-review and editing.
Supplementary Material
Acknowledgments
We thank K. Ota for technical assistance and Drs. Nagai and Tsuchimoto for critical reading of the manuscript.
This work was supported in part by Grant-in-Aid (KAKENHI) for Scientific Research B, 25293096 (to H. W.), Grant-in-Aid (KAKENHI) for Young Scientists B, 15K19561 (to A. O.), and Grant-in-Aid (KAKENHI) for Scientific Research C, 25460395 (to S. K.). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–S3 and Tables S1–S3.
The raw and normalized microarray data have been submitted to the GEO database at NCBI under accession number GSE104813.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party–hosted site.
- ECM
- extracellular matrix
- CNP
- conophylline
- αSMA
- alpha-smooth muscle actin
- HA
- hyaluronan
- TGFβ
- transforming growth factor β
- PCA
- Panther classification analysis
- AS
- antisense
- ITI
- inter-α-trypsin inhibitor
- MAPK
- mitogen-activated protein kinase
- PI3K
- phosphatidylinositol 3-kinase
- EGF
- epidermal growth factor
- GPCR
- G protein-coupled receptor
- ERK
- extracellular signal–regulated kinase
- CREB
- cAMP-response element-binding protein
- DMEM
- Dulbecco's modified Eagle's medium
- FBS
- fetal bovine serum
- cDNA
- complementary DNA
- cRNA
- complementary RNA
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- COL
- collagens
- GSEA
- gene set enrichment analysis
- GO
- gene ontology.
References
- 1. Birbrair A., Zhang T., Files D. C., Mannava S., Smith T., Wang Z. M., Messi M. L., Mintz A., and Delbono O. (2014) Type-1 pericytes accumulate after tissue injury and produce collagen in an organ-dependent manner. Stem Cell Res. Ther. 5, 122 10.1186/scrt512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Neary R., Watson C. J., and Baugh J. A. (2015) Epigenetics and the overhealing wound: the role of DNA methylation in fibrosis. Fibrogenesis Tissue Repair 8, 18 10.1186/s13069-015-0035-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Schrier D. J., Phan S. H., and McGarry B. M. (1983) The effects of the nude (nu/nu) mutation on bleomycin-induced pulmonary fibrosis: a biochemical evaluation 1–3. Am. Rev. Respir. Dis. 127, 614–617 10.1164/arrd.1983.127.5.614 [DOI] [PubMed] [Google Scholar]
- 4. Umezawa K., Ohse T., Yamamoto T., Koyano T., and Takahashi Y. (1994) Isolation of a new Vinca alkaloid from the leaves of Ervatamia microphylla as an inhibitor of ras functions. Anticancer Res. 14, 2413–2417 [PubMed] [Google Scholar]
- 5. Kojima I., and Umezawa K. (2006) Conophylline: a novel differentiation inducer for pancreatic β cells. Int. J. Biochem. Cell Biol. 38, 923–930 10.1016/j.biocel.2005.09.019 [DOI] [PubMed] [Google Scholar]
- 6. Ogata T., Li L., Yamada S., Yamamoto Y., Tanaka Y., Takei I., Umezawa K., and Kojima I. (2004) Promotion of β-cell differentiation by conophylline in fetal and neonatal rat pancreas. Diabetes 53, 2596–2602 10.2337/diabetes.53.10.2596 [DOI] [PubMed] [Google Scholar]
- 7. Saito R., Yamada S., Yamamoto Y., Kodera T., Hara A., Tanaka Y., Kimura F., Takei I., Umezawa K., and Kojima I. (2012) Conophylline suppresses pancreatic stellate cells and improves islet fibrosis in Goto-Kakizaki rats. Endocrinology 153, 621–630 10.1210/en.2011-1767 [DOI] [PubMed] [Google Scholar]
- 8. Nakade Y., Sakamoto K., Yamauchi T., Inoue T., Kobayashi Y., Yamamoto T., Ishii N., Ohashi T., Sumida Y., Ito K., Nakao H., Fukuzawa Y., Umezawa K., and Yoneda M. (2017) Conophylline inhibits non-alcoholic steatohepatitis in mice. PLoS ONE 12, e0178436 10.1371/journal.pone.0178436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Evanko S. P., Tammi M. I., Tammi R. H., and Wight T. N. (2007) Hyaluronan-dependent pericellular matrix. Adv. Drug Deliv. Rev. 59, 1351–1365 10.1016/j.addr.2007.08.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Blom A., Pertoft H., and Fries E. (1995) Inter-α-inhibitor is required for the formation of the hyaluronan-containing coat on fibroblasts and mesothelial cells. J. Biol. Chem. 270, 9698–9701 10.1074/jbc.270.17.9698 [DOI] [PubMed] [Google Scholar]
- 11. Salustri A., Garlanda C., Hirsch E., De Acetis M., Maccagno A., Bottazzi B., Doni A., Bastone A., Mantovani G., Beck Peccoz P., Salvatori G., Mahoney D. J., Day A. J., Siracusa G., Romani L., and Mantovani A. (2004) PTX3 plays a key role in the organization of the cumulus oophorus extracellular matrix and in in vivo fertilization. Development. 131, 1577–1586 10.1242/dev.01056 [DOI] [PubMed] [Google Scholar]
- 12. Tammi R. H., Passi A. G., Rilla K., Karousou E., Vigetti D., Makkonen K., and Tammi M. I. (2011) Transcriptional and post-translational regulation of hyaluronan synthesis. FEBS J. 278, 1419–1428 10.1111/j.1742-4658.2011.08070.x [DOI] [PubMed] [Google Scholar]
- 13. Monslow J., Williams J. D., Fraser D. J., Michael D. R., Foka P., Kift-Morgan A. P., Luo D. D., Fielding C. A., Craig K. J., Topley N., Jones S. A., Ramji D. P., and Bowen T. (2006) Sp1 and Sp3 mediate constitutive transcription of the human hyaluronan synthase 2 gene. J. Biol. Chem. 281, 18043–18050 10.1074/jbc.M510467200 [DOI] [PubMed] [Google Scholar]
- 14. Li L., Asteriou T., Bernert B., Heldin C. H., and Heldin P. (2007) Growth factor regulation of hyaluronan synthesis and degradation in human dermal fibroblasts: importance of hyaluronan for the mitogenic response of PDGF-BB. Biochem. J. 404, 327–336 10.1042/BJ20061757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ghatak S., Markwald R. R., Hascall V. C., Dowling W., Lottes R. G., Baatz J. E., Beeson G., Beeson C. C., Perrella M. A., Thannickal V. J., and Misra S. (2017) Transforming growth factor β1 (TGFβ1) regulates CD44V6 expression and activity through extracellular signal-regulated kinase (ERK)-induced EGR1 in pulmonary fibrogenic fibroblasts. J. Biol. Chem. 292, 10465–10489 10.1074/jbc.M116.752451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Maeda-Sano K., Gotoh M., Morohoshi T., Someya T., Murofushi H., and Murakami-Murofushi K. (2014) Cyclic phosphatidic acid and lysophosphatidic acid induce hyaluronic acid synthesis via CREB transcription factor regulation in human skin fibroblasts. Biochim. Biophys. Acta 1841, 1256–1263 10.1016/j.bbalip.2014.05.004 [DOI] [PubMed] [Google Scholar]
- 17. Porsch H., Bernert B., Mehic´ M., Theocharis A. D., Heldin C. H., and Heldin P. (2013) Efficient TGFβ-induced epithelial-mesenchymal transition depends on hyaluronan synthase HAS2. Oncogene 32, 4355–4365 10.1038/onc.2012.475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Schiller M., Dennler S., Anderegg U., Kokot A., Simon J. C., Luger T. A., Mauviel A., and Böhm M. (2010) Increased cAMP levels modulate transforming growth factor-β/Smad-induced expression of extracellular matrix components and other key fibroblast effector functions. J. Biol. Chem. 285, 409–421 10.1074/jbc.M109.038620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Makkonen K. M., Pasonen-Seppänen S., Törrönen K., Tammi M. I., and Carlberg C. (2009) Regulation of the hyaluronan synthase 2 gene by convergence in cyclic AMP response element-binding protein and retinoid acid receptor signaling. J. Biol. Chem. 284, 18270–18281 10.1074/jbc.M109.012492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Michael D. R., Phillips A. O., Krupa A., Martin J., Redman J. E., Altaher A., Neville R. D., Webber J., Kim M. Y., and Bowen T. (2011) The human hyaluronan synthase 2 (HAS2) gene and its natural antisense RNA exhibit coordinated expression in the renal proximal tubular epithelial cell. J. Biol. Chem. 286, 19523–19532 10.1074/jbc.M111.233916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Vigetti D., Deleonibus S., Moretto P., Bowen T., Fischer J. W., Grandoch M., Oberhuber A., Love D. C., Hanover J. A., Cinquetti R., Karousou E., Viola M., D'Angelo M. L., Hascall V. C., De Luca G., and Passi A. (2014) Natural antisense transcript for hyaluronan synthase 2 (HAS2-AS1) induces transcription of HAS2 via protein O-GlcNAcylation. J. Biol. Chem. 289, 28816–28826 10.1074/jbc.M114.597401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Vigetti D., Deleonibus S., Viola M., Karousou E., D'Angelo M. L., De Luca G., and Passi A. (2014) The long non-coding RNA HAS2-AS1 enhances the transcription of hyaluronan synthase 2. FASEB J. 28, 1005.1 [Google Scholar]
- 23. Li Y., Li L., Brown T. J., and Heldin P. (2007) Silencing of hyaluronan synthase 2 suppresses the malignant phenotype of invasive breast cancer cells. Int. J. Cancer 120, 2557–2567 10.1002/ijc.22550 [DOI] [PubMed] [Google Scholar]
- 24. Chao H., and Spicer A. P. (2005) Natural antisense mRNAs to hyaluronan synthase 2 inhibit hyaluronan biosynthesis and cell proliferation. J. Biol. Chem. 280, 27513–27522 10.1074/jbc.M411544200 [DOI] [PubMed] [Google Scholar]
- 25. Udabage L., Brownlee G. R., Waltham M., Blick T., Walker E. C., Heldin P., Nilsson S. K., Thompson E. W., and Brown T. J. (2005) Antisense-mediated suppression of hyaluronan synthase 2 inhibits the tumorigenesis and progression of breast cancer. Cancer Res. 65, 6139–6150 10.1158/0008-5472.CAN-04-1622 [DOI] [PubMed] [Google Scholar]
- 26. Suwan K., Choocheep K., Hatano S., Kongtawelert P., Kimata K., and Watanabe H. (2009) Versican/PG-M assembles hyaluronan into extracellular matrix and inhibits CD44-mediated signaling toward premature senescence in embryonic fibroblasts. J. Biol. Chem. 284, 8596–8604 10.1074/jbc.M806927200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Murasawa Y., Nakamura H., Watanabe K., Kanoh H., Koyama E., Fujii S., Kimata K., Zako M., Yoneda M., and Isogai Z. (2017) The Versican G1 fragment and serum-derived hyaluronan-associated proteins interact and form a complex in granulation tissue of pressure ulcers. Am. J. Pathol. 188, 432–449 [DOI] [PubMed] [Google Scholar]
- 28. Evanko S. P., Potter-Perigo S., Petty L. J., Workman G. A., and Wight T. N. (2015) Hyaluronan controls the deposition of fibronectin and collagen and modulates TGFβ1 induction of lung myofibroblasts. Matrix Biology 42, 74–92 10.1016/j.matbio.2014.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Etscheid M., Beer N., and Dodt J. (2005) The hyaluronan-binding protease upregulates ERK1/2 and PI3K/Akt signalling pathways in fibroblasts and stimulates cell proliferation and migration. Cell. Signal. 17, 1486–1494 10.1016/j.cellsig.2005.03.007 [DOI] [PubMed] [Google Scholar]
- 30. Bedi A., Chang X., Noonan K., Pham V., Bedi R., Fertig E. J., Considine M., Califano J. A., Borrello I., Chung C. H., Sidransky D., and Ravi R. (2012) Inhibition of TGFβ enhances the in vivo antitumor efficacy of EGF receptor-targeted therapy. Mol. Cancer. Ther. 11, 2429–2439 10.1158/1535-7163.MCT-12-0101-T [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Liu X., Hubchak S. C., Browne J. A., and Schnaper H. W. (2014) Epidermal growth factor inhibits transforming growth factor-β-induced fibrogenic differentiation marker expression through ERK activation. Cell. Signal. 26, 2276–2283 10.1016/j.cellsig.2014.05.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Atsumi S., Nagasawa A., Koyano T., Kowithayakornd T., and Umezawa K. (2003) Suppression of TGFβ signaling by conophylline via upregulation of c-Jun expression. Cell Mol. Life Sci. 60, 2516–2525 10.1007/s00018-003-3299-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kubo N., Saito R., Hamano K., Nagasawa M., Aoki F., Takei I., Umezawa K., Kuwano H., and Kojima I. (2014) Conophylline suppresses hepatic stellate cells and attenuates thioacetamide-induced liver fibrosis in rats. Liver Int. 34, 1057–1067 10.1111/liv.12328 [DOI] [PubMed] [Google Scholar]
- 34. Renzoni E. A., Abraham D. J., Howat S., Shi-Wen X., Sestini P., Bou-Gharios G., Wells A. U., Veeraraghavan S., Nicholson A. G., Denton C. P., Leask A., Pearson J. D., Black C. M., Welsh K. I., and du Bois R. M. (2004) Gene expression profiling reveals novel TGFβ targets in adult lung fibroblasts. Respiratory Res. 5, 24 10.1186/1465-9921-5-24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kolosova I., Nethery D., and Kern J. A. (2011) Role of Smad2/3 and p38 MAP kinase in TGFβ1-induced epithelial-mesenchymal transition of pulmonary epithelial cells. J. Cell Physiol. 226, 1248–1254 10.1002/jcp.22448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Moustakas A., and Heldin C. H. (2005) Non-Smad TGFβ signals. J. Cell Sci. 118, 3573–3584 10.1242/jcs.02554 [DOI] [PubMed] [Google Scholar]
- 37. Zhang Y. E. (2009) Non-Smad pathways in TGFβ signaling. Cell Res. 19, 128–139 10.1038/cr.2008.328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Seubwai W., Vaeteewoottacharn K., Kraiklang R., Umezawa K., Okada S., and Wongkham S. (2016) Inhibition of NF-κB activity enhances sensitivity to anticancer drugs in cholangiocarcinoma cells. Oncol. Res. 23, 21–28 10.3727/096504015X14424348426071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Huber W., von Heydebreck A., Sültmann H., Poustka A., and Vingron M. (2002) Variance stabilization applied to microarray data calibration and to the quantification of differential expression. Bioinformatics 18, S96–S104 10.1093/bioinformatics/18.suppl_1.S96 [DOI] [PubMed] [Google Scholar]
- 40. Knudson C. B. (1993) Hyaluronan receptor-directed assembly of chondrocyte pericellular matrix. J. Cell Biol. 120, 825–834 10.1083/jcb.120.3.825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hata R., Ninomiya Y., Nagai Y., and Tsukada Y. (1980) Biosynthesis of interstitial types of collagen by albumin-producing rat liver parenchymal cell (hepatocyte) clones in culture. Biochemistry 19, 169–176 10.1021/bi00542a026 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.