

Abstract
The transcriptional coactivator WW domain–binding protein 2 (WBP2) is an emerging oncogene and serves as a node between the signaling protein Wnt and other signaling molecules and pathways, including epidermal growth factor receptor, estrogen receptor/progesterone receptor, and the Hippo pathway. The upstream regulation of WBP2 is well-studied, but its downstream activity remains unclear. Here, we elucidated WBP2's role in triple-negative breast cancer (TNBC), in which Wnt signaling is predominantly activated. Using RNAi coupled with RNA-Seq and MS analyses to identify Wnt/WBP2- and WBP2-dependent targets in MDA-MB-231 TNBC cells, we found that WBP2 is required for the expression of a core set of genes in Wnt signaling. These included AXIN2, which was essential for Wnt/WBP2-mediated breast cancer growth and migration. WBP2 also regulated a much larger set of genes and proteins independently of Wnt, revealing that WBP2 primes cells to Wnt activity by up-regulating G protein pathway suppressor 1 (GPS1) and TRAF2- and NCK-interacting kinase (TNIK). GPS1 activated the c-Jun N-terminal kinase (JNK)/Jun pathway, resulting in a positive feedback loop with TNIK that mediated Wnt-induced AXIN2 expression. WBP2 promoted TNBC growth by integrating JNK with Wnt signaling, and its expression profoundly influenced the sensitivity of TNBC to JNK/TNIK inhibitors. In conclusion, WBP2 links JNK to Wnt signaling in TNBC. GPS1 and TNIK are constituents of a WBP2-initiated cascade that primes responses to Wnt ligands and are also important for TNBC biology. We propose that WBP2 is a potential drug target for JNK/TNIK-based precision medicine for managing TNBC.
Keywords: transcription coactivator, oncogene, axin, Wnt signaling, cell signaling, breast cancer, precision medicine, therapy, TNIK, WW domain–binding protein 2
Introduction
Breast cancer is the most common cancer in women and a leading cause of cancer mortality worldwide taking half a million lives annually (1). Despite the advancements in targeted therapies, many challenges remain (2). There is no specific treatment guideline for patients with triple-negative breast cancer (TNBC).5 Patients with TNBC are treated with standard chemotherapy that results in poor response and high relapse rate. A better understanding of the molecular etiology of this disease could improve management of TNBC.
The canonical Wnt signaling pathway plays an important role in embryonic development and tissue homeostasis (3). Aberrant Wnt signaling has been implicated in the etiology of multiple human malignancies (4). Transcription coactivator β-catenin is a key effector in this pathway (5). In the absence of Wnt, β-catenin is phosphorylated by GSK3 and targeted for β-TrCP-dependent ubiquitination and destruction in the cytoplasm. Upon Wnt stimulation, the GSK3 destruction complex is inhibited, leading to β-catenin stabilization and translocation into nucleus, where it potentiates transcription of target genes via TCF/LEF transcription factors (6, 7). Breast cancer subtypes exhibit heterogeneity in Wnt signaling. For example, high levels of nuclear β-catenin and Wnt target genes were strongly associated with TNBC, making the Wnt pathway a rational therapeutic target for this disease (8, 9).
New insights on the Wnt-signaling pathway are emerging. Genes associated with the Hippo pathway have been increasingly reported to regulate Wnt signaling. For example, activation of the Hippo/serine/threonine kinase pathway results in phosphorylation of TAZ at Ser-89 and YAP at Ser-127 by large tumor suppressor kinase, leading to the binding of TAZ/YAP to 14-3-3 proteins and sequestration of TAZ/YAP/β-catenin in the cytosol thereby preventing Wnt activation (10, 11). YAP and TAZ has also been shown to be stabilized by Wnt signaling and mediate gene transcription (12, 13). More recently, YAP and TAZ are found to be essential for the formation of the β-catenin destruction complex in the absence of Wnt ligand (14). WW domain–binding protein 2 (WBP2) transcription co-activator, which binds to YAP and TAZ, was recently implicated in the Hippo and Wnt signaling pathways.
WBP2 was initially cloned as a cognate ligand of the WW domain of YAP (15). It has been reported to bind to PAX8 transcription factor with unknown function (16). WBP2 was progressively revealed to be a transcription coactivator in estrogen receptor/progesterone receptor hormonal signaling (17–19), a novel tyrosine kinase substrate in epidermal growth factor receptor signaling (18, 20) and an oncogenic coactivator of Wnt signaling in breast cancer cells (18, 21). The ability of Yorkie (Drosophila ortholog of YAP) and TAZ (YAP paralog) to drive tissue growth and tumorigenesis (22, 23) was dependent on WBP2. Clinically, WBP2 is up-regulated in breast cancer compared with normal tissues. Elevated WBP2 expression is significantly associated with poor prognosis, overall, and disease-free survival (21). The expression of the WBP2 oncoprotein is reversibly controlled by tumor suppressors. WBP2 is degraded by itchy E3 ubiquitin protein ligase (ITCH E3 ligase) to prevent aberrant growth but is protected from ITCH and activated by Wnt oncogenic signaling to drive TCF/β-catenin–mediated transcription to promote breast cancer (21). Recent studies identified WBP2 as a key cofactor of YAP driving the clonal expansion of normal and neoplastic human epidermal stem cells via TEA domain transcription factor (TEAD) transcription factors (24), in modulating G1/S cell cycle transition in estrogen receptor+ breast cancer cells via a micro RNA-based mechanism (25) and crucial for normal glutamatergic synapses in the cochlea and hearing (26).
Although current evidences portray WBP2 to have pleotropic roles, knowledge on the mode of action of WBP2 remains confined to a limited set of genes and pathways. To better understand the molecular effects of WBP2, RNA-Seq and MS were performed to elucidate the Wnt/WBP2- and WBP2-dependent targets in MDA-MB-231 TNBC cells. Besides confirming the role of WBP2 as a mediator of Wnt signaling regulating known and novel gene targets including AXIN2, a new function for WBP2 as a primer of cellular response to Wnt ligand was discovered and validated through the elucidation of a signaling axis involving GPS1, JNK/Jun, and TNIK.
Results
WBP2 is frequently amplified in breast cancer
WBP2 protein was demonstrated to be overexpressed in breast cancer in a recent study of >400 clinical samples (21). To gain further insights into the deregulation of WBP2 in breast cancer, we analyzed the gene copy number alterations (CNA) and mRNA expression of the WBP2 gene in multiple large-scale breast cancer datasets such as TCGA and METABRIC. The results indicated that WBP2 is frequently amplified (4.1–25%) or gained (0–31.7%) in breast cancer patients, whereas deletion was barely present (Fig. 1A, Table 1). The TCGA dataset also reveals a positive association between WBP2 gene copy number and mRNA level (Fig. 1, B and C). On the other hand, the “genotype to outcome” Kaplan-Meier analysis was used to study the correlation of the WBP2 transcriptomic signature on patients' survival. This approach was applied because WBP2 has been known to act as a transcriptional coactivator and as such a network of genes could be affected upon its gain/amplification. Considering that the effect of WBP2 gain/amplification could be eventually leveraged by a set of genes, this genotype to outcome survival analysis is believed to be superior to the use of WBP2 expression alone. The data showed that the transcriptomic signature (defined either by the up or down-regulated genes) derived from patients with WBP2 amplification correlates with worse survival (Fig. 1D, i and ii) (27). These results indicate that WBP2 is a prognostic factor and its elevated levels in breast cancer may be caused by genomic amplification/gain in addition to the previously described epigenetic mechanism involving protein turnover (21).
Figure 1.

A, analysis of WBP2 amplification (multiplication of intra-chromosomal region of 0.5 to 10 Mb), gain (increase in larger chromosomal region or intact chromosome) and deletion in 6 studies of breast cancer. About 20–40% of breast cancer patients harbor WBP2 amplification/gain. B, a heat map showing the correlation of WBP2 mRNA expression and copy number alteration. y axis refers to individual clinical samples in the TCGA breast cancer database, whereas the x axis indicates the intensity of WBP2 gene expression (left panel) or copy number levels (right panel). C, dot plot of WBP2 mRNA expression in individual clinical breast cancer samples in the TCGA database categorized according to copy number alterations. D, Kaplan-Meier plot of breast cancer patient survival (n = 273) according to transcriptomic fingerprint of WBP2 amplification: (i) up- and (ii) down-regulated.
Table 1.
WBP2 CNAs in multiple breast cancer datasets
Exact numbers of total sample, WBP2 amplification, gain and deletion were shown (79–84).
WBP2 regulates a core set of WNT3A-induced genes in triple-negative MDA-MB-231 breast cancer cells
WBP2 plays a role in Wnt signaling by promoting TCF/β-catenin-mediated transcription (21). However, it is not clear to what extent the Wnt pathway is regulated by WBP2. To address this question, RNA-Seq was carried out on MDA-MB-231 TNBC cells because Wnt/β-catenin signaling is highly relevant to TNBC and MDA-MB-231 is one of the most responsive cell lines to WNT3A ligand (Fig. S1). As shown in Fig. 2A, MDA-MB-231 cells were infected with lentivirus expressing three shRNAs: scrambled (scr) shRNA, WBP2 shRNA1 targeting the 3′-UTR region, and WBP2 shRNA2 targeting the coding region, respectively. The cells were further treated with control serum-free medium (O) or serum-free medium containing 200 ng/ml of recombinant WNT3A for 12 h. Silencing of WBP2 and activation of the Wnt/β-catenin signaling were effectively achieved (Fig. 2B). Total RNA was extracted from the cells and used for RNA-Seq analysis. In total, 19,301 genes were mapped across the 18 replicates. About 16,000 genes were detected with an expression value. Comparison of the gene expression profiles between the O and WNT3A-treated samples within the control group produced a complete list of Wnt target genes in MDA-MB-231 cells. Next, lists of Wnt target genes independent on WBP2 were obtained by identifying those genes that displayed expression changes following WNT3A treatment despite WBP2 knockdown. The role of WBP2 in the WNT3A-induced transcriptional program was defined by subtracting the WNT3A-responsive, WBP2-independent targets from the complete list.
Figure 2.

A, schematic design of the RNA-Seq analysis, showing the strategy used for exploring the role of WBP2 in the WNT3A-induced transcriptional program. B, sample QC before RNA-Seq analysis. WBP2 was depleted by lentivirus-expressed shRNAs. MDA-MB-231 cells were treated with 200 ng/ml of rWNT3A for 12 h. C, heat map of the expression pattern of the 28 Wnt/WBP2 target genes. Color intensity refers to the fold-change in mRNA level between WNT3A treatment versus control. Each column represents the mean value generated from biological triplicates. D, percentages of the Wnt/WBP2 target genes under different fold change cut-offs.
The following criteria were used to identify the Wnt-induced differentially expressed genes: expression should be induced by at least 2-fold upon WNT3A stimulation; p value has to be less than 0.001, and false discovery rate (FDR) less than 0.01. Under this set of criteria, 34 genes were identified as Wnt/β-catenin target genes. The relatively small number of Wnt target genes is not surprising as various gene expression studies of the Wnt pathway in different cell lines identified target genes that range from 4 to about 200 in number (Table S1). It appears that Wnt/β-catenin activates distinct target genes in different cell types.
To determine which of the 34 Wnt/β-catenin target genes were dependent on WBP2, we defined a criterion that the fold-induction by WNT3A stimulation must be reduced by at least 30% upon WBP2 depletion. A fold-change cutoff from 1.3 to 2 has been widely adopted in transcriptomic studies (28–33). We found that 28 of the 34 (82%) Wnt target genes were significantly affected by WBP2 depletion (Fig. 2C), including the well-known AXIN2, BMP4, LGR5, and LEF1 (Student's t test, p < 0.05). Among the novel Wnt targets identified, some are highly related to cancer progression. For example, ADAMTS14 plays an important role in extracellular matrix assembly and degradation; SOX4 has been suggested as an oncogene in several cancers by previous studies; and HAS2, which regulated cell adhesion, migration, and proliferation, has been shown to be overexpressed in breast cancer (21, 34–37). When the fold-change criterion of Wnt target genes was set to 1.5- and 1.3-fold, 86 and 155 genes were identified as Wnt target genes, respectively. However, the numbers of WBP2-dependent Wnt target genes only increased to 41 for both cases (Fig. 2D). This suggests a specific role of WBP2 in regulating a core set of genes within the Wnt/β-catenin transcriptional program.
AXIN2 is a novel Wnt-induced WBP2 target gene essential for TNBC cancer biology
To shortlist candidates in the Wnt/WBP2 signaling axis that are critical to breast cancer for validation, we selected from the 28 Wnt/WBP2-dependent genes identified from RNA-seq study based on: (i) reported involvement in cancer to focus on oncogenes and (ii) whose transcripts displayed the highest fold-changes to identify the top ranking candidates. Eight candidates AXIN2, LGR5, HAS2, ADAMTS14, SPRY1, DCLK1, JDP2, and SLC1A3 satisfied these criteria. The effects of WBP2 depletion on the expression of these genes in MDA-MB-231 cells were examined via RT-qPCR. Although all 8 genes were validated to be induced by Wnt and dependent on WBP2, AXIN2, LGR5, and HAS2 were the most highly activated by WNT3A (>5-fold) and substantially inhibited upon WBP2 depletion by at least 60% (Fig. 3A).
Figure 3.

A, RT-qPCR validation of the Wnt/WBP2 target genes in MDA-MB-231 cells. B, RT-qPCR showing the expression of AXIN2 regulated by Wnt/WBP2 in MDA-MB-468 (i) and BT549 (ii). WBP2 manipulation was examined by Western blotting. C, transwell cell migration (i and ii) and clonogenic assay (iii and iv) in MDA-MB-231 cells were infected by lentiviruses expressing WBP2 shRNAs and AXIN2 plasmids as indicated and stimulated with 200 ng/ml of rWNT3A. Representative images (i and iii) and corresponding quantitative analysis (ii and iv) were shown. D, (i) RT-qPCR showed the effect of WBP2 depletion on AXIN2 expression and AXIN2 rescue by WBP2 expression in MDA-MB-231 cells. WBP2 knockdown-mediated down-regulation of active β-catenin (ii) and TCF reporter activity (iii) was rescued by AXIN2 overexpression in MDA-MB-231 cells.
Next, to study the pervasiveness of the Wnt/WBP2 regulation, we asked whether these genes could be regulated by Wnt/WBP2 in other WNT3A-responsive TNBC cell lines. We thus examined the expression of these genes under WNT3A stimulation and WBP2 manipulation in MDA-MB-468 and BT549 cells by RT-qPCR. Unexpectedly, the results indicated that AXIN2 is the only gene that could be consistently regulated by Wnt/WBP2 signaling in MDA-MB-468 (Fig. 3B, i) and BT549 cells (Fig. 3B, ii), suggesting heterogeneity of Wnt signaling even within the same subtype of breast cancer cells (Table S2). As shown in Fig. 3B, WBP2 depletion inhibited WNT3A-induced AXIN2 transcription and the inhibition could be rescued by WBP2 re-expression in both cell lines. Collectively, these findings indicated that AXIN2 is a novel downstream target gene of Wnt/WBP2 signaling in TNBC.
Identification of AXIN2 as Wnt/WBP2 target gene suggests that AXIN2 may be required for Wnt/WBP2-mediated cancer phenotypes. To test this hypothesis, cell migration and colony-formation assays were performed. MDA-MB-231 cells were transfected with WBP2 shRNAs and AXIN2 plasmid to investigate whether AXIN2 expression could rescue the inhibition effect caused by WBP2 depletion. As shown in Fig. 3C, i–iv, WNT3A-induced cell migration and colony growth were drastically inhibited by WBP2 depletion, and the phenotype can be significantly rescued by AXIN2 expression. Fig. 3D, i and ii, show that the AXIN2 mRNA, AXIN2 protein, and WBP2 protein levels were accordingly effected by the indicated exogenous expression and silencing.
Interestingly, expression of AXIN2 restored the level of active β-catenin (Fig. 3D, ii) and Wnt reporter activity (Fig. 3D, iii) in the WBP2 knockdown cells. This might account for rescue of the WBP2 knockdown phenotype by AXIN2. The antibody used detects the stabilized species of endogenous β-catenin that are not phosphorylated by GSK-3 at residues Ser-33, Ser-37, and Thr-41 and therefore represents the functionally active in the canonical Wnt signaling pathway (38–41). As the effect of WBP2 knockdown on the active β-catenin level was more prominent than on the total β-catenin level, active β-catenin will be used as the surrogate indicator for Wnt pathway activation in subsequent studies. Taken together, we conclude that AXIN2 is regulated at the mRNA level by Wnt/WBP2 and acts as an essential mediator in Wnt/WBP2-driven signaling and TNBC biology. This positions AXIN2 as a robust molecular and functional readouts for subsequent studies on the mode of action of WBP2 in Wnt signaling.
WBP2 regulates a diverse network of genes and proteins independently of Wnt ligand
The RNA-Seq in this study was performed under the context of chronic Wnt signaling (WNT3A stimulation for 12 h). In our previous gain-of-function study, WBP2 was demonstrated to be a transcriptional coactivator induced by WNT3A via epidermal growth factor receptor cross-talk to undergo nuclear translocation and interaction with β-catenin at the late-phase (4–8 h) of Wnt signaling, thus enhancing β-catenin/TCF transcription (21). To investigate whether WBP2 is required for the early events, the mRNA of AXIN2 was examined over a time course of WNT3A treatment. The results showed that AXIN2 mRNA expression could be induced by WNT3A as early as 3 h post-treatment (Fig. 4A, i) and that prior knockdown of WBP2 abolished this occurrence (Fig. 4A, ii). How WBP2 contributes to the early effect of WNT3A on AXIN2 mRNA is unclear. The reported Wnt-induced WBP2 nuclear translocation may not play a role in these early WNT3A events because the former peaked at 8 h (21). The above observations, coupled to the function of WBP2 as a transcription co-activator, supported the hypothesis that at the basal state, WBP2 functions to prepare the “molecular soil” and prime the cells for acute response to Wnt through transcriptional regulation of a set of target genes that are independent of Wnt.
Figure 4.

A, (i) RT-qPCR analysis of AXIN2 mRNA levels upon WNT3A stimulation at the indicated durations in MDA-MB-231 cells. (ii) RT-qPCR analysis of AXIN2 mRNA levels. MDA-MB-231 cells were transfected with WBP2 shRNAs for 48 h and treated with control serum-free medium (O) or 200 ng/ml of rWNT3A for 3 h to examine the early-phase transcription of AXIN2 (top). IB analysis of the depletion of WBP2 is shown in the bottom. B, (i) analysis strategy to identify WBP2-regulated genes. (ii) Heat map of the expression pattern of the 86 WBP2-regulated genes. (iii) Top enriched pathways of WBP2 target genes. The number in each bar represents number of WBP2 targets involved in this pathway. C, (i) heat map of the expression pattern of the 23 WBP2-regulated proteins. (ii) Protein-mRNA expression association analysis of the WBP2-regulated proteins, 7 proteins with significant mRNA change were indicated in black. GPS1 was highlighted in red.
To examine the constitution of the molecular soil provided by WBP2, we re-analyzed the RNA-Seq data to identify genes that are regulated solely by WBP2. The re-analysis strategy is shown in Fig. 4B, i. The criteria for classification as WBP2-regulated genes were: (i) the gene must be regulated in a same trend (either up-regulation or down-regulation) in both the WBP2 shRNA1 and shRNA2 samples; (ii) p value and FDR must be less than 0.001; and (iii) at least a 2-fold change must be observed between control and shRNA1, whereas at least a 1.3-fold change must be observed between control and shRNA2. The different fold-change implemented was due to the lower effectiveness of shRNA2 to deplete WBP2 expression compared with shRNA1. Under these criteria, a total of 86 genes were identified to be differentially expressed upon WBP2 depletion (Fig. 4B, ii). Forty genes were down-regulated upon WBP2 depletion and 46 genes were up-regulated upon WBP2 depletion. The WBP2-regulated gene list was much larger than the Wnt/WBP2 genes identified earlier, reiterating the notion that WBP2 function is not restricted to Wnt signaling.
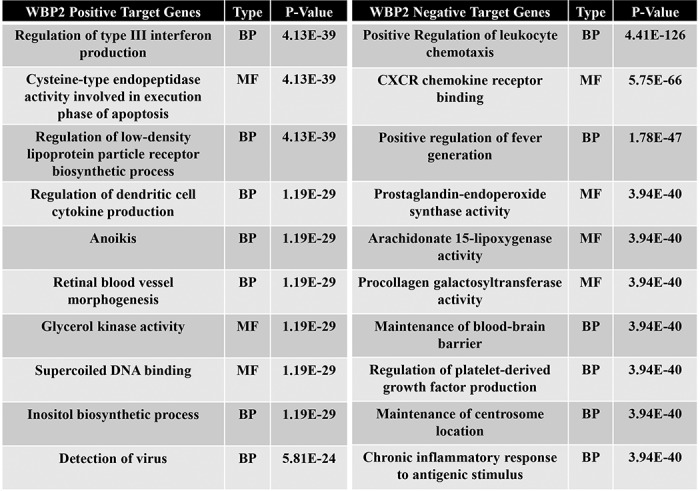
Bioinformatics analysis of the WBP2-regulated genes were performed to understand the molecular effects of WBP2 in breast cancer cells. First, gene ontology analysis using the PANTHER program indicated that WBP2-regulated genes were of diverse molecular functions and implicated in multiple biological processes (Fig. S2, A–C). Specifically, Table 2 shows that WBP2–up-regulated (positive) target genes are involved in antiviral responses, lipoprotein metabolism, and anoikis, whereas WBP2–down-regulated (negative) targets genes are involved in leukocyte biology, fever, inflammatory responses, and other critical processes. Additional analyses using multiple software revealed that the 4 major pathways that WBP2-regulated genes were involved are those related to (i) infection, (ii) inflammation, (iii) cancer, and (iv) metabolism (Fig. 4B, iii, Fig. S2, D and E).
Table 2.
Top molecular functions (MF) and biological processes (BP) enriched in WBP2 target genes (non-Wnt–dependent)
The above results proved that WBP2 alone regulates a sophisticated network of genes. This prompted us to examine the molecular soil at another level, the proteome, to unearth further evidence of WBP2 being a primer of Wnt signaling. To this end, a multiplex quantitative proteomics approach, iTRAQ-LC-MS, was used to analyze the control and WBP2-knocked down MDA-MB-231 cells. From the proteomics dataset, we compared WBP2 shRNA1 and shRNA2 to the control shRNA group to identify differentially expressed proteins upon WBP2 depletion. After selection, 23 differentially expressed proteins were identified, of which 16 and 7 proteins were down- and up-regulated by WBP2 depletion, respectively (Fig. 4C, i). These WBP2-target proteins were searched against the RNA-Seq data to understand how they are regulated. The mRNA-protein association analysis showed that 7 WBP2-regulated proteins displayed mRNA changes (>1.3-fold) in the same trend as the protein level, whereas the other 16 proteins showed no significant changes at the mRNA level (Fig. 4C, ii). This indicates that 30% of the WBP2-target proteins were regulated at the mRNA level, whereas the majority were regulated at post-transcriptional or post-translational levels. Collectively, proteomics and RNA-seq revealed that WBP2 regulates a considerable number of genes and proteins that might prime the cellular response to Wnt stimulation.
WBP2 primes Wnt signaling putatively via TNIK
From the Wnt-independent, WBP2-regulated RNA-seq gene list, TNIK (TRAF2 and NCK interacting kinase) was of particular interest and selected for further validation due to its previous implicated role in Wnt pathway regulation. TNIK is an essential activator of the Wnt-induced β-catenin/TCF transcriptional program that acts by binding directly to both TCF4 and β-catenin and phosphorylating TCF4 (42).
RNA-seq data shows that it is positively regulated by WBP2 (Table 3). To confirm this, RT-qPCR and Western blotting analyses were performed. WBP2 depletion in MDA-MB-231 cells resulted in a significant decrease of TNIK mRNA (Fig. 5A, i) and protein expression (Fig. 5A, ii), which could be rescued by WBP2 re-expression. To investigate whether there is correlation between WBP2 and TNIK expression in breast cancer specimens, we examined two published datasets as described under “Experimental procedures.” A significant co-occurrence of WBP2 and TNIK transcripts in breast cancer was observed (Fig. 5A, iii). These results raised a testable hypothesis that WBP2 primes cells, in part via up-regulation of TNIK expression to regulate the Wnt signaling induced transcriptional program, such as the increased AXIN2 expression, leading to potentiation of Wnt pathway activation.
Table 3.
WBP2 regulated genes identified from RNA-Seq dataset
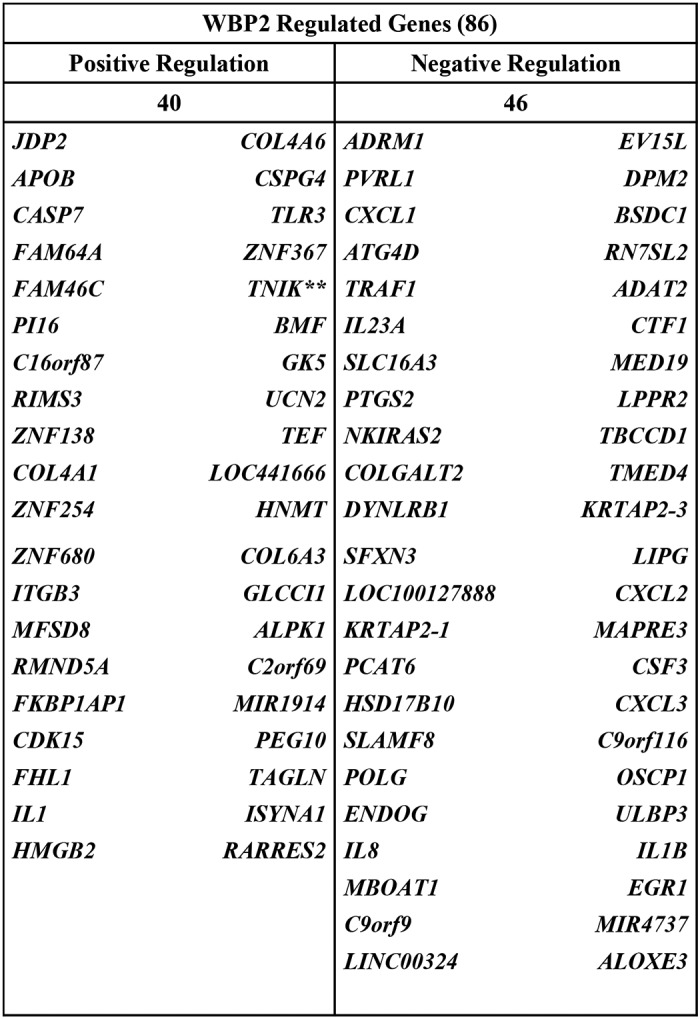
Figure 5.

A, qPCR (i) and IB (ii) analysis of TNIK expression upon WBP2 manipulation. MDA-MB-231 cells were infected with lentiviruses expressing WBP2 shRNAs and transfected with vector or WBP2 plasmid for 72 h before harvest. (iii) Analysis of WBP2 and TNIK up-regulation in breast cancer patients were performed on cBioPortal (www.cbioportal.org/) (77, 78). The odd ratios are originally generated as log odd ratios that were subsequently transformed by the natural exponential function. B, WBP2/WNT3A-induced up-regulation of TCF reporter activity (i) as well as AXIN2 expression and active β-catenin level (ii) were abolished by the TNIK inhibitor, NCB-0846 treatment in MDA-MB-231 cells. DMSO was used as the vehicle control.
As expected and shown in Fig. 5B, expression of WBP2 in MDA-MB-231 cells led to the increased WNT3A-induced Wnt reporter activity (Fig. 5B, i) with the accompanying up-regulation of TNIK and WNT3A-induced AXIN2 expression (Fig. 5B, ii). However, when TNIK was down-regulated by the TNIK inhibitor, NCB-0846 (43), both the basal and WBP2-induced Wnt reporter activity in MDA-MB-231 cells were significantly abolished (Fig. 5B, i). Concomitantly, WBP2-mediated up-regulation of AXIN2 protein expression and active β-catenin upon WNT3A stimulation were impaired in the presence of TNIK inhibitor (Fig. 5B, ii). Interestingly, immunoblotting shows TNIK protein expression to be induced upon WNT3A stimulation although it was not initially identified as a WNT3A-inducible gene from the RNA-Seq data. As expected, exogenous WBP2 expression positively regulated basal TNIK expression and overrode the WNT3A-induced expression. In other words, TNIK is a novel downstream mediator/effector of WBP2 in the positive regulation of Wnt-induced AXIN2 expression and pathway activation.
WBP2 may trigger a positive feedback loop involving TNIK and JNK/c-Jun pathway
Because TNIK is a target gene of WBP2, it would be pertinent to explore how WBP2 might contribute to the regulation of TNIK expression. A closer examination of the promoter region of TNIK revealed its enrichment with multiple c-Jun/AP-1 transcription factor-binding sites (Fig. 6A, i), suggesting that TNIK is likely to be a direct target gene of the c-Jun/AP-1 transcription factors and thus its expression may be functionally regulated by the JNK/c-Jun signaling pathway activity. However, the role of WBP2 in the JNK/c-Jun signaling pathway is unknown to date.
Figure 6.

A, (i) enriched c-Jun/AP-1–binding sites in the promoter region of TNIK. The image displays the most relevant transcription factor–binding sites in this gene promoter as predicted by SABiosciences Text Mining Application and the UCSC Genome Browser. IB analysis of JNK/c-Jun expression/phosphorylation upon WBP2 exogenous expression (ii) or depletion (iii) in MDA-MB-231 cells. (iv) WBP2/WNT3A-induced up-regulation of TCF reporter activity were abolished by the JNK inhibitor JNK-IN-8 treatment in MDA-MB-231 cells. DMSO was used as the vehicle control. (v) JNK1 + 2 siRNAs knockdown combined with JNK inhibitor JNK-IN-8 abolished the WBP2/WNT3A-induced TNIK and AXIN2 expression and active β-catenin level in MDA-MB-231 cells. (vi) TNIK inhibition via NCB-0846 treatment abolished the WBP2/WNT3A-induced phosphorylation of JNK/c-Jun in MDA-MB-231 cells. B, IB analysis of GPS1 protein expression upon WBP2 depletion (i) or overexpression (ii) in MDA-MB-231 cells. WBP2-induced up-regulation of TCF reporter activity (iii) and AXIN2/active β-catenin level (iv) were abolished by GPS1 siRNA knockdown in MDA-MB-231 cells. C, (i) IB analysis of JNK/c-Jun and TNIK expression/activity upon GPS1 siRNA knockdown in MDA-MB-231 cells. (ii) WBP2-induced up-regulation of TNIK and JNK/c-Jun expression/phosphorylation were abolished by GPS1 siRNA knockdown.
First, we investigated whether WBP2 regulates the JNK/c-Jun pathway activity. IB analysis showed that the levels of phospho-JNK and phospho-c-Jun were decreased upon WBP2 depletion, whereas the total level of JNK protein was not affected (Fig. 6A, ii). Conversely, increased phosphorylation of JNK/c-Jun and c-Jun expression but not total JNK were observed upon WBP2 overexpression (Fig. 6A, iii). This is consistent with the RNA-Seq results, which showed that JNK mRNA levels did not change upon WBP2 depletion. Putatively, activation of JNK phosphorylation/activity by WBP2 in turn regulates c-Jun expression, a well-established observation (44–47).
Next, we asked if the WBP2-induced JNK activity contributes to Wnt pathway activation. Fig. 6A, iv and v, showed that WBP2-driven TCF reporter activity and TNIK expression were significantly abolished in the presence of combined siRNA knockdown and inhibition of JNK via JNK-IN-8 (48). WBP2/WNT3A-induced AXIN2 expression and the active β-catenin level was concomitantly diminished upon double blocking of JNK (Fig. 6A, v). Altogether, the data infers the existence of the signaling axis: WBP2 → JNK/c-Jun → TNIK → AXIN2. Interestingly, expression of WBP2 was significantly down-regulated by JNK knockdown/inhibition. The mechanism is unclear, although JNK had been implicated in translation (49) and protein stability (50) control.
Conversely, several studies have demonstrated the reciprocal regulation between TNIK and the JNK/c-Jun signaling pathway (51–53), where TNIK is essential for activation of the JNK pathway via JNK1 and JNK2. We therefore hypothesized the existence of a positive feedback loop where the WBP2-induced TNIK expression leads to the reciprocal activation of JNK and c-Jun, which in turn further up-regulate its own expression. Indeed, TNIK inhibition by NCB-0846 abolished the WBP2-induced JNK/c-Jun activation/phosphorylation (Fig. 6A, vi). Interestingly, TNIK inhibition also down-regulated the c-Jun but not the JNK expression. The data supports the notion that TNIK and JNK/c-Jun reciprocally regulate each other in a positive feedback manner. To further rule out the possibility that WBP2 may regulate TNIK expression and/or JNK/c-Jun activity through the basal Wnt activity, we down-regulated the endogenous β-catenin or inhibited the β-catenin activity at the basal state. Our data showed that WBP2 overexpression could still up-regulate the basal TNIK expression and JNK/c-Jun phosphorylation even in the presence of β-catenin knockdown (Fig. S3A) or Wnt inhibitor-FH535 (Fig. S3B). This reconfirms the “priming” role of WBP2 by up-regulating the JNK/c-Jun activity and TNIK expression in an WNT3A-independent manner.
GPS1, a WBP2-induced protein, acts upstream of JNK/c-Jun and TNIK
GPS1, an essential component of the COP9 signalosome complex (CSN) involved in various cellular and developmental processes, was the top hit on the list of Wnt-independent, WBP2-regulated proteins from the proteomics dataset. One study demonstrated that GPS1 could enhance JNK activity (54) and this could be abolished by Asb-4, which reduces the cytosolic protein levels of GPS1. Thus GPS may mediate activation of the JNK/c-Jun pathway by WBP2. The mRNA–protein association analysis showed that GPS1 mRNA did not change upon WBP2 depletion, suggesting that WBP2 regulates GPS expression in a transcription-independent manner. Validation studies in Fig. 6B, i, showed that WBP2 knockdown down-regulates GPS1 protein expression. The converse is true when WBP2 was overexpressed (Fig. 6B, ii).
The above data led us to test the hypothesis that the WBP2-mediated increase in GPS1 contributes to activation of JNK/Jun. Prior to that, the potential role of GPS1 in WBP2-mediated Wnt pathway activation was investigated. The results showed that GPS1 knockdown significantly abolished the WBP2-driven TCF reporter activity in MDA-MB-231 cells, concomitant with the molecular down-regulation of WBP2/WNT3A-induced active β-catenin level and AXIN2 expression (Fig. 6B, iii and iv). Next, we explored if GPS1 acts along or in parallel with JNK/c-Jun and/or TNIK in WBP2-mediated Wnt pathway regulation. As shown in Fig. 6C, i, GPS1 depletion resulted in decreased phosphorylation of JNK and c-Jun as well as down-regulation of TNIK expression. Moreover, WBP2-induced up-regulation of TNIK expression and JNK/c-Jun phosphorylation could be significantly abolished by GPS1 knockdown (Fig. 6C, ii). Thus, GPS1, TNIK, and JNK/c-Jun may act together in the same regulatory axis in WBP2-mediated Wnt pathway activation.
WBP2 promotes breast cancer growth through JNK/TNIK and sensitizes TNBC to JNK/TNIK pharmacological inhibitors
It is conceivable that the function of WBP2 in breast cancer depends on TNIK. As shown in Fig. 7A, i, the increase in cell proliferation as a result of exogenous expression of WBP2 in MDA-MB-231 could be significantly abolished by TNIK knockdown. Similarly, the decrease in anchorage-dependent 2D (Fig. 7A, ii) and -independent 3D (Fig. 7A, iii) cell growth caused by WBP2 knockdown could be rescued by exogenous expression of TNIK.
Figure 7.

A, (i) WBP2-induced 2D proliferation of MDA-MB-231 could be partially abolished by TNIK siRNA knockdown. The anchorage-dependent 2D (ii) and -independent 3D (iii) cell proliferation of MDA-MB-231 were decreased by WBP2 siRNA knockdown and this could be partially rescued by exogenous TNIK expression. B, WBP2/WNT3A-induced 2D proliferation of MDA-MB-231 could be significantly abolished by JNK inhibition via (i) JNK-IN-8 and (ii) TNIK inhibition via NCB-0846. C, (i) dose-response 2D and 3D growth curve for TNIK inhibition via NCB-0846 in MDA-MB-231 upon WBP2 exogenous expression or siRNA/shRNA knockdown. (ii) Tabulated IC50 in summary for TNIK inhibitor, NCB-0846 for MDA-MB-231 with WBP2 OE or KD in 2D or 3D growth. The p value is calculated by the extra sum-of-squares F test.
Next, we surmise that pharmacological disruptions of JNK and TNIK would suppress growth of WBP2-positive TNBC. To this end, we investigated if WBP2/Wnt-mediated anchorage-dependent cell proliferation could be impaired by JNK or TNIK inhibitors. As shown in Fig. 7B, the increase in WNT3A-induced cell proliferation as a result of exogenous WBP2 expression was drastically abolished by the JNK inhibitor JNK-IN-8 (Fig. 7B, i) and TNIK inhibitor NCB-0846 (Fig. 7B, ii).
Activation of the JNK/TNIK pathway by WBP2 may lead to pathway addiction. We hypothesized that higher WBP2 expression sensitizes cells to the inhibition of JNK and/or TNIK. To this end, the dose-response curves for the JNK inhibitor JNK-IN-8 and TNIK inhibitor NCB-0846 were obtained by measuring the 2D and 3D cell growth/viability of MDA-MB-231 TNBC cells with (a) WBP2 stable expression, (b) WBP2 transient siRNA knockdown, and (c) WBP2 stable shRNA knockdown. The data for TNIK and JNK inhibitors are shown in Fig. 7C, i, and Fig. S3C, respectively. In general, the TNIK inhibitor displayed stronger inhibition than JNK inhibitor. In particular, the 3D rather than 2D viability of the TNBC cells was more prominently affected by the TNIK inhibitor than JNK inhibitor. A recent study (55) comparing the cancer cells in 2D versus 3D culture also revealed differences in their drug responses, where the 3D models displayed stronger anti-tumor responses to AKT or the mitogen-activated protein kinase pathway inhibitors compared with those in 2D models. This is attributed to the distinct rewiring of signaling in the 3D culture and during treatment. The better drug response in 3D culture compared with 2D was similarly observed in other studies also (56, 57). The IC50 calculated from their dose-response curves were summarized and tabulated in Fig. 7C, ii, and Fig. S3D. Overall, cells with higher WBP2 expression were more sensitive to the JNK/TNIK inhibition than their vector control by up to 15.65-fold, whereas cells with WBP2 knocked down were more resistant to JNK/TNIK inhibition compared with control by up to 6.91-fold.
Discussion
This study is an attempt to elucidate the molecular action of WBP2 signaling in Wnt signaling and TNBC by unraveling the intricate network of signaling pathways and genes that underlie the function of the WBP2 oncogene alone or in the presence of Wnt ligand. WBP2 is demonstrated to be involved in the Wnt-induced transcriptional program associated with a core set of genes in MDA-MB-231 TNBC cells, with up to 82% of the Wnt target genes being dependent on WBP2.
As the most pervasive Wnt target gene whose expression depends on WBP2, the biological function of AXIN2 has not been unambiguously recognized and it has been assumed to act as a negative regulator of Wnt signaling through a negative feedback loop targeting β-catenin, because its paralog AXIN1 is the core component of the β-catenin destruction complex (58–60). However, we identified AXIN2 as a robust target gene of Wnt/WBP2 signaling in TNBC cells, and it was essential for Wnt-induced cell migration and colony formation. Our findings are supported by several studies, which also showed that AXIN2 plays important roles in Wnt-induced cancer progression. Yook et al. (61) demonstrated that Wnt-induced AXIN2 functions as a nucleocytoplasmic chaperone for GSK3. It regulates the cytoplasmic translocation of GSK3, and leading to the stabilization of nuclear SNAIL1 protein, which plays a key role in Wnt-induced breast cancer cell migration and invasion. A similar phenomenon was observed by Wu et al. (62) in colorectal cancer, in which up-regulation of AXIN2 led to a marked increase in Snail1 activity and induction of EMT. These results suggest a pro-cancer function of AXIN2 other than a negative feedback regulator.
We postulated that WBP2 promotes Wnt signaling by priming cancer cells by conferring suitable molecular conditions independent of Wnt. TNIK and GPS1 were two targets from the transcriptomic and proteomic datasets, respectively, which were identified to be up-regulated by WBP2 in the basal condition. The lack of overlapping genes/proteins that are differentially expressed in both the RNA-Seq and proteomics studies is probably due to: 1) the relatively lower coverage of the proteomics data (2,035 proteins) compared with RNA-Seq data (19,301 genes) and 2) the proteins identified by proteomics could be regulated mainly at the post-transcriptional and/or translational levels as mRNA levels of the majority of proteins detected were not changed (refer to Fig. 4C, ii). These genes were not in the list of RNA-seq data because their fold-change, p value, or FDR did not meet the criteria during data processing. Nevertheless, our data suggest that WBP2 primes cellular response to Wnt by up-regulating GPS1 expression, which activates the JNK/Jun pathway, thus promoting transcription/expression of TNIK and activation of AXIN2 expression. Furthermore, JNK/c-Jun phosphorylation/activation generates a positive feedback loop through TNIK. This may explain how WBP2 contributes to the early-phase expression of Wnt target genes such as AXIN2. On the other hand, further investigations are required to confirm if c-Jun plays a direct role in transcriptional regulation of TNIK expression through its binding to the TNIK promoter and this regulation is modulated by the WBP2–GPS1–JNK signaling axis and Wnt stimulation.
The study identified the JNK/c-Jun pathway as a novel node through which WBP2 activates Wnt signaling. On the other hand, Wnt signaling activates and cross-talks to the noncanonical, β-catenin-independent pathways, including the JNK pathway (63–67). Interestingly, like GPS1 and TNIK, the WBP2 target gene AXIN2, being the essential component in both canonical and noncanonical Wnt pathways, was capable of inducing JNK activity through its MEKK1 binding and self-association domains (68). Although the latter was not tested in our study, these observations supports the notion that WBP2 is a key activator of the JNK pathway. Wnt and JNK pathways are intricately linked as they operate both in parallel and synergistically. Together, they cooperate to activate a subset of genes that are common targets for both pathways, such as c-myc and cyclin D1 (69, 70). The c-Jun gene itself is a well-characterized Wnt target gene (71, 72). JNK2, and to a much lesser extent JNK1 regulates canonical Wnt signaling via phosphorylating and inducing nuclear translocation of β-catenin (73). TCF4 is transcriptionally regulated through activated JNK signaling as the TCF4 promoter region is a target for phosphorylated c-Jun (74). It has also been observed that c-Jun interacts with both β-catenin (69) as well as TCF4 (75) in the canonical Wnt signaling. This study positions WBP2 as a part of a sophisticated signaling network that drives the Wnt/JNK function.
Wnt-independent WBP2 target genes that might contribute to the molecular etiology of TNBC through other pathophysiological processes may be discovered from the omics datasets. For example, inflammation-associated genes, such as IL11, TLR3, IL23A, IL8, IL1B, CSF3, CXCL1, CXCL3, and cell adhesion molecules including ADRM1, PVRL1, COL4A6, COL4A1, COL4A3, CSPG4, and ITGB3 are putative WBP2 targets. These multiple pathways and molecules regulated by WBP2 are likely to cooperate and contribute to the oncogenic function of WBP2. This creates opportunities for further investigation into the function and mechanism of WBP2 in cancer biology.
In conclusion, besides the Wnt-induced transcription co-activation function of WBP2 reported previously (21), WBP2 regulates Wnt signaling in TNBC cells via Wnt-independent priming effects. The study has successfully mapped a few key players in a WBP2-associated signaling axis that contribute to the molecular soil that primes the response of TNBC cells to Wnt signaling. The GPS1/JNK/Jun/TNIK pathway may be just one of the several signaling axes that WBP2 employs to facilitate a Wnt signaling pathway. The study has implications on precision medicine of TNBC, for which there is no clinically approved therapeutic target. The molecular soil provided by WBP2 is a potential source of new primers as drug targets and companion diagnostics.
Experimental procedures
Antibodies and reagents
WBP2 mAb (MABS441 clone 4C8H10) was purchased from EMD Millipore (Billerica, MA); anti-β-catenin and anti-TNIK mouse monoclonal antibodies were from BD Biosciences (San Diego, CA); anti-c-Jun (clone 60A8), anti-phospho-c-Jun (Ser-73, clone D47G9), anti-JNK, anti-phospho-JNK (Thr-183/Tyr-185), anti-nonphospho (active)-β-catenin (Ser-33/37/Thr-41, clone D13A1), and anti-AXIN2 (clone 76G6) rabbit antibodies were obtained from Cell Signaling Technology Inc. (Danvers, MA); anti-GPS1/CSN1 was obtained from Abcam (Cambridge, UK). Other antibodies, plasmids, reporters, siRNA, and shRNA sequences used are detailed in the supporting data.
TNIK inhibitor NCB-0846, and JNK inhibitor JNK-IN-8 were obtained from Selleck Chemicals (Houston, TX). Wnt/β-catenin inhibitor FH535 was obtained from Sigma.
Cell culture, treatments, and lysis
MCF10A, MCF7, MDA-MB-231, SKBR3, MDA-MB-468, BT549, HeLa, and HEK293 cells were obtained from American Type Culture Collection (Manassas, VA). The culture conditions for the cell lines, procedures for treatment of cells with inhibitors/ligands, transient/stable transfection/transduction, and cell lysis/immunoblotting are described in the supporting data.
Dual luciferase reporter assay and qPCR
The dual luciferase reporter assay (Promega) was performed according to the manufacturer's instructions and quantified using Luminoskan Ascent Microplate Luminometer (Thermo Scientific). Firefly signals were normalized to Renilla signals. RNA extraction and qPCR of mRNA are described in the supporting data.
In vitro cell-based assays
The colony-formation ability was assayed by clonogenic assay where 1000 cells were seeded in 6-well plates and incubated for 2–3 weeks until the colonies could be clearly observed by eye-sight. The plates were then fixed with methanol and stained with crystal violet to visualize the colonies (25% methanol, 0.5% crystal violet). The analysis was done using the ImageJ-plugin “ColonyArea,” which is optimized for rapid and quantitative analysis of focus-formation assays conducted in 6- to 24-well dishes (76).
For transwell-migration assays, 2 × 105 cells were seeded in serum-free medium with or without rWNT3A in the top chambers of a 96-transwell plate with polycarbonate membrane chambers (8-μm pore size, Cell Biolabs), and medium containing 10% fetal bovine serum was added to the bottom chambers as chemoattractant. After 12–16 h incubation, the top nonmigrated cells were removed; migrated cells on the bottom were subjected to H&E staining for analysis.
For cell-viability assay, 2500 cells were seeded in triplicate in 96-well plates and incubated overnight before being treated with various concentrations of JNK or TNIK inhibitor for 3 days. For assay involving WNT3A stimulation, the cells were seeded in culture medium with 0.5% fetal bovine serum. The cell viability was measured via CellTiter-GLO 2.0 assay (Promega, Madison, WI). For the 3D cell-viability assay, the cells were seeded in ultra-low-attachment plates and measured via CellTiter-Glo 3D Cell Viability Assay (Promega). All assays were performed according to the manufacturers' instructions. Results from the viability assays are expressed as percentage of the vehicle-treated control and error bars indicate standard deviation.
RNA-Seq analysis
Total RNA were extracted from cells and sent for next-generation RNA-Seq by BGI (Shenzhen, China). The gene expression level is calculated by the reads per kilobase/million mapped reads method. Each sample was prepared as biological triplicates to ensure the reliability of RNA-Seq data.
ITRAQ-LC-MS analysis
Proteins from each sample were subjected to iTRAQ labeling according to the manufacturer's protocol (Applied Biosystems, Framingham, MA). Sample preparation for iTRAQ labeling, MS and data analysis are described in the supporting data.
Analysis of WBP2 mRNA expression and CNA in clinical datasets
Two large volume clinical breast cancer tissue datasets: TCGA and METABRIC, were used to examine the expression and CNA of WBP2 and its association with patient survival and other phenotypes. These datasets were analyzed and visualized through cBioPortal (http://www.cbioportal.org/) (77, 78), UCSC Xena (http://xena.ucsc.edu/), and Kaplan-Meier Plotter (http://kmplot.com/analysis/).6 Analysis of the association between WBP2 and TNIK in breast cancer was detailed in the supporting data.
Statistical analysis
Data are presented as the mean ± S.E. from at least three independent experiments. Student's t test was conducted to analyze the data unless otherwise stated, and a difference was considered significant if p < 0.05.
Author contributions
Z. L., S. K. L., and X. L. data curation; Z. L., S. K. L., and X. L. formal analysis; Z. L. validation; Z. L. and S. K. L. investigation; Z. L., S. K. L., and X. L. methodology; Z. L. and S. K. L. writing-original draft; Y. P. L. conceptualization; Y. P. L. supervision; Y. P. L. funding acquisition; Y. P. L. project administration; Y. P. L. writing-review and editing.
Supplementary Material
Acknowledgment
We thank Prof. David M. Virshup for helpful discussions and comments.
This work was supported by Ministry of Education of Singapore Grant MOE2016-T2–2-007. The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Tables S1 and S2 and Figs. S1–S3.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
- TNBC
- triple-negative breast cancer
- β-TrCP
- β-transducin repeat containing E3 ubiquitin protein ligase
- ADAMTS14
- ADAM metallopeptidase with thrombospondin type 1 motif 14
- ADRM
- adhesion regulating molecule
- AP-1
- activator protein 1
- BMP4
- bone morphogenetic protein 4
- CNA
- copy number alterations
- COL4A6
- collagen type IV α6 chain
- CSF3
- colony-stimulating factor 3
- CSN1
- COP9 signalosome complex subunit 1
- CSPG4
- chondroitin sulfate proteoglycan 4
- CXCL1
- C-X-C motif chemokine ligand 1
- DCLK1
- double cortin-like kinase 1
- FDR
- false discovery rate
- GPS
- G protein pathway suppressor
- GSK
- glycogen synthase kinase
- HAS
- hyaluronan synthase
- IL
- interleukin
- ITCH
- itchy E3 ubiquitin protein ligase
- ITGB3
- integrin subunit β 3
- iTRAQ
- isobaric tags for relative and absolute quantitation
- JDP
- Jun dimerization protein
- JNK
- c-Jun NH2-terminal kinase
- LEF
- lymphoid enhancer binding factor
- LGR5
- leucine-rich repeat containing G protein-coupled receptor 5
- MEKK
- MAPK/ERK kinase kinase
- PANTHER
- Protein ANalysis THrough Evolutionary Relationships
- PAX8
- paired box 8
- PVRL
- poliovirus receptor-related protein
- qPCR
- quantitative polymerase chain reaction
- RNA-Seq
- RNA sequencing
- SCR
- scrambled
- shRNA
- short hairpin RNA
- SLC1A3
- solute carrier family 1 (glial high affinity glutamate transporter), member 3
- SOX4
- SRY-box 4
- SPRY1
- sprouty RTK signaling antagonist 1
- TAZ
- tafazzin
- TCF4
- transcriptional factor 4
- TLR3
- Toll-like receptor 3
- TNIK
- TRAF2 and NCK interacting kinase
- WBP2
- WW domain–binding protein
- YAP
- Yes-associated protein
- siRNA
- small interfering RNA
- IB
- immunoblot.
References
- 1. Ferlay J., Soerjomataram I., Ervik M., Dikshit R., Eser S., Mathers C., Rebelo M., Parkin D., Forman D., and Bray F. (2013) GLOBOCAN 2012 v1.0. Cancer incidence and mortality worldwide: IARC CancerBase, 2014, IARC, Lyon, France: [DOI] [PubMed] [Google Scholar]
- 2. Austreid E., Lonning P. E., and Eikesdal H. P. (2014) The emergence of targeted drugs in breast cancer to prevent resistance to endocrine treatment and chemotherapy. Expert Opin. Pharmacother. 15, 681–700 10.1517/14656566.2014.885952 [DOI] [PubMed] [Google Scholar]
- 3. Clevers H. (2006) Wnt/β-catenin signaling in development and disease. Cell 127, 469–480 10.1016/j.cell.2006.10.018 [DOI] [PubMed] [Google Scholar]
- 4. Clevers H., and Nusse R. (2012) Wnt/β-catenin signaling and disease. Cell 149, 1192–1205 10.1016/j.cell.2012.05.012 [DOI] [PubMed] [Google Scholar]
- 5. Valenta T., Hausmann G., and Basler K. (2012) The many faces and functions of β-catenin. EMBO J. 31, 2714–2736 10.1038/emboj.2012.150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Li V. S., Ng S. S., Boersema P. J., Low T. Y., Karthaus W. R., Gerlach J. P., Mohammed S., Heck A. J., Maurice M. M., Mahmoudi T., and Clevers H. (2012) Wnt signaling through inhibition of β-catenin degradation in an intact Axin1 complex. Cell 149, 1245–1256 10.1016/j.cell.2012.05.002 [DOI] [PubMed] [Google Scholar]
- 7. Zeng X., Huang H., Tamai K., Zhang X., Harada Y., Yokota C., Almeida K., Wang J., Doble B., Woodgett J., Wynshaw-Boris A., Hsieh J. C., and He X. (2008) Initiation of Wnt signaling: control of Wnt coreceptor Lrp6 phosphorylation/activation via frizzled, dishevelled and axin functions. Development 135, 367–375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. DiMeo T. A., Anderson K., Phadke P., Fan C., Feng C., Perou C. M., Naber S., and Kuperwasser C. (2009) A novel lung metastasis signature links Wnt signaling with cancer cell self-renewal and epithelial-mesenchymal transition in basal-like breast cancer. Cancer Res. 69, 5364–5373 10.1158/0008-5472.CAN-08-4135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Geyer F. C., Lacroix-Triki M., Savage K., Arnedos M., Lambros M. B., MacKay A., Natrajan R., and Reis-Filho J. S. (2011) β-Catenin pathway activation in breast cancer is associated with triple-negative phenotype but not with CTNNB1 mutation. Mod. Pathol. 24, 209–231 10.1038/modpathol.2010.205 [DOI] [PubMed] [Google Scholar]
- 10. Hergovich A., and Hemmings B. A. (2010) TAZ-mediated crosstalk between Wnt and Hippo signaling. Dev. Cell 18, 508–509 10.1016/j.devcel.2010.04.003 [DOI] [PubMed] [Google Scholar]
- 11. Imajo M., Miyatake K., Iimura A., Miyamoto A., and Nishida E. (2012) A molecular mechanism that links Hippo signalling to the inhibition of Wnt/β-catenin signalling. EMBO J. 31, 1109–1122 10.1038/emboj.2011.487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Azzolin L., Zanconato F., Bresolin S., Forcato M., Basso G., Bicciato S., Cordenonsi M., and Piccolo S. (2012) Role of TAZ as mediator of Wnt signaling. Cell 151, 1443–1456 10.1016/j.cell.2012.11.027 [DOI] [PubMed] [Google Scholar]
- 13. Rosenbluh J., Nijhawan D., Cox A. G., Li X., Neal J. T., Schafer E. J., Zack T. I., Wang X., Tsherniak A., Schinzel A. C., Shao D. D., Schumacher S. E., Weir B. A., Vazquez F., Cowley G. S., et al. (2012) β-Catenin-driven cancers require a YAP1 transcriptional complex for survival and tumorigenesis. Cell 151, 1457–1473 10.1016/j.cell.2012.11.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Azzolin L., Panciera T., Soligo S., Enzo E., Bicciato S., Dupont S., Bresolin S., Frasson C., Basso G., Guzzardo V., Fassina A., Cordenonsi M., and Piccolo S. (2014) YAP/TAZ incorporation in the β-catenin destruction complex orchestrates the Wnt response. Cell 158, 157–170 10.1016/j.cell.2014.06.013 [DOI] [PubMed] [Google Scholar]
- 15. Chen H. I., and Sudol M. (1995) The WW domain of Yes-associated protein binds a proline-rich ligand that differs from the consensus established for Src homology 3-binding modules. Proc. Natl. Acad. Sci. U.S.A. 92, 7819–7823 10.1073/pnas.92.17.7819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nitsch R., Di Palma T., Mascia A., and Zannini M. (2004) WBP-2, a WW domain binding protein, interacts with the thyroid-specific transcription factor Pax8. Biochem. J. 377, 553–560 10.1042/bj20031233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dhananjayan S. C., Ramamoorthy S., Khan O. Y., Ismail A., Sun J., Slingerland J., O'Malley B. W., and Nawaz Z. (2006) WW domain binding protein-2, an E6-associated protein interacting protein, acts as a coactivator of estrogen and progesterone receptors. Mol. Endocrinol. 20, 2343–2354 10.1210/me.2005-0533 [DOI] [PubMed] [Google Scholar]
- 18. Lim S. K., Orhant-Prioux M., Toy W., Tan K. Y., and Lim Y. P. (2011) Tyrosine phosphorylation of transcriptional coactivator WW-domain binding protein 2 regulates estrogen receptor α function in breast cancer via the Wnt pathway. FASEB J. 25, 3004–3018 10.1096/fj.10-169136 [DOI] [PubMed] [Google Scholar]
- 19. Buffa L., Saeed A. M., and Nawaz Z. (2013) Molecular mechanism of WW-domain binding protein-2 coactivation function in estrogen receptor signaling. IUBMB Life 65, 76–84 10.1002/iub.1105 [DOI] [PubMed] [Google Scholar]
- 20. Chen Y., Choong L. Y., Lin Q., Philp R., Wong C. H., Ang B. K., Tan Y. L., Loh M. C., Hew C. L., Shah N., Druker B. J., Chong P. K., and Lim Y. P. (2007) Differential expression of novel tyrosine kinase substrates during breast cancer development. Mol. Cell Proteomics 6, 2072–2087 10.1074/mcp.M700395-MCP200 [DOI] [PubMed] [Google Scholar]
- 21. Lim S. K., Lu S. Y., Kang S. A., Tan H. J., Li Z., Adrian Wee Z. N., Guan J. S., Reddy Chichili V. P., Sivaraman J., Putti T., Thike A. A., Tan P. H., Sudol M., Virshup D. M., Chan S. W., Hong W., and Lim Y. P. (2016) Wnt Signaling Promotes Breast Cancer by Blocking ITCH-Mediated Degradation of YAP/TAZ Transcriptional Coactivator WBP2. Cancer Res. 76, 6278–6289 10.1158/0008-5472.CAN-15-3537 [DOI] [PubMed] [Google Scholar]
- 22. Chan S. W., Lim C. J., Huang C., Chong Y. F., Gunaratne H. J., Hogue K. A., Blackstock W. P., Harvey K. F., and Hong W. (2011) WW domain-mediated interaction with Wbp2 is important for the oncogenic property of TAZ. Oncogene 30, 600–610 10.1038/onc.2010.438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhang X., Milton C., Poon C., Hong W., and Harvey K. (2011) Wbp2 cooperates with Yorkie to drive tissue growth downstream of the Salvador–Warts–Hippo pathway. Cell Death Differ. 18, 1346–1355 10.1038/cdd.2011.6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Walko G., Woodhouse S., Pisco A. O., Rognoni E., Liakath-Ali K., Lichtenberger B. M., Mishra A., Telerman S. B., Viswanathan P., Logtenberg M., Renz L. M., Donati G., Quist S. R., and Watt F. M. (2017) A genome-wide screen identifies YAP/WBP2 interplay conferring growth advantage on human epidermal stem cells. Nat. Commun. 8, 14744 10.1038/ncomms14744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ren Y. Q., Wang H. J., Zhang Y. Q., and Liu Y. B. (2017) WBP2 modulates G1/S transition in ER+ breast cancer cells and is a direct target of miR-206. Cancer Chemother. Pharmacol. 79, 1003–1011 10.1007/s00280-017-3302-0 [DOI] [PubMed] [Google Scholar]
- 26. Buniello A., Ingham N. J., Lewis M. A., Huma A. C., Martinez-Vega R., Varela-Nieto I., Vizcay-Barrena G., Fleck R. A., Houston O., Bardhan T., Johnson S. L., White J. K., Yuan H., Marcotti W., and Steel K. P. (2016) Wbp2 is required for normal glutamatergic synapses in the cochlea and is crucial for hearing. EMBO Mol. Med. 8, 191–207 10.15252/emmm.201505523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pongor L., Kormos M., Hatzis C., Pusztai L., Szab A., and Györffy B. (2015) A genome-wide approach to link genotype to clinical outcome by utilizing next generation sequencing and gene chip data of 6,697 breast cancer patients. Genome Med. 7, 104 10.1186/s13073-015-0228-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. McCarthy D. J., and Smyth G. K. (2009) Testing significance relative to a fold-change threshold is a TREAT. Bioinformatics 25, 765–771 10.1093/bioinformatics/btp053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hughes-Large J. M., and Borradaile N. M. (2016) Gene expression microarray data from human microvascular endothelial cells supplemented with a low concentration of niacin. Data Brief 6, 899–902 10.1016/j.dib.2016.01.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Railo A., Pajunen A., Itäranta P., Naillat F., Vuoristo J., Kilpeläinen P., and Vainio S. (2009) Genomic response to Wnt signalling is highly context-dependent–evidence from DNA microarray and chromatin immunoprecipitation screens of Wnt/TCF targets. Exp. Cell Res. 315, 2690–2704 10.1016/j.yexcr.2009.06.021 [DOI] [PubMed] [Google Scholar]
- 31. Labbé E., Lock L., Letamendia A., Gorska A. E., Gryfe R., Gallinger S., Moses H. L., and Attisano L. (2007) Transcriptional cooperation between the transforming growth factor-beta and Wnt pathways in mammary and intestinal tumorigenesis. Cancer Res. 67, 75–84 10.1158/0008-5472.CAN-06-2559 [DOI] [PubMed] [Google Scholar]
- 32. Maubant S., Tesson B., Maire V., Ye M., Rigaill G., Gentien D., Cruzalegui F., Tucker G. C., Roman-Roman S., and Dubois T. (2015) Transcriptome analysis of Wnt3a-treated triple-negative breast cancer cells. PLoS ONE 10, e0122333 10.1371/journal.pone.0122333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhuo Z., Lamont S. J., Lee W. R., and Abasht B. (2015) RNA-Seq analysis of abdominal fat reveals differences between modern commercial broiler chickens with high and low feed efficiencies. PLoS ONE 10, e0135810 10.1371/journal.pone.0135810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhang J., Liang Q., Lei Y., Yao M., Li L., Gao X., Feng J., Zhang Y., Gao H., Liu D. X., Lu J., and Huang B. (2012) SOX4 induces epithelial-mesenchymal transition and contributes to breast cancer progression. Cancer Res. 72, 4597–4608 10.1158/0008-5472.CAN-12-1045 [DOI] [PubMed] [Google Scholar]
- 35. Huang Y. W., Liu J. C., Deatherage D. E., Luo J., Mutch D. G., Goodfellow P. J., Miller D. S., and Huang T. H. (2009) Epigenetic repression of microRNA-129–2 leads to overexpression of SOX4 oncogene in endometrial cancer. Cancer Res. 69, 9038–9046 10.1158/0008-5472.CAN-09-1499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Udabage L., Brownlee G. R., Nilsson S. K., and Brown T. J. (2005) The over-expression of HAS2, Hyal-2 and CD44 is implicated in the invasiveness of breast cancer. Exp. Cell Res. 310, 205–217 10.1016/j.yexcr.2005.07.026 [DOI] [PubMed] [Google Scholar]
- 37. Kosaki R., Watanabe K., and Yamaguchi Y. (1999) Overproduction of hyaluronan by expression of the hyaluronan synthase Has2 enhances anchorage-independent growth and tumorigenicity. Cancer Res. 59, 1141–1145 [PubMed] [Google Scholar]
- 38. Bozatzi P., Dingwell K. S., Wu K. Z., Cooper F., Cummins T. D., Hutchinson L. D., Vogt J., Wood N. T., Macartney T. J., Varghese J., Gourlay R., Campbell D. G., Smith J. C., and Sapkota G. P. (2018) PAWS1 controls Wnt signalling through association with casein kinase 1α. EMBO Rep. 19, e44807 10.15252/embr.201744807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Huang P. Y., Kandyba E., Jabouille A., Sjolund J., Kumar A., Halliwill K., McCreery M., DelRosario R., Kang H. C., Wong C. E., Seibler J., Beuger V., Pellegrino M., Sciambi A., Eastburn D. J., and Balmain A. (2017) Lgr6 is a stem cell marker in mouse skin squamous cell carcinoma. Nat. Genet. 49, 1624–1632 10.1038/ng.3957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ndoye A., Budina-Kolomets A., Kugel C. H. 3rd, Webster M. R., Kaur A., Behera R., Rebecca V. W., Li L., Brafford P. A., Liu Q., Gopal Y. N. V., Davies M. A., Mills G. B., Xu X., Wu H., et al. (2017) ATG5 mediates a positive feedback loop between Wnt signaling and autophagy in melanoma. Cancer Res. 77, 5873–5885 10.1158/0008-5472.CAN-17-0907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hsu J. M., Xia W., Hsu Y. H., Chan L. C., Yu W. H., Cha J. H., Chen C. T., Liao H. W., Kuo C. W., Khoo K. H., Hsu J. L., Li C. W., Lim S. O., Chang S. S., Chen Y. C., Ren G. X., and Hung M. C. (2018) STT3-dependent PD-L1 accumulation on cancer stem cells promotes immune evasion. Nat. Commun. 9, 1908 10.1038/s41467-018-04313-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Mahmoudi T., Li V. S., Ng S. S., Taouatas N., Vries R. G., Mohammed S., Heck A. J., and Clevers H. (2009) The kinase TNIK is an essential activator of Wnt target genes. EMBO J. 28, 3329–3340 10.1038/emboj.2009.285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Masuda M., Uno Y., Ohbayashi N., Ohata H., Mimata A., Kukimoto-Niino M., Moriyama H., Kashimoto S., Inoue T., Goto N., Okamoto K., Shirouzu M., Sawa M., and Yamada T. (2016) TNIK inhibition abrogates colorectal cancer stemness. Nat. Commun. 7, 12586 10.1038/ncomms12586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kayahara M., Wang X., and Tournier C. (2005) Selective regulation of c-jun gene expression by mitogen-activated protein kinases via the 12-O-tetradecanoylphorbol-13-acetate-responsive element and myocyte enhancer factor 2 binding sites. Mol. Cell Biol. 25, 3784–3792 10.1128/MCB.25.9.3784-3792.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Eilers A., Whitfield J., Babij C., Rubin L. L., and Ham J. (1998) Role of the Jun kinase pathway in the regulation of c-Jun expression and apoptosis in sympathetic neurons. J. Neurosci. 18, 1713–1724 10.1523/JNEUROSCI.18-05-01713.1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Minet E., Michel G., Mottet D., Piret J. P., Barbieux A., Raes M., and Michiels C. (2001) c-JUN gene induction and AP-1 activity is regulated by a JNK-dependent pathway in hypoxic HepG2 cells. Exp. Cell Res. 265, 114–124 10.1006/excr.2001.5180 [DOI] [PubMed] [Google Scholar]
- 47. Nadruz W. Jr., Kobarg C. B., Kobarg J., and Franchini K. G. (2004) c-Jun is regulated by combination of enhanced expression and phosphorylation in acute-overloaded rat heart. Am. J. Physiol. Heart Circ. Physiol. 286, H760–H767 10.1152/ajpheart.00430.2003 [DOI] [PubMed] [Google Scholar]
- 48. Zhang T., Inesta-Vaquera F., Niepel M., Zhang J., Ficarro S. B., Machleidt T., Xie T., Marto J. A., Kim N., Sim T., Laughlin J. D., Park H., LoGrasso P. V., Patricelli M., Nomanbhoy T. K., Sorger P. K., Alessi D. R., and Gray N. S. (2012) Discovery of potent and selective covalent inhibitors of JNK. Chem. Biol. 19, 140–154 10.1016/j.chembiol.2011.11.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Gandin V., Brina D., Marchisio P. C., and Biffo S. (2010) JNK inhibition arrests cotranslational degradation. Biochim. Biophys. Acta 1803, 826–831 10.1016/j.bbamcr.2010.03.016 [DOI] [PubMed] [Google Scholar]
- 50. Bogoyevitch M. A., and Kobe B. (2006) Uses for JNK: the many and varied substrates of the c-Jun N-terminal kinases. Microbiol. Mol. Biol. Rev. 70, 1061–1095 10.1128/MMBR.00025-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Gui J., Yang B., Wu J., and Zhou X. (2011) Enormous influence of TNIK knockdown on intracellular signals and cell survival. Hum. Cell 24, 121–126 10.1007/s13577-011-0023-2 [DOI] [PubMed] [Google Scholar]
- 52. Shkoda A., Town J. A., Griese J., Romio M., Sarioglu H., Knöfel T., Giehler F., and Kieser A. (2012) The germinal center kinase TNIK is required for canonical NF-κB and JNK signaling in B-cells by the EBV oncoprotein LMP1 and the CD40 receptor. PLos Biol. 10, e1001376 10.1371/journal.pbio.1001376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Fu C. A., Shen M., Huang B. C., Lasaga J., Payan D. G., and Luo Y. (1999) TNIK, a novel member of the germinal center kinase family that activates the c-Jun N-terminal kinase pathway and regulates the cytoskeleton. J. Biol. Chem. 274, 30729–30737 10.1074/jbc.274.43.30729 [DOI] [PubMed] [Google Scholar]
- 54. Li J. Y., Chai B. X., Zhang W., Liu Y. Q., Ammori J. B., and Mulholland M. W. (2007) Ankyrin repeat and SOCS box containing protein 4 (Asb-4) interacts with GPS1 (CSN1) and inhibits c-Jun NH2-terminal kinase activity. Cell Signal. 19, 1185–1192 10.1016/j.cellsig.2006.12.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Riedl A., Schlederer M., Pudelko K., Stadler M., Walter S., Unterleuthner D., Unger C., Kramer N., Hengstschläger M., Kenner L., Pfeiffer D., Krupitza G., and Dolznig H. (2017) Comparison of cancer cells in 2D vs 3D culture reveals differences in AKT-mTOR-S6K signaling and drug responses. J. Cell Sci. 130, 203–218 10.1242/jcs.188102 [DOI] [PubMed] [Google Scholar]
- 56. Shannan B., Chen Q., Watters A., Perego M., Krepler C., Thombre R., Li L., Rajan G., Peterson S., Gimotty P. A., Wilson M., Nathanson K. L., Gangadhar T. C., Schuchter L. M., Weeraratna A. T., Herlyn M., and Vultur A. (2016) Enhancing the evaluation of PI3K inhibitors through 3D melanoma models. Pigment Cell Melanoma Res. 29, 317–328 10.1111/pcmr.12465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Aihara A., Abe N., Saruhashi K., Kanaki T., and Nishino T. (2016) Novel 3-D cell culture system for in vitro evaluation of anticancer drugs under anchorage-independent conditions. Cancer Sci. 107, 1858–1866 10.1111/cas.13095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Jho E. H., Zhang T., Domon C., Joo C. K., Freund J. N., and Costantini F. (2002) Wnt/β-catenin/Tcf signaling induces the transcription of Axin2, a negative regulator of the signaling pathway. Mol. Cell Biol. 22, 1172–1183 10.1128/MCB.22.4.1172-1183.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Leung J. Y., Kolligs F. T., Wu R., Zhai Y., Kuick R., Hanash S., Cho K. R., and Fearon E. R. (2002) Activation of AXIN2 expression by β-catenin-T cell factor: a feedback repressor pathway regulating Wnt signaling. J. Biol. Chem. 277, 21657–21665 10.1074/jbc.M200139200 [DOI] [PubMed] [Google Scholar]
- 60. Lustig B., Jerchow B., Sachs M., Weiler S., Pietsch T., Karsten U., van de Wetering M., Clevers H., Schlag P. M., Birchmeier W., and Behrens J. (2002) Negative feedback loop of Wnt signaling through upregulation of conductin/axin2 in colorectal and liver tumors. Mol. Cell. Biol. 22, 1184–1193 10.1128/MCB.22.4.1184-1193.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Yook J. I., Li X. Y., Ota I., Hu C., Kim H. S., Kim N. H., Cha S. Y., Ryu J. K., Choi Y. J., Kim J., Fearon E. R., and Weiss S. J. (2006) A Wnt-Axin2-GSK3β cascade regulates Snail1 activity in breast cancer cells. Nat. Cell Biol. 8, 1398–1406 10.1038/ncb1508 [DOI] [PubMed] [Google Scholar]
- 62. Wu Z. Q., Brabletz T., Fearon E., Willis A. L., Hu C. Y., Li X. Y., and Weiss S. J. (2012) Canonical Wnt suppressor, Axin2, promotes colon carcinoma oncogenic activity. Proc. Natl. Acad. Sci. U.S.A. 109, 11312–11317 10.1073/pnas.1203015109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Veeman M. T., Axelrod J. D., and Moon R. T. (2003) A second canon: functions and mechanisms of β-catenin-independent Wnt signaling. Dev. Cell 5, 367–377 10.1016/S1534-5807(03)00266-1 [DOI] [PubMed] [Google Scholar]
- 64. Kohn A. D., and Moon R. T. (2005) Wnt and calcium signaling: β-catenin-independent pathways. Cell Calcium 38, 439–446 10.1016/j.ceca.2005.06.022 [DOI] [PubMed] [Google Scholar]
- 65. Qiu W., Chen L., and Kassem M. (2011) Activation of non-canonical Wnt/JNK pathway by Wnt3a is associated with differentiation fate determination of human bone marrow stromal (mesenchymal) stem cells. Biochem. Biophys. Res. Commun. 413, 98–104 10.1016/j.bbrc.2011.08.061 [DOI] [PubMed] [Google Scholar]
- 66. Hwang S. G., Yu S. S., Lee S. W., and Chun J. S. (2005) Wnt-3a regulates chondrocyte differentiation via c-Jun/AP-1 pathway. FEBS Lett. 579, 4837–4842 10.1016/j.febslet.2005.07.067 [DOI] [PubMed] [Google Scholar]
- 67. Rosso S. B., Sussman D., Wynshaw-Boris A., and Salinas P. C. (2005) Wnt signaling through Dishevelled, Rac and JNK regulates dendritic development. Nat. Neurosci. 8, 34–42 10.1038/nn1374 [DOI] [PubMed] [Google Scholar]
- 68. Zhang Y., Neo S. Y., Wang X., Han J., and Lin S. C. (1999) Axin forms a complex with MEKK1 and activates c-Jun NH2-terminal kinase/stress-activated protein kinase through domains distinct from Wnt signaling. J. Biol. Chem. 274, 35247–35254 10.1074/jbc.274.49.35247 [DOI] [PubMed] [Google Scholar]
- 69. Toualbi K., Güller M. C., Mauriz J. L., Labalette C., Buendia M. A., Mauviel A., and Bernuau D. (2007) Physical and functional cooperation between AP-1 and β-catenin for the regulation of TCF-dependent genes. Oncogene 26, 3492–3502 10.1038/sj.onc.1210133 [DOI] [PubMed] [Google Scholar]
- 70. Yochum G. S., Cleland R., and Goodman R. H. (2008) A genome-wide screen for β-catenin binding sites identifies a downstream enhancer element that controls c-Myc gene expression. Mol. Cell Biol. 28, 7368–7379 10.1128/MCB.00744-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Mann B., Gelos M., Siedow A., Hanski M. L., Gratchev A., Ilyas M., Bodmer W. F., Moyer M. P., Riecken E. O., Buhr H. J., and Hanski C. (1999) Target genes of β-catenin-T cell-factor/lymphoid-enhancer-factor signaling in human colorectal carcinomas. Proc. Natl. Acad. Sci. U.S.A. 96, 1603–1608 10.1073/pnas.96.4.1603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Staal F. J., Weerkamp F., Baert M. R., van den Burg C. M., van Noort M., de Haas E. F., and van Dongen J. J. (2004) Wnt target genes identified by DNA microarrays in immature CD34+ thymocytes regulate proliferation and cell adhesion. J. Immunol. 172, 1099–1108 10.4049/jimmunol.172.2.1099 [DOI] [PubMed] [Google Scholar]
- 73. Wu X., Tu X., Joeng K. S., Hilton M. J., Williams D. A., and Long F. (2008) Rac1 activation controls nuclear localization of β-catenin during canonical Wnt signaling. Cell 133, 340–353 10.1016/j.cell.2008.01.052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Hayakawa J., Mittal S., Wang Y., Korkmaz K. S., Adamson E., English C., Ohmichi M., Omichi M., McClelland M., and Mercola D. (2004) Identification of promoters bound by c-Jun/ATF2 during rapid large-scale gene activation following genotoxic stress. Mol. Cell 16, 521–535 10.1016/j.molcel.2004.10.024 [DOI] [PubMed] [Google Scholar]
- 75. Nateri A. S., Spencer-Dene B., and Behrens A. (2005) Interaction of phosphorylated c-Jun with TCF4 regulates intestinal cancer development. Nature 437, 281–285 10.1038/nature03914 [DOI] [PubMed] [Google Scholar]
- 76. Guzmán C., Bagga M., Kaur A., Westermarck J., and Abankwa D. (2014) ColonyArea: an ImageJ plugin to automatically quantify colony formation in clonogenic assays. PLoS ONE 9, e92444 10.1371/journal.pone.0092444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Cerami E., Gao J., Dogrusoz U., Gross B. E., Sumer S. O., Aksoy B. A., Jacobsen A., Byrne C. J., Heuer M. L., Larsson E., Antipin Y., Reva B., Goldberg A. P., Sander C., and Schultz N. (2012) The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2, 401–404 10.1158/2159-8290.CD-12-0095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Gao J., Aksoy B. A., Dogrusoz U., Dresdner G., Gross B., Sumer S. O., Sun Y., Jacobsen A., Sinha R., Larsson E., Cerami E., Sander C., and Schultz N. (2013) Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 6, pl1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Ciriello G., Gatza M. L., Beck A. H., Wilkerson M. D., Rhie S. K., Pastore A., Zhang H., McLellan M., Yau C., Kandoth C., et al. (2015) Comprehensive molecular portraits of invasive lobular breast cancer. Cell 163, 506–519 10.1016/j.cell.2015.09.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Cancer Genome Atlas, N. (2012) Comprehensive molecular portraits of human breast tumours. Nature 490, 61–70 10.1038/nature11412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Martelotto L. G., De Filippo M. R., Ng C. K., Natrajan R., Fuhrmann L., Cyrta J., Piscuoglio S., Wen H. C., Lim R. S., Shen R., et al. (2015) Genomic landscape of adenoid cystic carcinoma of the breast. J. Pathol. 237, 179–189 10.1002/path.4573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Curtis C., Shah S. P., Chin S. F., Turashvili G., Rueda O. M., Dunning M. J., Speek D., Lynch A. G., Samarajiwa S., Yuan Y., Graf S., Ha G., et al. (2012) The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature 486, 346–352 10.1038/nature10983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Pereira B., Chin S. F., Rueda O. M., Vollan H. K., Provenzano E., Bardwell H. A., Pugh M., Jones L., Russell R., Sammut S. J., Tsui D. W., et al. (2016) The somatic mutation profiles of 2,433 breast cancers refines their genomic and transcriptomic landscapes. Nat. Commun. 7, 11479 10.1038/ncomms11479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Eirew P., Steif A., Khattra J., Ha G., Yap D., Farahani H., Gelmon K., Chia S., Mar C., Wan A., Laks E., Biele J., et al. (2015) Dynamics of genomic clones in breast cancer patient xenografts at single-cell resolution. Nature 518, 422–426 10.1038/nature13952 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.