

Abstract
The histone variant CENP-ACse4 is a core component of the specialized nucleosome at the centromere in budding yeast and is required for genomic integrity. Accordingly, the levels of Cse4 in cells are tightly regulated, primarily by ubiquitin-mediated proteolysis. However, structural transitions in Cse4 that regulate its centromeric localization and interaction with regulatory components are poorly understood. Using time-resolved fluorescence, NMR, and molecular dynamics simulations, we show here that soluble Cse4 can exist in a “closed” conformation, inaccessible to various regulatory components. We further determined that binding of its obligate partner, histone H4, alters the interdomain interaction within Cse4, enabling an “open” state that is susceptible to proteolysis. This dynamic model allows kinetochore formation only in the presence of H4, as the Cse4 N terminus, which is required for interaction with other centromeric components, is unavailable in the absence of H4. The specific requirement of H4 binding for the conformational regulation of Cse4 suggests a structure-based regulatory mechanism for Cse4 localization. Our data suggested a novel structural transition–based mechanism where conformational flexibility of the Cse4 N terminus can control Cse4 levels in the yeast cell and prevent Cse4 from interacting with kinetochore components at ectopic locations for formation of premature kinetochore assembly.
Keywords: centromere, histone, structural biology, molecular dynamics, fluorescence anisotropy, cell division, histone tails, histone variants, protein-protein interactions, kinetochore assembly, centromeric protein A, chromosome
Introduction
The basis of successful cell division is the faithful segregation of sister chromatids during mitosis and meiosis, a process driven by the formation of the kinetochore complex on the centromere. Centromeres in most eukaryotes are identified by the formation of a specialized nucleosome(s) where the histone 3 (H3)4 is replaced by a unique variant centromeric protein-A (CENP-A) (1). In budding yeast, this variant, known as Cse4 (2), forms the specialized nucleosome (3, 4) at a single centromere that mediates the segregation of chromosomes (5). The localization of Cse4 at the centromere and its level in the cells have to be tightly regulated as altered localization and expression of this protein are known to cause genetic instability (6). Two distinct pools of Cse4 are present in the cell: the core Cse4 at the centromere that mediates kinetochore formation and a pericentromeric reservoir that provides Cse4 molecules in case of eviction of core molecules from the centromere (7). How these populations are maintained in the cells and how the pericentromeric Cse4 is localized at the centromere are not clearly understood. Ubiquitin-mediated proteolysis is one of the key mechanisms known to regulate Cse4 levels and maintain kinetochore function (8). Psh1 was identified as the E3 ligase that specifically recognizes the CENP-A–targeting domain (CATD) in Cse4 and prevents its misincorporation in the chromatin (9, 10). Recently, an evolutionarily conserved protein, Pat1, was identified that protects Cse4 from Psh1-mediated degradation and was proposed to maintain the population of pericentromeric Cse4 molecules at the kinetochore (11). Interestingly, in the above studies, Cse4 was not completely stabilized when Psh1 was deleted, and a lysine-free mutant of Cse4 was still degraded in the cell. Some other proteins like Doa1/Ufd3 and Rcy1 are also implicated to be essential for Cse4 proteolysis (12, 13). However, none of the mechanisms have shown complete regulation of the Cse4 levels. Thus, it is possible that there are other, ubiquitin-independent mechanisms in the cell that regulate the Cse4 levels.
We asked whether the conformation of the soluble Cse4 itself could be a determining factor in mediating its interaction with regulators and with the kinetochore machinery. The C terminus of Cse4 is essential for centromere targeting (14, 15) and has been the focus of many studies. The structure of the histone fold domain (HFD) within the C terminus of Cse4 in complex with histone 4 (H4) and the chaperone Scm3 has been solved (16–18), but very little is known about the conformation of the N terminus of Cse4. Cse4 has a unique, long N terminus, which harbors the essential N-terminal domain (END) (residues 28–60), required for interaction with other kinetochore proteins (19). We hypothesized that the 129-amino-acid–long N-terminal domain (NTD) could have a more specialized function than other histone tails and that a longer tail could have a structural role in regulating the association of Cse4 with other proteins. Using time-resolved fluorescence, NMR, and molecular dynamics simulations, we found that in the native state the NTD interacts with the C-terminal domain (CTD). Such an interaction constitutes what we call a “closed” conformation of the Cse4 monomer and to our knowledge has not been reported for any histone so far. Such a conformation may hinder the association of regulators with the CATD and/or that of kinetochore proteins with the NTD, thereby negating the untimely mistargeting of Cse4 within the nucleosome and to other ectopic loci. In addition, we investigated whether the closed conformation of Cse4 was altered in the presence of H4, its obligate partner in the specialized nucleosome (4, 20). We observed that H4 binding indeed curtailed the interaction between the NTD and CTD of Cse4. The interaction with H4 allows the transition to an open conformation, permitting the interaction of Cse4 with kinetochore proteins to assemble a kinetochore and allowing the degradation machinery to access the CTD in case of any inadvertent mislocalization. Our results suggest a novel structure-based mechanism based on the conformational flexibility of the N terminus to regulate the levels of Cse4 in the cell and to resist premature kinetochore assembly.
Results
N terminus of Cse4 is conformationally restricted
To understand the effect of other centromeric components on the NTD of Cse4, it is vital to study its behavior in the context of the Cse4 monomer. Cse4 contains two Trp residues in its sequence, Trp-7 (in the NTD), which according to the modeled structure (21) is in a disordered region, and Trp-178 (part of the HFD), which is surrounded by the side chains of neighboring residues; hence, a clear difference in the fluorescence parameters of the residues is expected in the native state. The two Trp residues of Cse4 were used as intrinsic fluorescent probes by creating the single mutants W7L and W178L. In the Cse4 W178L mutant, the fluorescence signal will be derived solely from the N terminus (Trp-7), whereas the W7L mutant will report the behavior of the C terminus (Trp-178) (Fig. 1a). The secondary structure of both mutants (W7L and W178L) was comparable with the WT Cse4 (Fig. S1). Surprisingly, the fluorescence lifetime values for the Trp residues at the NTD and the CTD did not show a significant difference (Fig. 1b and Table 1), indicating that the two residues experience similar microenvironments. As an additional measure, we calculated the solvent accessibility of the Trp residues where the N terminus should show a higher value of the bimolecular rate constant for quenching (kq) (22) compared with the C terminus if Trp-7 is solvent-exposed. However, the difference in rate constants between W178L (3.8 × 109 m−1 s−1) and W7L (2.9 × 109 m−1 s−1) was not significant (Fig. 1b), but the kq for the respective Trp residues was higher in the denatured state (Fig. S1). Thus, it can be concluded that Trp-7 is not completely solvent-exposed, implying that the NTD is conformationally restricted in the Cse4 monomer.
Figure 1.

CENP-ACse4 N-terminal tail is restricted. a, strategy for the fluorescence assay to create single Trp mutants in Cse4 to study the two domains individually. The END and HFD are highlighted in red. NTD and CTD correspond to residues 1–129 and 130–229, respectively. Note that according to a proposed model, Trp-7 is in a disordered region, implying that it is expected to have more conformational freedom than Trp-178. b, comparison of fluorescence lifetimes (filled bars) and solvent accessibility (striped bars) of the two domains. c, conformational flexibility of the two Trp residues in the native state. Filled bars represent φ1, and the striped bars represent β1. d, gel-filtration profile of folded Cse4 protein (solid line). The dotted lines represent the protein markers ovalbumin (O; 44 kDa) and carbonic anhydrase (CA; 29 kDa). The Cse4 structure represented in all figures is the modeled structure by Bloom et al. (Protein Data Bank (PDB) code 2FSC; Ref. 21) and is rendered in PyMOL 1.8. The statistical significance was calculated by one-way analysis of variance: **, p < 0.01; NS (not significant), p > 0.05; error bars represent S.D. A.U., arbitrary units.
Table 1.
Fluorescence intensity decay parameters for N terminus (W178L) and C terminus (W7L) in native state and in presence of denaturant (D) (8 m urea)
| Mutant | τ1 (ns) (α1) | τ2 (ns) (α2) | τ3 (ns) (α3) | τm (ns) |
|---|---|---|---|---|
| N terminus | ||||
| W178L | 0.5 (0.37) | 1.7 (0.48) | 4.8 (0.15) | 1.7 ± 0.1 |
| W178L + D | 0.56 (0.26) | 1.58 (0.52) | 4.4 (0.22) | 1.9 ± 0.08 |
| C terminus | ||||
| W7L | 0.5 (0.37) | 1.7 (0.5) | 4.8 (0.13) | 1.7 ± 0.02 |
| W7L + D | 0.67 (0.31) | 1.8 (0.51) | 4.9 (0.18) | 2.0 ± 0.28 |
To gain further insight, the conformational flexibility of the Trp residues was probed using time-resolved fluorescence anisotropy. The local motion of the Trp in the protein (short correlation time; φ1) and the global tumbling motion of the entire protein (long correlation time; φ2) contribute to the fluorescence anisotropy decay (23). φ1 offers information about the site-specific conformational flexibility of the protein; higher values for φ1 and/or smaller values of its amplitude, β1, indicate reduced flexibility (0 implying completely rigid). According to the Cse4 modeled structure (21), Trp-7 is free to rotate, and Trp-178 is restricted by neighboring residues (Fig. 1a), but we observed no variation in the φ1. A slight change in β1 suggested that Trp-7 had more freedom to rotate than Trp-178, but it still did not explain the difference expected between a completely restricted Trp and a free Trp (Fig. 1c and Table 2). The microenvironment and conformational flexibility of the NTD indicate that the tail is not completely free as believed to be in the nucleosome structure. A single peak corresponding to the molecular weight of Cse4 in gel filtration (Fig. 1d) and the long component of the anisotropy decay for both mutants (∼15 ns) (Table 2) are consistent with a protein of molecular mass of ∼30 kDa, excluding the presence of Cse4 dimers or higher-order structures resulting from intermolecular interactions that may restrict the NTD. Thus, the observed behavior of Trp-7 is either due to local structural restrictions near Trp-7 or an interaction between the N terminus and C terminus in the native state. Both these states could conceivably hamper the interaction of the domains with regulators as well as other centromere components. Thus, both these possibilities were further investigated.
Table 2.
Parameters for fluorescence anisotropy decay for N terminus (W178L) and C terminus (W7L) in native state
| Mutant | φ1 (ns) | β1 | φ2 (ns) | β2 |
|---|---|---|---|---|
| N terminus | ||||
| W178L | 0.19 ± 0.01 | 0.75 ± 0.05 | 14.05 ± 1.5 | 0.25 ± 0.04 |
| C terminus | ||||
| W7L | 0.16 ± 0.02 | 0.55 ± 0.05 | 14.34 ± 0.8 | 0.45 ± 0.05 |
Monomeric Cse4 exhibits interdomain interaction
To understand whether conformational restriction of the NTD was caused by interdomain interactions between N and C termini or by local structures formed at the N terminus itself, the Leu residues were changed to Ala residues in the above-mentioned Trp mutants. In the event of local structural interactions within the NTD, the change in the amino acid residue at the CTD (W178L to W178A) would not affect the fluorescence parameters of Trp at the N terminus. In contrast, if the two domains interact, change at one terminus will have an effect on the other. It was observed that fluorescence lifetimes of the Ala mutants were significantly different from those of the Leu mutants at the C terminus (Fig. 2a and Table S4), indicating that the mutation had altered the microenvironment of the Trp residue. The mutation causes local structural changes at the C terminus as there is a difference in the side-chain lengths between Ala and Leu, which is reflected in the lifetime values. Similarly, there was a difference in the fluorescence anisotropy values for Trp in the two sets of mutants especially at the N terminus (Table S5) that showed higher conformational flexibility. The β1 for W178A showed a slightly smaller value than for W178L (Fig. 2b). A higher value of β1 indicates higher flexibility at the N terminus, suggesting that Trp-7 has more freedom to rotate than Trp-178, but it does not equate to the difference expected between a completely restricted and relatively free Trp residue. The Leu-to-Ala mutation caused a change in the microenvironment and the flexibility of the two Trp residues, suggesting that the two domains of Cse4 interact with each other. A significant difference was also observed in the kq values between the two mutants at the NTD, also pointing toward an interaction between the two termini (Fig. 2c).
Figure 2.

CENP-ACse4 N-terminal tail interacts with C terminus. a, comparison between the fluorescence lifetime values of the Ala and Leu mutants for the two domains. b, β1 associated with the short correlation time for each mutant. c, solvent accessibility of the two domains in the Ala and Leu mutants. d, overlap of the structure of Cse4 at the start (space-filled) and end (ribbon) of all four simulations. The conformation of the N terminus changes from extended to closed even though the four simulations end in different conformational basins. e, change in the conformation of Trp-7 throughout simulation 1. f, conformation of Trp-178 at the start and end of the simulation. g, 1H-15N HSQC spectrum of folded Cse4. In all graphs, X represents the mutant type with blue and green representing Leu and Ala mutants, respectively. Trp residues are shown in yellow, and N-terminal and C-terminal residues are shown in blue and red sticks, respectively. The statistical significance was calculated by one-way analysis of variance: *, p < 0.05; **, p < 0.01; NS (not significant), p > 0.05; error bars represent S.D.
Interdomain interaction was also observed in four independent atomistic molecular dynamics (MD) simulations of 300 ns each for monomeric Cse4 (Movie S1). Although the four simulations ended up in different conformations at the end of 300 ns (Fig. 2d and Fig. S2a), the rotational flexibility of the side chain of Trp-7 was not hindered as it interacted with different residues (Fig. 2e). Throughout the simulation, the Trp-7 side chain remained flexible in all four conformations (Fig. S2b); this can explain the increased β1 observed for the N-terminal Trp. However, the HFD residues in the vicinity (<4 Å) of Trp-178 did not change throughout the simulation, and in some simulations residues from the NTD were also observed in the proximity of Trp-178 (Fig. 2f). Thus, Trp-178 was not as flexible as Trp-7, which is reflected by a minimal change in the anisotropy values at the CTD. The interaction between the domains was also evident from the multiple contact points between the NTD and CTD (Fig. S3a) and reduced distance between the Cα atoms of Trp-7 and Trp-178 (Fig. S3, b and c) in all four simulations.
NMR spectroscopy was used to obtain insight into residue-specific dynamics and structure of the NTD. The 1H-15N HSQC spectrum of Cse4 showed fewer than the expected number of peaks (224 non-Pro peaks) (Fig. 2g). However, due to inadequate sample concentration, NMR assignments could not be completed (Fig. S4). We believe that the residues involved in the interaction between the two domains broadened due to exchange, leaving only the peaks of the noninteracting residues visible in the spectrum.
H4 binding stabilizes CENP-ACse4
Histone tails and their cores are known to have distinctive structures and functions. We wanted to determine whether the two domains of Cse4 also behave independently as this would affect their interactions with other proteins. To assess whether the domains showed distinctive behavior during folding, Cse4 was denatured, and changes in the residue-wise secondary structure and dynamics of the protein were monitored by NMR as the denaturant (8 m urea) was diluted. NMR signals were observed to shift considerably, and peak broadening was seen at lower urea concentrations (Fig. S5a). Resonances from the CTD started disappearing at 5 m urea, whereas those from NTD, although shifted, were still visible (Fig. 3a), signifying that CTD residues undergo conformational exchange earlier than those at the N terminus. At 4 m urea, the protein appeared to be in a molten globule state (Fig. S5a). The analysis of the peaks from the two domains also confirmed the absence of any oligomeric states formed during protein folding, which could have affected the calculated fluorescence parameters. To quantify the changes during folding, residue-wise secondary structural propensities were calculated. The propensities at 6 m urea (Fig. S5b) were similar to the protein in 8 m urea (24). But significant changes were seen at 5 m urea concentration where patches of the NTD showed helical propensities (Fig. 3b). Although the protein is not in the native conformation, the secondary structural propensities still give information about the structural rearrangements within the N-terminal residues. The results indicate that the NTD is not completely disordered but undergoes structural transitions independently of the CTD. It should be noted that these may or may not be the native propensities of the N-terminal residues, and further rearrangements are possible in the native state as observed earlier (25). The comparison between the 15N transverse relaxation rates (R2) of Cse4 in 8 and 6 m urea showed a considerable increase in the values for the residues 30–51 and 190–205 (Fig. S5c and Fig. 3c). The average difference in R2 in the NTD (residues 1–129) was 2.45 ± 0.1 s−1, and that for the region 30–51 was 3.1 ± 0.3 s−1 and for 190–205 was 5.1 ± 0.4 s−1. These residues belong to the critical END and HFD regions of Cse4 (Fig. 3d), demonstrating that biologically essential regions may serve as early nucleation sites for Cse4 folding. Cse4 contains five glycine residues, which are distributed throughout the 229-amino-acid sequence (Gly-18, Gly-29, Gly-79, Gly-196, and Gly-226). As a representative for both domains, these Gly residues were used as markers to map the dynamics of the different regions of Cse4 (Fig. 3, d and e). The increase in R2 for Gly-196 and the absence of peaks at 4 m urea reinforce the point that HFD is the early nucleation site for folding. The Gly residues at the NTD (Gly-18 and Gly-29) showed a consistent increase in R2 from 8 to 4 m urea, indicating increased rigidity and/or conformational exchange that may result from its interaction with the CTD; residue Gly-79 does not change significantly, suggesting that it is not involved in the interaction. This trend was seen in their intensity and positions where the peaks from NTD Gly residues were shifted, but those from CTD had broadened in the 5 m urea spectra (Fig. 3a, inset).
Figure 3.

H4 stabilizes CENP-ACse4. a, representative regions of 1H-15N HSQC spectra of Cse4 at different urea concentrations. Inset, peak positions of five Gly residues in similar conditions. b, structural propensities of Cse4 in 5 m urea. Plots of secondary chemical shifts from ΔδCα, ΔδCO, and ΔδHα of residues Asp-15 to Ser-18 (not marked), 30–42, and 75–78 show helical propensity (gray bars). c, difference between the residue-wise R2 rates of Cse4 in 8 and 6 m urea. Residues showing maximum difference for each domain are marked in red. d, modeled Cse4 structure (21) shows the position of residues showing higher R2 difference (red). Note that they are a part of the END and HFD regions. The Gly residues are shown in violet. e, R2 for Gly residues in different denaturant concentrations. f, overlapped regions of 1H-15N HSQC spectra of Cse4 and Cse4–H4 in 4 m urea buffer. Gly residues are marked to show broadening of the C-terminal residues. Inset, Trp side-chain peaks. g, 1H-15N HSQC spectrum of Cse4–H4 complex without denaturant. h, overlap between 1H-15N HSQC spectra of Cse4 and Cse4–H4 in the native state. The arrows indicate reappearance of the NTD Gly residues in the Cse4–H4 spectrum. i, overlap of the structure of Cse4–H4 at the start (space-filled) and end (ribbon) of simulation 1. The N terminus does not fold back on the C terminus. j, arrangement of the C-terminal helices with and without H4 binding at the start (Cse4, blue; H4, orange) and end (Cse4, cyan; H4, brown) of the simulation.
Next, we checked whether the presence of H4 stabilized any of the domains of Cse4. We observed an increase in the soluble fraction when uniformly 15N-labeled Cse4 was cofolded with unlabeled H4 upon complete removal of denaturant. The 15N-labeled Cse4–H4 complex was purified to yield the heterodimers (≈38 kDa), which require specialized experiments like transverse relaxation optimized spectroscopy (TROSY) and/or partial deuteration to observe sharper line widths. However, the sample concentration of Cse4 was a limiting factor. Nevertheless, important structural information about the two domains could still be acquired. At 4 m urea, the resolution of the Cse4 spectrum increased upon cofolding with H4 (Fig. 3f and Fig. S6). Gly-196 disappeared, and only one Trp side-chain peak was visible, whereas the NTD Gly resonances superimposed with the Cse4 monomer spectrum, suggesting that the C terminus was interacting with H4. This trend continued with complete removal of the denaturant, and the number of peaks increased in 15N-labeled Cse4–H4 (0 m urea) compared with Cse4 monomer (Figs. 3g and 2g). The NTD Gly resonances reappeared in the 15N-labeled Cse4–H4 sample, indicating a conformational change after H4 binding (Fig. 3h). The Cse4 C terminus has been shown to interact with H4 (16); thus, it is possible that CTD residues had broadened beyond detection, and the NTD residues were reappearing because of the “release” of the N terminus from the interaction with the C terminus when cofolded with H4.
The NTD did not interact with the CTD in any of the three MD simulations of the Cse4–H4 complex (Fig. 3i, Movie S2, and Fig. S7a). The distance between the Trp residues was >4.8 nm in the presence of H4, indicating a lower probability of contact between the C and N termini (Fig. S7b), which was also evident in the contact maps (Fig. S7c). The α-N and α-I helix that were dislocated at the C terminus in the monomeric Cse4 simulations remained rigid in Cse4–H4 simulations with slight displacement in α-N (Fig. 3j). The CTD of Cse4 was stabilized by the presence of H4. This is critical not only for nucleosome structure integrity but also for the various protein interactions where the Cse4 CTD is the binding interface. The structural rearrangement offers a plausible way of regulation at the centromere where the CTD is oriented correctly and the NTD is “free” to interact with other kinetochore proteins only upon H4 binding.
H4 binding alters the conformation of the NTD
Cofolding of Cse4 and H4 indicated that the NTD no longer interacted with the CTD, perhaps as a consequence of H4 binding at the C terminus, as shown by the MD and NMR experiments. Next, we investigated whether the binding of H4 to prefolded Cse4 could alter the conformation of the NTD, which may be relevant in its interaction with other kinetochore proteins. H4 forms a heterodimer with Cse4 in the specialized nucleosome, and its interaction with Cse4 CTD is well documented; however, the studies used truncated proteins (16–18). The binding affinity of the natively folded full-length constructs of Cse4 and H4 was determined using fluorescence spectroscopy (fluorescein isothiocyanate (FITC) labeling) and found to be 42 ± 9.1 nm (Fig. S8). Such high affinities are expected for histone interactions. Next, we probed the change in conformation when H4 was added to native Cse4 to assess whether it could free the NTD from the CTD. There was a change in fluorescence lifetime of Trp-178 at the CTD (Fig. 4a and Table 3), but the change at the NTD was not significant, suggesting that H4 binds to the CTD and that the residues of the NTD were not involved. The flexibility of the CTD decreased slightly (Fig. 4, b and c) as the side chains of surrounding residues within Cse4 restrict Trp-178 (Fig. 2f), and H4 binding caused only a small change in the degree of its rotational flexibility (Fig. S9). Surprisingly, the flexibility of the N-terminal Trp-7 decreased significantly as indicated by an increase in φ1 on H4 binding (Fig. 4b), signifying a local conformation change at the NTD upon H4 binding. The lower β1 for the NTD in the presence of H4 also indicates reduced rotational freedom, which did not vary significantly for Trp-178 (Fig. 4c and Table 4). Addition of H4 dramatically increased the solvent accessibility of Trp-7 (Fig. 4d), proving that H4 could free the NTD from interaction with the CTD. It should be noted that the dynamics of the Ala and Leu mutants differ slightly (Fig. S10). The Ala mutants did not show much reduction in the amplitude of the short correlation time (Table 4), suggesting the presence of some transient interactions between the two domains. H3 failed to induce a similar change in solvent accessibility of the NTD (Fig. 4e), indicating that the effect is specific for H4. This observation has important consequences regarding the regulation of Cse4 targeting and functioning at the centromere. Only the specific partner can alter the conformation of the NTD such that it will be available for interaction with other proteins.
Figure 4.

H4 binding alters NTD conformation. a, comparison between the fluorescence lifetime values of Cse4 and Cse4–H4 for the two domains. b, change in conformational flexibility of the Trp at the two domains with and without H4. c, β1 associated with the short correlation time for each mutant. d, solvent accessibility of Trp residues at the two domains in Cse4 and Cse4–H4. e, solvent accessibility of Trp residues at the two domains in Cse4 and Cse4–H3. f, overlap of 1H-15N HSQC spectra of Cse4–H4 (light blue) and Cse4(1–129) (purple). Inset, position of Trp side-chain peaks in Cse4, Cse4–H4, and Cse4ΔC. g, comparison of the residue-specific CSPs calculated for the NTD upon addition of H4 (orange histogram) and the change in intensity profile upon titration with CTD (blue scatter plot). Gray bars represent the residues involved in interaction with both the CTD and H4. Some residues show significant CSP but do not interact with the CTD (highlighted by red circles). In the graphs, blue, orange, and brown represent Cse4, Cse4–H4, and Cse4–H3, respectively. Mutants are specified at the x axis. NMR spectra are shown for Cse4–H4 (light blue), Cse4 (pink), and Cse4ΔC (residues 1–129) (purple). The statistical significance was calculated by one-way analysis of variance: *, p < 0.05; **, p < 0.01; NS (not significant), p > 0.05; error bars represent S.D.
Table 3.
Fluorescence intensity decay parameters of Cse4 mutants in presence of H4
| Mutant | τ1 (ns) (α1) | τ2 (ns) (α2) | τ3 (ns) (α3) | τm (ns) |
|---|---|---|---|---|
| W178L + H4 | 0.33 (0.4) | 1.5 (0.4) | 4 (0.2) | 1.7 ± 0.09 |
| W7L + H4 | 0.46 (0.4) | 1.8 (0.4) | 4.4 (0.2) | 1.8 ± 0.04 |
| W178A + H4 | 0.4 (0.45) | 1.7 (0.4) | 4.5 (0.15) | 1.6 ± 0.05 |
| W7A + H4 | 0.5 (0.5) | 2 (0.36) | 4.6 (0.14) | 1.6 ± 0.07 |
Table 4.
Fluorescence anisotropy decay parameters of Cse4 mutants in presence of H4
| Mutant | φ1 (ns) | β1 | φ2 (ns) | β2 |
|---|---|---|---|---|
| W178L + H4 | 0.33 ± 0.01 | 0.42 ± 0.06 | >20 | 0.58 ± 0.06 |
| W7L + H4 | 0.17 ± 0.03 | 0.49 ± 0.02 | 15 ± 2 | 0.51 ± 0.02 |
| W178A + H4 | 0.3 ± 0.06 | 0.45 ± 0.08 | >20 | 0.55 ± 0.08 |
| W7A + H4 | 0.2 ± 0.03 | 0.47 ± 0.03 | 18.6 ± 2.8 | 0.53 ± 0.05 |
Next, we recorded the 1H-15N HSQC spectrum for truncated Cse4 NTD (Cse4ΔC; 1–129) to examine whether the conformation of the NTD in full-length Cse4 actually shifted toward a state similar to free NTD upon addition of H4. The majority of the peaks were superimposable between the 1H-15N HSQC spectra of Cse4–H4 and Cse4ΔC (residues 130–229 deleted) (Fig. 4f), confirming that the peaks that had broadened out in the full-length Cse4 spectrum were from CTD and that the NTD was free in the Cse4–H4 complex. The Trp-7 side-chain peak for Cse4–H4 shifted closer to the Cse4ΔC than the full-length Cse4 (Fig. 4f, inset), showing that the NTD conformation was closer to its “open” state when H4 was added. Once the conformational plasticity was confirmed, we analyzed the residues involved in this interaction. Chemical shift perturbation (CSP) was observed for residues 20–28, 39–41, and 62–66 when Cse4ΔC was titrated against H4, indicating weak interaction between the two proteins. Residues 94–129 also show CSP, but the resonances of all residues could not be included because of overlap in the spectrum. Interestingly, the residues that interact with H4 were also found to be a subset of those interacting with the CTD (7–51, 63–65, and 108–129) as seen by significant intensity change in the resonances that indicate intermediate to strong interaction (Fig. 4g). Thus, the NTD residues that are involved in relatively strong binding with the CTD also have a weak affinity toward H4 that may help in “opening” the protein once H4 is added. This also explains the rigidity seen at the NTD upon addition of H4. The residue-wise interactions imply that the first step in the interaction between Cse4 and H4 would involve a weak affinity of H4 toward the Cse4 NTD, which may help to dislodge the NTD from CTD before H4 stably binds to the C terminus.
Discussion
The role of Cse4 as an epigenetic marker for centromere identity is well established (26, 27). A conserved CATD consisting of loop 1 and helix 2 of the histone fold domain in Cse4 is sufficient for maintaining centromere identity (14, 15). The N-terminal tail of Cse4 is dispensable for centromere targeting (28, 29), but the deletion of the first 50 residues of Cse4 is lethal to cells (30). A 33-residue stretch (28–60) that is required for interaction with other kinetochore proteins has been shown to be indispensable for cell survival (19), and its role in the regulation of Cse4 levels in the cell is increasingly becoming more apparent (12). The linear separation of the END from the HFD is not relevant as the END fused directly to the CTD has been shown to confer WT-like functions (19). However, some post-translational modifications are known to occur at the NTD that regulate chromosome segregation and kinetochore integrity (31, 32). Thus, the actual significance of the length of the NTD in Cse4 and its structural organization is not clear.
We demonstrated here that, in its soluble form, Cse4 exists in a closed conformation as a result of interaction between the NTD and CTD. This interaction causes a change in the HFD of the CTD where the α-N and α-I are dislocated. We propose that this change in the positions of helices of the CTD may interfere with Psh1 binding by changing the binding interface between the two proteins as Psh1 is known to interact with the CATD region of Cse4 and ubiquitinates four lysine residues in the CTD (9). A peptidyl-prolyl cis-trans isomerase, Fp3, is required to facilitate Psh1-mediaited degradation (33). It is hypothesized that in the cellular environment the interconversion of Pro-134 of Cse4 by this isomerase from the “cis” form to the “trans” form ensures that any soluble Cse4 is targeted toward Psh1-mediated degradation if not protected by the chaperone Scm3. Our data demonstrate a closed conformation of Cse4 where the target residues for Psh1 (Lys-131, Lys-155, Lys-163, and Lys-172) might be inaccessible (similar to the cis form), rendering Cse4 resistant to Psh1-mediated degradation. Our observations thus provide a rationale for the background levels of Cse4 observed in various cellular and biochemical assays. How this closed conformation subsequently interacts with the chaperone protein Scm3 will be interesting to study given the fact that Scm3 deposits the dimer/tetramer onto DNA (34). Crucially, in this conformation, the NTD will not be free to interact with any kinetochore components, and this could potentially negate the free monomeric Cse4 to nucleate kinetochore formation to any ectopic chromatin sites. Our data present structural insights on retention of “inert” soluble Cse4 molecule in the cell that is protected from proteolytic machinery and is also incapable to make any centromeric contacts. This indicates a novel mechanism of safeguarding Cse4 monomer, soon after its biogenesis, from proteolysis and mistargeting (Fig. 5).
Figure 5.
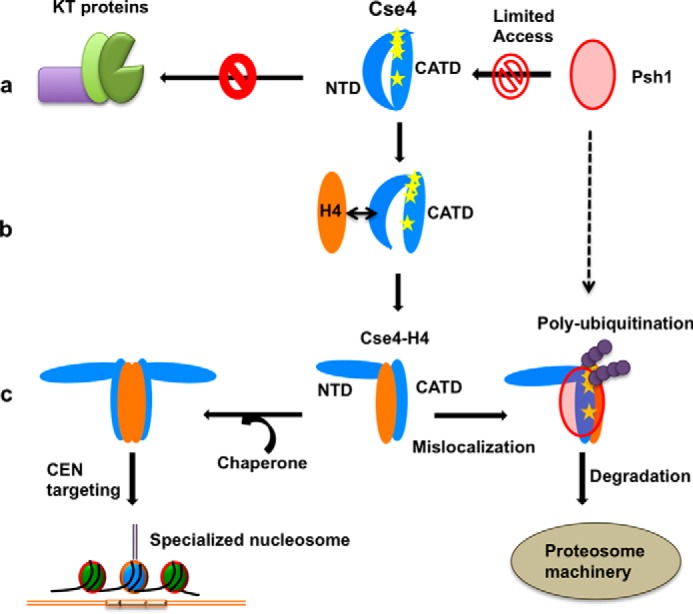
Proposed role of the NTD conformational change on regulation of Cse4. a, the closed state of the Cse4 NTD prevents interaction with kinetochore (KT) proteins. It also alters the positions of the helices in the C terminus, thereby masking the binding sites (Lys residues highlighted as yellow stars) for Psh1. b, a possible transient state when H4 comes in contact with Cse4. The affinity of NTD residues for H4 can help in dislodging the NTD from the CTD before H4 stably binds to the C terminus. c, upon H4 binding, the NTD adopts an open conformation. The Cse4–H4 dimer/tetramer can be deposited on the centromere by the chaperone Scm3 where the NTD is free to interact with the kinetochore. The C-terminal helices are reoriented in a manner that facilitates ubiquitination and further degradation if Cse4–H4 mislocalizes to an ectopic location.
We observed that the conformational flexibility of both domains of Cse4 is regulated by H4 binding. The positions of the α-N and α-I helices are retained close to their possible conformation in the nucleosome in H4-bound simulations. A recent study has shown that H4 facilitates the proteolysis of Cse4 by affecting its interaction with Psh1 (35). Our data are in agreement with this report; the dissociation of the NTD from the CTD due to H4 binding would expose Psh1-binding sites, and the complex would be targeted for degradation if localization to an ectopic site occurs. We report that the interaction of H4 at the CTD causes a structural change at the NTD, which is evident by an increase in solvent accessibility and the overlap of 15N-HN resonances of free NTD and Cse4–H4 complex spectra. Crucially, H3, which is structurally similar to H4, does not change the NTD conformation, indicating that this interaction is specific to H4. The interaction between the Cse4 CTD and H4 has been characterized in previous studies, and our own calculations with the full-length Cse4 and H4 show strong binding between the two proteins. Our MD data show a very stable NTD–CTD interaction in Cse4. Here, we show that in addition to the already characterized interaction with the CTD, H4 might also be involved in a transient interaction with the NTD that can facilitate the Cse4 open conformation. The structural rearrangement observed here could be one of the mechanisms of regulation of specialized nucleosome formation at the right time. Only upon H4 binding to inert Cse4 is the NTD released for interaction with the kinetochore proteins or possibly even with DNA. This release will allow the NTD to interact with various proteins at different time points in response to cell cycle cues. Overall, this process prevents premature nucleation of the kinetochore assembly in the absence of H4, although further experiments are required to verify this. In conclusion, this study reveals that conformational flexibility of the NTD may act as a regulator for the correct localization of Cse4 in cells, thereby preventing Cse4 from interacting with kinetochore components at ectopic locations.
Experimental procedures
Plasmids and mutagenesis
Yeast Saccharomyces cerevisiae Cse4 full-length protein cloned in pKS387 plasmid and histone 4 and histone 3 (S. cerevisiae) cloned in pET3a were kindly provided by Dr. K. Luger (University of Colorado Boulder). Single tryptophan mutants of Cse4 protein were created by site-directed mutagenesis using a Kppa HiFi PCR kit (Kapa Biosystems, Wilmington, MA). The mutants were selected by DpnI digestion (New England Biolabs). Plasmid DNA used for PCR was construct of pKS387 vector containing a full-length Cse4 insert. Tryptophan mutations were made at positions 7 and 178 (W7A, W7L, W178A, and W178L). The truncated Cse4 constructs Cse4ΔC (N terminus; 1–129) and Cse4ΔN (C terminus; 130–229) were cloned in pGEX6 vector. The clones have a GST tag with a PreScission protease cleavage site.
Protein purification
The purification of H4, H3, Cse4, and its mutants was carried out according to the protocol of Luger et al. (36). Briefly, the induced cells were harvested and incubated with lysozyme at room temperature. The lysate was sonicated and centrifuged at 20,000 × g for 20 min. The pellet obtained was washed twice with wash buffer (20 mm Tris, pH 7.5, 100 mm NaCl, 1 mm EDTA, 1 mm phenylmethylsulfonyl fluoride) containing 1% Triton X-100 and then washed again with wash buffer without Triton X-100. The remaining inclusion-body pellet was dissolved in guanidine hydrochloride (7 m). The sample was dialyzed against SAU-200 buffer (20 mm sodium acetate, pH 5.5, 8 m urea, 200 mm NaCl, 1 mm EDTA, 5 mm BME) and applied to an SP SepharoseTM Fast Flow column (GE Healthcare); the remaining impurities were removed by gel filtration (Sepharose 200). Protein purity was checked by SDS-PAGE. The proteins were dialyzed against distilled water with 10 mm BME, and the secondary structure of the proteins was checked by circular dichroism (CD) spectroscopy. For experiments with different urea concentrations, the proteins were dialyzed in sodium acetate buffer containing the required concentration of urea. The samples were maintained at pH 7 for all experiments and at pH 6.5 for NMR experiments.
The purification of GST-tagged N terminus (Cse4ΔC) and C terminus (Cse4ΔN) was done using the Sepharose 4B system (GE Healthcare). The cell supernatant was kept for binding with the beads for 3 h at 4 °C in lysis buffer (20 mm phosphate, pH 8, 100 mm NaCl, 1 mm EDTA). The beads were washed with wash buffers containing increasing amounts of salt (100, 300, and 500 mm NaCl). PreScission protease was used to cleave the GST tag from the protein. The purity was checked by SDS-PAGE. Cse4ΔC was maintained at pH 5.5 for NMR experiments.
Steady-state fluorescence experiments
Fluorescence measurements were performed on a PerkinElmer Life Sciences spectrofluorometer equipped with a data recorder and a temperature-controlled cell holder. The fluorescence spectra were measured at a protein concentration of 20 μm with a 1-cm–path-length cell and at constant temperature (25 °C). The samples were excited at 280 nm, and emission spectra were recorded in the 290–500-nm range. The excitation and emission slit width were set to 3 nm.
CD measurements
Secondary structure of the different proteins was analyzed using CD. Proteins (20–30 μm) in the respective buffers were used. Far-UV CD spectra of the proteins at 25 °C were recorded on a Jasco J-1500 CD spectrometer (Easton, MD). The samples were probed using 0.1-cm–path-length quartz cell (Starna, Hainault, London, UK) using a 1-nm bandwidth. For samples containing urea, scans were acquired from 210 to 260 nm; for all other samples, a 198–260-nm wavelength range was used. For signal averaging, three independent readings were taken. Raw data were processed by spectral smoothing and subtraction of respective buffers.
Time-resolved fluorescence measurements
The time-resolved fluorescence intensity measurements as well as anisotropy decay experiments were carried out using a rhodamine 6G dye laser (Spectra Physics, Mountain View, CA) pumped by a neodymium-doped yttrium aluminum garnet (Nd:YAG) laser (Millenia X, Spectra Physics) and a time-correlated single-photon counting setup coupled to microchannel plate photomultiplier (model R2809u, Hamamatsu Corp,). Pulses (1-ps duration) of 885 nm radiation from the rhodamine 6G dye laser were frequency-tripled to 295 nm by using a frequency doubler (GWU, Spectra Physics). The samples were excited at 295 nm, and the emission was measured at the emission maxima (λmax) of the respective proteins, determined from their steady-state fluorescence spectra. All the measurements were carried out on 30–50 μm samples. The instrument response function was obtained at a wavelength of 295 nm using a diluted colloidal solution of nondairy coffee whitener. For time-resolved fluorescence intensity decay experiments, peak counts of 10,000 were collected with the emission polarizer oriented at the magic angle (54.7°) with respect to excitation polarizer. In time-resolved fluorescence anisotropy decay experiments, peak counts of 10,000 were collected with emission polarizer oriented at 0° (parallel) and 90° (perpendicular) with respect to excitation polarizer.
Calculation of the mean fluorescence lifetime
The fluorescence lifetime was analyzed by a method based on the Levenberg–Marquardt algorithm (37). The observed decay was deconvoluted with the instrument response function to obtain the intensity decay function represented as a sum of three exponentials.
| (Eq. 1) |
where I(t) is the fluorescence intensity collected with the emission polarizer oriented at magic angle (54.7°) at time t, and αi is the amplitude of the ith lifetime τi such that ∑αi = 1.
The mean fluorescence lifetime is calculated as follows.
| (Eq. 2) |
The goodness of fits was assessed from the reduced χ2 values and from the randomness of the residuals obtained from analysis.
Fluorescence anisotropy decay kinetics
The anisotropy was calculated from experimentally obtained I‖(t) and I⊥(t) using the following equation.
| (Eq. 3) |
where r(t) is the time-dependent anisotropy, I‖(t) is the fluorescence intensity collected with emission polarizer at 0° (parallel) with respect to excitation polarizer, I⊥(t) is the fluorescence intensity collected with emission polarizer at 90° (perpendicular) with respect to excitation polarizer, and G(λ) is the geometry factor at the wavelength λ of emission. A 50 μm solution of N-acetyltryptophanamide was used to calculate the G(λ) for the optics.
The I‖(t) and I⊥(t) were fitted based on a model that assumes uniform motional dynamics in the sample with each protein molecule associated with two rotational correlation times (23).
| (Eq. 4) |
| (Eq. 5) |
| (Eq. 6) |
where r0 is the initial anisotropy, i.e. in the absence of any rotational diffusion (0.3), and βi is the amplitude associated with the ith rotational correlation times φi, such that ∑βi = 1. The two correlation times can be interpreted to be associated with the local motion (short correlation time; φ1) and the global motion (long correlation time; φ2). The goodness of fit was assessed from the χ2 values.
Acrylamide quenching of fluorescence
The protein concentration for quenching experiments was kept at 10 μm. The steady-state fluorescence setup was used for measurement. The protein aliquots were mixed with increasing concentrations of acrylamide (0–0.3 m), and Trp fluorescence spectra were recorded for each sample. The maximum fluorescence intensity (F) was noted for each sample, and the data were plotted according to the Stern–Volmer equation.
| (Eq. 7) |
where F0 is the maximum fluorescence intensity without acrylamide, KSV is the Stern–Volmer constant, and [Q] is the concentration of the acrylamide in m. The bimolecular rate constant kq was calculated using the following equation.
| (Eq. 8) |
where τ0 is the mean lifetime value of the protein in absence of acrylamide.
Determination of dissociation constant (Kd) using FITC dye
Cse4 was labeled using FITC as described previously (38). Briefly, Cse4 was incubated with a 4-fold excess of FITC at 4 °C for 4 h. Unbound FITC was removed by continuous buffer exchange using a 10-kDa concentrator (Amicon, Millipore). The incorporation ratio was determined by estimating the concentration of FITC-bound Cse4 to that of unlabeled Cse4 with the bound fraction calculated by using a molar extinction coefficient of 77,000 cm−1m−1 at 495 nm. 500 nm FITC-Cse4 was incubated with varying concentrations (0, 50, 100, 200, 400, 600, 800, 1000, 1500, and 2000 nm) of H4 at 25 °C for 30 min. The reaction mixture was then excited at 495 nm, and the emission spectrum was recorded from 500 to 620 nm on a Jasco FP-6500 spectrometer. The dissociation constant (Kd) of the interaction between Cse4 and H4 was determined by fitting the fluorescence data (using GraphPad Prism 5) into the following equation.
| (Eq. 9) |
where ΔF is the change in the fluorescence intensity of FITC-Cse4 in the presence of H4, ΔFmax is the change in the fluorescence intensity when Cse4 is saturated with H4, and L is the concentration of H4.
Statistical analysis
All the steady-state fluorescence data, Kd determinations, and time-resolved fluorescence data were independently collected with n = 3. The standard deviations are plotted as error bars in the graphs and also indicated in the supporting tables. The error associated with fluorescence lifetime measurement and its amplitude is ∼5–10%, and the standard deviation (S.D.) is indicated for τm in the tables. OriginPro software was used to calculate the statistical significance, which was calculated by one-way analysis of variance: *, p < 0.05; **, p < 0.01; NS (not significant), p > 0.05. The fitting of the time-resolved data was done as mentioned above. The time-resolved fluorescence data calculations were done using a home-built script as described in Saxena et al. (39).
NMR experiments
Uniform 15N- and/or 13C-labeled samples were prepared by culturing the cells expressing Cse4 (full length and truncated (1–129)) protein in minimal (M9) medium supplemented with 15NH4Cl or with 15NH4Cl and 13C-labeled glucose. The purification was done as described above. For the protein folding studies, the Cse4 full-length sample was buffer-exchanged to respective urea concentrations. The concentration of the samples used in urea denaturation studies varied from 600 to 800 μm depending on the urea dilutions. The folded Cse4 full-length protein was prepared by dialyzing the sample against distilled water containing 2 m arginine and 10 mm BME; the final concentration of the protein was 80–100 μm in different preparations. D2O was mixed in 90:10 (H2O/D2O) ratios before recording spectra. The pH of all Cse4 full-length samples was maintained at 6.5, and that of Cse4 truncated sample (Cse4ΔC) was maintained at pH 5.5. The proton chemical shifts were referenced using 2,2-dimethyl-2-silapentane-5-sulfonate as an external calibration agent at 0.0 ppm, whereas 15N and 13C were referenced indirectly according to the Biological Magnetic Resonance Bank (BMRB) protocol. The sample temperature was maintained at 25 °C.
NMR experiments were recorded on a Bruker Ascend 750-MHz spectrometer with a 5-mm triple-resonance inverse (TXI) probe with a Z-gradient. 2D 1H-15N HSQC was recorded for the full-length Cse4 and truncated Cse4 as well as Cse4–H4 (Cse4-labeled). The following experiments were recorded for assignment of Cse4 protein in 5 m urea sample buffer: 2D 1H-15N HSQC, 3D HNCACB, HNCOCACB, HNCO, HNCACO, and TOCSY-HSQC; the details are provided in Table S1. The Cse4ΔC sample (700 μm) was maintained in 20 mm phosphate buffer with 150 mm NaCl, and the following experiments were recorded for resonance assignment: 2D 1H-15N HSQC, 3D HNCA, HN(CO)CA, HNCO, HN(CA)CO, CBCA(CO)NH, TOCSY-HSQC, and H(CCO)NH (Table S2). The 1H-15N HSQC experiments (Table S3) with natively folded Cse4 and Cse4–H4 were recorded on samples at 75–100 μm, and similar contour levels were used for comparison. The D1 was set as 1 s for all experiments except the band-selective excitation short-transient (BEST) HSQC where the D1 was 0.1 s. All spectra were processed with Topspin version 2.1 and analyzed with CCPNMR 2.3.1 (40).
To analyze the secondary structure propensities of Cse4 protein in 6 and 5 m urea, the sequence-corrected secondary chemical shifts (Δδ) for Hα, Cα, and Cβ were calculated (41). The random-coil chemical shifts were taken from Schwarzinger et al. (42) who used 8 m urea, pH 2.3, at 20 °C for measurements on peptides to arrive at the random-coil chemical shifts. To investigate the backbone dynamics of Cse4, R2 data sets were recorded at 750-MHz frequency on a uniformly 15N-labeled denatured Cse4 as well as Cse4 equilibrated with 6 m urea. R2 values were measured using delays, 10, 25*, 50, 90, 120, 150, 180*, and 220 ms, where the asterisk indicates duplicate measurements. Duplicate measurements for two random points were carried out for the verification of the error estimates. The cross-peak intensities were measured as peak heights using CCPNMR 2.3.1, which was also used to fit the relaxation data. The fitting was done to a single-exponential decay function, I(t) = A + Be−R2t, to extract the R2 values.
For the interaction studies, various sets of 1H-15N HSQC spectra for Cse4ΔC sample were recorded at pH 5.5 with increasing equivalents of binding partner (CTD and H4). The extent of interaction with each component was analyzed by checking the change in intensity profile and CSP. The intensity profile of the amide cross-peaks affected during titration experiments was calculated by comparing their intensities (I) with those of the same cross-peaks (I0) without any addition; the data were normalized for dilution effect. The perturbation of amide cross-peaks chemical shifts during the interaction was calculated using the following formula.
| (Eq. 10) |
Residues showing CSPs greater than 2σ were considered significant (43).
Molecular dynamics
All the simulations were performed using the GROMACS 4.6 package (44–47) with Bloom et al. (21) as the source for the starting structures for monomeric Cse4 and Cse4–H4 simulations. Each protein system was placed in a dodecahedron box with a distance between the protein and the box surface of 1 nm. The Amber99sb force field was used for the protein (48). The simulation box was solvated using TIP3P water (49), and Na+ and Cl− ions were added to achieve a salt concentration of 150 mm. The total numbers of atoms in Cse4 and Cse4–H4 systems were 92,721 and 118,227, respectively. To enable the use of a 4-fs time step, all bond-angle hydrogens were treated using virtual sites (50). Each protein system was energy-minimized using a steepest-descent algorithm until the maximum force was less than 1000 kJ/mol/nm. Energy-minimized structures were subjected to 100-ps temperature equilibration to 298 K using a Berendsen thermostat (51) with a τt of 0.1 ps followed by pressure equilibration to 1 atm using a Berendsen barostat (51) with τt of 1 ps. The final structure from pressure equilibration was used as the starting structure for production run simulations where temperature was maintained using a velocity-rescaling thermostat (52) and a Parrinello–Rahman barostat (53) with τt of 1 ps and τp of 5 ps. Four independent simulations for Cse4 and three simulations for Cse4–H4 from the corresponding pressure-equilibrated structures were started with different starting velocities, and from each simulation, data were collected for 300 ns at 40-ps intervals. Analysis was done using tools from the GROMACS package and in-house Python programs. Contact maps were generated using g_contacts tools using data from the time window of 250–300 ns of each simulation (54).
Author contributions
N. M., S. K. G., and A. K. conceptualization; N. M. data curation; N. M. formal analysis; N. M. and S. S. validation; N. M. investigation; N. M. and S. C. D. visualization; N. M., S. C. D., S. S., G. K., and A. K. methodology; N. M. writing-original draft; N. M., S. C. D., S. S., S. K. G., G. K., and A. K. writing-review and editing; S. C. D. software; M. K., G. K., and A. K. resources; G. K. and A. K. supervision; A. K. funding acquisition; A. K. project administration.
Supplementary Material
Acknowledgments
We thank Prof. K. Luger (University of Colorado Boulder) for providing the plasmids for WT Cse4, H4, and H3. We acknowledge the HF-NMR facility funded by the Research Infrastructure Facility Committee, Industrial Research and Consultancy Center, Indian Institute of Technology (IIT) Bombay for NMR time; the Department of Chemical Sciences, Tata institute of Fundamental Research for access to the time-correlated single-photon counting setup; and the Centre for Development of Advanced Computing (CDAC) PARAM YUVA II Supercomputing facility for computational time. We are also grateful to Prof. N. Periasamy for sharing the fluorescence decay analysis software. We thank Dr. Rohit Mittal (Medical Research Council (MRC), Laboratory of Molecular Biology, Cambridge, UK) for critical feedback on the manuscript.
This work was supported by the Council of Scientific and Industrial Research, India (to N. M.) and Indian Institute of Technology Bombay and Department of Biotechnology, Government of India Seed Grant BT/PR21656/BRB/10/1562/2016 (to A. K.). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–S10, Tables S1–S5, and Movies S1 and S2.
- H3
- histone 3
- CENP-A
- centromeric protein-A
- CATD
- CENP-A–targeting domain
- HFD
- histone fold domain
- H4
- histone 4
- END
- essential N-terminal domain
- NTD
- N-terminal domain
- CTD
- C-terminal domain
- kq
- rate constant for quenching
- φ1
- short correlation time
- φ2
- long correlation time
- MD
- molecular dynamics
- HSQC
- heteronuclear single quantum coherence
- R2
- 15N transverse relaxation rate
- CSP
- chemical shift perturbation
- BME
- β-mercaptoethanol
- GST
- glutathione S-transferase
- Kd
- dissociation constant
- TOCSY
- total correlation spectroscopy.
References
- 1. Foltz D. R., Jansen L. E., Black B. E., Bailey A. O., Yates J. R. 3rd, and Cleveland D. W. (2006) The human CENP-A centromeric nucleosome-associated complex. Nat. Cell Biol. 8, 458–469 10.1038/ncb1397 [DOI] [PubMed] [Google Scholar]
- 2. Stoler S., Keith K. C., Curnick K. E., and Fitzgerald-Hayes M. (1995) A mutation in CSE4, an essential gene encoding a novel chromatin-associated protein in yeast, causes chromosome nondisjunction and cell cycle arrest at mitosis. Genes Dev. 9, 573–586 10.1101/gad.9.5.573 [DOI] [PubMed] [Google Scholar]
- 3. Furuyama S., and Biggins S. (2007) Centromere identity is specified by a single centromeric nucleosome in budding yeast. Proc. Natl. Acad. Sci. U.S.A. 104, 14706–14711 10.1073/pnas.0706985104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Meluh P. B., and Koshland D. (1997) Budding yeast centromere composition and assembly as revealed by in vivo cross-linking. Genes Dev. 11, 3401–3412 10.1101/gad.11.24.3401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zhang W., Mellone B. G., and Karpen G. H. (2007) A specialized nucleosome has a “point” to make. Cell 129, 1047–1049 10.1016/j.cell.2007.05.054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Heun P., Erhardt S., Blower M. D., Weiss S., Skora A. D., and Karpen G. H. (2006) Mislocalization of the Drosophila centromere-specific histone CID promotes formation of functional ectopic kinetochores. Dev. Cell 10, 303–315 10.1016/j.devcel.2006.01.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Haase J., Mishra P. K., Stephens A., Haggerty R., Quammen C., Taylor R. M. 2nd, Yeh E., Basrai M. A., and Bloom K. (2013) A 3D map of the yeast kinetochore reveals the presence of core and accessory centromere specific histone. Curr. Biol. 23, 1939–1944 10.1016/j.cub.2013.07.083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Collins K. A., Furuyama S., and Biggins S. (2004) Proteolysis contributes to the exclusive centromere localization of the yeast Cse4/CENP-A histone H3 variant. Curr. Biol. 14, 1968–1972 10.1016/j.cub.2004.10.024 [DOI] [PubMed] [Google Scholar]
- 9. Hewawasam G., Shivaraju M., Mattingly M., Venkatesh S., Martin-Brown S., Florens L., Workman J. L., and Gerton J. L. (2010) Psh1 is an E3 ubiquitin ligase that targets the centromeric histone variant Cse4. Mol. Cell 40, 444–454 10.1016/j.molcel.2010.10.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ranjitkar P., Press M. O., Yi X., Baker R., MacCoss M. J., and Biggins S. (2010) An E3 ubiquitin ligase prevents ectopic localization of the centromeric histone H3 variant via the centromere targeting domain. Mol. Cell 40, 455–464 10.1016/j.molcel.2010.09.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mishra P. K., Guo J., Dittman L. E., Haase J., Yeh E., Bloom K., and Basrai M. A. (2015) Pat1 protects centromere-specific histone H3 variant Cse4 from Psh1-mediated ubiquitination. Mol. Biol. Cell 26, 2067–2079 10.1091/mbc.e14-08-1335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Au W. C., Dawson A. R., Rawson D. W., Taylor S. B., Baker R. E., and Basrai M. A. (2013) A novel role of the N terminus of budding yeast histone H3 variant Cse4 in ubiquitin-mediated proteolysis. Genetics 194, 513–518 10.1534/genetics.113.149898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cheng H., Bao X., and Rao H. (2016) The F-box protein rcy1 is involved in the degradation of histone H3 variant Cse4 and genome maintenance. J. Biol. Chem. 291, 10372–10377 10.1074/jbc.M115.701813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Black B. E., Foltz D. R., Chakravarthy S., Luger K., Woods V. L. Jr., and Cleveland D. W. (2004) Structural determinants for generating centromeric chromatin. Nature 430, 578–582 10.1038/nature02766 [DOI] [PubMed] [Google Scholar]
- 15. Black B. E., Jansen L. E., Maddox P. S., Foltz D. R., Desai A. B., Shah J. V., and Cleveland D. W. (2007) Centromere identity maintained by nucleosomes assembled with histone H3 containing the CENP-A targeting domain. Mol. Cell 25, 309–322 10.1016/j.molcel.2006.12.018 [DOI] [PubMed] [Google Scholar]
- 16. Zhou Z., Feng H., Zhou B. R., Ghirlando R., Hu K., Zwolak A., Miller Jenkins L. M., Xiao H., Tjandra N., Wu C., and Bai Y. (2011) Structural basis for recognition of centromere histone variant CenH3 by the chaperone Scm3. Nature 472, 234–237 10.1038/nature09854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cho U. S., and Harrison S. C. (2011) Recognition of the centromere-specific histone Cse4 by the chaperone Scm3. Proc. Natl. Acad. Sci. U.S.A. 108, 9367–9371 10.1073/pnas.1106389108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hu H., Liu Y., Wang M., Fang J., Huang H., Yang N., Li Y., Wang J., Yao X., Shi Y., Li G., and Xu R. M. (2011) Structure of a CENP-A-histone H4 heterodimer in complex with chaperone HJURP. Genes Dev. 25, 901–906 10.1101/gad.2045111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chen Y., Baker R. E., Keith K. C., Harris K., Stoler S., and Fitzgerald-Hayes M. (2000) The N terminus of the centromere H3-like protein Cse4p performs an essential function distinct from that of the histone fold domain. Mol. Cell. Biol. 20, 7037–7048 10.1128/MCB.20.18.7037-7048.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Meluh P. B., Yang P., Glowczewski L., Koshland D., and Smith M. M. (1998) Cse4p is a component of the core centromere of Saccharomyces cerevisiae. Cell 94, 607–613 10.1016/S0092-8674(00)81602-5 [DOI] [PubMed] [Google Scholar]
- 21. Bloom K., Sharma S., and Dokholyan N. V. (2006) The path of DNA in the kinetochore. Curr. Biol. 16, R276–R278 10.1016/j.cub.2006.03.054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lakowicz J. R., and Masters B. R. (2008) Principles of fluorescence spectroscopy, third edition. J. Biomed. Opt. 13, 29901 10.1117/1.2904580 [DOI] [Google Scholar]
- 23. Jha A., Udgaonkar J. B., and Krishnamoorthy G. (2009) Characterization of the heterogeneity and specificity of interpolypeptide interactions in amyloid protofibrils by measurement of site-specific fluorescence anisotropy decay kinetics. J. Mol. Biol. 393, 735–752 10.1016/j.jmb.2009.08.053 [DOI] [PubMed] [Google Scholar]
- 24. Malik N., and Kumar A. (2016) Resonance assignment of disordered protein with repetitive and overlapping sequence using combinatorial approach reveals initial structural propensities and local restrictions in the denatured state. J. Biomol. NMR 66, 21–35 10.1007/s10858-016-0054-9 [DOI] [PubMed] [Google Scholar]
- 25. Kumar A., Srivastava S., Kumar Mishra R., Mittal R., and Hosur R. V. (2006) Residue-level NMR view of the urea-driven equilibrium folding transition of SUMO-1 (1–97): native preferences do not increase monotonously. J. Mol. Biol. 361, 180–194 10.1016/j.jmb.2006.06.003 [DOI] [PubMed] [Google Scholar]
- 26. Cleveland D. W., Mao Y., and Sullivan K. F. (2003) Centromeres and kinetochores: from epigenetics to mitotic checkpoint signaling. Cell 112, 407–421 10.1016/S0092-8674(03)00115-6 [DOI] [PubMed] [Google Scholar]
- 27. Henikoff S., and Furuyama T. (2010) Epigenetic inheritance of centromeres. Cold Spring Harb. Symp. Quant. Biol. 75, 51–60 10.1101/sqb.2010.75.001 [DOI] [PubMed] [Google Scholar]
- 28. Sullivan K. F., Hechenberger M., and Masri K. (1994) Human CENP-A contains a histone H3 related histone fold domain that is required for targeting to the centromere. J. Cell Biol. 127, 581–592 10.1083/jcb.127.3.581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Morey L., Barnes K., Chen Y., Fitzgerald-Hayes M, and Baker R. E. (2004) The histone fold domain of Cse4 is sufficient for CEN targeting and propagation of active centromere in budding yeast. Eukaryot. Cell 3, 1533–1543 10.1128/EC.3.6.1533-1543.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Keith K. C., Baker R. E., Chen Y., Harris K., Stoler S., and Fitzgerald-Hayes M. (1999) Analysis of primary structural determinants that distinguish the centromere-specific function of histone variant Cse4p from histone H3. Mol. Cell. Biol. 19, 6130–6139 10.1128/MCB.19.9.6130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Boeckmann L., Takahashi Y., Au W.-C., Mishra P. K., Choy J. S., Dawson A. R., Szeto M. Y., Waybright T. J., Heger C., McAndrew C., Goldsmith P. K., Veenstra T. D., Baker R. E., and Basrai M. A. (2013) Phosphorylation of centromeric histone H3 variant regulates chromosome segregation in Saccharomyces cerevisiae. Mol. Biol. Cell. 24, 2034–2044 10.1091/mbc.e12-12-0893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Samel A., Cuomo A., Bonaldi T., and Ehrenhofer-Murray A. E. (2012) Methylation of CenH3 arginine 37 regulates kinetochore integrity and chromosome segregation. Proc. Natl. Acad. Sci. U.S.A. 109, 9029–9034 10.1073/pnas.1120968109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ohkuni K., Abdulle R., and Kitagawa K. (2014) Degradation of centromeric histone H3 variant Cse4 requires the Fpr3 peptidyl-prolyl cis-trans isomerase. Genetics 196, 1041–1045 10.1534/genetics.114.161224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dechassa M. L., Wyns K., and Luger K. (2014) Scm3 deposits a (Cse4-H4)2 tetramer onto DNA through a Cse4-H4 dimer intermediate. Nucleic Acids Res. 42, 5532–5542 10.1093/nar/gku205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Deyter G. M., Hildebrand E. M., Barber A. D., and Biggins S. (2017) Histone H4 facilitates the proteolysis of the budding yeast CENP-ACse4 centromeric histone variant. Genetics 205, 113–124 10.1534/genetics.116.194027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Luger K., Mäder A. W., Richmond R. K., Sargent D. F., and Richmond T. J. (1997) Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature 389, 251–260 10.1038/38444 [DOI] [PubMed] [Google Scholar]
- 37. Bevington P. R. (1969) Data Reduction and Error Analysis for the Physical Sciences, McGraw-Hill, New York [Google Scholar]
- 38. Santra M. K., and Panda D. (2003) Detection of an intermediate during unfolding of bacterial cell division protein FtsZ: loss of functional properties precedes the global unfolding of FtsZ. J. Biol. Chem. 278, 21336–21343 10.1074/jbc.M301303200 [DOI] [PubMed] [Google Scholar]
- 39. Saxena A., Udgaonkar J. B., and Krishnamoorthy G. (2005) Protein dynamics and protein folding dynamics revealed by time-resolved fluorescence, in Fluorescence Spectroscopy in Biology: Advanced Methods and their Applications to Membranes, Proteins, DNA, and Cells (Hof M., Hutterer R., and Fidler V., eds) pp. 163–179, Springer, Berlin [Google Scholar]
- 40. Vranken W. F., Boucher W., Stevens T. J., Fogh R. H., Pajon A., Llinas M., Ulrich E. L., Markley J. L., Ionides J., and Laue E. D. (2005) The CCPN data model for NMR spectroscopy: development of a software pipeline. Proteins 59, 687–696 10.1002/prot.20449 [DOI] [PubMed] [Google Scholar]
- 41. Schwarzinger S., Kroon G. J., Foss T. R., Chung J., Wright P. E., and Dyson H. J. (2001) Sequence-dependent correction of random coil NMR chemical shifts. J. Am. Chem. Soc. 123, 2970–2978 10.1021/ja003760i [DOI] [PubMed] [Google Scholar]
- 42. Schwarzinger S., Kroon G. J., Foss T. R., Wright P. E., and Dyson H. J. (2000) Random coil chemical shifts in acidic 8 M urea: implementation of random coil shift data in NMRView. J. Biomol. NMR 18, 43–48 10.1023/A:1008386816521 [DOI] [PubMed] [Google Scholar]
- 43. Williamson M. P. (2013) Using chemical shift perturbation to characterise ligand binding. Prog. Nucl. Magn. Reson. Spectrosc. 73, 1–16 10.1016/j.pnmrs.2013.02.001 [DOI] [PubMed] [Google Scholar]
- 44. Berendsen H. J. C., Van der Spoel D., and Van Drunen R. (1995) GROMACS: a message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 91, 43–56 10.1016/0010-4655(95)00042-E [DOI] [Google Scholar]
- 45. Lindahl E., Hess B., and van der Spoel D. (2001) GROMACS 3.0: a package for molecular simulation and trajectory analysis. Mol. Model. Annu. 7, 306–317 10.1007/s008940100045 [DOI] [Google Scholar]
- 46. Hess B., Kutzner C., van der Spoel D., and Lindahl E. (2008) GROMACS 4: algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 4, 435–447 10.1021/ct700301q [DOI] [PubMed] [Google Scholar]
- 47. Pronk S., Páll S., Schulz R., Larsson P., Bjelkmar P., Apostolov R., Shirts M. R., Smith J. C., Kasson P. M., van der Spoel D., Hess B., and Lindahl E. (2013) GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 29, 845–854 10.1093/bioinformatics/btt055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hornak V., Abel R., Okur A., Strockbine B., Roitberg A., and Simmerling C. (2006) Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins 65, 712–725 10.1002/prot.21123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Mahoney M. W., and Jorgensen W. L. (2000) A five-site model for liquid water and the reproduction of the density anomaly by rigid, nonpolarizable potential functions. J. Chem. Phys. 112, 8910–8922 10.1063/1.481505 [DOI] [Google Scholar]
- 50. Feenstra K. A., Hess B., and Berendsen H. J. C. (1999) Improving efficiency of large time-scale molecular dynamics simulations of hydrogen-rich systems. J. Comput. Chem. 20, 786–798 [DOI] [PubMed] [Google Scholar]
- 51. Berendsen H. J. C., Postma J. P. M., Van Gunsteren W. F., DiNola A., and Haak J. R. (1984) Molecular dynamics with coupling to an external bath. J. Chem. Phys. 81, 3684–3690 10.1063/1.448118 [DOI] [Google Scholar]
- 52. Bussi G., Donadio D., and Parrinello M. (2007) Canonical sampling through velocity rescaling. J. Chem. Phys. 126, 014101 10.1063/1.2408420 [DOI] [PubMed] [Google Scholar]
- 53. Nosé S., and Klein M. L. (1983) Constant pressure molecular dynamics for molecular systems. Mol. Phys. 50, 1055–1076 10.1080/00268978300102851 [DOI] [Google Scholar]
- 54. Blau C., and Grubmuller H. (2013) g_contacts: fast contact search in bio-molecular ensemble data. Comput. Phys. Commun. 184, 2856–2859 10.1016/j.cpc.2013.07.018 [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.