

Abstract
Choroideremia is an X-linked inherited chorioretinal dystrophy leading to blindness by late adulthood. Choroideremia is caused by mutations in the CHM gene which encodes Rab escort protein 1 (REP1), an ubiquitously expressed protein involved in intracellular trafficking and prenylation activity. The exact site of pathogenesis remains unclear but results in degeneration of the photoreceptors, retinal pigment epithelium and choroid. Animal and stem cell models have been used to study the molecular defects in choroideremia and test effectiveness of treatment interventions. Natural history studies of choroideremia have provided additional insight into the clinical phenotype of the condition and prepared the way for clinical trials aiming to investigate the safety and efficacy of suitable therapies. In this review, we provide a summary of the current knowledge on the genetics, pathophysiology, clinical features and therapeutic strategies that might become available for choroideremia in the future, including gene therapy, stem cell treatment and small-molecule drugs with nonsense suppression action.
Keywords: choroideremia, gene therapy, nonsense suppression therapy, REP1, retinal dystrophy, stem cells
Introduction
Choroideremia (CHM, OMIM 303100) is a rare inherited chorioretinal dystrophy that manifests as a progressive degenerative disorder of the photoreceptor layer, retinal pigment epithelium (RPE) and choroid.1 It is estimated that the prevalence of CHM is between 1 in 50,000-100,000 people with a preponderance in the Finish population.1–5 The disease is inherited in an X-linked recessive pattern with male patients predominantly expressing the characteristic features of early night blindness that evolves into severe peripheral vision loss followed by legal blindness in late adulthood. Female carriers remain mostly asymptomatic although they can experience nyctalopia and exhibit pigmentary changes in the fundus with associated subnormal visual sensitivity.6–8
Historically, the Austrian ophthalmologist Ludwig Mauthner was the first to describe the disease in 1872.9 Mauthner named the disorder ‘Chorioideremie’ from the combination of the Greek derived words ‘erēmia’ (barren land or desert) and ‘chorioid’ (skin – ‘chorion’ and form/type – ‘eidos’) to define its association with the absent choroidal layer.9 Although originally it was suggested that the condition originates from the congenital absence of the choroid, further observations supported the progressive choroidal atrophy.10,11 The X-linked mode of inheritance was initially proposed by case observations and literature review by Goedbloed and Waardenburg, respectively, in 1942, while the causative gene was identified and further characterized by positional cloning in the 1990s.12–14 Recently, promising results have been published in relevance to the application of viral vectors as potential means of gene replacement in humans.9,15
Genetics of CHM
CHM is an X-linked recessive inherited disorder caused by mutations in the CHM gene (OMIM 303390). This gene is located on chromosome Xq21.2, spans a DNA genomic sequence of 186,382 base pairs (bp), consists of 15 exons and is ubiquitously expressed.1,16 CHM messenger RNA (mRNA) is approximately 5.6 kb long and produces a protein of 653 amino acids (95 kDa) called Rab escort protein 1 (REP1).1,16
Currently, there are 280 identified mutations that result in the CHM phenotype.17 A variety of mutations have been implicated including deletions, insertions, duplications, translocations, nonsense, splice site, frameshift, missense and deep intronic variants.1,18–20 Most of the mutations in the CHM gene are null, either through deletions (25–50%) or nonsense mutations (30%).1 Deletions may involve either a few kilobases or the entire length of the gene resulting in a dysfunctional or completely absent protein, while nonsense mutations introduce premature stop codons in the coding sequence signalling abrupt disruption of the translational process.1,21
Although missense mutations are rare, certain missense variants have been shown to be pathogenic.22–24 Sergeev and colleagues22 described a missense mutation (c.1679T>C, p.Leu550Pro) that results in decreased REP1 expression and predicted protein structure instability. Esposito and colleagues23 reported a missense variant (c.1520A>G, p.His507Arg) that also caused reduced REP1 expression and protein structure destabilisation. Torriano and colleagues24 identified a missense variant (c.1370T>C, p.Leu457Pro) resulting in a stable CHM transcript with decreased levels of REP1 expression.
A rare deep intronic variant (chrX: 85,220,593 T>C) was also recently identified in two unrelated men with CHM and it was hypothesized that splicing alteration could account for the dysfunctional protein.18 X-autosome translocations have been reported in female carriers presenting either with mild clinical features of CHM in association with ovarian dysgenesis or with a more severe disease phenotype along with ectodermal dysplasia.25,26 However, despite the increased insight into the mutational spectrum of the disease, there is no established genotype–phenotype correlation.27
Pathophysiology of CHM
REP1 has an essential role in intracellular trafficking of proteins, substrates and organelles.28,29 This process is regulated by small guanosine triphosphate (GTP)-binding proteins called Rab proteins. In order to bind to lipid membranes and mediate transportation, Rab proteins require appropriate lipid modification, which is called prenylation and entails the inclusion of geranyl–geranyl groups in their structure.30 The latter is accomplished through the geranyl–geranyl transferase 2 enzyme (GGTase2) and the REP1 binding to Rab proteins is the first crucial step.29 The prenylated Rab protein is then escorted by REP1 and delivered to the target intracellular compartment (Figure 1).30
Figure 1.
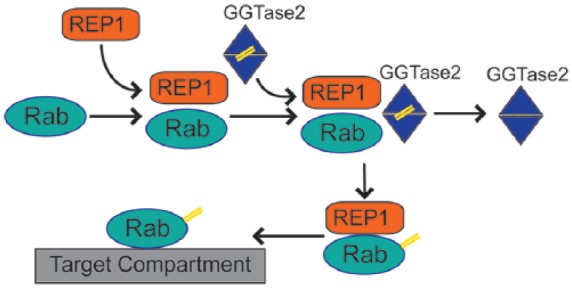
Rab protein prenylation pathway: Rab proteins require lipid modification (prenylation, highlighted as double yellow lines) to associate with membranes and organelles. REP1 mediates the initial step in the process by attaching to Rab proteins and facilitating interaction with geranyl–geranyl transferase 2 enzyme (GGTase2). GGTase 2 prenylates C-terminal cysteine motifs of Rabs and subsequently REP1 delivers modified Rab proteins to target intracellular compartment.
The preferential degeneration of the retina and choroid in patients with CHM led to the identification of CHM-Like (CHML) gene (OMIM 118825), which is located on chromosome 1q42 and encodes the homologue REP2. This protein is also ubiquitously expressed throughout the body, shares 75% sequence homology with REP1 and may partially counterbalance REP1 deficiency.31 A subset of Rab proteins compete for REP1 over REP2 for prenylation and in the absence of REP1 are underselected for modification leading to an accumulation of unprenylated Rabs in particular Rab27a, Rab27b, Rab38 and Rab42.31–33
The exact mechanism and site of pathogenesis remains under debate with some studies indicating that the inciting event originates in the RPE and gradually evolves into degeneration of the photoreceptors and the choroid, while other evidence suggests that the photoreceptors are affected first with subsequent RPE and choriocapillaris involvement, or that the affected cells degenerate simultaneously and autonomously.34–36 There are also histopathological data from patients supporting that choroidal atrophy secondary to uveal vessel disease is the primary defect in CHM.37
The cellular pathophysiology of the disease reveals dysfunctional phagocytosis with undigested photoreceptor outer segments found within mutated RPE cells in the zebrafish and mouse CHM models and furthermore abnormalities of melanosomes transport within the RPE.38,39 Chronic inflammatory changes might also contribute to CHM since T lymphocytes and retinal gliosis have also been histologically identified adjacent to sites of active disease.40
Clinical diagnosis
Clinical signs and symptoms
Affected male CHM patients experience symptoms of nyctalopia during the first decade of childhood.1,41 Progressive peripheral visual field loss continues through adulthood culminating in legal loss of central vision around the fifth to sixth decades.42 Some patients can still retain some degree of perception to light or hand motion although there is significant intra/interfamilial variability.41
Female carriers usually do not present with clinical symptoms and are able to retain good visual acuity throughout life.1 However, some can report nyctalopia and progressive sight loss with increasing age although the rate of decline is much slower than for men.42 There have also been reported cases of subretinal neovascularization and fibrosis in female carriers.6
In general, CHM patients display a bilateral and symmetrical pattern of eye involvement which has been exploited in genetic clinical trials since the contralateral eye can serve as an effective control.42,43
Although CHM is most commonly an isolated retinal disorder, chromosomal disruption of Xq21 has been associated with syndromic phenotypes.44 Rosenberg and colleagues45 described men with an interstitial deletion of the X chromosome (del[X][q13q21.3]) resulting in CHM features, severe mental retardation and cleft lip and palate. Van den Bosh46 reported two brothers with CHM phenotype that also shared skeletal deformities, cognitive impairment, acrokeratosis and anhidrosis and may be attributed to a small X-chromosome deletion. Female carriers with autosomal and X-chromosome translocations presented with a CHM phenotype, sensorineural deafness and ovarian dysgenesis, while translocation between chromosomes X, 1 and 3 resulted in CHM features and ectodermal dysplasia.25,26
Fundus examination
In the early disease stages, the fundus shows peripheral pigmentary clumping at the level of the RPE that progressively evolves into distinct areas of chorioretinal atrophy with scleral exposure and visible choroidal vessels (Figure 2(a)).1 This degenerative process begins at the equator following a centripetal distribution towards the anterior retina and posterior pole.42 Similar atrophic changes are also noted in the peripapillary region with some patients harbouring a central island of relatively preserved retinal tissue even in advanced stages.47 Furthermore, there is a gradual depletion of choriocapillaris, while the large choroidal and retinal vessels appear unaffected (Figure 2(a)).47
Figure 2.

Multimodal retinal imaging in a 27-year-old CHM male patient (c.715C>T, p.[Arg239*]). (a) Colour fundus photography shows extensive chorioretinal atrophy with visualisation of scleral vessels and a residual island of retinal tissue in the posterior pole. (b) Fundus autofluorescence of the same eye demonstrates distinct borders between the residual degenerating retinal island and atrophic tissue. (d–g) The green line delineates the level of the optical coherence tomography at the macula, orange and yellow boxes indicate locations for each high-resolution adaptive optics scanning light ophthalmoscopy (AOSLO) image. (c) Optical coherence tomography of the same eye shows disruption of the ellipsoid zone (EZ) (arrowheads) and outer retinal tubulations (ORTs) (arrows). (d–g) AOSLO confocal (D, F) and (e, g) split-detector imaging of the right eye illustrates locations of cone mosaic abrupt disruption at the border of RPE atrophy (yellow arrows in (d) and (e)) in contrast to (f and g) normal cone mosaic in anatomically preserved retinal loci.
Mild pigmentary changes can be detected on fundus examination of female carriers; however, more severely affected cases can present with atrophic areas similar to male patients (Figure 3(b)).1 Random X-inactivation could account for this phenotypic variation among female individuals but there may be specific genetic modifiers at play.
Figure 3.

Colour fundus photography and fundus autofluorescence of (a, c) an unaffected female subject versus (b, d) a CHM female carrier (c.715C>T, p.[Arg239*]). (b) Fundus photograph demonstrates peripapillary changes in CHM female carrier with accompanying peripheral pigmentary and atrophic areas similar to affected male patients. (d) Fundus autofluorescence shows a speckled pattern with intermixed areas of high- and low-density autofluorescence.
Additional ophthalmological findings in CHM include posterior subcapsular cataracts, macular oedema and choroidal neovascularization (CNV).48–50
Fundus autofluorescence
Fundus autofluorescence (FAF) has been shown to be a useful marker of disease progression in CHM and thus a potential outcome measure in prospective clinical trials.51,52 FAF provides information about lipofuscin distribution with most of the signal originating from the RPE (with the photoreceptors contributing in part).51,52 Progressive lipofuscin accumulation due to dysfunctional degradation and removal of photoreceptor outer segments underlines the pathogenic mechanism of several retinal diseases.52
Pathogenesis of CHM involves both the RPE and photoreceptors and the most common FAF pattern is characterised by decreased FAF with sharp demarcated borders of increased signal from residual degenerating retinal tissue (Figure 2(b)).1,47 The greater loss of FAF is mapped to the nasal retina around the optic nerve and to a lesser extent to the temporal retina and macular area possibly due to a different rod–cone density ratio in the affected areas.47,51 Jolly and colleagues51 estimated the rate of FAF loss to be 7.7% per year and described a CHM index which can be used as a tool to calculate the remaining FAF area based on age.
Female carriers demonstrate a unique pattern of speckled autofluorescence resembling a mosaic, where areas with granular low-density fluorescence are mixed with dots of high signal (Figure 3(d)).53 This finding is related to random inactivation of X-chromosome in females and has also been described in X-linked retinitis pigmentosa associated with mutations in RPGR gene.54 It is suggested that the hyperautofluorescence is due to phagocytosis of the degenerating rods from the RPE cells, whereas low-density fluorescence indicates RPE loss.53 Edwards and colleagues8 have suggested four different types of FAF in carriers, namely, fine, coarse, geographic and male pattern; they also found a correlation of hypofluorescent areas with decreased mean threshold sensitivities in microperimetry. Similar associations of FAF with abnormal multifocal electroretinogram (mfERG) recordings have been reported for carriers, but no correlation with age, severity and prognosis.53
Optical coherence tomography
Optical coherence tomography (OCT) in CHM patients reveals an asymptomatic increase in central retinal thickness from early childhood without other signs of retinal oedema.47 The central thickness remains normal until the fourth decade and then gradually decreases for most patients.55 Retrospective data from affected children demonstrate a progressive decrease in subfoveal choroidal thickness without obvious clinical implications suggesting that the choroid may be more critical in disease initiation than previously thought.47
Outer nuclear layer (ONL) thickness is also affected and the observed decrease has been correlated with borders of FAF degeneration.56 Attenuation of the ellipsoid zone (EZ) and external limiting membrane (ELM) was also identified and mapped to areas of decreased ONL thickness but it was not associated with abnormal FAF margins (Figure 2(c)).56,57
Outer retinal tubulations (ORTs), rosette-like structures, represent remodelling of degenerating photoreceptors and are common in affected men (Figure 2(c)).56 These lesions overlie directly Bruch’s membrane and are located outside the borders of retained retina (Figure 2(c)).56 Primary RPE dysfunction is thought to be the inciting event in the pathogenesis of ORTs in CHM with secondary effects on the photoreceptors.58 ORTs are also observed in other retinal degenerative disorders, remain stable after gene therapy manipulations and are unrelated to age and genetic mutation.55,56
Triangular ONL structures, previously termed interlaminar bridges, have been identified at the junction of normal and atrophic retina possibly representing hyperplastic Müller cells in response to the degenerative process.56,58 They might interfere with optical properties and proper imaging of neighbouring tissue.56,58
Heon and colleagues55 reported mild inner retinal microcysts in 20% of CHM patients and one case of frank cystoid macular oedema, while Genead and Fishman49 observed variable cystoid macular oedema in 65% of subjects. Macular cystic spaces were also identified on retrospective analysis of 57 CHM patients and correlated with progressive decrease in visual acuity and poor prognostic outcomes.57
In contrary to other inherited retinal disorders, macular hole formation was reported in 10% of advanced CHM cases, reflecting either developmental macular deficits or the adding effect of chronic structural and cystoid retinal changes.47
Optical coherence tomography angiography
Optical coherence tomography angiography (OCTA) is a noninvasive method to visualize retinal and choroidal microvasculature.59 The technique is based on the optical properties of blood and neuronal tissue. In short, areas of retina are sequentially imaged under short time scales. Neural tissue does not change its reflectance profile under short periods of time, while the signal derived from blood vessels does due to changes in contents moving through.60 OCTA allows visualization and quantification of four segmented zones: the superficial retinal plexus, the deep retinal plexus, the outer retina and the choriocapillaris.61 CHM affects photoreceptors, RPE and choriocapillaris and there is evidence that choriocapillaris atrophy is associated with RPE degeneration.62 Patel and colleagues63 described an association of CHM with decreased choriocapillaris density that was more pronounced in areas underlying abnormal photoreceptors. Abbouda and colleagues64 reported flow reduction in the superficial retinal plexus and choriocapillaris of CHM patients and an inverse relationship between age and choriocapillaris perfusion (Figure 4(b) and (d)). Further research is necessary to establish the use of OCTA as a tool to investigate CHM pathology and stratify disease progression.
Figure 4.

Optical coherence tomography angiography (OCTA) from the same 27-year-old CHM male patient (c.715C>T, p.[Arg239*]): Macular angiograms (3 × 3) of the superficial and choriocapillaris layers in (a, c) a control male subject versus (b, d) the CHM patient demonstrate decreased vascularity in the choriocapillaris layer of the affected male patient (orange arrows in (d)).
Fundus fluorescein angiography
Fundus fluorescein angiography (FFA) is rarely used in CHM patients except if CNV is suspected when leakage is the main diagnostic feature.65,66 It reveals a scalloped pattern of RPE loss with hypofluorescence in atrophic areas with missing choriocapillaris adjacent to hyperfluorescent areas of preserved choriocapillaris perfusion, narrow retinal vessels and delayed choroidal and retinal flow during the late stages in affected men.67 In female carriers, there is more variability in CNV cases with either complete absence of pathological features or observed areas of pigment irregularity, choroidal sclerosis and slow choroidal flow.6,67 More advanced cases can present with signs similar to affected male individuals.6,67
Cellular imaging techniques
Confocal adaptive optics scanning light ophthalmoscopy (AOSLO) allows effective photoreceptor cellular structure characterisation by correcting optical aberrations of the eye.68 AOSLO has been used in parallel with other imaging modalities to identify pathogenesis and morphological features of several retinal degenerative disorders.58,69 Morgan and colleagues35 reported normal cone mosaic in anatomically preserved retinal loci and persistence of mosaic integrity up to the border of degeneration (Figure 2(d)–(g)). Abnormalities of the mosaic were also noted including bubble-like hyperreflective spots with dark edges representing choroidal atrophy and hyporeflective inner retinal microcysts that correlate with the microcysts on OCT.58 Cone spacing patterns in men include either normal foveal cone distribution with peripheral abnormalities or increased foveal cone spacing with normal cone mosaic in retinal eccentricities with OCT-documented disrupted photoreceptor bands.58 Those features are identified within pathological retinal loci and are likely indicative of advanced disease stages.35 Nabholz and colleagues70 compared two groups of young male patients (6- to 15-year olds and 16- to 25-year olds) with observable cone mosaic with normal age-matched controls and demonstrated decreased cone density around the fovea, suggesting that cone loss could be quantified even during the asymptomatic disease stage reflecting the early pathogenic effect of CHM mutation on cellular function. Morgan and colleagues35 suggested that the RPE is the primary layer to be affected since decreased FAF was observed in areas with normal cone mosaic, but further studies are necessary to fully answer whether the RPE or photoreceptors are affected autonomously or simultaneously.
AOSLO imaging in asymptomatic CHM carriers reveals normal cone mosaic and cone spacing measures, while symptomatic women demonstrate patchy areas of abnormal cone spacing integrated with morphologically intact cones in unaffected retinal loci.58 Pathological features similar to male patients have also been described for CHM carriers and they typically correlate with abnormal findings on FAF and OCT scanning.35
Visual fields/microperimetry
Visual field testing confirms symmetric vision loss that corresponds to the observed areas of degeneration.71 Several studies have reported perimetry data for CHM patients, but there is no evidence of genotype–phenotype correlation.71 Heon and colleagues55 using Goldmann kinetic visual field have demonstrated age-related changes relevant to specific isopters (I-4e, III-4e and V-4e) with the earliest changes noted for the I-4e isopter and a predicted rate of visual field constriction by 8.3% per year for the V-4e target. The progressive atrophy will result in scotomas and gradual loss of central and peripheral vision; however, islands of retinal tissue with retained visual function have been documented in advanced disease stages.71
Microperimetry (macular perimetry) is a static retinal sensitivity test that can provide useful information about the central retinal function by mapping threshold sensitivities at specific macular loci. Investigation in CHM patients showed significant test–retest repeatability with a mean threshold sensitivity of 1.45 dB between different tests and interocular symmetry especially during the early stages of the disease.43 The latter finding provides utility as an outcome measure in clinical trials to monitor and quantify treatment response.
Electroretinography
Full-field electroretinograms (ERGs) demonstrate abnormal findings in male patients during early disease.1 Abnormal responses are recorded first for the scotopic component reflecting increased rod vulnerability and then for the photopic component when cones are also involved.1 As disease progresses, ERG findings deteriorate even when visual acuity and visual fields are preserved.72 However, a negative ERG has been reported in a young CHM male patient with a nonsense mutation (13 years old, c.838C>T, p.[Arg270*]), normal visual acuity (6/7.5) and visual field (III-4e) in both eyes with associated peripheral RPE abnormalities on fundus imaging.72 This finding might indicate signal transmission failure either due to photoreceptor dysfunction or because of an isolated bipolar cell defect.1,72 Furthermore, the intrafamilial and interfamilial variabilities noted preclude ERG use as a prognostic tool.1,72
Most carriers have normal ERGs; however, symptomatic women could present with subtle changes like subnormal 30-Hz flicker photopic response on full-field ERG, delayed latencies and decreased amplitudes for a and b wave on scotopic testing and significant elevation of 650-nm fully dark-adapted thresholds especially at extramacular sites.36,53 Due to the patchy retinal involvement in women, other modalities like the mfERG should be sought.72
Colour vision
Despite retaining good central visual acuity until advanced stages of disease, functional defects have also been reported for colour vision.55 The dysfunction of rod photoreceptors precedes cone photoreceptors, which closely follow as they are dependent on surrounding rod photoreceptors and underlying RPE for structural support.36,73 Early functional impairment of cone photoreceptors, and resulting confusion in the circuitry underlying colour vision, could explain subtle changes in colour vision even in subjects with normal visual acuity.73 In CHM, similar to other retinal disorders with retained central fixation, tritan discrimination is predominantly affected due to increased parafoveal density of S cones, while centrally located M- and L-cones remain relatively preserved.74,75 Male patients and often female carriers had poor colour discrimination on the Farnsworth–Munsell 100-hue test with tritanopia.76 Seitz and colleagues74 using the Cambridge Colour Test (CCT) reported that tritanopic defects were more common than red-green colour discrimination in CHM patients. They have also reported a statistically significant association of red-green colour discrimination with visual acuity and to a lesser degree between best corrected visual acuity (BCVA) and blue colour discrimination.74 Natural history studies are underway that could help to delineate defects using the CCT.
Differential diagnosis
Despite the characteristic genetic and clinical phenotype, CHM shares similar features with other ophthalmological conditions that need further differentiation. Examples include gyrate atrophy, retinitis pigmentosa (X-linked and RPE65 autosomal dominant phenotypes), Kearns–Sayre syndrome (KSS), Bietti’s crystalline dystrophy and thioridazine hydrochloride retinal toxicity.44,77,78
Gyrate atrophy can also present with nyctalopia, peripheral visual field defects and atrophic chorioretinal areas on funduscopy, but the disease is inherited as an autosomal-recessive trait, is associated with increased plasma ornithine levels due to ornithine-delta-aminotransferase (OAT) deficiency and disease manifestations include early cataracts, myopia and distinctive scalloped borders of centripetal retinal degeneration.77,79
Retinitis pigmentosa shares nyctalopia, visual field restriction and in some cases choroidal involvement, but differentiating clinical findings include a bone spicule pigment migration pattern, optic disc pallor, retinal vessel narrowing, cystoid macular oedema and epiretinal membrane formation.78,80,81
KSS is due to mitochondrial DNA deletions and fundal imaging may reveal diffuse chorioretinal atrophy similar to advanced CHM stages. Additional findings that help differential diagnosis include progressive external ophthalmoplegia, cerebellar ataxia, cardiac conduction abnormalities, elevated protein in the cerebrospinal fluid and microscopic mitochondrial abnormalities in the muscle biopsy.77,82
Bietti’s crystalline dystrophy is a rare autosomal-recessive degenerative chorioretinal disorder and affected individuals may develop symptoms similar to CHM between the second and fourth decades of life, but the distinctive presence of cholesterol crystals in the posterior pole of the retina and corneal stroma facilitates accurate disease characterization.77,83
Thioridazine hydrochloride retinal toxicity is associated with decreased visual acuity, nyctalopia, pigmentary changes and chorioretinal degeneration, but there is also a history of psychiatric disease and medication intake in high doses.77
Therapeutic approaches
Despite the progress in genetic and clinical characterisation of CHM, currently there is no effective treatment. Gene therapy has reached phase 3 clinical trials. However, pharmacological compounds and nonviral gene delivery are still in preclinical stages using animal and stem cell disease models.
Gene therapy
CHM is amenable to gene therapy approaches as the disease is monogenic, the complementary DNA (cDNA) is within the size capacity of the commonly used adeno-associated virus (AAV) vector capacity, diagnosis is made early due to unique retinal degeneration pattern, progression is slow allowing a broader therapeutic window for intervention and its pathologic effects are targeted to the eye.84 The eye represents an ideal target for gene augmentation strategies due to ocular immune privilege that decreases the risk of an immune response to injected material (although patients are still given prophylactic topical and oral steroids to reduce any inflammation), tight blood–ocular barrier that decreases systemic particle penetration and treatment response monitoring using the contralateral eye as a control.85
Previous and current works with gene therapy in preclinical studies for inherited retinal diseases have employed a variety of vectors.9,86 Both AAV and lentivirus (LV) vectors have been used for RPE65 associated Leber congenital amaurosis.87–89 In CHM, LV vectors were used to introduce CHM cDNA in a mouse model, but their limited affinity to photoreceptors hindered wider applicability.90,91 Appropriate vector selection is based on tissue of interest, cloning capacity and safety profile.85 AAVs are small, single-stranded DNA viruses of the parvovirus family and represent the most common viral vector used in retinal diseases due to their nonintegrating capacity and favourable immunologic, inflammatory and toxicity profile.85,92 AAV serotypes 2, 5 and 7–9 are used to transduce photoreceptors and RPE (but are not specific for these cell types and can also target retinal ganglion cells or Müller cells).92 Induced pluripotent stem cell (iPSC)-derived RPE cells have been used to study different AAV serotypes. Cereso and colleagues93 demonstrated superiority of AAV2/5 viral vector over AAV2/2 in restoring normal phenotype when they were transduced with the normal CHM gene. Torriano and colleagues24 also used the AAV2/5 vector to study the pathogenic effect of a novel CHM missense variant and moreover documented restoration of prenylation function after gene replacement strategies similar to nonsense variants. Duong and colleagues94 suggested optimal transduction of RPE and photoreceptor cells with a recombinant AAV2/2(7m8) vector and demonstrated restoration of prenylation, phagocytosis and trafficking defects in the iPSC-RPEs with the same vector. In CHM, AAV2/2 and AAV2/8 have been used in animal models and preclinical studies to explore efficacy and therapeutic benefit in humans.9,84
The first proof-of-concept study used a recombinant adenovirus vector in cell cultures of fibroblasts and lymphocytes derived from CHM patients and demonstrated REP1 expression and functional restoration.95 Tolmachova and colleagues91 produced lentiviral vectors to transduce CHM knock out mouse retinal cells (in vitro and in vivo) and CHM human fibroblasts with CHM/REP1 cDNA and showed increase in prenylation activity. The same group developed an AAV2 vector (AAV2/2-CBA-REP1) and successfully achieved REP1 transgene expression in human and mouse photoreceptors and RPE cells.96 They have also reported potential therapeutic benefit by demonstrating increased a- and b-wave ERG responses in CHM mouse retinal models.96
The first multicentre nonrandomised open-label dose escalation phase 1/2 clinical trial (NCT01461213) in CHM patients reported encouraging results using an AAV2.REP1 vector.15 In total, 14 patients were recruited at different disease stages and underwent pars plana vitrectomy (PPV) followed by retinotomy and injection of 0.1 ml of 1 × 1010 AAV2.REP1 particles [two patients were treated off protocol due to surgically induced retinal stretching in one case and due to intraocular inflammation (vitritis and choroiditis) in the second case] into the subretinal space.15,97,98 Besides CHM/REP1 cDNA, the expression cassette also included a chicken β-actin promoter for RPE long-term transduction and a woodchuck hepatitis virus posttranslational regulatory element (WPRE) which facilitates transgene expression.15,97,99 Clinical assessment comprised visual acuity testing using Early Treatment Diabetic Retinopathy Study (ETDRS) letters and retinal sensitivity evaluation with dark adapted microperimetry.15,97 After 2 years, the median visual acuity increased by 4.5 letters in the treated eyes versus 1.5 letters loss in the untreated eyes.98 For the 12 patients who followed the protocol without complications, at 2 years, 5.5 letters gain above their baseline level was reported for the treated eyes compared to the untreated eyes, while the 2 patients who developed complications lost 15 and 14 letters, respectively.98 In order to decrease the incidence of the reported complications, the protocol was modified during the trial by introduction of an automated subretinal injection system, intra-operative OCT and an extended course of postoperative immune suppression.98
Despite the promising results, several challenges remain to be addressed such as defining the ideal therapeutic window to intervene before irreversible degeneration, ensuring that the necessary cell types are adequately transduced, minimizing viral toxicity, deciphering long-term integration effects, affinity to target tissues, ideal delivery method, long-term transgenic potential and need to repeat treatment.85 The genetic variants represent an important consideration since patients with missense mutations who express a residual mutated protein could also respond favourably to gene therapy.24 Nonviral DNA vectors could also be used autonomously or in combination with scaffold matrix attachment region (S/MAR) motifs, as shown in mouse models of Leber Congenital Amaurosis.100 Alternative delivery methods like intravitreal injections have been used experimentally in mouse models of retinitis pigmentosa and laser-induced CNV, but efficient cell penetration and possible humoral immune response represent possible limitations.101,102
Stem cell models
Stem cells and regenerative medicine aim to restore degenerative tissue and improve visual function. In CHM, iPSC models have been used to study pathogenesis, confirm disease-specific molecular deficits and verify REP1 expression following gene therapy techniques.,24,93,94 Trans-plantation of either RPE or photoreceptors is the cornerstone of stem cell treatments and has been used to identify potential therapeutic benefit in patients with age-related macular degeneration (AMD) and other inherited retinal disorders (retinitis pigmentosa, Stargardt disease).103 Besides ethical considerations related to embryonic stem cells (ESCs), further research is necessary to address potential risks including hyperproliferation, tumour formation, recipient rejection and delivery method optimisation.
Cell-based approaches could also have therapeutic applications to CHM and factors that need to be considered include definition of the appropriate cell type (RPE, photoreceptors), selection of the most efficient stem cell category [human iPSCs, ESCs, retinal progenitor cells (RPCs)] to achieve tissue regeneration, establishment of the most efficient delivery method and therapeutic window determination.90 Fibroblast-derived iPSCs that were differentiated into RPE cells have been used in clinical trials of wet AMD in Japan and RPCs were injected intravitreally in a Phase IIb Clinical Trial for Retinitis Pigmentosa (NCT02320812).104 Recently, hESC-derived RPE cells layered on synthetic membrane have been reported to improve visual acuity (visual acuity gain of 29 and 21 letters, respectively) in two patients with acute wet AMD and rapid deterioration in visual acuity.105 Long-term results from these clinical trials will be required to establish safety profile and clinical effectiveness and establish them as an approved potential clinical therapeutic.
Small-molecule and other drug compounds
A significant number of CHM cases ~30% are attributed to in-frame nonsense mutations, resulting in premature termination codons (PTCs).1 Application of small-molecule drugs that are able to promote ribosomal read-through of PTCs and bypass abnormal stop signals is possible through competitive binding of near-cognate aminoacyl-tRNAs (transfer RNAs) to eukaryotic release factors (eRFs).1,106 The end result could be production of ~25% wild-type levels of functional proteins that are necessary to halt disease progression.106 This approach has been employed in cystic fibrosis (CF) and Duchenne muscular dystrophy (DMD).106 In the CHM zebrafish model and a CHM patient derived fibroblast cell line treatment with PTC124 (ataluren) and the optimised agent PTC414 demonstrated REP1 expression restoration resulting in increased survival, normal eye morphology, prevention of retinal degeneration and increased prenylation function.106,107
Among the compounds with proven read-through activity are traditional aminoglycosides [gentamicin, paromomycin, Geneticin (G418), streptomycin], the less toxic next-generation designer aminoglycoside-derivatives (NB84, NB74 and NB54), nonaminoglycoside small-molecule drugs (PTC124 and PTC414) and small-molecule read-through (SMRT) compounds (RTC13, RTC14, GJ071 and GJ072).106,108 PTC124 (also known as ataluren or Translarna™) has received NICE (National Institute for Health and Care Excellence) approval for DMD treatment caused by nonsense mutation in the dystrophin gene. Ataluren has been proven to be a potent nonsense suppression agent across many different genes, can be administered orally, has limited toxicity and does not override normal stop codons.106,109 This read-through specificity is due to differences in termination efficiency between normal stop codons and PTCs which could be attributed to the closed-loop configuration of mRNA and the different sites of PTCs termination (usually not close to poly(A) tail as for normal stop codons).110 As a result, the interaction with factors that normally mediate protein release [eRF3, poly(A)-binding protein (PABP)] is changed and this leads to ribosomal pausing at PTCs.109,110
Potential limitations include expression of unwanted nonsense mutations present in other genes and decreased availability of transcripts with incorporated PTCs due to a natural occurring nonsense-mediated decay (NMD) pathway.106 It is possible that combining read-through agents with NMD pathway inhibitors (NMDI1, VG1) or dual combination agents (amlexanox) could enhance therapeutic benefit.108,111,112
Lutein supplementation
Lutein supplements (either alone or in combination with vitamin A) have been studied in inherited retinal disorders to identify potential association with delayed disease progression and visual acuity improvement.113,114 Lutein is a carotenoid that augments proper macular pigment function through short-wavelength filtration and reactive oxygen species stabilisation.115 Oral supplementation with lutein for 6 months has been studied in CHM patients; although an increase in macular pigment levels and serum lutein levels was reported, there was no measurable gain in terms of absolute foveal sensitivity and short-term central visual acuity.115
General supportive measures
Supportive measures include strategies to enhance performance in daily activities, improve quality of life and address ophthalmological comorbidities. All patients should have access to low vision rehabilitation (mobility training, educational support and occupational therapy) and devices to support visual performance (low-vision aids, magnifiers, spectacles, high-intensity lamps and contrast-enhancing filters). General recommendations to prevent retinal damage by avoiding smoking, excessive sun exposure and toxic medications should also be provided.116
Subclinical cystoid macular oedema (CMO) in CHM patients can result in severe visual acuity deterioration.49 Treatment options include topical carbonic anhydrase inhibitors (e.g. acetazolamide) and intravitreal injections of anti-VEGF agents based on efficacy of those agents for other inherited dystrophies like retinitis pigmentosa.117,118
Specific considerations regarding cataract treatment in CHM patients include retinal phototoxicity risk from the operating microscope, postoperative CMO and increased rates of postoperative capsular fibrosis.119 A case series of cataract surgical outcomes did not show significant postoperative complications other than early capsular phimosis and posterior capsular opacification. Cataract surgery could be offered to CHM patients after appropriate consultation regarding risks and potential visual outcomes.119
Current and future research
CHM represents a genetic eye disease that has been extensively studied since its first description in 1872 and after CHM gene cloning in the 1990s. Its monogenic nature enables gene therapy approaches and future research aims to fill gaps in our knowledge and explore novel treatment strategies.58,106
Different animal models have been used to study CHM. Tolmachova and colleagues30,96 produced a conditional knockout mouse model to study disease pathogenesis and efficacy of gene therapies. The group showed a progressive retinal degeneration in the mouse model similar to human subjects and suggested that photoreceptors/RPE are affected autonomously and RPE degeneration may not always lead to photoreceptor loss.30
Zebrafish have only one rep isoform and this makes testing therapies aiming to boost REP1 activity very useful as there is no compensation from a REP2.11 The chm (−/−) zebrafish has a nonsense mutation (c.96C>T, p.Gln33*) that leads to severe loss of retinal lamination and multisystem failure resulting in embryonic lethality after 4.5 days postfertilisation (dpf).11,38,120
Traditionally, CHM has been considered an isolated eye disorder. However, Zhou and colleagues121 reported an increased prevalence of systemic disease in patients based on a cross-sectional Internet survey completed by 256 subjects (CHM patients, carriers and unaffected brothers). Affected men were classified into two groups based on visual function (with or without functional loss).121 The most common systemic diseases reported from CHM patients without functional loss were hypertension, diabetes and hypercholesterolaemia, but after age adjustment, there was no significant difference from patients with visual loss.121 More recently, Zhang and colleagues122 reported intracellular crystals (rod and needle like) within lymphocytes in a CHM family (two male patients and one carrier) with c.936delT, p.(Y312*) in CHM and further replicated their findings using electron microscopy in four CHM patients, while no crystals were found in the three controls of the cohort. They had also discovered fatty acid abnormalities in serum and red blood cell (RBC) membrane after obtaining fasting blood samples from five CHM patients compared with controls.122 Among the lipid abnormalities noted, increased plasmalogen levels in RBC membrane could be associated with functional and structural defects as well as oxidative stress damage.123 Oxidative stress has been linked to AMD, diabetic retinopathy and glaucoma and one possible mechanism is through autophagy dysregulation and mitochondrial DNA damage.124–126 The retina has the highest metabolic profile and increased mitochondrial density. Further research is needed to explore whether the retinal degeneration associated with CHM could be attributed to accumulative mitochondrial pathology and furthermore whether it is a systemic condition.125–127
Ongoing natural history studies of CHM aim to provide additional insight into disease pathogenesis and progression and also define valid outcome measures to augment future clinical trials design. Visual acuity seems to be a weak indicator of disease progression based on retrospective analysis of 60 CHM male patients where statistically significant changes were reported only for those >30 years.55 Furthermore, the results published by Dimopoulos and colleagues128 after 2 years of a phase 1 clinical trial in six CHM patients where an AAV2.REP1 vector was used imply to that visual acuity may not represent a sensitive outcome measure due to test–retest variability. Quantification of preserved EZ length and FAF is highly reproducible which is very important for disease severity stratification and treatment response monitoring.129 Recently, a deep learning platform has also been used to automatically identify areas of EZ loss from OCT images and augment progress classification.130 A high accuracy (~90%) of segmentation match to expert grader in preserved photoreceptor area was reported.130 Additional data from natural history studies are needed to identify suitable candidates for clinical trials, to determine reliable clinical trial endpoints for treatment efficiency and to decrease variability of reported outcomes.
The ongoing clinical trials for CHM include the initiation of a phase 3 multicentre gene therapy trial (NCT03496012). The aim of the study is to evaluate safety and efficiency of the AAV2-REP1 vector through single-dose subretinal injection in 140 CHM patients. Participants will be randomised to three study arms (high dose, low dose and no treatment) and they will be followed up for 12 months. Primary endpoint will be the BCVA, while secondary outcomes include FAF, OCT, microperimetry, contrast sensitivity and colour vision. Retinal prosthetic devices have been tested in clinical trials to explore ways to improve visual function in CHM patients with severe sight impairment (light perception or less). The devices used so far include the Intelligent Retinal Implant System (IRIS V1-NCT01864486) and a 44-channel suprachoroidal Bionic Eye Device (NCT03406416) and results are expected to provide more insight into surgical procedure safety, potential serious adverse events and clinical benefit. Future clinical trials with small-molecule drugs for nonsense suppression may represent an alternative treatment option for early intervention or in combination with gene therapy. It has the potential to be widely applicable to other inherited retinal diseases resulting from nonsense mutations. Finally, with all those efforts to develop a suitable therapy for CHM, patients are close to welcoming an approved treatment to potentially prevent further sight loss.
Footnotes
Conflict of interest statement: The author(s) declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Funding: The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: We gratefully acknowledge funding from the Wellcome Trust 205174/Z/16/Z, National Institute of Health Research, Moorfields Eye Charity and the Choroi-deremia Research Foundation, USA.
ORCID iD: Mariya Moosajee  https://orcid.org/0000-0003-1688-5360
https://orcid.org/0000-0003-1688-5360
Contributor Information
Andreas Mitsios, Institute of Ophthalmology, University College London, London, UK; Moorfields Eye Hospital, London, UK.
Adam M. Dubis, Institute of Ophthalmology, University College London, London, UK NIHR Biomedical Research Centre, Moorfields Eye Hospital, London, UK.
Mariya Moosajee, Institute of Ophthalmology, University College London, London, UK; NIHR Biomedical Resource Centre at Moorfields Eye Hospital, London, UK; Great Ormond Street Hospital for Children NHS Foundation Trust, London, UK.
References
- 1. Moosajee M, Ramsden SC, Black GC, et al. Clinical utility gene card for: choroideremia. Eur J Hum Genet. Epub ahead of print 21 August 2013. DOI: 10.1038/ejhg.2013.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. MacDonald IM, Sereda C, McTaggart K, et al. Choroideremia gene testing. Expert Rev Mol Diagn 2004; 4: 478–484. [DOI] [PubMed] [Google Scholar]
- 3. U.S. National Library of Medicine, National Institutes of Health, Department of Health & Human Services. Choroideremia, https://ghr.nlm.nih.gov/condition/choroideremia#statistics (accessed 2 January 2018).
- 4. Sankila EM, Chapelle ADLA, Karna J, et al. Choroideremia: close linkage to DXYS1 and DXYS12 demonstrated by segregation analysis and historical-genealogical evidence. Clin Genet 1987; 31: 315–322. [DOI] [PubMed] [Google Scholar]
- 5. Sankila EM, Tolvanen R, van den Hurk JAJM, et al. Aberrant splicing of the CHM gene is a significant cause of choroideremia. Nat Genet 1992; 1: 109–113. [DOI] [PubMed] [Google Scholar]
- 6. MacDonald IM, Hume S, Chan S, et al. Choroideremia (Gene reviews) https://www.ncbi.nlm.nih.gov/books/NBK1337/?report=reader#_NBK1337_pubdet (accessed 21 January 2018).
- 7. Bonilha VL, Trzupek KM, Li Y, et al. Choroideremia: analysis of the retina from a female symptomatic carrier. Ophthalmic Genet 2008; 29: 99–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Edwards TL, Groppe M, Jolly JK, et al. Correlation of retinal structure and function in choroideremia carriers. Ophthalmology 2015; 122: 1274–1276. [DOI] [PubMed] [Google Scholar]
- 9. Barnard AR, Groppe M, Maclaren RE. Gene therapy for choroideremia using an adeno-associated viral (AAV) vector. Cold Spring Harb Perspect Med 2015; 5: a017293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sanchez-Alcudia R, Garcia-Hoyos M, Lopez-Martinez MA, et al. A comprehensive analysis of choroideremia: from genetic characterization to clinical practice. PLoS ONE 2016; 11: e0151943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Moosajee M, Tulloch M, Baron RA, et al. Single choroideremia gene in nonmammalian vertebrates explains early embryonic lethality of the zebrafish model of choroideremia. Invest Ophthalmol Vis Sci 2009; 50: 3009–3016. [DOI] [PubMed] [Google Scholar]
- 12. Sorsby A, Franceschetti A, Joseph R, et al. Choroideremia; clinical and genetic aspects. Br J Ophthalmol 1952; 36: 547–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cremers FPM, van de Pol DJR, van Kerkhoff LPM, et al. Cloning of a gene that is rearranged in patients with choroideraemia. Nature 1990; 347: 674–677. [DOI] [PubMed] [Google Scholar]
- 14. Van Bokhoven H, Van Den, Hurk JAJM, Bogerd L, et al. Cloning and characterization of the human choroideremia gene. Hum Mol Genet 1994; 3: 1041–1046. [DOI] [PubMed] [Google Scholar]
- 15. MacLaren RE, Groppe M, Barnard AR, et al. Retinal gene therapy in patients with choroideremia: initial findings from a phase 1/2 clinical trial. Lancet 2014; 383: 1129–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Furgoch MJB, Mewes-Arès J, Radziwon A, et al. Molecular genetic diagnostic techniques in choroideremia. Mol Vis 2014; 20: 535–544. [PMC free article] [PubMed] [Google Scholar]
- 17.https://grenada.lumc.nl/LOVD2/Usher_montpellier/home.php?select_db=CHM (accessed 20 February 2018).
- 18. Carss K, Arno G, Erwood M, et al. Comprehensive rare variant analysis via whole-genome sequencing to determine the molecular pathology of inherited retinal disease. Am J Hum Genet 2017; 100: 75–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chi JY, MacDonald IM, Hume S. Copy number variant analysis in CHM to detect duplications underlying choroideremia. Ophthalmic Genet 2013; 34: 229–233. [DOI] [PubMed] [Google Scholar]
- 20. Edwards TL, Williams J, Patrício MI, et al. Novel non-contiguous exon duplication in choroideremia. Clin Genet 2018; 93: 144–148. [DOI] [PubMed] [Google Scholar]
- 21. Van den Hurk JA, Schwartz M, Van Bokhoven H, et al. Molecular basis of choroideremia (CHM): mutations involving the Rab escort protein-1 (REP-1) gene. Hum Mutat 1997; 9: 110–117. [DOI] [PubMed] [Google Scholar]
- 22. Sergeev YV, Smaoui N, Sui R, et al. The functional effect of pathogenic mutations in Rab escort protein 1. Mutat Res 2009; 665: 44–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Esposito G, De Falco F, Tinto N, et al. Comprehensive mutation analysis (20 families) of the choroideremia gene reveals a missense variant that prevents the binding of REP1 with rab geranylgeranyl transferase. Hum Mutat 2011; 32: 1460–1469. [DOI] [PubMed] [Google Scholar]
- 24. Torriano S, Erkilic N, Faugère V, et al. Pathogenicity of a novel missense variant associated with choroideremia and its impact on gene replacement therapy. Hum Mol Genet 2017; 26: 3573–3584. [DOI] [PubMed] [Google Scholar]
- 25. Lorda-Sanchez I, Ibañez A, Sanz R, et al. Choroideremia, sensorineural deafness, and primary ovarian failure in a woman with a balanced X-4 translocation. Ophthalmic Genet 2000; 21: 185–189. [PubMed] [Google Scholar]
- 26. Mukkamala K, Gentile RC, Willner J, et al. Choroideremia in a woman with ectodermal dysplasia and complex translocations involving chromosomes X, 1, and 3. Ophthalmic Genet 2010; 31: 178–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Simunovic MP, Jolly JK, Xue K, et al. The spectrum of CHM gene mutations in choroideremia and their relationship to clinical phenotype. Invest Ophthalmol Vis Sci 2016; 57: 6033–6039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Olkkonen VM, Ikonen E. Genetic defects of intracellular-membrane transport. N Engl J Med 2000; 343: 1095–1104. [DOI] [PubMed] [Google Scholar]
- 29. Preising MN, Ayuso C. Rab escort protein 1 (REP1) in intracellular traffic: a functional and pathophysiological overview. Ophthalmic Genet 2004; 25: 101–110. [DOI] [PubMed] [Google Scholar]
- 30. Tolmachova T, Anders R, Abrink M, et al. Independent degeneration of photoreceptors and retinal pigment epithelium in conditional knockout mouse models of choroideremia. J Clin Invest 2006; 116: 386–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cremers FPM, Armstrong SA, Seabra MC, et al. REP-2, a Rab escort protein encoded by the choroideremia-like gene. J Biol Chem 1994; 269: 2111–2117. [PubMed] [Google Scholar]
- 32. Seabra MC, Ho YK, Anant JS. Deficient geranylgeranylation of Ram/Rab27 in choroideremia. J Biol Chem 1995; 270: 24420–24427. [DOI] [PubMed] [Google Scholar]
- 33. Köhnke M, Delon C, Hastie ML, et al. Rab GTPase prenylation hierarchy and its potential role in choroideremia disease. PLoS ONE 2013; 8: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Flannery JG, Bird AC, Farber DD, et al. A histopathologic study of a choroideremio carrier. Invest Ophthalmol Vis Sci 1990; 31: 229–236. [PubMed] [Google Scholar]
- 35. Morgan JIW, Han G, Klinman E, et al. High-resolution adaptive optics retinal imaging of cellular structure in choroideremia. Invest Opthalmol Vis Sci 2014; 55: 6381–6397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mura M, Sereda C, Jablonski MM, et al. Clinical and functional findings in choroideremia due to complete deletion of the CHM gene. Arch Ophthalmol 2007; 125: 1107–1113. [DOI] [PubMed] [Google Scholar]
- 37. Cameron JD, Fine BS, Shapiro I. Histopathologic observations in choroideremia with emphasis on vascular changes of the uveal tract. Ophthalmology 1987; 94: 187–196. [DOI] [PubMed] [Google Scholar]
- 38. Krock BL, Bilotta J, Perkins BD. Noncell-autonomous photoreceptor degeneration in a zebrafish model of choroideremia. Proc Natl Acad Sci U S A 2007; 104: 4600–4605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wavre-Shapton ST, Tolmachova T, da Silva ML, et al. Conditional ablation of the choroideremia gene causes age-related changes in mouse retinal pigment epithelium. PLoS ONE 2013; 8: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. MacDonald IM, Russell L, Chan CC. Choroideremia: new findings from ocular pathology and review of recent literature. Surv Ophthalmol 54: 401–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Shankar SP. Hereditary retinal and choroidal dystrophies. In: Emery and Rimoin’s principles and practice of medical genetics. Elsevier, 2013, pp. 1–18, 10.1016/B978-0-12-383834-6.00147-6 [DOI]
- 42. Coussa RG, Kim J, Traboulsi EI. Choroideremia: effect of age on visual acuity in patients and female carriers. Ophthalmic Genet 2012; 33: 66–73. [DOI] [PubMed] [Google Scholar]
- 43. Jolly JK, Xue K, Edwards TL, et al. Characterizing the natural history of visual function in choroideremia using microperimetry and multimodal retinal imaging. Invest Ophthalmol Vis Sci 2017; 58: 5575–5583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Coussa RG, Traboulsi EI. Choroideremia: a review of general findings and pathogenesis. Ophthalmic Genet 2012; 33: 57–65. [DOI] [PubMed] [Google Scholar]
- 45. Rosenberg T, Schwartz M, Niebuhr E, et al. Choroideremia in interstitial deletion of the X chromosome. Ophthalmic Paediatr Genet 2009; 7: 205–210. [DOI] [PubMed] [Google Scholar]
- 46. Van den Bosch J. A new syndrome in three generations of a Dutch family. Ophthalmologica 1959; 137: 422–423. [Google Scholar]
- 47. Khan KN, Islam F, Moore AT, et al. Clinical and genetic features of choroideremia in childhood. Ophthalmology 2016; 123: 2158–2165. [DOI] [PubMed] [Google Scholar]
- 48. MacDonald IM, Hume S, Chan S, et al. Choroideremia. In: Pagon RA, Adam MP, Ardinger HH, et al. (eds) GeneReviews. Seattle, WA: University of Washington; 2003 Feb 21 [Updated 2015 Feb 26]; 1993–2018. [Google Scholar]
- 49. Genead MA, Fishman GA. Cystic macular oedema on spectral-domain optical coherence tomography in choroideremia patients without cystic changes on fundus examination. Eye 2011; 25: 84–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Campos-Pavon J, Torres-Pena JL. Choroidal neovascularization secondary to choroideremia. Arch Soc Esp Oftalmol 2015; 90: 289–291. [DOI] [PubMed] [Google Scholar]
- 51. Jolly JK, Edwards TL, Moules J, et al. A qualitative and quantitative assessment of fundus autofluorescence patterns in patients with choroideremia. Invest Opthalmol Vis Sci 2016; 57: 4498–4503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Schmitz-Valckenberg S, Holz FG, Bird AC, et al. Fundus autofluorescence imaging: review and perspectives. Retina 2008; 28: 385–409. [DOI] [PubMed] [Google Scholar]
- 53. Preising MN, Wegscheider E, Friedburg C, et al. Fundus autofluorescence in carriers of choroideremia and correlation with electrophysiologic and psychophysical data. Ophthalmology 2009; 116: 1201–1209.e2. [DOI] [PubMed] [Google Scholar]
- 54. Wegscheider E, Preising MN, Lorenz B. Fundus autofluorescence in carriers of X-linked recessive retinitis pigmentosa associated with mutations in RPGR, and correlation with electrophysiological and psychophysical data. Graefes Arch Clin Exp Ophthalmol 2004; 242: 501–511. [DOI] [PubMed] [Google Scholar]
- 55. Heon E, Alabduljalil T, McGuigan DB, et al. Visual function and central retinal structure in choroideremia. Invest Opthalmol Vis Sci 2016; 57: 377–387. [DOI] [PubMed] [Google Scholar]
- 56. Xue K, Oldani M, Jolly JK, et al. Correlation of optical coherence tomography and autofluorescence in the outer retina and choroid of patients with choroideremia. Invest Ophthalmol Vis Sci 2016; 57: 3674–3684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Abbouda A, Lim W, Sprogyte L, et al. Quantitative and qualitative features of spectral-domain optical coherence tomography provide prognostic indicators for visual acuity in patients with choroideremia. Ophthalmic Surg Lasers Imaging Retina 2017; 48: 711–716. [DOI] [PubMed] [Google Scholar]
- 58. Syed R, Sundquist SM, Ratnam K, et al. High-resolution images of retinal structure in patients with choroideremia. Invest Ophthalmol Vis Sci 2013; 54: 950–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Fingler J, Readhead C, Schwartz DM, et al. Phase-contrast OCT imaging of transverse flows in the mouse retina and choroid. Invest Ophthalmol Vis Sci 2008; 49: 5055–5059. [DOI] [PubMed] [Google Scholar]
- 60. Spaide RF, Fujimoto JG, Waheed NK, et al. Optical coherence tomography angiography. Prog Retin Eye Res 2018; 64: 1–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Koustenis A, Harris A, Gross J, et al. Optical coherence tomography angiography: an overview of the technology and an assessment of applications for clinical research. Br J Ophthalmol 2017; 101: 16–20. [DOI] [PubMed] [Google Scholar]
- 62. Jain N, Jia Y, Gao SS, et al. Optical coherence tomography angiography in choroideremia: correlating choriocapillaris loss with overlying degeneration. JAMA Ophthalmol 2016; 134: 697–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Patel RC, Gao SS, Zhang M, et al. Optical coherence tomography angiography of choroidal neovascularization in four inherited retinal dystrophies. Retina 2016; 36: 2339–2347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Abbouda A, Dubis AM, Webster AR, et al. Identifying characteristic features of the retinal and choroidal vasculature in choroideremia using optical coherence tomography angiography. Eye 2017; 32: 563–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Sawa M, Tamaki Y, Klancnik JM, et al. Intraretinal foveal neovascularization in choroideremia. Retina 2006; 26: 585–588. [DOI] [PubMed] [Google Scholar]
- 66. Endo K, Yuzawa M, Ohba N. Choroideremia associated with subretinal neovascular membrane. Acta Ophthalmol Scand 2000; 78: 483–486. [DOI] [PubMed] [Google Scholar]
- 67. Forsius H, Hyvärinen L, Nieminen H, et al. Fluorescein and indocyanine green fluorescence angiography in study of affected males and in female carriers with choroidermia: a preliminary report. Acta Ophthalmol 1977; 55: 459–470. [DOI] [PubMed] [Google Scholar]
- 68. Dubra A, Sulai Y, Norris JL, et al. Noninvasive imaging of the human rod photoreceptor mosaic using a confocal adaptive optics scanning ophthalmoscope. Biomed Opt Express 2011; 2: 1864–1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Duncan JL, Zhang Y, Gandhi J, et al. High-resolution imaging with adaptive optics in patients with inherited retinal degeneration. Invest Ophthalmol Vis Sci 2007; 48: 3283–3291. [DOI] [PubMed] [Google Scholar]
- 70. Nabholz N, Lorenzini MC, Bocquet B, et al. Clinical evaluation and cone alterations in choroideremia. Ophthalmology 2016; 123: 1830–1832. [DOI] [PubMed] [Google Scholar]
- 71. Dimopoulos IS, Radziwon A, St. Laurent CD, et al. Choroideremia. Curr Opin Ophthalmol 2017; 28: 410–415. [DOI] [PubMed] [Google Scholar]
- 72. Renner AB, Kellner U, Cropp E, et al. Choroideremia: variability of clinical and electrophysiological characteristics and first report of a negative electroretinogram. Ophthalmology 2006; 113: 2066.e1–2066.e10. [DOI] [PubMed] [Google Scholar]
- 73. Jolly JK, Groppe M, Birks J, et al. Functional defects in color vision in patients with choroideremia. Am J Ophthalmol 2015; 160: 822–831.e3. [DOI] [PubMed] [Google Scholar]
- 74. Seitz IP, Jolly JK, Fischer MD, et al. Colour discrimination ellipses in choroideremia. Graefes Arch Clin Exp Ophthalmol 2018; 1: 665–673. [DOI] [PubMed] [Google Scholar]
- 75. Simunovic MP. Acquired color vision deficiency. Surv Ophthalmol 2016; 61: 132–155. [DOI] [PubMed] [Google Scholar]
- 76. McCulloch C. Choroideremia: a clinical and pathologic review. Trans Am Ophthalmol Soc 1969; 67: 142–195. [PMC free article] [PubMed] [Google Scholar]
- 77. Genead MA, Fishman GA, Grover S. Hereditary choroidal diseases. In: Retina. 5th ed. Amsterdam: Elsevier, 2012, pp. 891–898, 10.1016/B978-1-4557-0737-9.00043-6 [DOI] [Google Scholar]
- 78. Bowne SJ, Humphries MM, Sullivan LS, et al. A dominant Mutation in RPE65 identified by whole-exome sequencing causes retinitis pigmentosa with choroidal involvement. Eur J Hum Genet 2011; 19: 1074–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Sergouniotis PI, Davidson AE, Lenassi E, et al. Retinal structure, function, and molecular pathologic features in gyrate atrophy. Ophthalmology 2012; 119: 596–605. [DOI] [PubMed] [Google Scholar]
- 80. Lee TKM, McTaggart KE, Sieving PA, et al. Clinical diagnoses that overlap with choroideremia. Can J Ophthalmol 2003; 38: 364–372. [DOI] [PubMed] [Google Scholar]
- 81. Hartong DT, Berson EL, Dryja TP. Retinitis pigmentosa. Lancet 2006; 368: 1795–1809. [DOI] [PubMed] [Google Scholar]
- 82. Kabunga P, Lau AK, Phan K, et al. Systematic review of cardiac electrical disease in Kearns-Sayre syndrome and mitochondrial cytopathy. Int J Cardiol 2015; 181: 303–310. [DOI] [PubMed] [Google Scholar]
- 83. Halford S, Liew G, MacKay DS, et al. Detailed phenotypic and genotypic characterization of bietti crystalline dystrophy. Ophthalmology 2014; 121: 1174–1184. [DOI] [PubMed] [Google Scholar]
- 84. Black A, Vasireddy V, Chung DC, et al. Adeno-associated virus 8-mediated gene therapy for choroideremia: preclinical studies in in vitro and in vivo models. J Gene Med 2014; 16: 122–130. [DOI] [PubMed] [Google Scholar]
- 85. Moore NA, Morral N, Ciulla TA, et al. Gene therapy for inherited retinal and optic nerve degenerations. Expert Opin Biol Ther 2018; 18: 37–49. [DOI] [PubMed] [Google Scholar]
- 86. Kumar SR, Markusic DM, Biswas M, et al. Clinical development of gene therapy: results and lessons from recent successes. Mol Ther Methods Clin Dev 2016; 3: 16034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Bainbridge JWB, Mehat MS, Sundaram V, et al. Long-term effect of gene therapy on Leber’s congenital amaurosis. N Engl J Med 2015; 372: 1887–1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Jacobson SG, Cideciyan AV, Roman AJ, et al. Improvement and decline in vision with gene therapy in childhood blindness. N Engl J Med 2015; 372: 1920–1926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Bemelmans AP, Kostic C, Crippa SV, et al. Lentiviral gene transfer of RPE65 rescues survival and function of cones in a mouse model of leber congenital amaurosis. PLoS Med 2006; 3: 1892–1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Chan SC, Bubela T, Dimopoulos IS, et al. Choroideremia research: report and perspectives on the second international scientific symposium for choroideremia. Ophthalmic Genet 2016; 37: 267–275. [DOI] [PubMed] [Google Scholar]
- 91. Tolmachova T, Tolmachov O, Wavre Shapton ST, et al. CHM/REP1 cDNA delivery by lentiviral vectors provides functional expression of the transgene in the retinal pigment epithelium of choroideremia mice. J Gene Med 2012; 14: 158–168. [DOI] [PubMed] [Google Scholar]
- 92. Bueler H. Adeno-associated viral vectors for gene transfer and gene therapy. Biol Chem 1999; 380: 613–622. [DOI] [PubMed] [Google Scholar]
- 93. Cereso N, Pequignot MO, Robert L, et al. Proof of concept for AAV2/5-mediated gene therapy in iPSC-derived retinal pigment epithelium of a choroideremia patient. Mol Ther Methods Clin Dev 2014; 1: 14011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Duong TT, Vasireddy V, Ramachandran P, et al. Use of induced pluripotent stem cell models to probe the pathogenesis of choroideremia and to develop a potential treatment. Stem Cell Res 2018; 27: 140–150. [DOI] [PubMed] [Google Scholar]
- 95. Anand V, Barral DC, Zeng Y, et al. Gene therapy for choroideremia: in vitro rescue mediated by recombinant adenovirus. Vision Res 2003; 43: 919–926. [DOI] [PubMed] [Google Scholar]
- 96. Tolmachova T, Tolmachov OE, Barnard AR, et al. Functional expression of Rab escort protein 1 following AAV2-mediated gene delivery in the retina of choroideremia mice and human cells ex vivo. J Mol Med 2013; 91: 825–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Xue K, Groppe M, Salvetti AP, et al. Technique of retinal gene therapy: delivery of viral vector into the subretinal space. Eye 2017; 31: 1308–1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. MacLaren RE, Xue K, Barnard A, et al. Gene therapy for choroideremia in a multicenter dose escalation phase I/II clinical trial. Invest Ophthalmol Vis Sci 2018; 59: 1195. [Google Scholar]
- 99. Loeb JE, Cordier WS, Harris ME, et al. Enhanced expression of transgenes from adeno-associated virus vectors with the woodchuck hepatitis virus posttranscriptional regulatory element: implications for gene therapy. Hum Gene Ther 1999; 10: 2295–2305. [DOI] [PubMed] [Google Scholar]
- 100. Koirala A, Makkia RS, Conley SM, et al. S/MAR-containing DNA nanoparticles promote persistent RPE gene expression and improvement in RPE65-associated LCA. Hum Mol Genet 2013; 22: 1632–1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Chalovich JM, Eisenberg E. NIH public access. Magn Reson Imaging 2013; 31: 477–479.23102945 [Google Scholar]
- 102. Li Q, Miller R, Han P-Y, et al. Intraocular route of AAV2 vector administration defines humoral immune response and therapeutic potential. Mol Vis 2008; 14: 1760–1769. [PMC free article] [PubMed] [Google Scholar]
- 103. MacLaren RE, Bennett J, Schwartz SD. Gene therapy and stem cell transplantation in retinal disease: the new Frontier. Ophthalmology 2016; 123(10Suppl.): S98–S106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Mandai M, Watanabe A, Kurimoto Y, et al. Autologous induced stem-cell-derived retinal cells for macular degeneration. N Engl J Med 2017; 376: 1038–1046. [DOI] [PubMed] [Google Scholar]
- 105. Da Cruz L, Fynes K, Georgiadis O, et al. Phase 1 clinical study of an embryonic stem cell–derived retinal pigment epithelium patch in age-related macular degeneration. Nat Biotechnol 2018; 36: 328–337. [DOI] [PubMed] [Google Scholar]
- 106. Richardson R, Smart M, Tracey-White D, et al. Mechanism and evidence of nonsense suppression therapy for genetic eye disorders. Exp Eye Res 2017; 155: 24–37. [DOI] [PubMed] [Google Scholar]
- 107. Moosajee M, Tracey-White D, Smart M, et al. Functional rescue of REP1 following treatment with PTC124 and novel derivative PTC- 414 in human choroideremia fibroblasts and the nonsense-mediated zebrafish model. Hum Mol Genet 2015; 25: 3416–3431. [DOI] [PubMed] [Google Scholar]
- 108. Guerin K, Gregory-Evans CY, Hodges MD, et al. Systemic aminoglycoside treatment in rodent models of retinitis pigmentosa. Exp Eye Res 2008; 87: 197–207. [DOI] [PubMed] [Google Scholar]
- 109. Welch EM, Barton ER, Zhuo J, et al. PTC124 targets genetic disorders caused by nonsense mutations. Nature 2007; 447: 87–91. [DOI] [PubMed] [Google Scholar]
- 110. Keeling KM, Xue X, Gunn G, et al. Therapeutics based on stop codon readthrough. Annu Rev Genomics Hum Genet 2014. Aug 31; 15: 371–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Gotham VJB, Hobbs MC, Burgin R, et al. Synthesis and activity of a novel inhibitor of nonsense-mediated mRNA decay. Org Biomol Chem 2016; 14: 1559–1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Gonzalez-Hilarion S, Beghyn T, Jia J, et al. Rescue of nonsense mutations by amlexanox in human cells. Orphanet J Rare Dis 2012; 7: 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Aleman TS, Cideciyan AV, Windsor EAJ, et al. Macular pigment and lutein supplementation in ABCA4-associated retinal degenerations. Invest Ophthalmol Vis Sci 2007; 48: 1319–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Rayapudi S, Schwartz SG, Wang X, et al. Vitamin A and fish oils for retinitis pigmentosa. Cochrane Database Syst Rev 2013; 12: CD008428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Duncan JL, Aleman TS, Gardner LM, et al. Macular pigment and lutein supplementation in choroideremia. Exp Eye Res 2002; 74: 371–381. [DOI] [PubMed] [Google Scholar]
- 116. Sahel JA, Marazova K, Audo I. Clinical characteristics and current therapies for inherited retinal degenerations. Cold Spring Harb Perspect Med 2014; 5: a017111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Yuzbasioglu E, Artunay O, Rasier R, et al. Intravitreal bevacizumab (Avastin) injection in retinitis pigmentosa. Curr Eye Res 2009; 34: 231–237. [DOI] [PubMed] [Google Scholar]
- 118. Melo GB, Farah ME, Aggio FB. Intravitreal injection of bevacizumab for cystoid macular edema in retinitis pigmentosa. Acta Ophthalmol Scand 2007; 85: 461–463. [DOI] [PubMed] [Google Scholar]
- 119. Edwards TL, Groppe M, MacLaren RE. Outcomes following cataract surgery in choroideremia. Eye 2015; 29: 460–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Torriano S, Erkilic N, Baux D, et al. The effect of PTC124 on choroideremia fibroblasts and iPSC-derived RPE raises considerations for therapy. Sci Rep 2018; 8: 8234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Zhou Q, Weis E, Ye M, et al. An internet-based health survey on the co-morbidities of choroideremia patients. Ophthalmic Physiol Opt 2013; 33: 157–163. [DOI] [PubMed] [Google Scholar]
- 122. Zhang AY, Mysore N, Vali H, et al. Choroideremia is a systemic disease with lymphocyte crystals and plasma lipid and RBC membrane abnormalities. Invest Ophthalmol Vis Sci 2015; 56: 8158–8165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Braverman NE, Moser AB. Functions of plasmalogen lipids in health and disease. Biochim Biophys Acta 2012; 1822: 1442–1452. [DOI] [PubMed] [Google Scholar]
- 124. Barot M, Gokulgandhi MR, Mitra AK. Mitochondrial dysfunction in retinal diseases. Curr Eye Res 2011; 36: 1069–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Jarrett SG, Lin H, Godley BF, et al. Mitochondrial DNA damage and its potential role in retinal degeneration. Prog Retin Eye Res 2008; 27: 596–607. [DOI] [PubMed] [Google Scholar]
- 126. Mitter SK, Song C, Qi X, et al. Dysregulated autophagy in the RPE is associated with increased susceptibility to oxidative stress and AMD. Autophagy 2014; 10: 1989–2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Payne AJ, Kaja S, Naumchuk Y, et al. Antioxidant drug therapy approaches for neuroprotection in chronic diseases of the retina. Int J Mol Sci 2014; 15: 1865–1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Dimopoulos IS, Hoang SC, Radziwon A, et al. Two-year results after AAV2-mediated gene therapy for choroideremia: the Alberta experience. Am J Ophthalmol 2018; 193: 130–142. [DOI] [PubMed] [Google Scholar]
- 129. Hariri AH, Velaga SB, Girach A, et al. Measurement and reproducibility of preserved ellipsoid zone area and preserved retinal pigment epithelium area in eyes with choroideremia. Am J Ophthalmol 2017; 179: 110–117. [DOI] [PubMed] [Google Scholar]
- 130. Camino A, Wang Z, Wang J, et al. Deep learning for the segmentation of preserved photoreceptors on en face optical coherence tomography in two inherited retinal diseases. Biomed Opt Express 2018; 9: 3092. [DOI] [PMC free article] [PubMed] [Google Scholar]