

Abstract
Inflammasomes are supramolecular signaling platforms integral to innate immune defense against invading pathogens. The NOD-like receptor (NLR) family apoptosis inhibitory protein (NAIP)·NLR family caspase-recruiting domain (CARD) domain-containing 4 (NLRC4) inflammasome recognizes intracellular bacteria and induces the polymerization of the caspase-1 protease, which in turn executes maturation of interleukin-1β (IL-1β) and pyroptosis. Several high-resolution structures of the fully assembled NAIP·NLRC4 complex are available, but these structures do not resolve the architecture of the CARD filament in atomic detail. Here, we present the cryo-EM structure of the filament assembled by the CARD of human NLRC4 (NLRC4CARD) at 3.4 Å resolution. The structure revealed that the helical architecture of the NLRC4CARD filament is essentially identical to that of the downstream filament assembled by the CARD of caspase-1 (casp1CARD), but deviates from the split washer-like assembly of the NAIP·NLRC4 oligomer. Our results suggest that architectural complementarity is a major driver for the recognition between upstream and downstream CARD assemblies in inflammasomes. Furthermore, a Monte Carlo simulation of the NLRC4CARD filament assembly rationalized why an (un)decameric NLRC4 oligomer is optimal for assembling the helical base of the NLRC4CARD filament. Together, our results explain how symmetric and asymmetric supramolecular assemblies enable high-fidelity signaling in inflammasomes.
Keywords: innate immunity, cryo-electron microscopy, nod-like receptor (NLR), inflammasome, biophysics, caspase, Monte Carlo simulation, NLRC4, pyroptosis
Introduction
Inflammasomes are supramolecular signaling platforms integral to the metazoan innate immune system (1). The outcome of inflammasome pathways is the activation of the caspase-1 protease, which executes two key innate immune processes (1). First, caspase-1 cleaves pro–IL-1β into mature IL-1β (2), and second, the protease activates pore-forming gasdermin-D, which provides the secretory channel to IL-1β and also promotes pyroptosis (3). Inflammasomes are essential for host defense against pathogen invasion (e.g. HIV, Herpesviridae, Listeria monocytogene, and Francisella tularemia) (4–8). Moreover, they are implicated in regulating various human maladies (e.g. rheumatoid arthritis and systemic lupus erythematosus (9, 10), sepsis (11), metabolic disorders (12, 13), and various types of cancer (14, 15)).
The vast majority of inflammasome receptors such as AIM2 and NLRP3 signal through ASC (Fig. 1A, left); AIM2, absent in melanoma 2; NLRP3, nucleotide-binding oligomerization domain (NOD)-like receptor (NLR) containing pyrin-domain (PYD) 3; ASC, apoptosis-associated speck-forming protein containing CARD (1). That is, incoming signals induce oligomerization of upstream receptors, resulting in filament assembly of their PYDs (16). Upstream PYD filaments then nucleate the polymerization of ASCPYD (PYD of ASC), dramatically accelerating the assembly of the downstream filament (16, 17)). The ASCPYD filament in turn promotes the filament assembly of its CARD (ASCCARD) (16, 18), which then nucleates the filament assembly of the CARD of caspase-1 (casp1CARD). This final polymerization step activates the protease via proximity-induced dimerization and auto-proteolysis (1, 16, 18). However, not all inflammasome receptors require ASC to activate caspase-1. Most notably, the NAIP-NLRC4 signaling axis represents a unique ASC-independent pathway (19) (Fig. 1, A and B, NAIP, NLRC4). For instance, NAIP directly recognizes bacterial rod proteins such as PrgJ and FliC, subsequently undergoing a conformational change from a closed inactive state to an active open state (20–24). Activated NAIP then nucleates the oligomerization of NLRC4, resulting in a split washer-like structure composed of 10 to 11 NLRC4 subunits (20–24) (Fig. 1, A and B). This oligomerization allows NLRC4CARD to cluster into a helical filament at the center, providing a scaffold for the assembly of casp1CARD filament (23, 24) without requiring ASC (19) (Fig. 1, A and B).
Figure 1.
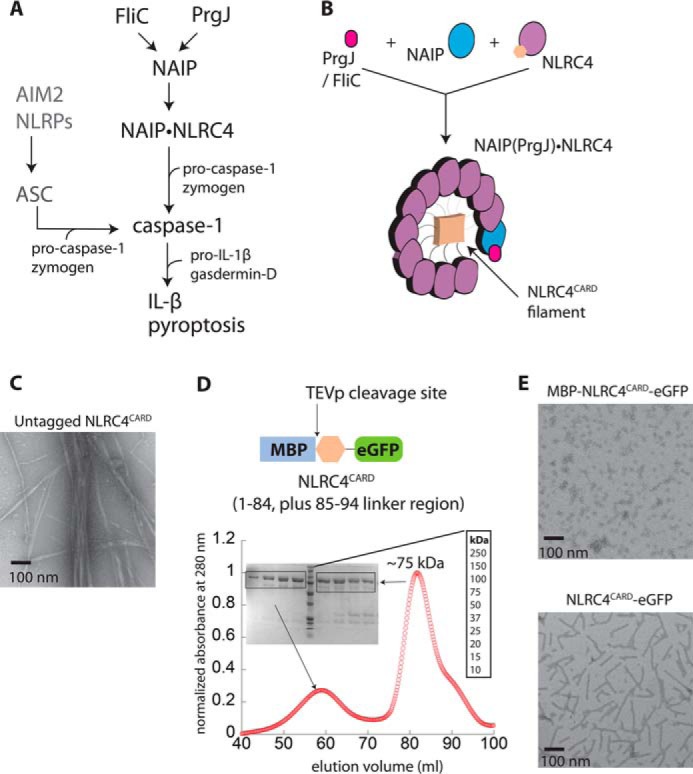
Preparation of the NLRC4CARD for cryo-EM reconstruction. A, a scheme describing inflammasome signaling pathways. B, a cartoon describing the assembly of the NAIP·NLRC4 complex. Upon binding of bacterial proteins, NAIP undergoes a conformational change and consequently nucleates the assembly of (un)decameric NLRC4 oligomer that resembles a split washer. C, nsEM micrographs showing the filament assembly by NLRC4CARD constructs. D, top, a scheme describing the NLRC4CARD construct used for cryo-EM reconstruction. Bottom, SEC (Superdex 200 16/600) and SDS-PAGE of MBP-NLRC4CARD-eGFP. E, nsEM micrographs showing the filament assembly by NLRC4CARD-eGFP.
Currently, multiple near-atomic resolution structures of the fully assembled NAIP·NLRC4 complex are available (21–24). However, these structures either omit or fail to resolve the architecture of the CARD filament in atomic detail. Such a gap in knowledge leaves two key questions unanswered in understanding the mechanisms by which the NAIP·NLRC4 inflammasome operates: What is the structural framework that allows NLRC4 to recognize caspase-1, and why is the split washer-like assembly composed of 10 to 11 NLRC4 subunits necessary to assemble the CARD filament?
Here, we present the cryo-EM structure of NLRC4CARD filament at 3.4 Å resolution, which is a 1-start, left-handed helix with 3.6 subunits per helical turn. The overall architecture of the NLRC4CARD filament is remarkably similar to that of the casp1CARD filament (18), indicating that the CARD·CARD interaction between upstream NLRC4 and downstream caspase-1 is based on congruent helical assemblies. Side-chains that mediate filament assembly are also similar between casp1CARD and NLRC4CARD, indicating that conserved interactions underpin the assembly of homologous suprastructures. A Monte Carlo simulation of the CARD filament assembly suggests that the (un)decameric assembly of the NLRC4 oligomer maximizes the probability of assembling the helical base of the NLRC4CARD filament, which provides the cognate signaling interface for caspase-1. Together, our studies provide insights into how symmetric and asymmetric supramolecular structures might maximize the signal efficiency and fidelity in inflammasome pathways.
Results
Preparation of the NLRC4CARD filament for structural studies
NAIP and NLRC4 co-assemble into a split washer-like structure in which NLRC4CARD clusters into a helical filament in the middle (21–24) (Fig. 1B). To understand the architectural organization of the NLRC4CARD filament, we generated C-terminally His6-tagged NLRC4CARD. The size-exclusion chromatography (SEC)4 profile during purification indicated that this construct behaves as multiple oligomeric species (Fig. S1A). Moreover, negative stain EM (nsEM) imaging revealed that NRLC4CARD self-assembles into filaments (Fig. 1C). Our results support the notion that monomeric NLRC4 assumes a closed conformation to suppress the self-polymerization activity of its CARD (20). Nevertheless, this construct was not conducive to structural studies, as the filaments were prone to aggregation and bundling (Fig. 1C).
While screening for a more suitable sample for structural studies, we found an NLRC4CARD construct with an N-terminal maltose-binding protein (MBP) and a C-terminal enhanced green fluorescence protein (eGFP) to be an ideal candidate for structural determination (Fig. 1D, top). For instance, the resulting protein purified mostly as a monomer with some oligomeric fraction (likely a dimer) (Fig. 1D). Under nsEM, the sample showed nonfilamentous oligomers, and cleaving MBP using the tobacco etch virus protease (TEVp) induced filament assembly (Fig. 1E). Importantly, it appeared that the presence of C-terminal eGFP greatly reduced the aggregation of NLRC4CARD filaments (Fig. 1, C versus E). These observations indicated that using N- and C-terminal protein tags helps control the oligomerization activity of NLRC4CARD, resulting in a suitable sample for cryo-EM reconstruction.
Cryo-EM reconstruction of the NLRC4CARD filament
To determine the architecture of the NLRC4CARD filament, we collected the cryo-EM images of NLRC4CARD-eGFP filaments (Fig. 2A). The average power spectrum of the 384-pixel-long overlapping filament segments showed that the NLRC4CARD filament displays a one-start helical symmetry of 100.6° rotation (3.6 subunit per helical turn) and an axial rise of 5.0 Å (Fig. 2B). The ∼3.4 Å resolution of the reconstruction allowed for an unambiguous determination of the hand of the structure. The reconstruction showed that the one-start helix must be left-handed, as this yielded clearly right-handed α-helices. The alternative reconstruction, assuming that the one-start helix is right-handed, yielded left-handed α-helices, which would be impossible for a protein. (Fig. 2D). Of note, these parameters are remarkably consistent with other CARD filaments such as casp1CARD (100.21° and 5.1 Å) (18), MAVSCARD (mitochondria antiviral signaling protein; 101.1° and 5.13Å) (25), and BCL10CARD (B-cell lymphoma 10; 100.8° and 5.1 Å) (26) filaments. Our observations not only highlight the shared assembly theme among CARD suprastructures but also indicate that our reconstruction represents a biologically relevant assembly. The resolution of the final model was 3.4 Å according to the gold standard method (Fig. 2C and Fig. S1B). The high-resolution map then allowed us to model in most bulky and aliphatic side-chains (Fig. 2D). Of note, the C-terminal eGFP was not visible in our reconstruction, indicating that the tag is flexibly attached to the filament and does not prevent polymerization (Fig. 2D). The final model showed that four NLRC4CARD protomers constitute the base of the filament (i.e. 3.6 subunits per helical turn) (Fig. 2E). The diameter of the outer rim is about 70 Å and that of the inner cavity is about 15 Å (Fig. 2E).
Figure 2.

The cryo-EM structure of NLRC4CARD. A, a cryo-electron micrograph of NLRC4CARD-eGFP filaments. B, an average power spectrum of the NLRC4CARD-eGFP filament generated from 299,537 overlapping segments. C, the FSC curve of the cryo-EM map of the NLRC4CARD filament obtained from 299,537 segments. The dotted lines indicate the 0.143 threshold for the resolution. D, the NLRC4CARD filament model docked into the EM map. The monomer with visible side-chains is also shown. E, the model of NLRC4CARD filament. In the top and bottom views, the chains (subunits) located at either pole are colored differently from the rest of the filament. The numbers indicate the four subunits that compose the horizontal plane of the filament.
As seen from other CARD filaments (18, 25, 26), our structure showed that each NLRC4CARD subunit contributes six unique surfaces to filament assembly, resulting in three distinct interfaces (Fig. 3A). Examining each interface of the filament indicated that the type 1a:1b interface is largely mediated by salt bridges and polar interactions (Fig. 3B). On the other hand, the type 2a:2b and 3a:3b interfaces are mediated by both polar and hydrophobic interactions (Fig. 3B). Aligning the NLRC4CARD filament to the casp1CARD filament revealed remarkable structural similarity between the upstream and downstream assemblies with all subunits placed in nearly identical positions along the helical axis (Fig. 3C). Such near perfect architectural complementarity supports the notion that upstream oligomers provide templates for assembling downstream oligomers in signaling supramolecular structures (25, 27, 28).
Figure 3.

Deciphering NLRC4CARD side-chains that mediate three unique filament interfaces. A, a cartoon representation of three unique filament interfaces and “type” of participating surfaces per subunit. B, side-chains that mediate each unique protein-protein interactions around each filament interface are shown as a stick configuration. C, an overlay of the NLRC4CARD and casp1CARD (PDB ID: 5FNA) filaments. The root mean square deviation of aligned chain A is 1.2 Å. The hexagon representing a NLRC4CARD subunit is shown with six unique protein-protein interaction surfaces following the convention used in Ref. 18. D, an amino acid sequence alignment of NLRC4CARD and casp1CARD. Side-chains that participate in each filament interface is indicated. Boxed side-chains indicate those we mutated, and equivalent caspase-1 residues are also boxed with the same color. Asn-34 (N) and Pro-70 (P) are unique to NLRC4 (colored in light orange).
Conserved side-chain interactions underpin the assembly of homologous filaments
Each NLRC4CARD subunit makes the three unique interactions in the filament (Fig. 3, A–C), using the same interface pairs as in the casp1CARD filament (18). Moreover, we noted that the interface residues are similar between the upstream and downstream proteins (Fig. 3D). To determine the energetic contribution of NLRC4CARD side-chains in assembly, we mutated several conserved and unconserved side-chains on each of three unique interfaces and tested whether they compromise the filament assembly of NLRC4CARD-eGFP in vitro and also in cells (Fig. 4; see also Fig. 3, C and D, for their positions). All NLRC4CARD mutants purified predominantly as monomers (Fig. 4A) and did not show any significant oligomers under nsEM (Fig. S1C). Upon cleaving MBP via TEVp, most mutants did not produce filaments (only P79R showed filaments, but much less than WT) (Fig. 4B). These observations indicated that mutations compromised the oligomerization activity of NLRC4CARD. To establish the physiological relevance of our in vitro studies, we also transfected WT and mutant NLRC4CARD-eGFP into HEK293T cells and monitored the filament assembly via confocal microscopy. WT NLRC4CARD-eGFP showed filaments in cells (Fig. 4, C and D). No filaments were observed from R52E and D25R, and significantly fewer filaments were detected from the rest of the mutants (Figs. 4, C and D). Overall, our in vitro and cell-based experiments consistently agree with our reconstruction, and also suggest that conserved side-chain interactions in CARD filaments underpin their common helical architecture.
Figure 4.

Testing the energetic contribution of surface side-chains of the NLRC4CARD filament. A, SEC profiles of NLRC4CARD-eGFP mutants (Superdex 200 16/600). The dotted arrows indicate expected elution peak volumes from WT (see also Fig. 1D). B, negative stain electron micrographs of NLRC4CARD-eGFP variants. The WT image from Fig. 1E is shown for comparison. C, confocal microscope images HEK293T cells transfected with various NLRC4CARD-eGFP variants. D, summary of confocal imaging studies (number of cells showing NLRC4CARD-eGFP filaments per eight random fields, 20× objective zoom). Error bar indicates standard deviation.
Asymmetric assembly between the NLRC4CARD filament and NAIP·NLRC4 oligomer
The congruent architecture between the NLRC4CARD filament and the casp1CARD filament is expected, as such structural complementarity along the helical axis would facilitate recognition between upstream and downstream supramolecular structures (25, 27, 28). For instance, the casp1CARD filament would assemble from either end of the NLRC4CARD filament following the same helical trajectory (3.6 subunit per helical turn, ∼4 subunits at the base of both filaments) (Fig. 2E). However, it is not clear why NAIP and NLRC4 co-assemble into an 11- to 12-membered split washer-like structure (20–24) to assemble a filament that entails a tetrameric signaling interface (Figs. 1B and 2E). Also of note, the helical symmetry of the NLRC4CARD filament (left-handed helix) is different from the rest of the assembly (mostly right-handed helix) (23, 24). On the other hand, it is noteworthy that a CARD tetramer resulting from the dimerization of retinoic acid–inducible gene-I (RIG-I) is sufficient to induce the polymerization of downstream MAVS (each RIG-I contains two CARDs in tandem, RIG-I2CARD) (25, 29). Thus, in principle, tetramerization of upstream receptor with a single CARD should be sufficient for assembling a signal-competent complex, yet NLRC4 assembles into an (un)decamer (20–24).
In other inflammasomes that signal through ASC, the recognition between upstream and downstream PYD filaments is also based on their congruent architecture (16, 28). For instance, the filaments assembled by the PYDs of AIM2 and ASC (AIM2PYD and ASCPYD) display the same helical symmetry (e.g. 6 subunits at the helical base with C3 symmetry) (16, 28). Using Monte Carlo simulation, we previously demonstrated that dsDNA length, akin to the number of bound AIM2 molecules per duplex, regulates the probability of assembling the hexameric base of the AIM2PYD filament (17). A key phenotype of this assembly mechanism is that both self-assembly and signaling activity of AIM2 nonlinearly increase with the length of dsDNA up to where it would provide the maximal probability for assembling the hexameric base (>200 bp, ∼20 AIM2 molecules per duplex) (17, 28, 30). Unlike the AIM2PYD·ASCPYD complex, the recognition between the NLRC4CARD·casp1CARD complex occurs via a tetrameric interface (Fig. 2E, top and bottom views). We thus hypothesized that a possible reason why NLRC4 assembles into an (un)decamer is to maximize the probability of assembling the tetrameric base of its CARD filament. Of note, the linker between the CARD and the rest of NLRC4 will introduce some degree of freedom even when the CARDs are placed in proximity (∼12 amino acids). Moreover, each CARD has six protein-protein interaction surfaces instead of one or two well-defined tetrameric interfaces. Thus, simple tetramerization of NLRC4 might not be sufficient to arrange the CARDs in a correct architecture.
To test our hypothesis, we conducted a Monte Carlo simulation that determines the probability of contiguously positioning four CARD protomers on a filament with a tetrameric base (Fig. 5A). First, we created a butterflied view of an empty shell that represented the NLRC4CARD filament (each side was still connected in the simulation). We then randomly placed NLRC4CARD protomers to fill the shell, with the stipulation that subsequent protomers must contact existing ones via one of the six protein-protein interaction surfaces. We then monitored the number of protomers required to contiguously fill two, three, and four lateral positions along the helical axis. Here, our simulation results suggested that there was only 5% chance for creating the intact tetrameric base with four available protomers, and the probabilities of aligning two and three protomers along the helical axis were 100 and 50%, respectively (Fig. 5B). By contrast, with 11 available protomers, the probability of filling the four lateral positions (i.e. creating a tetrameric interface) was nearly 90%. These results explain why simple tetramerization of NLRC4 might not be sufficient to assemble its CARD filament. Moreover, our simulation results provide a rationale for why an oligomer composed of 10 to 11 NLRC4 molecules can be ideal for generating a cognate signaling interface for downstream caspase-1.
Figure 5.
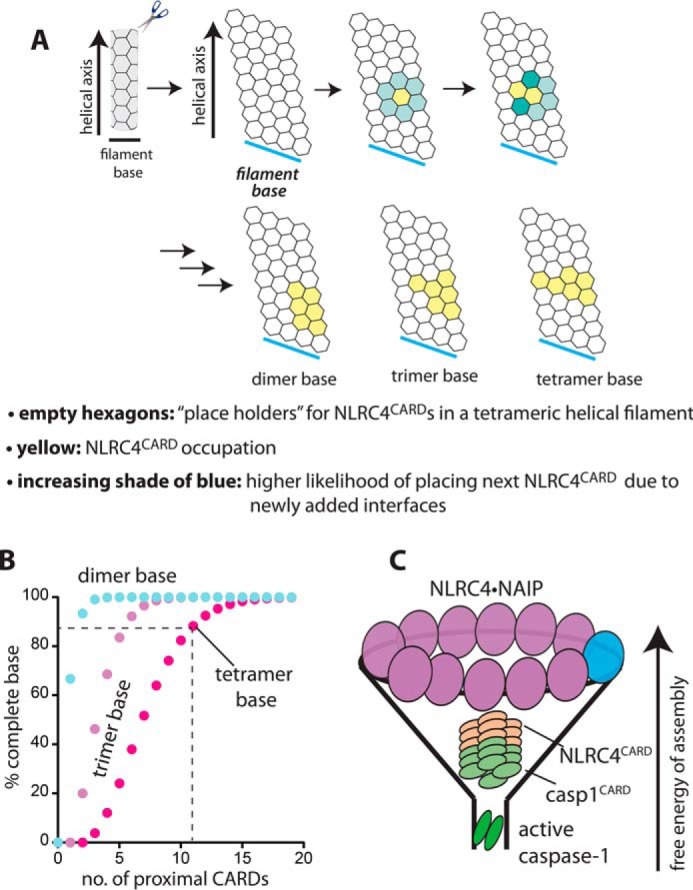
The higher-order split washer–like assembly of NLRC4 maximizes the probability of forming the tetrameric base of its CARD filament. A, a scheme describing the Monte Carlo simulation for the NLRC4CARD filament assembly. The filament shell is shown as a butterflied view. In the simulation, the horizontal sides are still connected and the length of the shell is infinite. Each arrow indicates stepwise progression of the simulation (i.e. individual CARD protomers are added sequentially). B, a plot of the probability of base assembly (2, 3, and 4 subunits per intact base) versus available proximal NLRC4CARD molecules. C, a funnel-like assembly of inflammasome signaling platforms.
Discussion
Inflammasomes are essential for host innate defense against pathogen infection, and are also implicated in cancer formation and auto-inflammatory responses (1, 10, 12). The NAIP·NLRC4 complex represents a unique axis in inflammasome pathways as it directly signals through caspase-1 without ASC. Although there are multiple high-resolution structures of the NAIP·NLRC4 complex, that of the CARD oligomer has been absent. Here, we provide the cryo-EM structure of the NLRC4CARD filament at 3.4 Å resolution, providing the last missing high-resolution piece of the holo-complex. Of note, while our study was under review, Li et al. (31) reported a cryo-EM structure of the NLRC4CARD filament at 3.6 Å resolution. Our reconstruction and that of Li et al. are essentially identical with the same helical parameters (root mean square deviation = 0.6 Å; rise, 5 Å versus 4.9 Å; rotation, 100.6° versus 100.5°), further supporting the biological relevance of the present study (31). Combined, here we address two key issues in understanding the signaling mechanisms of inflammasomes: What is the structural framework for the recognition between NLRC4 and caspase-1 and, why is the split washer-like assembly composed of 10 to 11 NLRC4 subunits necessary to assemble the CARD filament?
The structural framework for the signal transduction of the NAIP·NLRC4 inflammasome
A unique aspect of inflammasome pathways is that signal is transduced by the sequential assembly of filamentous oligomers, instead of more ubiquitously found enzyme-mediated chemical reactions (16, 27). In enzymatic cascades, a specific motif at the target/active site or a remote docking site allows the recognition between upstream and downstream signaling partners (32–34). Whether the recognition occurs via the active sites or exo-sites, local structural complementarity is critical for distinguishing cognate partners from others (i.e. lock and key) (32–34). It has become increasingly apparent that shape complementarity is also important for signal transduction by supramolecular platforms, only at a larger scale (16, 25–28, 35). For instance, we reported previously that the helical architecture of the upstream AIM2PYD filament is consistent with that of the downstream ASCPYD filament (28). Moreover, the helical architecture of the upstream RIG-I2CARD tetramer is also identical to that of the downstream MAVSCARD filament (25), and such a shape complementarity is also evident in the assembly of PIDDosome (p53-induced protein containing death domain) (35). Our cryo-EM structure of NLRC4CARD further underscores that architectural complementarity underpins the recognition between supramolecular signaling platforms.
Despite the large divergence in primary sequences (e.g. Fig. 3D), the NLRC4CARD filament shares the common helical architecture with all other known CARD filaments: ∼4 subunits per helical turn, left-handed one-start helix with ∼100 ± 0.1° twist and 5.0 ± 0.1 Å rise (18, 25, 26). Moreover, all CARDs share very high structural similarity (e.g. Fig. 3C), the same six interfaces are utilized in the same manner in the filament assembly, and each subunit does not undergo any significant conformational change upon oligomerization (18, 25, 26). Additionally, it appears that similar side-chain interactions are utilized in each interface (Fig. 3D) (18, 25, 26). Nevertheless, these filaments do not cross-interact and exclusively signal through their cognate partners (18, 25–27). Future studies will illuminate how homologous filaments recognize and distinguish their cognate and noncognate signaling partners.
(Un)decamerization of NLRC4 and signal funneling by assembly
In inflammasomes that employ the ASC hub, upstream receptors can assemble into infinitely long filaments (e.g. AIM2) (16, 28). However, the signal transmission occurs along the helical axis between upstream and downstream assemblies (i.e. perpendicular filament base to the helical axis) (16, 28). Previously, we found that dsDNA long enough to accommodate at least 20 AIM2 molecules maximizes the probability of assembling the hexameric filament base ideal for the recognition of ASC (17). Here, our structure shows that even though NLRC4 assembles into an (un)decamer, the signal transduction to caspase-1 occurs via a tetrameric interface between two CARD filaments. Of note, our Monte Carlo simulation suggests that the assembly of the tetrameric base can be maximized by 10 or more proximal protomers. Our results provide an explanation for why the (un)decamerization of NLRC4 would be ideal for signal propagation. On the other hand, the final step in all inflammasome pathways is the dimerization of caspase-1 (1, 18), again requiring a lower-order oligomeric signaling interface than the tetrameric interface between CARD filaments. Thus, it appears that the oligomeric states required to carry out each signaling step progressively decrease in inflammasome cascades. Together, considering that kinetic and thermodynamic barriers would be greater for assembling higher-order suprastructures than lower-order assemblies, it is tempting to speculate that such a strategy would allow inflammasomes to maximize their signaling fidelity and efficiency in a funnel-like manner (Fig. 5C).
Experimental procedures
Protein expression and purification
WT and mutant human NLRC4CARD constructs (amino acids 1–96) were cloned into either unmodified pET21b vector or a pET21b containing an N-terminal MBP followed by a recognition site for the TEVp and C-terminal eGFP. Resulting NLRC4CARD constructs were transformed into Escherichia coli BL21 Rosetta2DE3 cells and protein expression was induced using 200 μm isopropyl 1-thio-β-d-galactopyranoside at 18 °C for 16–18 h. The cells were lysed by sonication in 25 mm NaPO4, 400 mm NaCl, 40 mm imidazole, and 8 mm β-mercaptoethanol at pH 8. NLRC4CARD constructs were then purified by nickel-nitrilotriacetic acid chromatography via a C-terminal His6-tag followed by SEC. Proteins were stored in 20 mm HEPES at pH 7.4, 400 mm NaCl, 10% glycerol, 1 mm EDTA, and 1 mm DTT and frozen at −80 °C.
Negative stain EM
Samples were diluted to 750 nm into a buffer containing 40 mm HEPES, 160 mm NaCl, 1 mm EDTA, 5 mm DTT, at pH 7. To induce polymerization, 3 μm TEVp was added to each sample and incubated for 1.5 h at room temperature (24 ± 2 °C). nsEM imaging experiments were performed using glow-discharged carbon-coated copper grids and 0.75% uranyl formate staining solution as described previously (17, 28).
Cryo-EM sample preparation
5 μl sample was applied to Lacey grids, followed by automatically blotting for 1.5 s and plunge freezing using the FEI Vitrobot Mark IV operated at 100% humidity and room temperature (Johns Hopkins University). Cryo-EM data were collected at the National Cancer Institute National cryo-EM facility (NCI NCEF, Frederick, MD) using the FEI Titan equipped with the Gatan K2 direct electron detector operating at 300 keV using the super-resolution mode (0.66 Å/pixel). A total of 1,690 micrographs were collected from one grid at a defocus range from −1.0 to −2.5 μm. Each micrograph was equally fractioned into 40 frames with a total exposure time of 15 s and a total dose of ∼40 electrons/Å2.
Helical reconstruction and model building
The 1,690 micrographs were binned by two sections (to 1.32 Å/pixel) and frames were aligned using MotionCor2 (36). The defocus values and astigmatism of the micrographs were determined by CTFFIND3 for the aligned full-dose micrographs (37). A total of 1,486 micrographs were selected (images with good contrast transfer function (CTF) determination and defocus <3 μm) for subsequent image processing. CTF was corrected by multiplying the micrographs (only first 20 frames were aligned with a total dose of ∼20 electrons/Å2) with the theoretical CTF, which both corrects the phases and improves the signal-to-noise ratio. The e2helixboxer program in EMAN2 software package (38) was used for boxing long filaments from the full-dose images. The CTF-corrected micrographs were used for the segment extraction, with a total of 402,078 384-pixel-long overlapping segments (with a shift of 1.5 times of axial rise) generated. The SPIDER software package (39) was used for subsequent processing and reconstruction. Using a featureless cylinder as an initial reference, 299,537 segments were used in IHRSR program for the final reconstruction after the helical parameters (a rotation of 100.6° and an axial rise of 5.0 Å per subunit) converged. The resolution of the final reconstruction was estimated by the Fourier Shell Correlation (FSC) between two independent half maps, which shows 3.4 Å at FSC = 0.143.
We used the crystal structure of a homologous protein CARD8 (PDB ID: 4IKM) (40) as an initial template to dock into the NLRC4 cryo-EM map by rigid body fitting, and then manually edited the model in Chimera (41) and Coot (42). We then used the modified model as the starting template to further refine against the segmented cryo-EM map using RosettaCM using default values (43). The refined monomeric model of NLRC4 was then rebuilt by RosettaCM with helical symmetry and real-space refined using Phenix (44) to improve the stereochemistry as well as the model-map correlation coefficient. The NLRC4 model was validated with MolProbity (45) and the coordinates deposited to the Protein Data Bank with the accession code 6MKS. The corresponding cryo-EM map was deposited in the EMDB with accession code EMD-9137.
Confocal microscopy
NLRC4CARD-eGFP variants were cloned into pCMV6 vector. Plasmids were then transiently transfected into HEK293T cells using Lipofectamine (Invitrogen). Cells were fixed with 4% paraformaldehyde, permeabilized in acetone, and mounted on glass slides using ProLong Gold Antifade Mountant with DAPI (Thermo Fisher). Cells were imaged using a Zeiss Axioskop 50 with a Zeiss Axiocam HRC camera and an Axio Observer inverted microscope with LSM700 confocal module.
Monte Carlo simulation
The overall strategy is based on our previously published protocol (17), with specific modifications for NLRC4CARD architecture. The code has been rewritten in Rust and is available at https://gitlab.com/matyszm/pydnode-nlrc4 (commit 7f17c4db was used to generate the values present in the paper)5. Each point is an average of 1,000,000 runs.
Author contributions
M. M. and W. Z. software; M. M., E. H. E., and J. S. validation; M. M., W. Z., J. L., B. A., and J. S. investigation; M. M., W. Z., and J. S. writing-original draft; M. M., W. Z., J. L., and E. H. E. writing-review and editing; B. A. data curation; B. A. methodology; E. H. E. and J. S. supervision; E. H. E. and J. S. project administration; J. S. conceptualization; J. S. formal analysis; J. S. funding acquisition.
Supplementary Material
Acknowledgments
We thank the Sohn lab members for discussion and the staff at NCI NCEF for cryo-EM data collection. The Johns Hopkins Microscope Facility was supported by National Institutes of Health Grant S10 OD016734.
This work was supported by Research Scholars Grant RG-15–224 from American Cancer Society (to J. S.), National Institutes of Health Grant R35GM0352569 (to E. H. E.), and Rheumatology Research Foundation and Cupid Foundation (to B.A.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Fig. S1.
The atomic coordinates and structure factors (code 6MKS) have been deposited in the Protein Data Bank (http://wwpdb.org/).
The corresponding cryo-EM map was deposited in the Electron Microscopy Data Bank with accession code EMD-9137.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
- SEC
- size-exclusion chromatography
- nsEM
- negative stain EM
- eGFP
- enhanced GFP
- TEVp
- tobacco etch virus protease
- CTF
- contrast transfer function
- FSC
- Fourier Shell Correlation.
References
- 1. Guo H., Callaway J. B., and Ting J. P. (2015) Inflammasomes: Mechanism of action, role in disease, and therapeutics. Nat. Med. 21, 677–687 10.1038/nm.3893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Thornberry N. A., Bull H. G., Calaycay J. R., Chapman K. T., Howard A. D., Kostura M. J., Miller D. K., Molineaux S. M., Weidner J. R., and Aunins J. (1992) A novel heterodimeric cysteine protease is required for interleukin-1β processing in monocytes. Nature 356, 768–774 10.1038/356768a0 [DOI] [PubMed] [Google Scholar]
- 3. Liu X., Zhang Z., Ruan J., Pan Y., Magupalli V. G., Wu H., and Lieberman J. (2016) Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 535, 153–158 10.1038/nature18629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bandera A., Masetti M., Fabbiani M., Biasin M., Muscatello A., Squillace N., Clerici M., Gori A., and Trabattoni D. (2018) The NLRP3 inflammasome is upregulated in HIV-infected antiretroviral therapy-treated individuals with defective immune recovery. Front. Immunol. 9, 214 10.3389/fimmu.2018.00214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Maruzuru Y., Ichinohe T., Sato R., Miyake K., Okano T., Suzuki T., Koshiba T., Koyanagi N., Tsuda S., Watanabe M., Arii J., Kato A., and Kawaguchi Y. (2018) Herpes simplex virus 1 VP22 inhibits AIM2-dependent inflammasome activation to enable efficient viral replication. Cell Host Microbe 23, 254–265.e257 10.1016/j.chom.2017.12.014 [DOI] [PubMed] [Google Scholar]
- 6. Kim S., Bauernfeind F., Ablasser A., Hartmann G., Fitzgerald K. A., Latz E., and Hornung V. (2010) Listeria monocytogenes is sensed by the NLRP3 and AIM2 inflammasome. Eur. J. Immunol. 40, 1545–1551 10.1002/eji.201040425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Reyes Ruiz V. M., Ramirez J., Naseer N., Palacio N. M., Siddarthan I. J., Yan B. M., Boyer M. A., Pensinger D. A., Sauer J. D., and Shin S. (2017) Broad detection of bacterial type III secretion system and flagellin proteins by the human NAIP/NLRC4 inflammasome. Proc. Natl. Acad. Sci. U.S.A. 114, 13242–13247 10.1073/pnas.1710433114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Fernandes-Alnemri T., Yu J. W., Juliana C., Solorzano L., Kang S., Wu J., Datta P., McCormick M., Huang L., McDermott E., Eisenlohr L., Landel C. P., and Alnemri E. S. (2010) The AIM2 inflammasome is critical for innate immunity to Francisella tularensis. Nat. Immunol. 11, 385–393 10.1038/ni.1859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yang C. A., and Chiang B. L. (2015) Inflammasomes and human autoimmunity: A comprehensive review. J. Autoimmun. 61, 1–8 10.1016/j.jaut.2015.05.001 [DOI] [PubMed] [Google Scholar]
- 10. Yi Y. S. (2018) Role of inflammasomes in inflammatory autoimmune rheumatic diseases. Korean J Physiol. Pharmacol. 22, 1–15 10.4196/kjpp.2018.22.1.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lee S., Nakahira K., Dalli J., Siempos I. I., Norris P. C., Colas R. A., Moon J. S., Shinohara M., Hisata S., Howrylak J. A., Suh G. Y., Ryter S. W., Serhan C. N., and Choi A. M. K. (2017) NLRP3 inflammasome deficiency protects against microbial sepsis via increased lipoxin B4 synthesis. Am. J. Respir. Crit. Care Med. 196, 713–726 10.1164/rccm.201604-0892OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Henao-Mejia J., Elinav E., Thaiss C. A., and Flavell R. A. (2014) Inflammasomes and metabolic disease. Annu. Rev. Physiol. 76, 57–78 10.1146/annurev-physiol-021113-170324 [DOI] [PubMed] [Google Scholar]
- 13. De Nardo D., and Latz E. (2011) NLRP3 inflammasomes link inflammation and metabolic disease. Trends Immunol. 32, 373–379 10.1016/j.it.2011.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Man S. M., Karki R., and Kanneganti T. D. (2016) AIM2 inflammasome in infection, cancer, and autoimmunity: Role in DNA sensing, inflammation, and innate immunity. Eur. J. Immunol. 46, 269–280 10.1002/eji.201545839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lin C., and Zhang J. (2017) Inflammasomes in inflammation-induced cancer. Front Immunol. 8, 271 10.3389/fimmu.2017.00271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lu A., Magupalli V. G., Ruan J., Yin Q., Atianand M. K., Vos M. R., Schröder G. F., Fitzgerald K. A., Wu H., and Egelman E. H. (2014) Unified polymerization mechanism for the assembly of ASC-dependent inflammasomes. Cell 156, 1193–1206 10.1016/j.cell.2014.02.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Matyszewski M., Morrone S. R., and Sohn J. (2018) Digital signaling network drives the assembly of the AIM2-ASC inflammasome. Proc. Natl. Acad. Sci. U.S.A. 115, E1963–E1972 10.1073/pnas.1712860115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lu A., Li Y., Schmidt F. I., Yin Q., Chen S., Fu T. M., Tong A. B., Ploegh H. L., Mao Y., and Wu H. (2016) Molecular basis of caspase-1 polymerization and its inhibition by a new capping mechanism. Nat. Struct. Mol. Biol. 23, 416–425 10.1038/nsmb.3199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Vance R. E. (2015) The NAIP/NLRC4 inflammasomes. Curr. Opin. Immunol. 32, 84–89 10.1016/j.coi.2015.01.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hu Z., Yan C., Liu P., Huang Z., Ma R., Zhang C., Wang R., Zhang Y., Martinon F., Miao D., Deng H., Wang J., Chang J., and Chai J. (2013) Crystal structure of NLRC4 reveals its autoinhibition mechanism. Science 341, 172–175 10.1126/science.1236381 [DOI] [PubMed] [Google Scholar]
- 21. Hu Z., Zhou Q., Zhang C., Fan S., Cheng W., Zhao Y., Shao F., Wang H. W., Sui S. F., and Chai J. (2015) Structural and biochemical basis for induced self-propagation of NLRC4. Science 350, 399–404 10.1126/science.aac5489 [DOI] [PubMed] [Google Scholar]
- 22. Zhang L., Chen S., Ruan J., Wu J., Tong A. B., Yin Q., Li Y., David L., Lu A., Wang W. L., Marks C., Ouyang Q., Zhang X., Mao Y., and Wu H. (2015) Cryo-EM structure of the activated NAIP2-NLRC4 inflammasome reveals nucleated polymerization. Science 350, 404–409 10.1126/science.aac5789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tenthorey J. L., Haloupek N., López-Blanco J. R., Grob P., Adamson E., Hartenian E., Lind N. A., Bourgeois N. M., Chacón P., Nogales E., and Vance R. E. (2017) The structural basis of flagellin detection by NAIP5: A strategy to limit pathogen immune evasion. Science 358, 888–893 10.1126/science.aao1140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Diebolder C. A., Halff E. F., Koster A. J., Huizinga E. G., and Koning R. I. (2015) Cryoelectron tomography of the NAIP5/NLRC4 inflammasome: Implications for NLR activation. Structure 23, 2349–2357 10.1016/j.str.2015.10.001 [DOI] [PubMed] [Google Scholar]
- 25. Wu B., Peisley A., Tetrault D., Li Z., Egelman E. H., Magor K. E., Walz T., Penczek P. A., and Hur S. (2014) Molecular imprinting as a signal-activation mechanism of the viral RNA sensor RIG-I. Mol. Cell 55, 511–523 10.1016/j.molcel.2014.06.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. David L., Li Y., Ma J., Garner E., Zhang X., and Wu H. (2018) Assembly mechanism of the CARMA1-BCL10-MALT1-TRAF6 signalosome. Proc. Natl. Acad. Sci. U.S.A. 115, 1499–1504 10.1073/pnas.1721967115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kagan J. C., Magupalli V. G., and Wu H. (2014) SMOCs: Supramolecular organizing centres that control innate immunity. Nat. Rev. Immunol. 14, 821–826 10.1038/nri3757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Morrone S. R., Matyszewski M., Yu X., Delannoy M., Egelman E. H., and Sohn J. (2015) Assembly driven activation of the AIM2 foreign-dsDNA sensor provides a polymerization template for downstream ASC. Nat. Commun. 6, 7827 10.1038/ncomms8827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Peisley A., Wu B., Xu H., Chen Z. J., and Hur S. (2014) Structural basis for ubiquitin-mediated antiviral signal activation by RIG-I. Nature 509, 110–114 10.1038/nature13140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Roberts T. L., Idris A., Dunn J. A., Kelly G. M., Burnton C. M., Hodgson S., Hardy L. L., Garceau V., Sweet M. J., Ross I. L., Hume D. A., and Stacey K. J. (2009) HIN-200 proteins regulate caspase activation in response to foreign cytoplasmic DNA. Science 323, 1057–1060 10.1126/science.1169841 [DOI] [PubMed] [Google Scholar]
- 31. Li Y., Fu T. M., Lu A., Witt K., Ruan J., Shen C., and Wu H. (2018) Cryo-EM structures of ASC and NLRC4 CARD filaments reveal a unified mechanism of nucleation and activation of caspase-1. Proc. Natl. Acad. Sci. U.S.A. 115, 10845–10852 10.1073/pnas.1810524115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sohn J., Kristjánsdóttir K., Safi A., Parker B., Kiburz B., and Rudolph J. (2004) Remote hot spots mediate protein substrate recognition for the Cdc25 phosphatase. Proc. Natl. Acad. Sci. U.S.A. 101, 16437–16441 10.1073/pnas.0407663101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Schulman B. A., Lindstrom D. L., and Harlow E. (1998) Substrate recruitment to cyclin-dependent kinase 2 by a multipurpose docking site on cyclin A. Proc. Natl. Acad. Sci. U.S.A. 95, 10453–10458 10.1073/pnas.95.18.10453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Li J., and Yuan J. (2008) Caspases in apoptosis and beyond. Oncogene 27, 6194–6206 10.1038/onc.2008.297 [DOI] [PubMed] [Google Scholar]
- 35. Park H. H., Logette E., Raunser S., Cuenin S., Walz T., Tschopp J., and Wu H. (2007) Death domain assembly mechanism revealed by crystal structure of the oligomeric PIDDosome core complex. Cell 128, 533–546 10.1016/j.cell.2007.01.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zheng S. Q., Palovcak E., Armache J. P., Verba K. A., Cheng Y., and Agard D. A. (2017) MotionCor2: Anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332 10.1038/nmeth.4193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mindell J. A., and Grigorieff N. (2003) Accurate determination of local defocus and specimen tilt in electron microscopy. J. Struct. Biol. 142, 334–347 10.1016/S1047-8477(03)00069-8 [DOI] [PubMed] [Google Scholar]
- 38. Tang G., Peng L., Baldwin P. R., Mann D. S., Jiang W., Rees I., and Ludtke S. J. (2007) EMAN2: An extensible image processing suite for electron microscopy. J. Struct. Biol. 157, 38–46 10.1016/j.jsb.2006.05.009 [DOI] [PubMed] [Google Scholar]
- 39. Frank J., Radermacher M., Penczek P., Zhu J., Li Y., Ladjadj M., and Leith A. (1996) SPIDER and WEB: Processing and visualization of images in 3D electron microscopy and related fields. J. Struct. Biol. 116, 190–199 10.1006/jsbi.1996.0030 [DOI] [PubMed] [Google Scholar]
- 40. Jin T., Huang M., Smith P., Jiang J., and Xiao T. S. (2013) The structure of the CARD8 caspase-recruitment domain suggests its association with the FIIND domain and procaspases through adjacent surfaces. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 69, 482–487 10.1107/S1744309113010075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., and Ferrin T. E. (2004) UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 10.1002/jcc.20084 [DOI] [PubMed] [Google Scholar]
- 42. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 10.1107/S0907444910007493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wang R. Y., Song Y., Barad B. A., Cheng Y., Fraser J. S., and DiMaio F. (2016) Automated structure refinement of macromolecular assemblies from cryo-EM maps using Rosetta. Elife 5, e17219 10.7554/eLife.17219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., and Zwart P. H. (2010) PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 10.1107/S0907444909052925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Chen V. B., Arendall W. B. 3rd, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., and Richardson D. C. (2010) MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 10.1107/S0907444909042073 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.