

Abstract
Monoallelic point mutations in the gene encoding the cytosolic, NADP+-dependent enzyme isocitrate dehydrogenase 1 (IDH1) cause increased production of the oncometabolite 2-hydroxyglutarate (2-HG) in multiple cancers. Most IDH1 mutant tumors retain one wildtype (WT) IDH1 allele. Several studies have proposed that retention of this WT allele is protumorigenic by facilitating substrate channeling through a WT–mutant IDH1 heterodimer, with the WT subunit generating a local supply of α-ketoglutarate and NADPH that is then consumed by the mutant subunit to produce 2-HG. Here, we confirmed that coexpression of WT and mutant IDH1 subunits leads to formation of WT–mutant hetero-oligomers and increases 2-HG production. An analysis of a recently reported crystal structure of the WT–R132H IDH1 heterodimer and of in vitro kinetic parameters for 2-HG production, however, indicated that substrate channeling between the subunits is biophysically implausible. We also found that putative carbon-substrate flux between WT and mutant IDH1 subunits is inconsistent with the results of isotope tracing experiments in cancer cells harboring an endogenous monoallelic IDH1 mutation. Finally, using a mathematical model of WT–mutant IDH1 heterodimers, we estimated that the NADPH:NADP+ ratio is higher in the cytosol than in the mitochondria, suggesting that NADPH is unlikely to be limiting for 2-HG production in the cytosol. These findings argue against supply of either substrate being limiting for 2-HG production by a cytosolic IDH1 mutant and suggest that the retention of a WT allele in IDH1 mutant tumors is not due to a requirement for carbon or cofactor flux between WT and mutant IDH1.
Keywords: cancer biology, glioblastoma, isotopic tracer, mathematical modeling, systems biology, tumor metabolism, isocitrate dehydrogenase, metabolic compartmentalization, oncometabolite, substrate channeling
Introduction
The metabolic enzymes isocitrate dehydrogenase 1 (IDH1)4 and 2 (IDH2) are mutated in numerous cancers, including glioma, acute myeloid leukemia (AML), chondrosarcoma, and cholangiocarcinoma (1–6). These mutations confer a neomorphic activity for the NADPH-dependent reduction of α-ketoglutarate (α-KG) to R(−)-2-hydroxyglutarate (2-HG), an oncometabolite that is proposed to competitively inhibit a range of α-KG–dependent enzymes involved in epigenetic regulation (4, 7, 8). Small-molecule inhibitors of mutant IDH1 and IDH2 were recently approved for treatment of IDH-mutant AML, and additional clinical trials are ongoing for other cancers (9–12). 2-HG is also a biomarker of disease burden in multiple IDH mutant malignancies (13, 14).
With few exceptions, mutations in IDH1 are monoallelic with retention of a wildtype (WT) allele. Explanations for retention of the WT allele in tumors remain controversial. One possible reason is that mutant IDH might deplete cellular NADPH levels (15, 16). In this context, retention of a WT IDH allele could mitigate NADPH depletion, minimizing toxic accumulation of reactive oxygen species or impairment of biosynthetic capacity. As the kinetics of the NADPH-consuming reaction of mutant IDH proteins are slow relative to the NADPH-producing reactions of WT IDH and other enzymes (4, 7, 8), however, 2-HG production may not be sufficient to deplete NADPH except in engineered cell systems with mutant IDH1 overexpressed.
Alternatively, a retained WT subunit could be critical for optimum 2-HG production by a mutant subunit as part of a WT–mutant IDH heterodimer. Ward et al. (17) showed previously that for the cytosolic IDH1 isoform, but not the mitochondrial isoform IDH2, retention of the WT allele in cells is indeed important for maximizing 2-HG production by the mutant. Others have disputed these findings based on experiments performed with transfected cells in culture (18). There exist, however, rare human glioblastoma samples with IDH1 mutations in which the WT IDH1 allele is eventually lost. In these patient-derived samples, there is a nearly 10-fold decrease in intratumor 2-HG concomitant with loss of the WT allele (19). In vitro assays of α-KG and NADPH consumption in IDH mutant cell lysates have also suggested synergy between the WT and mutant IDH activities (7).
Several explanations have been proposed for the dependence of mutant IDH1 activity on WT IDH1. It is possible that the enzymatic activity of WT–mutant heterodimers is intrinsically superior to that of mutant–mutant homodimers as suggested by some studies of recombinant IDH1 proteins (20, 21). Others, however, have found no advantage for the recombinant IDH1 heterodimer (22). Alternatively, the WT dependence might be due to substrate channeling or some other form of cooperativity between the WT and mutant subunits as proposed previously (17, 23). Pietrak et al. (24) demonstrated that both the WT activity for oxidative decarboxylation of isocitrate and the mutant activity for reduction of α-KG to 2-HG remain intact in a WT–mutant IDH1 heterodimer, suggesting that channeling could be involved. The dependence of 2-HG production on WT IDH1 might also reflect differences in substrate availability in the cytosol and mitochondria as levels of α-KG and NADPH may differ between compartments.
Here, we report a lack of evidence for substrate channeling or metabolic flux between WT and mutant IDH1. We first considered the possibility that α-KG and NADPH are produced and maintained in a protected pool around the heterodimer by the WT subunit. On the basis of existing structural evidence and in vitro IDH1 enzyme kinetics, we calculated that such channeling is biophysically implausible, even for densely packed IDH1 heterodimers. After ruling out substrate channeling, we then characterized the extent of all carbon flux through WT IDH1. Using stable isotope tracing, we showed that 2-HG is predominantly derived from glutamine in IDH1 mutant cancer cells in culture with no need to invoke flux of α-KG between subunits. Finally, through quantitative modeling of IDH1 WT–mutant heterodimers, we estimated the mitochondrial to cytosolic NADPH:NADP+ ratio, which suggests that NADPH is unlikely to be limiting for the production of 2-HG by mutant IDH1 in the cytosol.
Results
Coexpression of WT and mutant IDH1 increases 2-HG production and is associated with the formation of WT-mutant IDH1 hetero-oligomeric species
WT IDH1 in the cytosol can produce α-KG and NADPH. Mutant IDH1 consumes α-KG and NADPH, producing the oncometabolite 2-HG and regenerating NADP+ (Fig. 1A). Most cancer-associated IDH1 mutations occur in monoallelic fashion with retention of one WT IDH1 allele in the tumor cells. It was shown previously that coexpression of WT and R132H IDH1 in cells results in greater 2-HG production than expression of equimolar amounts of mutant IDH1 alone (17). In these prior experiments, however, the generation of WT–mutant IDH1 heterodimers was not formally demonstrated. Here, we confirm previous findings that the coexpression of WT and mutant IDH1 increases 2-HG production (Fig. 1B). In HEK 293T cells already expressing endogenous WT IDH1, expression of increasing amounts of FLAG-tagged R132H IDH1 resulted in increasing 2-HG production. Coexpression of Myc-tagged WT IDH1 further enhanced the production of 2-HG, consistent with previous findings (17). In addition, we found that the exogenous coexpression of WT and mutant IDH1 resulting in enhanced 2-HG production is associated with the formation of WT–mutant IDH1 hetero-oligomeric species in cells. Cells were transfected with either empty vector, a mutant IDH1 construct with a FLAG epitope tag (R132H-FLAG), or R132H-FLAG in combination with a WT IDH1 construct containing a Myc epitope tag. 48 h after transfection, cells were quenched rapidly in 80% MeOH and subsequently analyzed for 2-HG levels by GC-MS. A parallel set of transfected cells was lysed and used for FLAG immunoprecipitation and Western blotting. Myc-tagged WT IDH1 was found to coprecipitate with FLAG-tagged R132H IDH1 in cells in which they were coexpressed (Fig. 1B).
Figure 1.

WT IDH1 activity enhances 2-HG production in the cytoplasm. A, schematic of the WT and neomorphic reactions catalyzed by the WT–mutant IDH1 heterodimer. WT IDH1 catalyzes the NADP+-dependent oxidative decarboxylation of isocitrate to α-KG. Mutant IDH1 reduces α-KG to the oncometabolite 2-HG. The dashed arrows indicate potential opportunities for substrate channeling between the WT and mutant subunits of the IDH1 heterodimer. B, FLAG-tagged R132H IDH1 (at various doses), FLAG-tagged R132H IDH1 plus Myc-tagged WT IDH1, or empty vector was transfected into HEK 293T cells. Cells were harvested 48 h post-transfection and assayed for 2-HG accumulation by GC-MS (top) or protein expression by immunoprecipitation (IP) and Western blotting (bottom). Absolute 2-HG levels were determined by normalization of the 2-HG GC-MS signal intensity to that of a deuterated 2-HG internal standard and then normalized to total cellular protein. The upper band detected by the anti-Myc tag antibody is nonspecific. Error bars denote one S.D.
In vitro kinetic characterization of IDH1
A number of recent studies have reported in vitro kinetic parameters for WT IDH1 and various oncogenic mutants (7, 21, 23–26). There is substantial variability in the values reported; for instance, measurements of kcat for the WT enzyme range from 8.6 to 44,000 s−1 and from 0.19 to 1,000 s−1 for R132H IDH1. To investigate this discrepancy and to obtain values for mathematical modeling, we measured the in vitro kinetics of recombinant WT and R132H IDH1 (Table 1). The parameters obtained are in close agreement with the prior values reported in two studies of the enzymatic mechanism of IDH1 (24, 26) and are used for all subsequent calculations.
Table 1.
In vitro kinetics of WT and R132H IDH1
All uncertainty values are standard errors.
| WT | |
| Km, isocitrate (μm) | 6.88 ± 0.97 |
| Km, NADP+ (μm) | 6.79 ± 0.31 |
| kcat (s−1) | 3.10 ± 0.14 |
| R132H | |
| Km, α-KG (μm) | 988 ± 77 |
| Km, NADPH (μm) | 1.36 ± 0.39 |
| kcat (s−1) | 0.0666 ± 0.0016 |
Substrate channeling is biophysically implausible
As discussed above, the neomorphic reaction catalyzed by mutant IDH1 requires α-KG and NADPH, both of which are produced in the WT reaction (Fig. 1A). As such, it has been suggested that cytoplasmic 2-HG synthesis might be enhanced by the local production of α-KG and NADPH in WT–mutant heterodimers and that this effect might explain the dramatic reduction of 2-HG levels following loss of the WT enzyme (17). As an initial test of this hypothesis, we performed a biophysical analysis of putative substrate channeling between the WT and mutant subunits.
A crystal structure of the IDH1 WT–R132H heterodimer at 3.2-Å resolution has been reported previously (Fig. 2A) (25). The structure reveals that there is no physical association between the active sites of the WT and the mutant, suggesting that any interaction would be due to a local elevation of [α-KG] and [NADPH] around each heterodimer. We first examined diffusion of α-KG and NADPH away from the WT active site to assess the feasibility of achieving local enrichment. From inspection of the crystal structure, we estimated that the straight-line distance between the two active sites is ∼37.3 Å. Assuming Dα-KG = 6.7 × 10− 10 m2/s and DNADPH = 4.1 × 10−10 m2/s (see “Experimental procedures”) and three-dimensional diffusion governed by
| (Eq. 1) |
it would take just 3.5 and 5.7 ns for α-KG and NADPH, respectively, to travel the distance between the active sites. In 15 s, the time required for one turnover of the mutant enzyme, both substrates could travel over 3000 times further than the intersite distance. This simple calculation suggests that the local and bulk cytoplasmic concentrations of α-KG and NADPH are likely equilibrated on catalytically relevant timescales. As our estimate for intersite distance is based on a crystal structure, it does not account for possible dynamic changes in the conformation of the two subunits, which could bring the active sites into closer proximity. It is plausible that such shifts do occur in the IDH1 heterodimer as certain inhibitors of the mutant enzyme are known to bind to an allosteric pocket on the WT subunit, and the activity of WT IDH1 is controlled in part by the highly flexible α10 regulatory element (27, 28). Any dynamic change, however, would have to reduce the straight-line distance by at least several orders of magnitude for local elevation of substrate concentration to be relevant.
Figure 2.
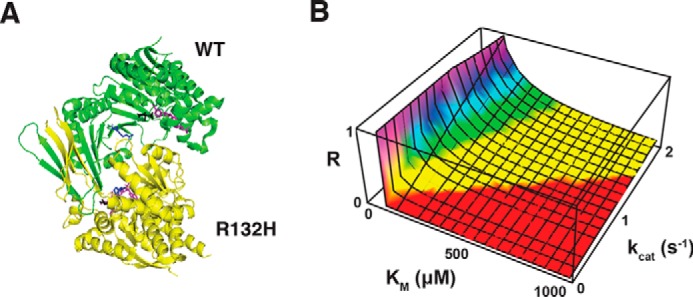
Biophysical evaluation of putative substrate channeling between WT and R132H IDH1 subunits. A, crystal structure of the WT–R132H IDH1 heterodimer. The WT subunit is colored green; the mutant is yellow. Residue 132 is colored blue in both subunits. Isocitrate (black) and NADP(H) (purple) are bound in the respective active sites. The structure was drawn from data reported in Yang et al. (25) (Protein Data Bank (PDB) code 3MAS). B, channeling by enzyme agglomeration is infeasible for the IDH1 WT–R132H heterodimer. Shown is a plot of the ratio of the processing rate to the escape rate as a function of Km (for either α-KG or NADPH) and kcat for N = 4000 heterodimers clustered in a sphere with radius r = 40 nm.
Packing of enzymes into dense clusters has been suggested to enable channeling even in the absence of physical association of active sites. Castellana et al. (29) reported a detailed theoretical analysis of such channeling by agglomeration. Using their framework, we investigated whether N = 4000 IDH1 heterodimers confined in a sphere with radius r = 40 nm (i.e. at the dense-packing limit nmax = 25 mm) could efficiently channel α-KG and NADPH. Efficient channeling requires that the processing rate
| (Eq. 2) |
exceed the escape rate
| (Eq. 3) |
where κ is the catalytic efficiency of the enzyme. For this calculation, we do not need to make any assumptions about the relative positions of the WT and mutant active sites. The RP/RE ratio for carbamoyl-phosphate synthetase and aspartate carbamoyltransferase in Escherichia coli, a system for which Castellana et al. (29) obtained experimental evidence of channeling by agglomeration, is ∼9.5. In contrast, we calculated ratios of 4.0 × 10−8 and 4.7 × 10−3 for the processing of α-KG and NADPH, respectively, by the dense IDH1 cluster. Further analysis indicated that submicromolar Km values would be required for both substrates to achieve parity given the relatively slow kcat for R132H IDH1 (Fig. 2B).
2-HG production in IDH1 mutant cancer cells is predominantly from glutamine and can be explained without carbon flux between WT and mutant subunits
The most direct route of 2-HG production from glucose involves production of cytosolic α-KG by WT IDH1 followed by consumption of this α-KG by mutant IDH1 to generate 2-HG. In contrast, 2-HG production by mutant IDH1 from glutamine-derived α-KG can occur without production of α-KG by WT IDH (Fig. 3A). We grew HT1080 fibrosarcoma cells, which harbor an endogenous monoallelic IDH1 mutation, either in glutamine-containing, glucose-free medium supplemented with uniformly labeled glucose ([13C6]glucose) or in glucose-containing, glutamine-free medium supplemented with uniformly labeled glutamine ([13C5]glutamine). As shown in Fig. S1, [13C6]glucose can be converted via glycolysis to pyruvate with three 13C atoms (M + 3 pyruvate), which subsequently can enter the TCA cycle as M + 2 acetyl-CoA with further metabolism to M + 2 citrate and M + 2 isocitrate. Cytosolic pools of M + 2 isocitrate can be converted by WT IDH1 to M + 2 α-KG, which finally can be reduced to M + 2 2-HG. When we followed the fate of glucose carbon by growing HT1080 cells in medium with [13C6]glucose (and unlabeled glutamine), we observed only 10% incorporation of glucose-derived carbon into 2-HG (Fig. 3B). In contrast, [13C5]glutamine can be converted via glutaminolysis to fully labeled M + 5 α-KG, which can then be reduced to fully labeled M + 5 2-HG. Comparing 2-HG production from labeled glucose and labeled glutamine therefore provides information about the extent of carbon flux between the WT and mutant subunits in the IDH1 heterodimer (Fig. 3A). For HT1080 cells grown in medium with [13C5]glutamine (and unlabeled glucose), we found that nearly the entire 2-HG pool was labeled with carbon derived from glutamine (Fig. 3B), suggesting minimal importance of intersubunit flux.
Figure 3.

In vivo evaluation of 2-HG production kinetics. A, schematics showing predicted labeling when cells are grown in medium either with uniformly 13C-labeled glucose ([13C6]glucose) and unlabeled glutamine (left) or with uniformly 13C-labeled glutamine ([13C5]glutamine) and unlabeled glucose (right). [13C6]Glucose is converted to doubly labeled isocitrate, α-KG, and 2-HG. [13C5]Glutamine is converted to fully labeled α-KG and 2-HG. Dashed arrows indicate that one or more intermediate reactions have been omitted from the diagram for clarity. The red arrow indicates possible flux of labeled α-KG derived from [13C6]glucose. B, kinetics of 2-HG labeling with 13C over 24 h when cells are cultured in complete medium containing [13C6]glucose (with unlabeled glutamine) or [13C5]glutamine (with unlabeled glucose). M0–M6 refer to the number of labeled carbon atoms. C and D, kinetics of citrate, α-KG, and succinate labeling with 13C over 24 h of growth in complete medium containing [13C6]glucose (with unlabeled glutamine) (C) or [13C5]glutamine (with unlabeled glucose) (D). The color scheme is the same as in B.
Evidence for a branched TCA cycle in cancer cells harboring an IDH1 mutation
Further analysis of the labeling data from IDH1 mutant HT1080 cells revealed that TCA cycle metabolites were not labeled in a pattern consistent with a complete traditional TCA cycle. As predicted, HT1080 cells cultured with [13C6]glucose and unlabeled glutamine exhibited rapid labeling of the citrate pool, evidenced by significant accumulation of M + 2 citrate (Fig. 3C). There was, however, low (<10%) labeling of the α-KG and succinate pools from [13C6]glucose even after 24 h. Cells cultured with [13C5]glutamine and unlabeled glucose exhibited robust production of fully labeled α-KG (M + 5 α-KG) from glutaminolysis as predicted. They also exhibited further oxidative metabolism through part of the TCA cycle to M + 4 succinate and then to M + 4 citrate (Fig. 3D). M + 4 citrate was not further metabolized to α-KG through aconitase and IDH, however, as indicated by minimal accumulation of M + 3 α-KG or M + 2 succinate (these labeled species would be expected if the TCA cycle turned multiple times). Of note, we also observed <5% labeling of the M + 5 citrate pool of cells cultured in [13C5]glutamine, consistent with low net flux from α-KG to citrate through reductive carboxylation. Overall, these data suggest minimal net flux from citrate to α-KG. Oxidative metabolism of either glucose- or glutamine-derived carbon can generate citrate, but significant metabolism of citrate through aconitase and IDH does not appear to occur in these cells under standard culture conditions. Instead, most citrate is likely exported to the cytosol where it can be metabolized by ATP-citrate lyase to produce cytosolic acetyl-CoA and oxaloacetate (Fig. S1A). This cytosolic oxaloacetate could potentially be converted to cytosolic malate and then re-enter the mitochondria through the malate/α-KG antiporter (Fig. 1B). As this model invokes export of mitochondrial α-KG to the cytosol, it also accounts for our observation of significant flux from M + 5 glutamine to M + 5 α-KG and then to M + 5 2-HG in cells harboring an IDH1 mutation despite glutaminolysis being primarily an intramitochondrial process (30, 31).
Estimation of compartment-specific NADPH levels
In addition to converting isocitrate to α-KG, WT IDH1 also produces cytosolic NADPH, which could potentially be limiting for 2-HG production by mutant IDH1. Rigorous determination of compartment-specific NADPH levels and NADPH:NADP+ ratios is technically challenging. We sought therefore to estimate [NADPH] in the cytosol and mitochondria using quantitative modeling of IDH heterodimers. To this end, we developed a biochemically realistic mathematical model of the IDH1 WT–mutant heterodimer. The model involves 11 species, IDH1 (denoted E), isocitrate (denoted I), α-KG, NADP+, NADPH, 2-HG, and five enzyme–substrate complexes, and 16 reactions, each with a corresponding kinetic parameter. Fig. 4 shows the full reaction network diagram, and Table 2 lists numerical estimates for the rate constants (see “Experimental procedures” for details of parameter estimation). Following recent experimental studies of the IDH1 enzyme mechanism (26), we model the WT reaction as random sequential so that either isocitrate or NADP+ may bind first to form an E·I·NADP+ ternary complex (module 1) and the neomorphic reaction as ordered sequential with NADPH and then α-KG binding to the enzyme to form an E·KG·NADPH ternary complex (module 2). The production of 2-HG by the IDH1 heterodimer occurs in vivo as part of a larger metabolic network. To make the network an open system, we include reactions for the zero-order synthesis of isocitrate and first-order degradation of 2-HG by processes not explicitly modeled (module 3). For simplicity, any reactions involving α-KG, NADP+, or NADPH that occur independently of the IDH1 heterodimer are excluded from the model.
Figure 4.
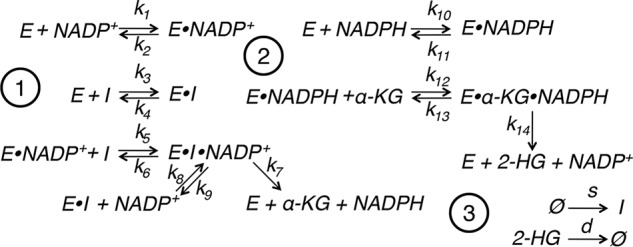
Biochemically realistic mathematical modeling of IDH1 heterodimers. Shown is the reaction network diagram for a detailed mechanistic model of WT IDH1 (1) and mutant IDH1 (2). The WT conversion of isocitrate to α-KG is assumed to follow a random-order, ternary complex mechanism. The mutant conversion of α-KG to 2-HG is assumed to follow an ordered sequential mechanism with NADPH binding before α-KG. The reactions in group 3 reflect the synthesis of isocitrate and α-KG and degradation of 2-HG by processes not explicitly modeled. E denotes the WT–mutant IDH1 heterodimer, and I denotes isocitrate.
Table 2.
Kinetic parameters for reaction network in Fig. 4
| Parameter class | Rate constants | Value |
|---|---|---|
| kon, NADP+ (μm−1 s−1) | k1, k8 | 1.00 × 108 |
| kon, isocitrate (μm−1 s−1) | k3, k5 | 1.00 × 108 |
| koff, NADP+ (s−1) | k2, k9 | 676 |
| koff, isocitrate (s−1) | k4, k9 | 685 |
| kcat, WT (s−1) | k7 | 3.10 |
| kon, NADPH (μm−1 s−1) | k10 | 1.00 × 108 |
| kon, α-KG (μm−1 s−1) | k12 | 1.00 × 108 |
| koff, NADPH (s−1) | k11 | 136 |
| koff, α-KG (s−1) | k13 | 9.88 × 104 |
| kcat, R132H (s−1) | k14 | 0.0666 |
Following mass-action kinetics, the reaction network in Fig. 4 gives rise to a system of 11 ordinary differential equations, which reduces at steady state to a polynomial system. The goal of this section is to derive a steady-state expression for [NADPH] in terms of just [I], [α-KG], [NADP+], and the rate constants, which, in combination with literature values for metabolite concentrations, enable estimation of the ratio of mitochondrial to cytosolic NADPH ([NADPH]m/[NADPH]c). At steady state, production and consumption of each of the binary enzyme–substrate complexes involved in the WT reaction (E·NADP+ and E·I) must balance, so that we have the following equations.
| (Eq. 4) |
and
| (Eq. 5) |
Similarly, production and consumption of the ternary complex E·NADP·I must balance as follows.
| (Eq. 6) |
Using Equations 4 and 5 in Equation 6 yields an expression for the ternary complex in terms of [E], [I], and [NADP+].
| (Eq. 7) |
The mutant reactions can be analyzed in an analogous fashion. From steady-state balance of the binary ([E·NADPH]) and ternary ([E·α-KG·NADPH]) complexes, we have
| (Eq. 8) |
and
| (Eq. 9) |
that together give Equation 10.
| (Eq. 10) |
Finally, balance of production and consumption of [NADPH] gives Equation 11.
| (Eq. 11) |
Using Equations 7 and 10 in Equation 11 yields
| (Eq. 12) |
as desired. Equation 12 can also be obtained directly from a Gröbner basis calculation (see “Experimental procedures” for details). Crucially, the expression is independent of [E] and of the concentrations of the various enzyme–substrate complexes, which are inaccessible to measurement.
In contrast to IDH1, the enzymatic mechanism of the mitochondrial isoform IDH2 has not been characterized in detail. It is plausible, however, that WT–R172K IDH2 heterodimers, which are structurally analogous to WT–R132H IDH1, can be modeled by the reaction network in Fig. 4. Making this assumption, [NADPH]m/[NADPH]c can be estimated from just Equation 12 and mitochondrial and cytosolic values for [I], [α-KG], and [NADP+]. Using a novel technique for absolute quantification of mitochondrial matrix metabolites, Chen et al. (32) recently reported compartment-specific concentrations of cis-aconitate (25 μm matrix and 800 μm whole cell), α-KG (150 μm matrix and 1200 μm whole cell), and NADP+ (30 μm matrix and 75 μm whole cell) but not of NADPH. Using the cis-aconitate values as a proxy for [I] and assuming that cytosolic metabolite concentrations are well-approximated by the whole-cell measurements, we calculated that [NADPH]m/[NADPH]c = 0.10. As Chen et al. (32) reported that [NADP+]m/[NADP+]c = 0.40, this estimate implies that ([NADPH]m/[NADP+]m)/([NADPH]c/[NADP+]c) = 0.25.
Discussion
A nearly universal feature of IDH1 mutant cancers is the retention of a WT IDH1 allele. The monoallelic nature of IDH1 mutations was initially difficult to reconcile with the observation that IDH mutations result in loss of function for WT activity (conversion of isocitrate to α-KG) (2). An initial explanation for this paradox invoked a dominant-negative mechanism whereby a mutant IDH1 subunit would heterodimerize with and inhibit the activity of a WT IDH1 subunit. This mechanism was suggested to promote tumorigenesis by decreasing α-KG levels and increasing HIF-1α expression (33). In assays of isocitrate-dependent NADPH production in lysates from HEK 293T cells coexpressing WT and mutant IDH1, however, no appreciable inhibition of WT enzyme activity was found (7). In these same lysates, α-KG–dependent NADPH consumption consistent with a gain of function for a reductive activity was observed. This mutant IDH1 activity was also enhanced with coexpression of WT IDH1 (7), suggesting that it facilitates a novel reaction rather than a mere reversal of the normal WT oxidative decarboxylation of isocitrate. The neomorphic activity of mutant IDH1 was found to be the NADPH-dependent reduction of α-KG to the oncometabolite 2-HG, which is a common feature of the vast majority of recurrent IDH1/2 mutations in cancer (4, 7, 8). Explanations for the observed synergism between WT and mutant IDH1, however, are controversial.
It was confirmed that coexpression of WT and mutant IDH1 greatly enhanced 2-HG production compared with mutant IDH1 expression alone (17). The importance of retained WT IDH1 to 2-HG production by mutant IDH1 was also demonstrated in studies of IDH1 mutant glioma samples exhibiting spontaneous loss of the WT allele (19). Frequently invoked models to explain the retention of a WT IDH1 allele involve either substrate channeling or bulk metabolic flux between WT and mutant subunits in an IDH1 heterodimer (17, 23, 24).
The data presented here argue against both of the above models. It remains critical, however, to understand how WT and mutant IDH do synergize in vivo in human cancers. We present evidence that the primary carbon source for 2-HG production in a cancer cell line harboring an endogenous IDH1 mutation is glutamine. In a direct metabolic pathway involving glutamine → glutamate → α-KG → 2-HG, there is no need to invoke flux through a WT IDH subunit to account for 2-HG production. Consistent with our results, Salamanca-Cardona et al. (34) recently reported that the majority of 2-HG produced by IDH both in cultured cancer cells and in vivo is derived from glutamine, facilitating in vivo monitoring approaches for IDH mutant solid tumors with hyperpolarized [1-13C]glutamine. In contrast to our present study and to Salamanca-Cardona et al. (34), Gelman et al. (16) reported that both glucose and glutamine carbon contribute substantially to 2-HG production by mutant IDH1. Gelman et al. (16) relied on cells that were engineered to express the IDH1 mutation artificially, whereas we and Salamanca-Cardona et al. (34) performed metabolic tracing experiments in cell lines harboring naturally occurring IDH1 mutations at the endogenous locus, possibly accounting for the divergent results. Also consistent with our results, Fack et al. (35) observed negligible labeling of 2-HG from [13C6]glucose in orthotopic patient-derived xenographs of IDH1 mutant glioma.
13C labeling approaches do not address the source of required redox cofactors. It may still be possible that WT IDH1 is necessary for generating an optimum local supply of NADPH for 2-HG production by mutant IDH1 in the cytosol. We estimate here, however, that the ratio of NADPH:NADP+ is higher in the cytosol than in the mitochondrial matrix, which suggests that, if NADPH production by a WT IDH subunit were essential for a mutant IDH subunit to produce 2-HG, it would be at least as important for mitochondrial IDH2 as for cytosolic IDH1. Arguing against this prediction is a prior report that the WT IDH2 allele is dispensable for mutant IDH2-dependent 2-HG production in cells (17). Several groups have also reported loss of the WT IDH2 allele in IDH2 mutant tumors, suggesting that WT IDH2 is dispensable in vivo (36, 37). We conclude that metabolic flux of either carbon substrate or redox cofactors between WT and mutant IDH does not contribute substantially to 2-HG production in cancer.
An alternative, not mutually exclusive, explanation for the retention of one WT IDH1 allele in IDH1 mutant tumors is that the WT–mutant IDH1 heterodimer may harbor a structural or biochemical advantage over mutant–mutant IDH1 homodimers in vivo. Initial studies with recombinant IDH proteins were not universally supportive of this hypothesis (22), although subsequent data have prompted reconsideration. In particular, Brooks et al. (21) demonstrated a lower Km for α-KG in the WT–mutant IDH1 heterodimer than in the R132H–R132H homodimer. In light of the evidence we present against intersubunit flux, the possibility of intrinsic biochemical superiority of the heterodimer, perhaps involving allosteric modulation of the mutant subunit by the WT (28), warrants further consideration. This possibility is of particular importance given that many recently developed inhibitors of mutant IDH act at the dimerization interface. For IDH1, it has been shown that the R132H mutation destabilizes a regulatory segment (the α10 helix) at the interface where several inhibitors bind (28). Moreover, data were reported recently from three AML patients enrolled in phase I/II clinical trials of IDH inhibitors that bind at the dimer interface (38). Each of these patients, after initially responding to IDH inhibition, eventually experienced disease relapse and recurrence of elevated 2-HG levels that were associated with the emergence of second-site IDH mutations conferring resistance to the IDH inhibitor. In two of these patients, the second-site resistance mutation occurred in trans on the WT subunit not containing the mutation responsible for 2-HG production. With further trials of IDH inhibitors ongoing in a variety of malignancies, one might predict that additional IDH mutant tumors will evolve therapeutic resistance through mechanisms involving both the mutant and WT IDH alleles, potentially at residues impacting the dimer interface.
Experimental procedures
Cell culture
HT1080 fibrosarcoma cells (which harbor an endogenous, monoallelic R132C IDH1 mutation) and HEK 293T cells (which lack endogenous IDH mutations) were cultured routinely in Dulbecco's modified Eagle's medium (DMEM), 10% fetal bovine serum, 25 mm glucose, and 6 mm glutamine. For metabolite tracing experiments, DMEM without glutamine or pyruvate (Gibco) and with 10% dialyzed fetal calf serum was supplemented with 4 mm l-[13C5]glutamine (Aldrich, 605166). DMEM without glucose or pyruvate (Gibco) was supplemented with 10 mm d-[13C6]glucose (Cambridge Isotope Laboratories, CLM-1396). All cells were in logarithmic growth phase during the entire period of labeling.
Metabolite extraction and GC-MS analysis
Following gentle removal of culture medium from proliferating cells, cells were rapidly quenched with 80% methanol prechilled to −80 °C. For experiments in which absolute quantification of 2-HG was performed, this 80% methanol was spiked with an M + 5 internal standard of R(−)-2-HG containing five deuterium atoms (D-2-hydroxyglutaric-2,3,3,4,4-d5 acid; details of synthesis are provided in Ref. 17). Following incubation at −80 °C for at least 30 min, cell extracts in 80% MeOH were collected, sonicated, and centrifuged at 14,000 × g for 20 min at 4 °C to remove precipitated protein. Supernatants were then dried under nitrogen gas, redissolved in 20 μl of methoxamine reagent (Thermo Scientific), and heated at 37 °C for 90 min followed by addition of 25 μl of tert-butyldimethylchlorosilane (t-BDMS; Regis Technologies) and heating at 60 °C for 60 min. Derivatized samples were analyzed by GC-MS using a DB-35MS column (30.25-mm inner diameter; Agilent J&W Scientific) installed in an Agilent 7890A gas chromatograph interfaced with an Agilent 5975C mass spectrometer. Mass isotopomer distributions were determined by integrating metabolite ion fragments and corrected for natural abundance using in-house algorithms adapted from Fernandez et al. (39) and described in further detail in Lewis et al. (40). Absolute 2-HG levels were obtained by quantifying the peak area of the ion at m/z 433, formed through the loss of a t-butyl group (−57 atomic mass unit) from the molecular ion tri-t-BDMS–2-HG, and normalizing to the peak area of the m/z 438 ion (representing the analogous derivative of the d5-2-HG internal standard spiked at known concentration) and to the total cellular protein as measured by BCA.
Protein harvest and quantitation, Western blotting, and immunoprecipitation
Cells were lysed 48 h following transfection with mammalian protein extraction reagent (Pierce) supplemented with protease inhibitor mixture (Complete Mini, EDTA-free, Roche Applied Science, 11-836-170-001) and phosphatase inhibitor mixtures 2 and 3 (Sigma). Lysates were sonicated with 2 × 30-s pulses and then centrifuged at 14,000 × g for 20 min at 4 °C. Supernatants were subsequently collected and assessed for protein concentration with a BCA protein assay (Pierce). For Western blotting, lysates were separated by SDS-PAGE on 10% polyacrylamide gels, transferred to polyvinylidene difluoride membranes, and blocked in 5% nonfat milk in PBS containing 0.2% Tween 20. Primary antibodies used were: anti-IDH1 (Santa Cruz Biotechnology, sc-49996; 1:200 dilution), anti-IDH2 (Abcam, ab55271; 1:500), anti-HSP70 (Cell Signaling Technology, 48729; 1:1000), anti-FLAG (Cell Signaling Technology, 2368; 1:10,000), and anti-Myc tag (Babco, clone 9e10; 1:1000). Detection was performed with horseradish peroxidase–conjugated anti-rabbit, anti-mouse, or anti-goat antibodies (GE Healthcare, NA934V; GE Healthcare, NA931V; and Santa Cruz Biotechnology, sc-2020, respectively; all 1:10,000 dilution).
For immunoprecipitation experiments, concentrated cell lysates in Mammalian Protein Extraction Reagent (M-PER) lysis buffer containing protease inhibitors as above was diluted to 500-μl total volume with hypotonic lysis buffer (20 mm HEPES, 5 mm KCl, 1 mm MgCl2, pH 7.0) and supplemented with dithiothreitol (DTT) to 5 mm. Anti-FLAG M2 affinity gel resin (Sigma, A220, lot SLBG5784V; 20 μl of beads) was washed in hypotonic lysis buffer plus protease inhibitor. 40 μl of washed bead suspension was mixed with 500 μl of diluted whole-cell lysate, centrifuged for 3 min at 1000 rpm at 4 °C, and then washed twice with hypotonic lysis buffer and protease inhibitor. Elution was performed by resuspending beads gently in 3 μl of 5 mg/ml 3× FLAG peptide (Sigma, F4799) and 97 μl of hypotonic lysis buffer plus protease inhibitor and DTT, rotating for 30 min at 4 °C, and then centrifuging for 1 min at 21,000 × g. The supernatant from this final spin was saved as the eluate and separated by SDS-PAGE as above.
Plasmid construction and transfection
The cDNA clone of human IDH1 (BC012846.1) was obtained from American Type Culture Collection. R132H IDH1 point mutation was generated as described previously (21). FLAG or Myc tags were added to the C termini of the open reading frames by standard PCR techniques as detailed previously (17). The integrity of constructs was confirmed by direct sequencing prior to transfection into HEK 293T cells in pCMV-Sport6 expression vector using Lipofectamine 2000 according to the manufacturer's instructions.
In vitro kinetics of WT and R132H IDH1
N-terminally His6-tagged recombinant IDH1 (WT or R132H) was expressed from pET28a(+) in BL21 E. coli by inducing an A600 = 0.7 culture with 0.5 mm isopropyl β-d-1-thiogalactopyranoside for 6 h at room temperature. IDH1 was batch-purified using nickel-nitrilotriacetic acid-agarose beads (Qiagen), and purified protein was dialyzed against 50 mm Tris, pH 7.5, 10 mm MgCl2, 25 mm NaCl, 20% glycerol, 0.15% β-mercaptoethanol. Enzyme concentration was determined by Bradford assay using a BSA standard. Enzymatic activity was monitored by a change in absorbance at 340 nm, corresponding to production or consumption of NADPH. Each reaction was carried out at room temperature in a 100-μl volume containing buffer (100 mm Tris-HCl, pH 7.5, 1.3 mm MnCl2) and varying concentrations of substrates. To assay WT IDH1 activity, 10 ng of enzyme was included in each reaction, and either NADP+ was titrated in the presence of 1 mm dl-isocitrate, or dl-isocitrate was titrated in the presence of 1 mm NADP+. (The enzyme is specific to d-isocitrate, so the effective concentration of d-isocitrate is half that of total dl-isocitrate.) To assay R132H IDH1 activity, 10 μg of enzyme was included in each reaction, and either α-KG was titrated in the presence of 500 μm NADPH, or NADPH was titrated in the presence of 10 mm α-KG. For each assay, activity was compared with that of a “no-enzyme” control reaction. Km and kcat values were determined by fitting the initial rate data to the Michaelis–Menten model in GraphPad Prism (GraphPad Software, Inc.).
Parameter estimation
Kinetic parameters for the mathematical model of 2-HG production (k1···k14) were estimated from the data in Table 1. The parameters fall into 10 major classes, on-rate and off-rate (for isocitrate and NADP+) and catalytic rate for the WT reaction, and on-rate and off-rate (for α-KG and NADPH) and catalytic rate for the neomorphic reaction. Within a particular class, all parameters are assumed to have an identical value. We set the on-rates for both reactions to 1 × 108 m−1 s−1, a standard value for diffusion-limited interactions (41). We set the catalytic rate constants for WT and R132H equal to the kcat values that we determined experimentally (Table 1). Finally, we calculated off-rates from the on-rates, catalytic rates, and experimentally determined Michaelis–Menten constants according to the following formula.
| (Eq. 13) |
Estimation of diffusion coefficients
Diffusion coefficients for α-KG and NADPH were computed using the Stokes–Einstein equation.
| (Eq. 14) |
where T is the temperature, η is the dynamic viscosity, r is the molecular radius, and kB is the Boltzmann constant. We set T = 310 K and η = 1.0 kg m−1 s−1. To determine r, we computed molecular volume using the formula of Abraham and McGowan (42).
| (Eq. 15) |
where Va is the volume of constituent atoms and Nb is the number of covalent bonds. This method yielded radii of 3.4 × 10−10 and 5.5 × 10−10 m for α-KG and NADPH, respectively, corresponding to Dα-KG = 6.7 × 10−10 m2/s and DNADPH = 4.1 × 10−10 m2/s. The calculated values are close to experimentally determined literature values for the diffusion coefficients of related molecules such as citrate and NADH (43–45).
Algebraic calculations
All algebraic calculations were done in Mathematica 9.0 (Wolfram Research). Gröbner basis calculations on the steady-state polynomial system were performed following the procedure described previously (46) and applied to a prior mathematical analysis of bacterial IDH (47). In brief, we computed a lexicographically ordered Gröbner basis for the 11 polynomials using the built-in Mathematica function GroebnerBasis. The variable ordering was [E·NADPH·α-KG], [E·NADP·I], [E·I], [E·NADPH], [E·NADP+], [E], [I], [α-KG], [2-HG], [NADP+], and [NADPH]. Equation 12 follows directly from one of the terms of the Gröbner basis.
Author contributions
J. P. D., P. S. W., T. D., J. G., and M. G. V. H. conceptualization; J. P. D. and P. S. W. data curation; J. P. D., P. S. W., T. D., and A. M. H. formal analysis; J. P. D. and P. S. W. validation; J. P. D., P. S. W., and A. M. H. investigation; J. P. D., P. S. W., T. D., and A. M. H. methodology; J. P. D., P. S. W., J. G., and M. G. V. H. writing-original draft; J. P. D., P. S. W., J. G., and M. G. V. H. writing-review and editing; A. M. H. resources; J. G. and M. G. V. H. supervision; J. G. and M. G. V. H. funding acquisition; J. G. and M. G. V. H. project administration.
Supplementary Material
Acknowledgments
We thank members of the Vander Heiden laboratory, especially Jared Mayers and Caroline Lewis, for technical assistance.
This work was supported by National Science Foundation (NSF) Graduate Research Fellowship DGE1144152 (to J. P. D.); the University of Pennsylvania Medical Scientist Training Program (to P. S. W.); NSF Grant 1462629 (to J. G.); and Stand Up To Cancer (SU2C), the Massachusetts Institute of Technology (MIT) Center for Precision Cancer Medicine, the Ludwig Center at MIT, the NCI, National Institutes of Health, and a faculty scholars award from the Howard Hughes Medical Institute (to M. G. V. H.). M. G. V. H. is a consultant for and member of the scientific advisory board of Agios Pharmaceuticals and Aeglea Biotherapeutics. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Fig. S1.
- IDH
- isocitrate dehydrogenase
- 2-HG
- 2-hydroxyglutarate
- AML
- acute myeloid leukemia
- α-KG
- α-ketoglutarate
- TCA
- tricarboxylic acid
- I
- isocitrate
- DMEM
- Dulbecco's modified Eagle's medium
- t-BDMS
- tert-butyldimethylchlorosilane.
References
- 1. Parsons D. W., Jones S., Zhang X., Lin J. C., Leary R. J., Angenendt P., Mankoo P., Carter H., Siu I. M., Gallia G. L., Olivi A., McLendon R., Rasheed B. A., Keir S., Nikolskaya T., et al. (2008) An integrated genomic analysis of human glioblastoma multiforme. Science 321, 1807–1812 10.1126/science.1164382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Yan H., Parsons D. W., Jin G., McLendon R., Rasheed B. A., Yuan W., Kos I., Batinic-Haberle I., Jones S., Riggins G. J., Friedman H., Friedman A., Reardon D., Herndon J., Kinzler K. W., et al. (2009) IDH1 and IDH2 mutations in gliomas. N. Engl. J. Med. 360, 765–773 10.1056/NEJMoa0808710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mardis E. R., Ding L., Dooling D. J., Larson D. E., McLellan M. D., Chen K., Koboldt D. C., Fulton R. S., Delehaunty K. D., McGrath S. D., Fulton L. A., Locke D. P., Magrini V. J., Abbott R. M., Vickery T. L., et al. (2009) Recurring mutations found by sequencing an acute myeloid leukemia genome. N. Engl. J. Med. 361, 1058–1066 10.1056/NEJMoa0903840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ward P. S., Patel J., Wise D. R., Abdel-Wahab O., Bennett B. D., Coller H. A., Cross J. R., Fantin V. R., Hedvat C. V., Perl A. E., Rabinowitz J. D., Carroll M., Su S. M., Sharp K. A., Levine R. L., et al. (2010) The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting α-ketoglutarate to 2-hydroxyglutarate. Cancer Cell 17, 225–234 10.1016/j.ccr.2010.01.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Amary M. F., Bacsi K., Maggiani F., Damato S., Halai D., Berisha F., Pollock R., O'Donnell P., Grigoriadis A., Diss T., Eskandarpour M., Presneau N., Hogendoorn P. C., Futreal A., Tirabosco R., et al. (2011) IDH1 and IDH2 mutations are frequent events in central chondrosarcoma and central and periosteal chondromas but not in other mesenchymal tumours. J. Pathol. 224, 334–343 10.1002/path.2913 [DOI] [PubMed] [Google Scholar]
- 6. Borger D. R., Tanabe K. K., Fan K. C., Lopez H. U., Fantin V. R., Straley K. S., Schenkein D. P., Hezel A. F., Ancukiewicz M., Liebman H. M., Kwak E. L., Clark J. W., Ryan D. P., Deshpande V., Dias-Santagata D., et al. (2012) Frequent mutation of isocitrate dehydrogenase (IDH)1 and IDH2 in cholangiocarcinoma identified through broad-based tumor genotyping. Oncologist 17, 72–79 10.1634/theoncologist.2011-0386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dang L., White D. W., Gross S., Bennett B. D., Bittinger M. A., Driggers E. M., Fantin V. R., Jang H. G., Jin S., Keenan M. C., Marks K. M., Prins R. M., Ward P. S., Yen K. E., Liau L. M., et al. (2009) Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 462, 739–744 10.1038/nature08617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gross S., Cairns R. A., Minden M. D., Driggers E. M., Bittinger M. A., Jang H. G., Sasaki M., Jin S., Schenkein D. P., Su S. M., Dang L., Fantin V. R., and Mak T. W. (2010) Cancer-associated metabolite 2-hydroxyglutarate accumulates in acute myelogenous leukemia with isocitrate dehydrogenase 1 and 2 mutations. J. Exp. Med. 207, 339–344 10.1084/jem.20092506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yen K., Travins J., Wang F., David M. D., Artin E., Straley K., Padyana A., Gross S., DeLaBarre B., Tobin E., Chen Y., Nagaraja R., Choe S., Jin L., Konteatis Z., et al. (2017) AG-221, a first-in-class therapy targeting acute myeloid leukemia harboring oncogenic IDH2 mutations. Cancer Discov. 7, 478–493 10.1158/2159-8290.CD-16-1034 [DOI] [PubMed] [Google Scholar]
- 10. Stein E. M., DiNardo C. D., Pollyea D. A., Fathi A. T., Roboz G. J., Altman J. K., Stone R. M., DeAngelo D. J., Levine R. L., Flinn I. W., Kantarjian H. M., Collins R., Patel M. R., Frankel A. E., Stein A., et al. (2017) Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood 130, 722–731 10.1182/blood-2017-04-779405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Popovici-Muller J., Lemieux R. M., Artin E., Saunders J. O., Salituro F. G., Travins J., Cianchetta G., Cai Z., Zhou D., Cui D., Chen P., Straley K., Tobin E., Wang F., David M. D., et al. (2018) Discovery of AG-120 (ivosidenib): a first-in-class mutant IDH1 inhibitor for the treatment of IDH1 mutant cancers. ACS Med. Chem. Lett. 9, 300–305 10.1021/acsmedchemlett.7b00421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. DiNardo C. D., Stein E. M., de Botton S., Roboz G. J., Altman J. K., Mims A. S., Swords R., Collins R. H., Mannis G. N., Pollyea D. A., Donnellan W., Fathi A. T., Pigneux A., Erba H. P., Prince G. T., et al. (2018) Durable remissions with ivosidenib in IDH1-mutated relapsed or refractory AML. N. Engl. J. Med. 378, 2386–2398 10.1056/NEJMoa1716984 [DOI] [PubMed] [Google Scholar]
- 13. Borger D. R., Goyal L., Yau T., Poon R. T., Ancukiewicz M., Deshpande V., Christiani D. C., Liebman H. M., Yang H., Kim H., Yen K., Faris J. E., Iafrate A. J., Kwak E. L., Clark J. W., et al. (2014) Circulating oncometabolite 2-hydroxyglutarate is a potential surrogate biomarker in patients with isocitrate dehydrogenase-mutant intrahepatic cholangiocarcinoma. Clin. Cancer Res. 20, 1884–1890 10.1158/1078-0432.CCR-13-2649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. de la Fuente M. I., Young R. J., Rubel J., Rosenblum M., Tisnado J., Briggs S., Arevalo-Perez J., Cross J. R., Campos C., Straley K., Zhu D., Dong C., Thomas A., Omuro A. A., Nolan C. P., et al. (2016) Integration of 2-hydroxyglutarate-proton magnetic resonance spectroscopy into clinical practice for disease monitoring in isocitrate dehydrogenase-mutant glioma. Neuro Oncol. 18, 283–290 10.1093/neuonc/nov307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Li S., Chou A. P., Chen W., Chen R., Deng Y., Phillips H. S., Selfridge J., Zurayk M., Lou J. J., Everson R. G., Wu K. C., Faull K. F., Cloughesy T., Liau L. M., and Lai A. (2013) Overexpression of isocitrate dehydrogenase mutant proteins renders glioma cells more sensitive to radiation. Neuro Oncol. 15, 57–68 10.1093/neuonc/nos261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gelman S. J., Naser F., Mahieu N. G., McKenzie L. D., Dunn G. P., Chheda M. G., and Patti G. J. (2018) Consumption of NADPH for 2-HG synthesis increases pentose phosphate pathway flux and sensitizes cells to oxidative stress. Cell Rep. 22, 512–522 10.1016/j.celrep.2017.12.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ward P. S., Lu C., Cross J. R., Abdel-Wahab O., Levine R. L., Schwartz G. K., and Thompson C. B. (2013) The potential for isocitrate dehydrogenase mutations to produce 2-hydroxyglutarate depends on allele specificity and subcellular compartmentalization. J. Biol. Chem. 288, 3804–3815 10.1074/jbc.M112.435495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Robinson G. L., Philip B., Guthrie M. R., Cox J. E., Robinson J. P., VanBrocklin M. W., and Holmen S. L. (2016) In vitro visualization and characterization of wild type and mutant IDH homo- and heterodimers using bimolecular fluorescence complementation. Cancer Res. Front. 2, 311–329 10.17980/2016.311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jin G., Reitman Z. J., Duncan C. G., Spasojevic I., Gooden D. M., Rasheed B. A., Yang R., Lopez G. Y., He Y., McLendon R. E., Bigner D. D., and Yan H. (2013) Disruption of wild-type IDH1 suppresses D-2-hydroxyglutarate production in IDH1-mutated gliomas. Cancer Res. 73, 496–501 10.1158/0008-5472.CAN-12-2852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bralten L. B., Kloosterhof N. K., Balvers R., Sacchetti A., Lapre L., Lamfers M., Leenstra S., de Jonge H., Kros J. M., Jansen E. E., Struys E. A., Jakobs C., Salomons G. S., Diks S. H., Peppelenbosch M., et al. (2011) IDH1 R132H decreases proliferation of glioma cell lines in vitro and in vivo. Ann. Neurol. 69, 455–463 10.1002/ana.22390 [DOI] [PubMed] [Google Scholar]
- 21. Brooks E., Wu X., Hanel A., Nguyen S., Wang J., Zhang J. H., Harrison A., and Zhang W. (2014) Identification and characterization of small-molecule inhibitors of the R132H/R132H mutant isocitrate dehydrogenase 1 homodimer and R132H/wild-type heterodimer. J. Biomol. Screen. 19, 1193–1200 10.1177/1087057114541148 [DOI] [PubMed] [Google Scholar]
- 22. Leonardi R., Subramanian C., Jackowski S., and Rock C. O. (2012) Cancer-associated isocitrate dehydrogenase mutations inactivate NADPH-dependent reductive carboxylation. J. Biol. Chem. 287, 14615–14620 10.1074/jbc.C112.353946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Avellaneda Matteo D., Grunseth A. J., Gonzalez E. R., Anselmo S. L., Kennedy M. A., Moman P., Scott D. A., Hoang A., and Sohl C. D. (2017) Molecular mechanisms of isocitrate dehydrogenase 1 (IDH1) mutations identified in tumors: the role of size and hydrophobicity at residue 132 on catalytic efficiency. J. Biol. Chem. 292, 7971–7983 10.1074/jbc.M117.776179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Pietrak B., Zhao H., Qi H., Quinn C., Gao E., Boyer J. G., Concha N., Brown K., Duraiswami C., Wooster R., Sweitzer S., and Schwartz B. (2011) A tale of two subunits: how the neomorphic R132H IDH1 mutation enhances production of αHG. Biochemistry 50, 4804–4812 10.1021/bi200499m [DOI] [PubMed] [Google Scholar]
- 25. Yang B., Zhong C., Peng Y., Lai Z., and Ding J. (2010) Molecular mechanisms of “off-on switch” of activities of human IDH1 by tumor-associated mutation R132H. Cell Res. 20, 1188–1200 10.1038/cr.2010.145 [DOI] [PubMed] [Google Scholar]
- 26. Rendina A. R., Pietrak B., Smallwood A., Zhao H., Qi H., Quinn C., Adams N. D., Concha N., Duraiswami C., Thrall S. H., Sweitzer S., and Schwartz B. (2013) Mutant IDH1 enhances the production of 2-hydroxyglutarate due to its kinetic mechanism. Biochemistry 52, 4563–4577 10.1021/bi400514k [DOI] [PubMed] [Google Scholar]
- 27. Xu X., Zhao J., Xu Z., Peng B., Huang Q., Arnold E., and Ding J. (2004) Structures of human cytosolic NADP-dependent isocitrate dehydrogenase reveal a novel self-regulatory mechanism of activity. J. Biol. Chem. 279, 33946–33957 10.1074/jbc.M404298200 [DOI] [PubMed] [Google Scholar]
- 28. Xie X., Baird D., Bowen K., Capka V., Chen J., Chenail G., Cho Y., Dooley J., Farsidjani A., Fortin P., Kohls D., Kulathila R., Lin F., McKay D., Rodrigues L., et al. (2017) Allosteric mutant IDH1 inhibitors reveal mechanisms for IDH1 mutant and isoform selectivity. Structure 25, 506–513 10.1016/j.str.2016.12.017 [DOI] [PubMed] [Google Scholar]
- 29. Castellana M., Wilson M. Z., Xu Y., Joshi P., Cristea I. M., Rabinowitz J. D., Gitai Z., and Wingreen N. S. (2014) Enzyme clustering accelerates processing of intermediates through metabolic channeling. Nat. Biotechnol. 32, 1011–1018 10.1038/nbt.3018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gao P., Tchernyshyov I., Chang T. C., Lee Y. S., Kita K., Ochi T., Zeller K. I., De Marzo A. M., Van Eyk J. E., Mendell J. T., and Dang C. V. (2009) c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 458, 762–765 10.1038/nature07823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cassago A., Ferreira A. P., Ferreira I. M., Fornezari C., Gomes E. R., Greene K. S., Pereira H. M., Garratt R. C., Dias S. M., and Ambrosio A. L. (2011) Mitochondrial localization and structure-based phosphate activation mechanism of glutaminase C with implications for cancer metabolism. Proc. Natl. Acad. Sci. U.S.A. 109, 1092–1097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chen W. W., Freinkman E., Wang T., Birsoy K., and Sabatini D. M. (2016) Absolute quantification of matrix metabolites reveals the dynamics of mitochondrial metabolism. Cell 166, 1324–1337.e11 10.1016/j.cell.2016.07.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhao S., Lin Y., Xu W., Jiang W., Zha Z., Wang P., Yu W., Li Z., Gong L., Peng Y., Ding J., Lei Q., Guan K. L., and Xiong Y. (2009) Glioma-derived mutations in IDH1 dominantly inhibit IDH1 catalytic activity and induce HIF-1α. Science 324, 261–265 10.1126/science.1170944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Salamanca-Cardona L., Shah H., Poot A. J., Correa F. M., Di Gialleonardo V., Lui H., Miloushev V. Z., Granlund K. L., Tee S. S., Cross J. R., Thompson C. B., and Keshari K. R. (2017) In vivo imaging of glutamine metabolism to the oncometabolite 2-hydroxyglutarate in IDH1/2 mutant tumors. Cell Metab. 26, 830–841.e3 10.1016/j.cmet.2017.10.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Fack F., Tardito S., Hochart G., Oudin A., Zheng L., Fritah S., Golebiewska A., Nazarov P. V., Bernard A., Hau A. C., Keunen O., Leenders W., Lund-Johansen M., Stauber J., Gottlieb E., et al. (2017) Altered metabolic landscape in IDH-mutant gliomas affects phospholipid, energy, and oxidative stress pathways. EMBO Mol. Med. 9, 1681–1695 10.15252/emmm.201707729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pansuriya T. C., van Eijk R., d'Adamo P., van Ruler M. A., Kuijjer M. L., Oosting J., Cleton-Jansen A.-M., van Oosterwijk J. G., Verbeke S. L., Meijer D., van Wezel T., Nord K. H., Sangiorgi L., Toker B., Liegl-Atzwanger B., et al. (2011) Somatic mosaic IDH1 and IDH2 mutations are associated with enchondroma and spindle cell hemangioma in Ollier disease and Maffucci syndrome. Nat. Genet. 43, 1256–1261 10.1038/ng.1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pichler M. M., Bodner C., Fischer C., Deutsch A. J., Hiden K., Beham-Schmid C., Linkesch W., Guelly C., Sill H., and Wölfler A. (2011) Evaluation of mutations in the isocitrate dehydrogenase genes in therapy-related and secondary acute myeloid leukaemia identifies a patient with clonal evolution to IDH2 R172K homozygosity due to uniparental disomy. Br. J. Haematol. 152, 669–672 10.1111/j.1365-2141.2010.08404.x [DOI] [PubMed] [Google Scholar]
- 38. Intlekofer A. M., Shih A. H., Wang B., Nazir A., Rustenburg A. S., Albanese S. K., Patel M., Famulare C., Correa F. M., Takemoto N., Durani V., Liu H., Taylor J., Farnoud N., Papaemmanuil E., et al. (2018) Acquired resistance to IDH inhibition through trans or cis dimer-interface mutations. Nature 559, 125–129 10.1038/s41586-018-0251-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Fernandez C. A., Des Rosiers C., Previs S. F., David F., and Brunengraber H. (1996) Correction of 13C mass isotopomer distributions for natural stable isotope abundance. J. Mass Spectrom. 31, 255–262 10.1002/(SICI)1096-9888(199603)31:3<255::AID-JMS290>3.0.CO%3B2-3 [DOI] [PubMed] [Google Scholar]
- 40. Lewis C. A., Parker S. J., Fiske B. P., McCloskey D., Gui D. Y., Green C. R., Vokes N. I., Feist A. M., Vander Heiden M. G., and Metallo C. M. (2014) Tracing compartmentalized NADPH metabolism in the cytosol and mitochondria of mammalian cells. Mol. Cell 55, 253–263 10.1016/j.molcel.2014.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Cornish-Bowden A. (2004) Fundamentals of Enzyme Kinetics, 3rd Ed., p. 169, Portland Press Ltd., London [Google Scholar]
- 42. Abraham M. H., and McGowan J. C. (1987) The use of characteristic volumes to measure cavity terms in reversed phase liquid chromatography. Chromatographia 23, 243–246 10.1007/BF02311772 [DOI] [Google Scholar]
- 43. Southard M. Z., Dias L. J., Himmelstein K. J., and Stella V. J. (1991) Experimental determinations of diffusion coefficients in dilute aqueous solution using the method of hydrodynamic stability. Pharm. Res. 8, 1489–1494 10.1023/A:1015886131198 [DOI] [PubMed] [Google Scholar]
- 44. Hasinoff B. B., and Chishti S. B. (1983) Viscosity dependence of the kinetics of the diffusion-controlled reaction of carbon monoxide with the separated α and β chains of hemoglobin. Biochemistry 22, 58–61 10.1021/bi00270a008 [DOI] [PubMed] [Google Scholar]
- 45. Samec Z., Trojanek A., and Samcova E. (1994) Evaluation of ion transport parameters in a Nafion membrane from ion-exchange measurements. J. Phys. Chem. 98, 6352–6358 10.1021/j100076a020 [DOI] [Google Scholar]
- 46. Manrai A. K., and Gunawardena J. (2008) The geometry of multisite phosphorylation. Biophys. J. 95, 5533–5543 10.1529/biophysj.108.140632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Dexter J. P., and Gunawardena J. (2013) Dimerization and bifunctionality confer robustness to the isocitrate dehydrogenase regulatory system in Escherichia coli. J. Biol. Chem. 288, 5770–5778 10.1074/jbc.M112.339226 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.