

Abstract
Mitochondrial inner membrane protein MPV17 is a protein of unknown function that is associated with mitochondrial DNA (mtDNA)-depletion syndrome (MDS). MPV17 loss-of-function has been reported to result in tissue-specific nucleotide pool imbalances, which can occur in states of perturbed folate-mediated one-carbon metabolism (FOCM), but MPV17 has not been directly linked to FOCM. FOCM is a metabolic network that provides one-carbon units for the de novo synthesis of purine and thymidylate nucleotides (e.g. dTMP) for both nuclear DNA (nuDNA) and mtDNA replication. In this study, we investigated the impact of reduced MPV17 expression on markers of impaired FOCM in HeLa cells. Depressed MPV17 expression reduced mitochondrial folate levels by 43% and increased uracil levels, a marker of impaired dTMP synthesis, in mtDNA by 3-fold. The capacity of mitochondrial de novo and salvage pathway dTMP biosynthesis was unchanged by the reduced MPV17 expression, but the elevated levels of uracil in mtDNA suggested that other sources of mitochondrial dTMP are compromised in MPV17-deficient cells. These results indicate that MPV17 provides a third dTMP source, potentially by serving as a transporter that transfers dTMP from the cytosol to mitochondria to sustain mtDNA synthesis. We propose that MPV17 loss-of-function and related hepatocerebral MDS are linked to impaired FOCM in mitochondria by providing insufficient access to cytosolic dTMP pools and by severely reducing mitochondrial folate pools.
Keywords: folate, one-carbon metabolism, mitochondria, thymidine kinase 2, nucleoside/nucleotide transport, mitochondrial transport, nucleoside/nucleotide biosynthesis, mitochondrial disease, mitochondrial thymidylate synthesis, MPV17, mitochondrial DNA-depletion syndrome (MDS), deoxythymidine monophosphate (dTMP)
Introduction
Mitochondrial inner membrane protein MPV17 is a ubiquitously expressed protein of unknown function (1–3). Over 30 distinct mutations throughout the MPV17 gene have been associated with mitochondrial DNA-depletion syndrome (MDS),2 which is characterized by a tissue-specific reduction in mitochondrial DNA (mtDNA) copy number (4–6). Despite a lack of knowledge of its biochemical function(s), MPV17 has been shown to protect against mitochondrial dysfunction and apoptosis and to regulate reactive oxygen species (8, 9). It has been proposed to function as a nonselective mitochondrial channel protein that opens under conditions characteristic of damaged mitochondria to preserve mitochondrial homeostasis by decreasing the membrane potential and thus preventing the formation of reactive oxygen species (10).
Loss-of-function mutations in genes involved in regulation or synthesis of nucleotides can result in MDS. Mice that lack MPV17 function exhibit a 35 and 30% reduction in dTTP and dGTP pools, respectively, in liver mitochondria, but not in other tissues tested (11). Maintenance and regulation of cellular dTTP synthesis and pool size for mtDNA replication are critical, as both depleted and expanded dTTP pool sizes have been associated with mtDNA depletion (12).
Folate-mediated one-carbon metabolism (FOCM) is a network of interconnected metabolic pathways that use tetrahydrofolate (THF) cofactors to carry and chemically activate single carbon units for remethylation of homocysteine to methionine and the de novo synthesis of purine nucleotides and dTMP (13) (Fig. 1). FOCM functions in the mitochondria, nucleus, and cytosol. The synthesis of dTMP occurs in the mitochondria (14) and the cytosol/nucleus (15) both through salvage pathway synthesis catalyzed by thymidine kinase (TK1 and TK2) and through folate-dependent de novo synthesis. Mitochondrial folate-dependent dTMP synthesis involves the enzymes serine hydroxymethyltransferase 2 (SHMT2), dihydrofolate reductase 2 (DHFR2, formally known as DHFRL1), and thymidylate synthase (TYMS). SHMT2 transfers one-carbon (1C) units from serine to THF to synthesize glycine and 5,10-methylene THF, the 1C donor for the conversion of dUMP to dTMP in a reaction catalyzed by TYMS. In this reaction, the folate cofactor serves as both a 1C donor and a source of 2 electrons, generating dihydrofolate (DHF) as a product. THF is regenerated from DHF by DHFR2 to complete the dTMP cycle (14).
Figure 1.

FOCM. One-carbon metabolism is required for the synthesis of purines, dTMP, and methionine. The hydroxymethyl group of serine is a major source of 1C units, which are generated in the mitochondria in the form of formate via SHMT2 or in the cytoplasm through the activity of SHMT1 or SHMT2α. Mitochondria-derived formate can enter the cytoplasm and function as a one-carbon unit for folate metabolism. The synthesis of dTMP occurs in the nucleus and mitochondria. At S phase, the enzymes of the dTMP synthesis pathway undergo SUMO-dependent translocation to the nucleus. The remethylation of homocysteine to methionine by methionine synthase (MTR) requires vitamin B12. The one carbon is labeled in boldface type. The inset shows the thymidylate synthesis cycle, which involves the enzymes SHMT1, SHMT2α, TYMS, and DHFR. MTHFR, methylenetetrahydrofolate reductase; MTHFD1, methylene THF dehydrogenase; GAR Tfase, glycinamide ribonucleotide transformylase; AICAR Tfase, aminoimidazolecarboxamide ribonucleotide transformylase; GCS, glycine cleavage system.
FOCM in mitochondria is the primary source of formate, which when translocated to the cytosol serves as the primary source of 1Cs for homocysteine remethylation and de novo purine and dTMP biosynthesis. Nuclear dTMP biosynthesis via FOCM requires isoforms of each of the mitochondrial dTMP synthesis enzymes (SHMT1/SHMT2α, TYMS, and DHFR). These enzymes are SUMOylated and translocated from the cytosol to the nucleus during S phase, where they form a multienzyme complex for nuclear dTMP synthesis at sites of nuclear DNA (nuDNA) replication and repair (14–16). Folate-dependent de novo dTMP synthesis is compromised in states of perturbed FOCM and folate deficiency, leading to increased uracil misincorporation into DNA (17, 18).
Under folate-deficient conditions, lack of folate-activated 1C units for dTMP synthesis results in dUMP accumulation leading to dUTP synthesis, which can be misincorporated into DNA by DNA polymerases, which do not distinguish between dUTP and dTTP during DNA synthesis (19). Uracil-DNA glycosylases cleave the misincorporated U base, leaving an abasic site; multiple rounds of repair can result in DNA strand breaks, genomic instability, and cell death (20). These mechanisms have been studied more extensively in nuDNA than in mtDNA. In this study, the effect of MPV17 expression on markers of impaired FOCM, including nucleotide synthesis and uracil misincorporation, was investigated.
Results
Impact of MPV17 expression on mitochondrial folate-dependent nucleotide synthesis
HeLa cells with reduced MPV17 expression generated by shRNA (MPV17 knockdown, KD) exhibited an 80–85% reduction in MPV17 protein levels when compared with cell lines treated with scrambled shRNA (Fig. 2). The deoxyuridine (dU) suppression assay measures the cellular capacity to synthesize dTMP via the de novo ([14C]dU) and salvage ([3H]thymidine, dT) pathways for DNA synthesis. The decreased MPV17 expression did not affect the relative contribution of de novo dTMP synthesis and salvage dTMP synthesis to mtDNA synthesis, as indicated by the dU suppression assay (Fig. S1); the activity of both the salvage and de novo dTMP synthesis were elevated similarly in mitochondria of MPV17 knockdown cell lines compared with mitochondria of control lines (Fig. 3, A–C). Mitochondrial TK2 and TYMS protein levels were unaffected by reduced MPV17 expression when compared with control MPV17-expressing cell lines (Fig. 3H). Separation of DNA bases by HPLC showed that both dTMP precursors, [14C]dU and [3H]dT, were incorporated into mtDNA primarily as dTTP and not dUTP (Fig. S2). In contrast, incorporation of [3H]dT, via the salvage pathway, and [14C]dU, via the folate-dependent de novo dTMP pathway, into nuDNA were not affected by reduced MPV17 expression (Fig. 3 (D and E) and Fig. S3). De novo purine synthesis capacity in the cytosol was also quantified in MPV17-deficient cells via the formate suppression assay. Reduced MPV17 expression did not affect the relative ratio of de novo to salvage purine synthesis capacity in HeLa cells, as demonstrated by the ratio of [14C]formate (incorporated via folate-dependent de novo synthesis) and [3H]hypoxanthine (incorporated via salvage synthesis) present in nuDNA (Fig. S4). Both salvage and de novo purine synthesis were reduced in MPV17 KD cells when compared with control lines (Fig. 3, F and G), but the magnitude of the difference is small and unlikely to be biologically important.
Figure 2.

MPV17 stable KD cell lines. MPV17 KD cell lines were generated by clonal selection using shRNA in a HeLa cell background. MPV17 protein expression was reduced by 80–85% in KD lines. Western blotting was quantified using ImageJ.
Figure 3.

MPV17 KD increased mitochondrial dTMP synthesis. A, [3H]thymidine incorporation into newly synthesized mtDNA via salvage pathway: KD, n = 3; control, n = 2. B, [14C]deoxyuridine incorporation into mtDNA via folate-dependent de novo dTMP pathway: KD, n = 3; control, n = 2. C, mtDNA was extracted from isolated mitochondria. Representative Western blotting of protein extracts of mitochondrial fractions and respective whole-cell extracts is shown. Mitochondrial fractions were free of nuclear contamination (proliferating cell nuclear antigen (PCNA) (nuclear marker) and hexokinase I (mitochondrial marker)). D and E, [3H]thymidine incorporation into newly synthesized nuDNA via salvage pathway (D) and [14C]deoxyuridine incorporation into nuDNA via folate-dependent de novo dTMP pathway (E): n = 4 each group, not significant (NS). F and G, newly synthesized nuDNA containing purines made via the salvage pathway (F) and the folate-dependent de novo synthesis pathway (G), as quantified by [3H]hypoxanthine and [14C]formate incorporation, respectively; n = 6 each group. A, B, and D–G, 3H and 14C channels were counted in dual DPM mode on a scintillation counter. H and I, Western blots of mitochondrial protein extracts from different biological replicates probed for TK2, TYMS, and FPGS. Densitometry was performed using ImageJ. The intensities of nonsaturated bands were quantified and normalized to CoxIV, which served as a mitochondrial marker and mitochondrial protein-loading control. Data are shown as mean ± S.D. (error bars) (A–H) or means ± S.E. (error bars) (I). Statistical significance was determined by a two-tailed Student's t test (NS, p > 0.05). Ctr., scrambled shRNA control.
Impact of MPV17 on cellular folate levels
Intracellular folates are compartmentalized in the mitochondria, nucleus, and cytosol as discrete pools (13, 18, 21–23). Folate cofactors enter the cell in a monoglutamate form and are converted to a polyglutamate form by the enzyme folylpoly-γ-glutamate synthetase (FPGS). The polyglutamate moiety serves to retain the cofactors within the cell and subcellular compartments and increase the affinity of the cofactor for enzymes (24, 25). Mitochondrial FPGS levels were reduced (30%) in MPV17 KD cell lines, with a trend toward significance (p = 0.068) (Fig. 3, H and I). Total cellular folate levels were similar for control and MPV17 KD lines (Table 1). However, mitochondrial folate levels in MPV17 KD cells were 43% lower than mitochondrial folate levels in control cell lines (Table 1, p ≤ 0.0001).
Table 1.
Cells deficient in MPV17 have 43% less mitochondrial folate
Total intracellular folate in whole-cell lysates and isolated mitochondria were quantified in a L. casei microbiological assay. Whole-cell lysates: shRNA control, n = 3; MPV17 KD, n = 6; NS (p > 0.05). Mitochondria: n = 4 each group; ‡, p ≤ 0.0001. Data are shown as mean ± S.E. Statistical significance was determined by two-tailed Student's t test.
| Folates in whole cellNS | Folates in mitochondrial fraction‡ | |
|---|---|---|
| fmol/μg protein | fmol/μg protein | |
| MPV17 KD | 107.64 ± 17.98 | 74.08 ± 6.11 |
| shRNA control | 100.95 ± 14.18 | 129.63 ± 5.29 |
The effect of MPV17 on folate accumulation and turnover was investigated in MPV17 KD and control lines cultured with labeled (6S)-[3H]5-formyl-THF. Cells with reduced MPV17 expression accumulated 15% less labeled folate than control lines (Fig. 4A). Similarly, the rate of (6S)-[3H]5-formyl-THF uptake was lower for MPV17 KD cells than for control lines over time (Fig. 4B). Whole-cell folate turnover rates were similar in MPV17-deficient cells and control cell lines (Fig. 4C).
Figure 4.

MPV17 and folate accumulation. A, MPV17 KD cells incubated with (6S)-[3H]5-formyl-THF for 12 h accumulated 15% less of the label than control lines; n = 3 each group. B, accumulation of 3H-labeled folates in viable cells over 48 h. Folate uptake in cells deficient in MPV17 is less than control lines; n = 3 each group. C, the rate of 3H-labeled folate depletion in cells pulsed with (6S)-[3H]5-formyl-THF for 13 h was similar between MPV17 KD cells and control lines; n = 3 each group. Data are shown as mean ± S.D. (error bars). Statistical significance was determined by two-tailed Student's t test (NS, p > 0.05). Ctr., scrambled shRNA control.
Effect of reduced MPV17 levels on mitochondrial DNA integrity from HeLa cells
Uracil misincorporation in DNA is a marker of low-folate status (20, 26). In folate deficiency, the dUTP/dTTP ratio is increased, leading to uracil misincorporation in DNA, double-strand breaks, and DNA instability (27). Uracil levels in mtDNA and nuDNA from MPV17 KD and control cells were quantified by GC-MS. HeLa cells cultured in folate-depleted medium for at least four doublings exhibited an 84 ± 14% increase in uracil levels in mtDNA when compared with mtDNA from cells cultured in folate-replete medium (Table 2). Similarly, uracil levels in mtDNA were 3-fold higher in cells with reduced MPV17 expression than in control lines (Fig. 5A). However, reduced MPV17 expression did not affect uracil levels in nuDNA (Fig. 5B). Decreased MPV17 expression did not change mtDNA copy number or mitochondrial mass in HeLa cells as compared with control cell lines (Fig. 6, A and B).
Table 2.
Uracil levels in mtDNA from HeLa cells
HeLa cells grown in folate-depleted medium for four doublings or more exhibited at least 55% more uracil in their mitochondrial genome than HeLa cells grown in folate-replete medium. The table shows uracil levels quantified by GC-MS for three different biological replicates; each biological replicate was the average of three independent measurements except for replicate 3 (n = 1). Data are shown as percentage increase for each replicate and the mean ± S.E.
| Uracil in mtDNA from folate-depleted HeLa cells |
|||
|---|---|---|---|
| Replicate 1 | Replicate 2 | Replicate 3 | |
| % | % | % | |
| Percentage increase | 99 | 55 | 97 |
| Mean ± S.E. | 84 ± 14 | ||
Figure 5.

MPV17 deficiency increases uracil levels in mtDNA. A, uracil levels were 3-fold higher in mtDNA extracted from isolated mitochondria of MPV17 KD cells than in that from control lines. n = 2 each group. B, there was no difference in uracil levels from nuDNA with changes in MPV17 expression. n = 4 each group. Data are shown as mean ± S.D. (error bars). Statistical significance was determined by two-tailed Student's t test (NS, p > 0.05). Ctr., scrambled shRNA control.
Figure 6.

Mitochondrial content and mitochondrial mass are not affected by MPV17 expression in HeLa cells. A, mitochondrial copy number was not affected in HeLa cells with reduced MPV17 expression; mtDNA content in MPV17 KD lines is expressed relative to mtDNA content in control lines: KD, n = 5; control, n = 4; each measurement done in triplicates. B, mitochondrial mass was not affected by lack of MPV17 expression; mitochondrial mass was determined by citrate synthase activity assay and normalized to total protein: KD, n = 5; control, n = 2; each measurement done in duplicates. Data shown as mean ± S.E. (error bars) (A) or mean ± S.D. (error bars) (B). Statistical significance was determined by two-tailed Student's t test (NS, p > 0.05). Ctr., scrambled shRNA control.
Discussion
The interaction between MPV17 and FOCM was investigated to provide insight into the role of MPV17 in mitochondrial nucleotide synthesis and mtDNA depletion. These studies demonstrate that MPV17 function is critical to maintain mitochondrial folate levels and prevent uracil accumulation in mtDNA, providing new insights into the role of MPV17 in MDS.
Mpv17 has been hypothesized to be a nonselective mitochondrial transporter (10, 28). Liver mitochondria with reduced MPV17 expression have been reported to have lower levels of dTTP and dGTP pools (11). In this study, we focused on the role of MPV17 depletion on dTMP synthesis and uracil accumulation in mtDNA because of the effect of MPV17 depletion on mitochondrial folate pools. Our data demonstrate that that MPV17-deficient cells synthesized and incorporated more dTMP from the de novo and salvage pathways into mtDNA (Fig. 3 (A–E) and Fig. S2) without an increase in the expression levels of dTMP synthesis enzymes (Fig. 3H). However, the 3-fold increase in uracil levels in the mitochondrial genome (Fig. 5A) of MPV17-deficient cells indicates that dTMP pools are compromised even though biosynthesis capacity is increased. These data suggest that MPV17 is essential to prevent uracil accumulation in mtDNA, independent of dTMP synthesis capacity. The elevated levels of uracil in mtDNA are also independent of uracil misincorporation in the nuclear genome and nuclear dTMP synthesis capacity, which were not affected (Figs. 3 (D and E) and 5B).
Nucleosides and nucleotides are transported into the mitochondria for mtDNA synthesis. The mitochondrial pyrimidine nucleotide transporter SLC25A33 (PNC1) preferentially transports uracil, in addition to thymine, and cytosine (deoxy) nucleoside di- and triphosphates by an antiport mechanism (29, 30). Similarly, SLC25A36 (PNC2) transports cytosine and uracil (deoxy)nucleoside mono-, di-, and triphosphates by uniport and antiport mechanisms (29, 30). There is evidence in mouse liver for an unidentified, highly selective mitochondrial dTMP transporter, with lower affinity for other thymine forms (base and di- and triphosphate) (31). Hence, there are three potential sources of dTMP for mtDNA replication: 1) endogenous folate-dependent de novo biosynthesis from dUMP; 2) salvage pathway synthesis from thymidine, and 3) import of dTMP nucleotides from the cytosol. Given the elevated levels of uracil in the mtDNA (Fig. 5A) from cells with reduced MPV17 expression without evidence for disturbed dTMP biosynthesis capacity from both the salvage and de novo synthesis pathways (Fig. 3), as well as the reported cases of dysregulation of mitochondrial dTTP pools in MPV17-deficient cells (11), we hypothesize that MPV17 is an important source of dTMP by serving as a dTMP transporter from the cytosol into the mitochondria for mtDNA synthesis (Fig. 7). The apparent increase in both mitochondrial de novo and salvage dTMP biosynthesis (Fig. 3, A and B) suggests that these pathways are partially compensating for other mitochondrial sources of dTTP, namely dTMP transport from the cytoplasm by MPV17.
Figure 7.
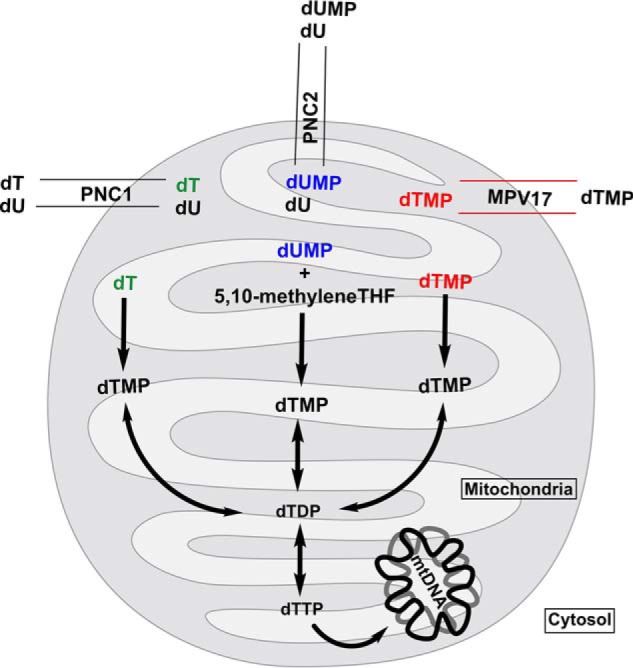
Proposed model. Three primary sources of the mitochondrial dTMP pool are shown. Red, source 1: salvage dTMP synthesis using dT from cytosolic and mitochondrial pools. Salvage synthesis of dTMP is independent of folate status; it relies on TK2 activity to convert dT to dTMP. Mitochondrial pyrimidine nucleotide transporter PNC1 facilitates the import of dT from cytosolic pools. Blue, source 2: de novo dTMP synthesis using dUMP from mitochondrial and cytosolic pools. Cytosolic dUMP is imported into the mitochondria via PNC2, where it participates in folate-dependent de novo dTMP biosynthesis. Purple, source 3: transport dTMP from cytosolic pools, to be incorporated into the mitochondrial dTMP pool. dTMP synthesized in the cytosol via the salvage pathway or de novo pathway is imported into the mitochondria to help sustain mtDNA synthesis; the mitochondrial import of synthesized dTMP is perturbed under MPV17-deficient conditions.
Mitochondria contain about 50% of total cellular folate, the nucleus contains ∼10%, and the remaining folates are present in the cytosol (32, 33). The 43% reduction in mitochondrial folates (Table 1) correlates with the 15% decrease in 3H cellular folate accumulation (Fig. 4A). This suggests that the mitochondrial folate pool is the main folate pool affected by MPV17 expression and that the decrease in the accumulation of the labeled folate by MPV17 KD cells may be a reflection of the depletion of mitochondrial folate pools. There was no observed change in total folate levels from whole-cell lysates of MPV17 KD cells compared with control cell lines (Table 1) or in overall whole-cell folate turnover (Fig. 4C), indicating that accumulation of cytoplasmic folate levels is not affected by reduced MPV17 expression. The spatial compartmentalization of folate pools is possible by the addition of polyglutamate chains to folate substrates by FPGS. The polyglutamate chains limit the translocation of folates across organelles and increase selectivity for several folate-dependent enzymes (24, 25). Reduced FPGS protein levels in mitochondria in cells with reduced MPV17 expression may contribute to lower folate levels in that compartment.
The mechanism for reduced mitochondrial folate pools in MPV17 deficiency is not clear. Cells deficient in MPV17 cultured with labeled (6S)-[3H]5-formyl-THF have reduced uptake of the tritium label as shown by the quantitation of the label accumulation into the cell over time (Fig. 4B). It is not clear from these data if the lower folate uptake levels represent lower uptake by the cell as a whole or lower uptake of folates exclusively by mitochondria. The expression of FPGS in MPV17-deficient cells may be the important determinate of reduced folate accumulation in mitochondria (Fig. 3 (H and I)). The pulse-chase data (Fig. 4C) indicate that folate stability was not affected by the MPV17 expression, suggesting that the turnover of folate is the same for both MPV17 KD and control lines.
Dietary folate has been shown to affect on mtDNA damage, specifically in protecting against mtDNA deletions by unknown mechanisms (34–36). mtDNA from HeLa cells grown in folate-deplete medium for at least four doublings had 84 ± 14% more uracil than cells grown in folate-replete medium (Table 2), suggesting that mtDNA is highly sensitive to uracil misincorporation under folate-deficient conditions. A potential mechanism by which folate deficiency may be associated with mtDNA depletion is by generation of double-strand breaks as a result of unrepaired uracil misincorporation. Uracil in mtDNA because of lack of MPV17 expression may be accompanied by mtDNA instability and may be the foundation for MPV17-related MDS.
Mitochondrial mass and copy number are important indicators of mitochondria integrity. Depletion of mtDNA by MPV17 is tissue-dependent. Others have also shown that changes in mtDNA copy number are also affected by cellular state, regardless of the tissue type. In human fibroblasts, MPV17 deficiency results in mtDNA depletion only in quiescent cells and not in proliferating cells (11). This may explain why there was no difference in mtDNA copy number or mitochondrial mass in HeLa cells deficient in MPV17 (Fig. 6).
In summary, this study reports the impacts of reduced MPV17 expression in HeLa cells on FOCM. These data show that reduced MPV17 expression depletes mitochondrial folate pools and that mitochondrial dTMP synthesis capacity is not sufficient to prevent uracil misincorporation, suggesting that the mitochondrial genome requires access to cytosolic dTMP pools. We postulate that there are three important sources of dTMP for mtDNA replication: de novo synthesis, salvage synthesis, and transport from the cytoplasm. We predict that MPV17 may be involved in the transport of dTMP from the cytosol to the mitochondria to maintain the dTMP pool (Fig. 7) for mtDNA synthesis. It is also likely responsible for transport of dGMP, as MPV17 loss-of-function mutations depress mitochondrial dGTP and dTTP pools (11). In the absence of the cytosolic sources of dTMP, mtDNA replication becomes dependent on increased rates of de novo and salvage dTMP synthesis as well as misincorporation of dUTP. Whereas the impact of reduced MPV17 expression (and the resulting severe reduction in mitochondrial folate pools) did not impact mitochondrial de novo dTMP synthesis in HeLa cells, the relationship between MPV17 expression, mitochondrial folate pools, and mitochondrial folate metabolism should be investigated in animal models. This report provides evidence that MPV17-related hepatocerebral MDS may be linked to altered mitochondrial folate accumulation, dTMP synthesis, and impaired FOCM in mitochondria.
Experimental procedures
Stable cell lines
HeLa cells with reduced MPV17 expression were generated using an MPV17 shRNA construct (Origene Technologies) with the Mirus Ingenio electroporation kit and NucleofectorTM technology (Lonza). Clonal populations were selected using puromycin and transferred to 96-well plates. Initial puromycin concentrations were 0.25 μg/ml and were slowly increased up to 1 μg/ml. Cells were isolated and expanded as individual cell lines while always maintaining puromycin selection. Control lines were generated under the same conditions but using scrambled shRNA construct (Qiagen). MPV17 knockdown was validated by Western blotting normalized to protein concentration using a Lowry protein assay.
Culture conditions
Cells were cultured in HyClone minimum essential medium α (αMEM) supplemented with 10% fetal bovine serum (FBS), 1% penicillin/streptomycin, and 0.50 μg/ml puromycin to maintain selection. Modified αMEM (HyClone) lacking glycine, serine, methionine, folate, and nucleosides was used in experiments as indicated; modified αMEM was supplemented with 10% dialyzed FBS, 200 μm methionine, and 20 nm (6S)-5-formyl-THF (folate-replete cells only).
dU suppression assay
This assay quantifies de novo dTMP synthesis capacity based on incorporation of [14C]deoxyuridine, the precursor of folate-dependent de novo synthesis of dTMP, compared with [3H]thymidine incorporation from the salvage pathway, into DNA. Cells were plated 1:8 in modified αMEM containing 2 μm 2′-[14C]deoxyuridine (American Radiochemicals) and 25 nm [3H]thymidine (PerkinElmer Life Sciences). Cells were rinsed twice with 1× PBS and detached from the tissue culture plates with 1× 0.25% trypsin-EDTA (Corning, Inc.); trypsin was neutralized with unlabeled medium (for nuclear dU suppression) or with 10% FBS in 1× PBS (for mitochondrial dU suppression). For nuclear dTMP biosynthesis capacity, genomic DNA was extracted using the DNAeasy Tissue and Blood Kit (Qiagen). For mitochondrial dTMP biosynthesis capacity, mitochondria were isolated, followed by mtDNA purification using the Zyppy Plasmid Miniprep kit (Zymo Research). 3H and 14C channels were counted in dual DPM mode on an LS6500 scintillation counter (Beckman Instruments).
Mitochondrial extraction
Mitochondrial extraction was carried out on ice. Solutions and equipment were maintained 4 °C or ice before and during the procedure. A total of 35 T175 flasks, 90–100% confluent, were collected per sample; cells were rinsed twice with 1× PBS and detached with 0.25% trypsin-EDTA 1× (Corning); trypsin was neutralized with 10% FBS, 1% penicillin/streptomycin in 1× PBS. Cell pellets were washed with 1× PBS and centrifuged (1000 × g, 5 min) before proceeding with mitochondrial extraction. Extraction of mitochondria was performed with an OptiPrep (60% iodixanol, Sigma-Aldrich) discontinuous gradient with some modifications.
OptiPrep discontinuous gradient
Cell pellets were washed with homogenization medium (0.25 m sucrose, 1 mm EDTA, 20 mm HEPES-NaOH, pH 7.4, and freshly added protease inhibitor), followed by centrifugation. Pellet was resuspended in 5 ml of homogenization medium and homogenized in a Dounce homogenizer pestle A, followed by centrifugation (1000 × g for 10 min in a fixed-angle rotor) to pellet nuclei. Pellet was suspended using a Dounce homogenizer pestle B, and the mitochondria was pelleted by centrifugation. Supernatants from both homogenization steps were combined and centrifuged at 17,000 × g for 10 min to obtain a crude mitochondrial pellet. The crude mitochondrial pellet was resuspended with a type B Dounce homogenizer. The crude fraction was adjusted to 36% (w/v) iodixanol with 50% iodixanol (diluted in 0.25 m sucrose, 6 mm EDTA, 120 mm Hepes-NaOH, pH 7.4, and fresh protease inhibitor). The mitochondrial fraction + iodizanol solution was loaded on the bottom of the ultracentrifuge tube and layered with equal parts of 25 and 20% iodixanol gradient solutions. One ml of the homogenization solution was added to the top layer. The sample was centrifuged at 100,000 × g for 4 h (Beckman SW41TI). The mitochondrial fraction was collected and diluted 3 times with homogenization buffer. The diluted mitochondrial fraction was centrifuged at 30,000 × g for 30 min to pellet mitochondria.
Formate suppression assay
This assay quantifies the relative rate of de novo purine synthesis capacity as a ratio of [14C]formate, the precursor of folate-dependent de novo synthesis, and [3H]hypoxanthine, a precursor for salvage purine biosynthesis. MPV17 stable knockdown and control cells were plated 1:6 in modified αMEM supplemented with 10% dialyzed fetal bovine serum, 200 μm methionine, and 20 nm (6S)-5-formyl-THF, 0.4 nm [3H]hypoxanthine, and 4 μm [14C]formate. At confluence, cells were rinsed twice with 1× PBS and detached with 1× 0.25% trypsin-EDTA (Corning). Nuclear DNA was isolated using the DNAeasy Tissue and Blood Kit (Qiagen) according to the manufacturer's protocols. 3H and 14C channels were counted in dual DPM mode on an LS6500 scintillation counter (Beckman Instruments).
Western blotting analyses and densitometry
All primary antibodies were diluted in 5% BSA and 0.02% sodium azide as indicated: proliferating cell nuclear antigen (nuclear marker; Cell Signaling, 1:1000, mouse); hexokinase I and CoxIV (mitochondrial markers; Cell Signaling, 1:1000, rabbit); FPGS (mouse, 1:1000, Zuckerman laboratory (37)); MPV17 (Abcam, 1:80, rabbit or mouse); anti-thymidine kinase 2 (Abcam, 1:100, rabbit); TYMS (Cell Signaling, 1:2000, rabbit). Secondary antibodies were diluted in 10% nonfat milk made in PBS. Densitometry was performed using ImageJ. Background was subtracted, and intensities of nonsaturated bands were quantified and normalized to loading control (CoxIV served as mitochondrial marker and mitochondrial loading control). Due to the size similarity, protein expressions of TK2 and TYMS were quantified on different Western blots.
Microbiological Lactobacillus casei assay
Folates in whole-cell samples as well as in isolated mitochondria were quantified as described (38, 39). Total folates were normalized to protein concentration (quantified with the Lowry protein assay) for each given sample.
Folate accumulation
Cells were plated in triplicates in 6-well plates in αMEM until 50% confluence. Growth medium was replaced with modified MEM containing 25 nm (6S)-[3H]5-formyl-THF, and the cells were cultured for 12 h. Cells were harvested, rinsed twice with 1× PBS, and counted with an automated cell counter (Bio-Rad, TC-20). Cells were lysed with 0.2 m ammonium hydroxide. Tritium in the cells was quantified in an LS6500 scintillation counter (Beckman Instruments) and normalized to the number of cells.
Folate uptake
Uptake of folate over time was determined. Cells were plated in triplicates in 6-well plates in αMEM. Once cells reached about 50% confluence, they were labeled with 25 nm (6S)-[3H]5-formyl-THF present in modified MEM for different time periods as shown in the graph. Cells were detached with 0.25% trypsin-EDTA (Corning) at the given time points, rinsed twice with 1× PBS, and counted with an automated cell counter (Bio-Rad, TC-20). Cells were lysed in 0.2 m ammonium hydroxide. Tritium in the cells was quantified in a scintillation counter and normalized to the number of cells.
Measurements of folate turnover by pulse-chase
Cells were plated in 15-cm plates and cultured in αMEM. When cells reached 70–80% confluence, medium was replaced with radioactive 25 nm (6S)-[3H]5-formyl-THF–modified MEM. After 13 h in labeled medium, cells were rinsed twice with 1× PBS, detached with 0.25% trypsin-EDTA (Corning), and counted with an automated cell counter (Bio-Rad, TC-20). About one-fifth of the cells were harvested (time 0); the rest of the cells were passaged into 10 6-well plates to be harvested under the same conditions at different time points. Each measurement was performed in triplicate. Harvested cells were lysed with 0.2 m ammonium hydroxide. Tritium remaining inside the cells was quantified in a scintillation counter and normalized to the number of cells.
Uracil content in DNA
Uracil present in both nuDNA and mtDNA was quantified by GC-MS and normalized to μg of DNA. For isolating nuDNA, whole cells were harvested by trypsinizing. nuDNA was extracted using the Roche High Pure PCR Template Preparation Kit and eluted in DNase/RNase-free water. nuDNA was briefly sonicated and treated with RNase A for 30 min. For isolation of mtDNA, mitochondria were isolated as described above, followed by mtDNA extraction using the Zyppy Plasmid Miniprep kit (Zymo Research), and eluted in DNase/RNase-free water. mtDNA was briefly sonicated and treated with RNase A for 30 min. DNA from nuclear and mitochondrial samples were quantified as described previously (40) with the following modification; 50 pg of 15N6 uracil was added as an internal standard to all samples and standards.
Mitochondrial content and mass
Mitochondrial DNA content, the number of mitochondrial genomes per cell, was quantified by real-time quantitative PCR (Roche LightCycler® 480) as described previously (41), using LightCycler® 480 SYBR Green I Master (Roche Applied Science) and 3 μg of DNA/reaction. Mitochondrial mass, number of mitochondria per cell, was quantified with the Citrate Synthase Activity Assay Kit (Invitrogen) according to the manufacturer's instructions. Citrate synthase activity is a marker for mitochondrial mass. Samples consisted of whole-cell extracts normalized to total cellular protein. Protein was quantified by the Lowry protein assay.
Isolation of nucleotides in mtDNA
This procedure confirms that dTMP precursors, dU and dT, were incorporated into mtDNA as dTTP. Cells were cultured and harvested in the presence of [14C]deoxyuridine and [3H]thymidine, as described for dU suppression. Mitochondria were isolated, followed by mtDNA purification using the Zyppy Plasmid Miniprep kit (Zymo Research). DNA was digested to nucleosides as described previously (7). Nucleosides were separated by HPLC with a Synergi Fusion-RP column (Phenomenex) using a binary buffer system with a flow rate of 1 ml/min as described previously (18). Radioactivity in each fraction was quantified using a Beckman LS-6500 liquid scintillation counter in dual disintegrations/min mode.
Author contributions
J. R. A., M. S. F., and P. J. S. designed the research and wrote the manuscript. C. V. generated the MPV17 shRNA knockdown stable cell lines used for this research. J. R. A. conducted the research. M. S. F. separated nucleotide bases by HPLC (Fig. S2). All authors analyzed the results and approved the final version of the manuscript.
Supplementary Material
Acknowledgment
We thank Dr. Rick Moran (Virginia Commonwealth University) for the generous gift of monoclonal anti-FPGS human antibody.
This work was supported by Public Health Service Grant R37DK58144 (to P. J. S.). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–S4.
- MDS
- mitochondrial DNA-depletion syndrome
- SHMT
- serine hydroxymethyltransferase
- DHFR
- dihydrofolate reductase
- TYMS
- thymidylate synthase
- mtDNA
- mitochondrial DNA
- FOCM
- folate-mediated one-carbon metabolism
- THF
- tetrahydrofolate
- 1C
- one-carbon
- DHF
- dihydrofolate
- nuDNA
- nuclear DNA
- KD
- knockdown
- FPGS
- folylpoly-γ-glutamate synthetase
- αMEM
- minimum essential medium α
- FBS
- fetal bovine serum
- SUMO
- small ubiquitin-like modifier
- TK
- thymidine kinase
- AdoHcy
- S-adenosylhomocysteine
- AdoMet
- S-adenosylmethionine.
References
- 1. Spinazzola A., Viscomi C., Fernandez-Vizarra E., Carrara F., D'Adamo P., Calvo S., Marsano R. M., Donnini C., Weiher H., Strisciuglio P., Parini R., Sarzi E., Chan A., DiMauro S., Rötig A., et al. (2006) MPV17 encodes an inner mitochondrial membrane protein and is mutated in infantile hepatic mitochondrial DNA depletion. Nat. Genet. 38, 570–575 10.1038/ng1765 [DOI] [PubMed] [Google Scholar]
- 2. Weiher H., Noda T., Gray D. A., Sharpe A. H., and Jaenisch R. (1990) Transgenic mouse model of kidney disease: insertional inactivation of ubiquitously expressed gene leads to nephrotic syndrome. Cell 62, 425–434 10.1016/0092-8674(90)90008-3 [DOI] [PubMed] [Google Scholar]
- 3. Fagerberg L., Hallström B. M., Oksvold P., Kampf C., Djureinovic D., Odeberg J., Habuka M., Tahmasebpoor S., Danielsson A., Edlund K., Asplund A., Sjöstedt E., Lundberg E., Szigyarto C. A., Skogs M., et al. (2014) Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol. Cell. Proteomics 13, 397–406 10.1074/mcp.M113.035600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. AlSaman A., Tomoum H., Invernizzi F., and Zeviani M. (2012) Hepatocerebral form of mitochondrial DNA depletion syndrome due to mutation in MPV17 gene. Saudi J. Gastroenterol. 18, 285–289 10.4103/1319-3767.98439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. El-Hattab A. W., Scaglia F., Craigen W. J., and Wong L. J. C. (2012) MPV17-related hepatocerebral mitochondrial DNA depletion syndrome. in GeneReviews (Adam M. P., Ardinger H. H., Pagon R. A., Wallace S. E., Amemiya A., Bean L. J. H., Bird T. D., Ledbetter N., Mefford H. C., Smith R. J. H., and Stephens K., eds) University of Washington, Seattle [Google Scholar]
- 6. Bijarnia-Mahay S., Mohan N., Goyal D., and Verma I. C. (2014) Mitochondrial DNA depletion syndrome causing liver failure. Indian Pediatr. 51, 666–668 10.1007/s13312-014-0475-z [DOI] [PubMed] [Google Scholar]
- 7. Friso S., Choi S. W., Girelli D., Mason J. B., Dolnikowski G. G., Bagley P. J., Olivieri O., Jacques P. F., Rosenberg I. H., Corrocher R., and Selhub J. (2002) A common mutation in the 5,10-methylenetetrahydrofolate reductase gene affects genomic DNA methylation through an interaction with folate status. Proc. Natl. Acad. Sci. U.S.A. 99, 5606–5611 10.1073/pnas.062066299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zwacka R. M., Reuter A., Pfaff E., Moll J., Gorgas K., Karasawa M., and Weiher H. (1994) The glomerulosclerosis gene Mpv17 encodes a peroxisomal protein producing reactive oxygen species. EMBO J. 13, 5129–5134 10.1002/j.1460-2075.1994.tb06842.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Casalena G., Krick S., Daehn I., Yu L., Ju W., Shi S., Tsai S. Y., D'Agati V., Lindenmeyer M., Cohen C. D., Schlondorff D., and Bottinger E. P. (2014) Mpv17 in mitochondria protects podocytes against mitochondrial dysfunction and apoptosis in vivo and in vitro. Am. J. Physiol. Renal Physiol. 306, F1372–F1380 10.1152/ajprenal.00608.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Antonenkov V. D., Isomursu A., Mennerich D., Vapola M. H., Weiher H., Kietzmann T., and Hiltunen J. K. (2015) The human mitochondrial DNA depletion syndrome gene MPV17 encodes a non-selective channel that modulates membrane potential. J. Biol. Chem. 290, 13840–13861 10.1074/jbc.M114.608083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dalla Rosa I., Cámara Y., Durigon R., Moss C. F., Vidoni S., Akman G., Hunt L., Johnson M. A., Grocott S., Wang L., Thorburn D. R., Hirano M., Poulton J., Taylor R. W., Elgar G., et al. (2016) MPV17 loss causes deoxynucleotide insufficiency and slow DNA replication in mitochondria. PLoS Genet. 12, e1005779 10.1371/journal.pgen.1005779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. González-Vioque E., Torres-Torronteras J., Andreu A. L., and Martí R. (2011) Limited dCTP availability accounts for mitochondrial DNA depletion in mitochondrial neurogastrointestinal encephalomyopathy (MNGIE). PLoS Genet. 7, e1002035 10.1371/journal.pgen.1002035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fox J. T., and Stover P. J. (2008) Folate-mediated one-carbon metabolism. Vitam. Horm. 79, 1–44 10.1016/S0083-6729(08)00401-9 [DOI] [PubMed] [Google Scholar]
- 14. Anderson D. D., Quintero C. M., and Stover P. J. (2011) Identification of a de novo thymidylate biosynthesis pathway in mammalian mitochondria. Proc. Natl. Acad. Sci. U.S.A. 108, 15163–15168 10.1073/pnas.1103623108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Woeller C. F., Anderson D. D., Szebenyi D. M. E., and Stover P. J. (2007) Evidence for small ubiquitin-like modifier-dependent nuclear import of the thymidylate biosynthesis pathway. J. Biol. Chem. 282, 17623–17631 10.1074/jbc.M702526200 [DOI] [PubMed] [Google Scholar]
- 16. Anderson D. D., Woeller C. F., Chiang E.-P., Shane B., and Stover P. J. (2012) Serine hydroxymethyltransferase anchors de novo thymidylate synthesis pathway to nuclear lamina for DNA synthesis. J. Biol. Chem. 287, 7051–7062 10.1074/jbc.M111.333120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Herbig K., Chiang E.-P., Lee L.-R., Hills J., Shane B., and Stover P. J. (2002) Cytoplasmic serine hydroxymethyltransferase mediates competition between folate-dependent deoxyribonucleotide and S-adenosylmethionine biosyntheses. J. Biol. Chem. 277, 38381–38389 10.1074/jbc.M205000200 [DOI] [PubMed] [Google Scholar]
- 18. Field M. S., Kamynina E., Agunloye O. C., Liebenthal R. P., Lamarre S. G., Brosnan M. E., Brosnan J. T., and Stover P. J. (2014) Nuclear enrichment of folate cofactors and methylenetetrahydrofolate dehydrogenase 1 (MTHFD1) protect de novo thymidylate biosynthesis during folate deficiency. J. Biol. Chem. 289, 29642–29650 10.1074/jbc.M114.599589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Das K. C., and Herbert V. (1989) In vitro DNA synthesis by megaloblastic bone marrow: effect of folates and cobalamins on thymidine incorporation and de novo thymidylate synthesis. Am. J. Hematol. 31, 11–20 10.1002/ajh.2830310103 [DOI] [PubMed] [Google Scholar]
- 20. Blount B. C., Mack M. M., Wehr C. M., MacGregor J. T., Hiatt R. A., Wang G., Wickramasinghe S. N., Everson R. B., and Ames B. N. (1997) Folate deficiency causes uracil misincorporation into human DNA and chromosome breakage: implications for cancer and neuronal damage. Proc. Natl. Acad. Sci. U.S.A. 94, 3290–3295 10.1073/pnas.94.7.3290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tibbetts A. S., and Appling D. R. (2010) Compartmentalization of mammalian folate-mediated one-carbon metabolism. Annu. Rev. Nutr. 30, 57–81 10.1146/annurev.nutr.012809.104810 [DOI] [PubMed] [Google Scholar]
- 22. Trent D. F., Seither R. L., and Goldman I. D. (1991) Compartmentation of intracellular folates: failure to interconvert tetrahydrofolate cofactors to dihydrofolate in mitochondria of L1210 leukemia cells treated with trimetrexate. Biochem. Pharmacol. 42, 1015–1019 10.1016/0006-2952(91)90283-B [DOI] [PubMed] [Google Scholar]
- 23. Ye Y.-L., Chan Y.-T., Liu H.-C., Lu H.-T., and Huang R.-F. S. (2010) Depleted folate pool and dysfunctional mitochondria associated with defective mitochondrial folate proteins sensitize Chinese ovary cell mutants to tert-butylhydroperoxide-induced oxidative stress and apoptosis. J. Nutr. Biochem. 21, 793–800 10.1016/j.jnutbio.2009.05.005 [DOI] [PubMed] [Google Scholar]
- 24. Lin B. F., Huang R. F., and Shane B. (1993) Regulation of folate and one-carbon metabolism in mammalian cells. III. Role of mitochondrial folylpoly-γ-glutamate synthetase. J. Biol. Chem. 268, 21674–21679 [PubMed] [Google Scholar]
- 25. Lowe K. E., Osborne C. B., Lin B. F., Kim J. S., Hsu J. C., and Shane B. (1993) Regulation of folate and one-carbon metabolism in mammalian cells. II. Effect of folylpoly-γ-glutamate synthetase substrate specificity and level on folate metabolism and folylpoly-γ-glutamate specificity of metabolic cycles of one-carbon metabolism. J. Biol. Chem. 268, 21665–21673 [PubMed] [Google Scholar]
- 26. Basten G. P., Duthie S. J., Pirie L., Vaughan N., Hill M. H., and Powers H. J. (2006) Sensitivity of markers of DNA stability and DNA repair activity to folate supplementation in healthy volunteers. Br. J. Cancer 94, 1942–1947 10.1038/sj.bjc.6603197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Stover P. J., and Weiss R. S. (2012) Sensitizing cancer cells: is it really all about U? Cancer Cell 22, 3–4 10.1016/j.ccr.2012.06.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Löllgen S., and Weiher H. (2015) The role of the Mpv17 protein mutations of which cause mitochondrial DNA depletion syndrome (MDDS): lessons from homologs in different species. Biol. Chem. 396, 13–25 [DOI] [PubMed] [Google Scholar]
- 29. Di Noia M. A., Todisco S., Cirigliano A., Rinaldi T., Agrimi G., Iacobazzi V., and Palmieri F. (2014) The human SLC25A33 and SLC25A36 genes of solute carrier family 25 encode two mitochondrial pyrimidine nucleotide transporters. J. Biol. Chem. 289, 33137–33148 10.1074/jbc.M114.610808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Blakely E. L., Butterworth A., Hadden R. D. M., Bodi I., He L., McFarland R., and Taylor R. W. (2012) MPV17 mutation causes neuropathy and leukoencephalopathy with multiple mtDNA deletions in muscle. Neuromuscul. Disord. 22, 587–591 10.1016/j.nmd.2012.03.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ferraro P., Nicolosi L., Bernardi P., Reichard P., and Bianchi V. (2006) Mitochondrial deoxynucleotide pool sizes in mouse liver and evidence for a transport mechanism for thymidine monophosphate. Proc. Natl. Acad. Sci. U.S.A. 103, 18586–18591 10.1073/pnas.0609020103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Neuburger M., Rébeillé F., Jourdain A., Nakamura S., and Douce R. (1996) Mitochondria are a major site for folate and thymidylate synthesis in plants. J. Biol. Chem. 271, 9466–9472 10.1074/jbc.271.16.9466 [DOI] [PubMed] [Google Scholar]
- 33. Bailey L. B. (2009) Folate in Health and Disease, 2nd Ed., CRC Press, Inc., Boca Raton, FL [Google Scholar]
- 34. Chou Y.-F., and Huang R.-F. S. (2009) Mitochondrial DNA deletions of blood lymphocytes as genetic markers of low folate-related mitochondrial genotoxicity in peripheral tissues. Eur. J. Nutr. 48, 429–436 10.1007/s00394-009-0031-0 [DOI] [PubMed] [Google Scholar]
- 35. Branda R. F., Brooks E. M., Chen Z., Naud S. J., and Nicklas J. A. (2002) Dietary modulation of mitochondrial DNA deletions and copy number after chemotherapy in rats. Mutat. Res. 501, 29–36 10.1016/S0027-5107(02)00014-3 [DOI] [PubMed] [Google Scholar]
- 36. Crott J. W., Choi S.-W., Branda R. F., and Mason J. B. (2005) Accumulation of mitochondrial DNA deletions is age, tissue and folate-dependent in rats. Mutat. Res. 570, 63–70 10.1016/j.mrfmmm.2004.09.009 [DOI] [PubMed] [Google Scholar]
- 37. Dotzlaf J., Carpenter J., Luo S., Miles R. R., Fisher D., Qian Y. W., Ehsani M., Wang X., Lin A., McClure D. B., Chen V. J., and Zuckerman S. H. (2007) Derivation and characterization of monoclonal antibodies against human folypolyglutamate synthetase. Hybridoma 26, 155–161 10.1089/hyb.2007.004 [DOI] [PubMed] [Google Scholar]
- 38. Horne D. W. (1997) Microbiological assay of folates in 96-well microtiter plates. Methods Enzymol. 281, 38–43 [DOI] [PubMed] [Google Scholar]
- 39. Molloy A. M., and Scott J. M. (1997) Microbiological assay for serum, plasma, and red cell folate using cryopreserved, microtiter plate method. Methods Enzymol. 281, 43–53 10.1016/S0076-6879(97)81007-5 [DOI] [PubMed] [Google Scholar]
- 40. MacFarlane A. J., Liu X., Perry C. A., Flodby P., Allen R. H., Stabler S. P., and Stover P. J. (2008) Cytoplasmic serine hydroxymethyltransferase regulates the metabolic partitioning of methylenetetrahydrofolate but is not essential in mice. J. Biol. Chem. 283, 25846–25853 10.1074/jbc.M802671200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Venegas V., Wang J., Dimmock D., and Wong L. J. (2011) Real-time quantitative PCR analysis of mitochondrial DNA content. Curr. Protoc. Hum. Genet., Chapter 19, Unit 19.17 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.