

Abstract

Tuberculosis and parasitic diseases, such as giardiasis, amebiasis, leishmaniasis, and trypanosomiasis, all urgently require improved treatment options. Recently, it has been shown that antitubercular bicyclic nitroimidazoles such as pretomanid and delamanid have potential as repurposed therapeutics for the treatment of visceral leishmaniasis. Here, we show that pretomanid also possesses potent activity against Giardia lamblia and Entamoeba histolytica, thus expanding the therapeutic potential of nitroimidazooxazines. Synthetic analogues with a novel nitroimidazopyrazin-one/-e bicyclic nitroimidazole chemotype were designed and synthesized, and structure–activity relationships were generated. Selected derivatives had potent antiparasitic and antitubercular activity while maintaining drug-like properties such as low cytotoxicity, good metabolic stability in liver microsomes and high apparent permeability across Caco-2 cells. The kinetic solubility of the new bicyclic derivatives varied and was found to be a key parameter for future optimization. Taken together, these results suggest that promising subclasses of bicyclic nitroimidazoles containing different core architectures have potential for further development.
Introduction
Infectious diseases are an enormous global health burden. The nitroimidazole class of antibiotics, exemplified by metronidazole (1, Figure 1), has a long history of use to treat bacterial and parasitic infections.1 The mode of action of nitroimidazoles involves partial reduction of the nitro group and subsequent decomposition of the compound to give toxic radical species that cause DNA and protein damage.1 The nitro group is activated by different mechanisms, with multiple enzymes involved in electron transfer reactions in different target organisms, which accounts for the remarkably broad spectra of action of the class. More recently, there have been issues with clinical efficacy that are accompanied, in some cases, by development of resistance, which has prompted re-examination of this old, but still widely used drug class.
Figure 1.

Monocyclic nitroimidazoles 1–4 and the bicyclic variants delamanid (5), pretomanid (6), CGI 17341 (7), and (R)-PA-824 (8).
New clinical applications of “old” nitroimidazoles and the development of novel nitroimidazoles with a bicyclic core scaffold architecture have significant potential to address the emergent unmet medical need imparted by resistant bacteria and parasites. For example, secnidazole (2, Figure 1) was recently approved in the USA for the treatment of bacterial vaginosis, despite being available earlier as a generic in many jurisdictions.2,3 A pediatric formulation of benznidazole 3 (Figure 1), a 2-nitroimidazole used for the treatment of Chagas disease caused by the parasite Trypanosoma cruzi, gained FDA approval in 2017 for use in children.4 The Drugs for Neglected Diseases initiative (DNDi) is currently investigating fexinidazole (4, Figure 1) as a candidate for oral treatment of human African trypanosomiasis (HAT), a parasitic infection caused by Trypanosoma brucei spp. that is a public health threat to ∼70 million people in Africa.5 In November 2018 the European Medicines Agency recommended approval of fexinidazole as the first all-oral treatment for sleeping sickness. Furthermore, bicyclic nitroimidazoles, such as delamanid (5, Figure 1) and pretomanid (6, PA-824), are promising new antimicrobials being developed for the treatment of tuberculosis (TB),6 the number one cause of death from infectious diseases and the ninth leading cause of death worldwide.7 The nitroimidazooxazole 5 gained conditional approval in the European Union in 2014 for the treatment of drug-resistant TB. This agent was derived from CGI 17341 (7)8,9 and overcame the mutagenic liability of 7 (Figure 1).10,11 Meanwhile, 6 was developed concurrently with 5 and is currently in phase III trials. The PK profile of 6 is superior to 5 and this permits once daily dosing, although 6 is less potent.12
Investigations on how 5 and 6 inhibit Mycobacterium tuberculosis under aerobic and anaerobic growth conditions revealed an interesting dual mode of action. Transcriptional profiling of M. tuberculosis treated with 6 under aerobic growth conditions gave a response consistent with both the inhibition of cell wall mycolic acid biosynthesis and also respiratory poisoning.13 Additionally, it was shown that deazaflavin-dependent nitroreductase (Ddn) catalyzed reduction of 6 to the des-nitro 9 metabolite and that this process generated nitric oxide.14 Nitric oxide could be detected in Mycobacterium bovis cells treated with 6 under both aerobic and anaerobic growth conditions and the rate of NO release in M. bovis cells correlated with the anaerobic activities for a series of analogues, supporting the premise that this is the mode of action of this class under anaerobic growth conditions in M. tuberculosis.14 In comparison, 1 is only active against non-replicating M. tuberculosis under anaerobic growth conditions (<0.06% oxygen).15 Nitroimidazooxazine 6 was also investigated for activity against other kinetoplastid organisms in multiple developmental life stages including Leishmania donovani (promastigote and amastigote), T. brucei brucei (procyclic and bloodstream), and T. cruzi (epimastigote and amastigote),16 with the findings prompting further mode of action studies in L. donovani. Compound 8, the R-enantiomer of 6, was shown to be more effective than the S-enantiomer in an in vivo model of visceral leishmaniasis.16 Recently, an NAD(P)H oxidase was identified as the activating nitroreductase (NTR2) for the nitroimidazo-oxazole/-oxazine.17 However, the monocyclic sulfone metabolite of 4, known to be activated by a type I nitroreductase,18 was only marginally activated by NTR2.17 These results support the hypothesis that 6 is activated by an alternative mechanism of action in L. donovani under these culture conditions and illustrates the biological complexity of the mode of action of various nitroimidazoles.
In the course of developing 5 and 6 as treatments for TB, over 1000 derivatives were prepared.19 The structure–activity relationships (SARs) disclosed to date mostly include compounds with modifications to the aryl side chain, with a smaller number of variants that alter the bicyclic core structure (Figure 2A). The nitro group and stereochemistry of the side chain have been shown to be critical for activity as both 8 and S-des-nitro-PA-824 (9) derivatives were inactive in in vitro cultures at 50 μM.16 An analogue with the nitro group at the 3′ position was also shown to be inactive, clarifying that the 2′ position of the nitro group on the 6 bicyclic scaffold is important for activity.20 Replacement of the benzylic oxygen with nitrogen led to an amino-linked series (10a–b) with favorable in vitro activity and solubility properties.21,22 Modification of the imidazole ring to a pyrazole (11a) or triazole (11b) was detrimental to M. tuberculosis activity, as was replacement of the oxygen heteroatom in the oxazine ring with sulfoxide (11c), sulfone (11d), amino (11e), or methylene (11f) groups, although a sulfur (11g) heteroatom in the ring was tolerated.23
Figure 2.

(A) Structural variation of bicyclic nitroimidazoles described in the literature and (B) novel nitroimidazopyrazinones and nitroimidazopyrazines developed by cyclizing the 4(5)-nitroimidazole framework, described here.
More recent efforts have explored the activity of bicyclic nitroimidazoles against the kinetoplastids Leishmania and trypanosomes. DNDi is currently investigating additional nitroimidazooxazines for development against visceral leishmaniasis,24 including DNDI-8219 (12) and DNDI-0690 (13), 6- and 7-substituted nitroimidazooxazines, as two promising backup candidates.25,26 Interestingly, nitroimidazothiazine oxides 14a–d were found to display favorable activity against T. b. brucei, an animal infective strain that is commonly used as a model of HAT.19 Moreover, a “thio-delamanid” derivative (15) proved to be efficacious against T. cruzi, although it was inactive against Leishmania.27 These studies demonstrate that the selectivity profile toward different parasites can be altered by structural modifications of the bicyclic scaffold and that different subclasses of the bicyclic nitroimidazoles expand the potential therapeutic scope of this antimicrobial class.
We reasoned that bicyclic nitroimidazole derivatives could have potential against an even wider range of organisms than Leishmania and trypanosomes. Previously, we found that 4(5)-nitroimidazoles (16)28 had potent activity against Giardia lamblia, Entamoeba histolytica, Trichomonas vaginalis, and Clostridium difficile (Figure 2B), and therefore, it was hypothesized that 6 might also have activity against these organisms. Anaerobic protozoa such as G. lamblia and E. histolytica and the anaerobic bacteria C. difficile occupy the gut under reduced oxygen tension and cause diarrheal infections. These organisms spread by the fecal oral route through stable cyst forms for the protozoa or through spores for C. difficile. Metronidazole 1 can be used therapeutically for infections caused by these organisms, but alternative treatment options are desirable. Furthermore, cyclizing the 4(5)-nitroimidazoles 16 from the 1′ imidazole ring position to the carboxamide nitrogen could produce new bicyclic nitroimidazopyrazin-one/-e scaffolds. These could potentially extend the spectra of biological activity to include M. tuberculosis and result in altered SARs against a panel of parasitic organisms. Previous studies have shown that compounds containing the imidazopyrazinone scaffold have a range of different biological activities, including agonism of the GABAA receptor,29 antagonism of the ionotropic transmembrane receptor,30,31 modulation of ion channels to control arrhythmia,32 and inhibition of M. tuberculosis glutamine synthetase.33 However, no studies have investigated the antimicrobial activity of a nitrated derivative of this class.
We now report the findings from an evaluation of 6 against a wide range of organisms, and the subsequent design, synthesis, and biological evaluation of novel bicyclic nitroimidazoles, nitroimidazopyrazinones (17) and nitroimidazopyrazines (18) (Figure 2B), derived from the 4(5)-nitroimidazole scaffold 16 previously reported.28 Given the potential for activity against many different organisms, compounds were screened against a wide range of microorganisms including: G. lamblia, E. histolytica, T. b. brucei, L. donovani, M. tuberculosis, C. difficile, Cryptococcus neoformans, Candida albicans, and representative ESKAPE bacteria: Escherichia coli, Staphylococcus aureus, Klebsiella pneumoniae, Pseudomonas aeruginosa, and Acinetobacter baumannii. Derivatives were counter-screened for cytotoxicity against mammalian liver and kidney cell lines. Preferred derivatives with favorable antimicrobial or antiparasitic activity were then evaluated for therapeutic properties including metabolic stability, plasma protein binding (PPB), and intestinal permeability. Most of the compounds were also assessed for aqueous kinetic solubility. This report presents the first description of SARs and evaluation of nitroimidazopyrazin-ones/-es for antimicrobial and antiparasitic activity with therapeutic potential.
Chemistry
Design and Synthesis of Bicyclic Nitroimidazoles with Variation at R1, R2 = R3 = H
In previous work,28 a set of amide-substituted monocyclic nitroimidazoles were prepared, and it was hypothesized for this study that linking the amide and imidazole nitrogen could form a bicyclic nitroimidazopyrazinone system, similar to the pretomanid 6 core (Figure 2B). A library of nitroimidazopyrazinones 17a–t were prepared with different functional groups at R1 designed to explore a range of physicochemical properties (Scheme 1). Initial biological results indicated that benzylic R1 groups were favorable for antimicrobial and antiparasitic activity. The library was therefore tailored to contain analogues with a number of different benzyl substituted R1 groups (17b–j), including derivative 17c with a 4-OCF3-substituted benzyl group similar to pretomanid, and analogues with substitutions at the 3′ and 2′ positions. Compound 17k was prepared with R1 = CH2-(2-pyridyl) to introduce a hydrogen bond acceptor and to impart more polarity and improve aqueous solubility. A methyl group was introduced at the benzylic position (17l) to increase the lipophilicity, bulk, and potentially restrict the conformation of the side chain. Two phenethylene derivatives (17m–n) were prepared to investigate the effect of spacing the aromatic ring further from the bicyclic core. Derivatives with polar side chain R1 groups were also synthesized, including ethyl acetate 17o, ethyl hydroxyl 17p, morpholine derivatives 17q–r, and the carboxamide group (17s). Compound 17t with R1 = cyclohexyl group was prepared to investigate the effect of a bulky aliphatic group at this position.
Scheme 1. Synthesis of Nitroimidazopyrazin-ones/-es 17a–t, 18i, and 18m by Route (1) Condensation and Dehydration via Nitroimidazole Carboxamide Intermediates or Route (2) Alkylation of 17a.

(i) Bromoacetaldehyde diethyl acetal, K2CO3, μW 150−180 °C, 69%—quant. yield, (ii) 2 M aq HCl (10 vol), 1,4-dioxane (10 vol), μW 120 °C, 42–87%, (17a: 5% aq HCl, reflux, 66%); (iii) alkyl/benzyl halide, K2CO3/Cs2CO3, DMF, rt—μW 120 °C or Ag2CO3/toluene, 80 °C as detailed in the experimental, 3–84%, and (iv) K2CO3, MeOH, rt, 45%. Compounds 18g–h and 18q were detected but not isolated in pure form.
Initially, nitroimidazopyrazinones 17a–f, 17k–l, 17n, and 17t were prepared from 4(5)-nitroimidazole carboxamides 16a–f, 16k–l, 16n, and 16t(28) by cyclizing the 1′ imidazole to the 2′ free amide nitrogen (route 1, Scheme 1). This was achieved by alkylating 4(5)-nitroimidazoles with bromoacetaldehyde diethyl acetal under basic conditions (K2CO3) which strongly favored the 4-nitro regioisomer product.34 Second, microwave heating (μW 120 °C) of 19a–f, 19k–l, 19n, and 19t under acidic conditions afforded the bicyclic products 17a–f, 17k–l, 17n, and 17t. This synthesis was based on the preparation of des-nitro-imidazopyrazinone 20a, as previously described by Prévot and Leumann;35 however, microwave heating at higher temperatures and for shorter periods than conventional reflux facilitated analogue generation for both steps. Second, inclusion of a cosolvent (aq 2 M HCl/1,4-dioxane 1:1) was necessary to solubilize the secondary amide starting material and achieve conversion to the desired products 17b–f, 17k–l, 17n, and 17t. With this approach, products often precipitated from the reaction mixture and could be isolated in high purity by filtration and washes (H2O) alone, especially if the intermediate had been purified by chromatography. Alternatively, the products were purified by recrystallization. In the 1H NMR, the imidazopyrazinone R2 and R3 proton groups were typically two doublets (J = ∼5.9 Hz), each integrating for 1 proton at ∼δ 7.4 ppm and ∼δ 7.6 ppm, respectively. Furthermore, both NMR (2D HMBC) experiments and an X-ray crystal structure of 17a confirmed the 2-position of the nitro group (Supporting Information, Figure S1).
A second approach to prepare bicyclic nitroimidazoles 17g–j, 17m, 17o, and 17q–s was developed as shown in route 2, Scheme 1. This method was more convergent as it eliminated the need to prepare 4(5)-nitroimidazole intermediates of each derivative. Route 2 involved alkylation of 17a with alkyl/aryl halides under basic conditions (carbonate base) to form 17g–j, 17m, 17o, and 17q–s. This approach also produced the minor O-alkylated regioisomer in some cases. This was considered advantageous because the alternative pyrazine ring was anticipated to have different biological activities and properties. In all cases, use of K2CO3 or Cs2CO3 in dimethylformamide (DMF) resulted in >10:1 ratio of the pyrazinone to pyrazine derivatives. Reaction with Ag2CO3 in toluene and heating as described in the literature36,37 was briefly explored and found to increase the ratio of the minor imidazopyrazine isomer, with pyrazinone/pyrazine ratios of 2:1 for 17g/18g and 17i/18i. Unfortunately, the Ag2CO3/toluene method resulted in unreacted starting material and a greater number of side products compared to K2CO3 or Cs2CO3 in DMF. This complicated the separation of 18g to the required >95% purity for biological assay. Therefore, isomers 17m and 18m were prepared using K2CO3 in DMF.
Nitroimidazopyrazines were readily distinguished from nitroimidazopyrazinones as they were more lipophilic, and thus were more strongly retained on C18 silica and more weakly retained on normal phase silica. Furthermore, in the 1H NMR, the resonance due to the OCH2 of 18i and 18m was 0.4–0.6 ppm further downfield than the corresponding resonance due to the NCH2 group in the pyrazinone scaffold (e.g., 18i/17i δ 5.64 vs 5.21 ppm). In the 13C NMR spectrum, the corresponding OCH2 resonance was also shifted 17–19 ppm downfield for 18i and 18m compared to the corresponding NCH2 resonance (e.g., 18i/17i δ 62.7 vs 45.7 ppm).
Des-Nitro Derivatives
Des-nitro derivatives 20b–c were prepared as negative control compounds, essentially as described for the nitrated series above (Scheme 2). It was hypothesized that these compounds should be biologically inactive if the parent compounds had a mechanism of action involving reduction of the nitro group.
Scheme 2. Synthesis of Des-nitro Imidazopyrazinones 20b–c.

(i) (COCl)2, cat. DMF, DCM, 0 °C → rt, then conc. NH4OH, 73%28 (ii) SOCl2, reflux, then benzyl amine, 69%; (iii) bromoacetaldehyde diethyl acetal, K2CO3, μW 120–180 °C, 81–92%; (iv) for 20a: 5% aq HCl, reflux, 50%; for 20b: 5% aq HCl, 80 °C, 39%; (v) 4-fluorobenzylamine, K2CO3, DMF, rt, 19%.
Design and Synthesis of Nitroimidazopyrazin-ones/-es with Modifications at R2, R3
We then explored different variations at the R2 and R3 positions, established a shortened route to synthesize the nitroimidazopyrazinone core, and further investigated the activity of the nitroimidazopyrazine scaffold (Scheme 3). Both core scaffolds were prepared with three different combinations of R2 and R3 groups in order to assess how functionalization at these sites could affect activity against different organisms. Because benzyl groups at R1 were favorable for biological activity, derivatives 24a–e containing R2 = Ph and R3 = H, but including polar substituents (CH2CH2OAc or CH2CH2OH) at R1 to counteract the increase in compound lipophilicity, were prepared. The effect of a methyl group at R2 = CH3, as a small, nonpolar substituent (derivatives 25a–m) was also assessed. For the series with R2 = CH3, both polar and nonpolar substituents at R1, including R1 = CH2CH2OAc, CH2CH2OH, and several preferred benzyl derivatives were investigated. Lastly, matched pair analogues with R2 = R3 = CH3 (26a–i) with the preferred benzyl substituents at R1 were prepared.
Scheme 3. Synthesis of Nitroimidazopyrazinon-one/-es 24a–e, 25a–m, and 26a–i with Variations at R2 and R3.
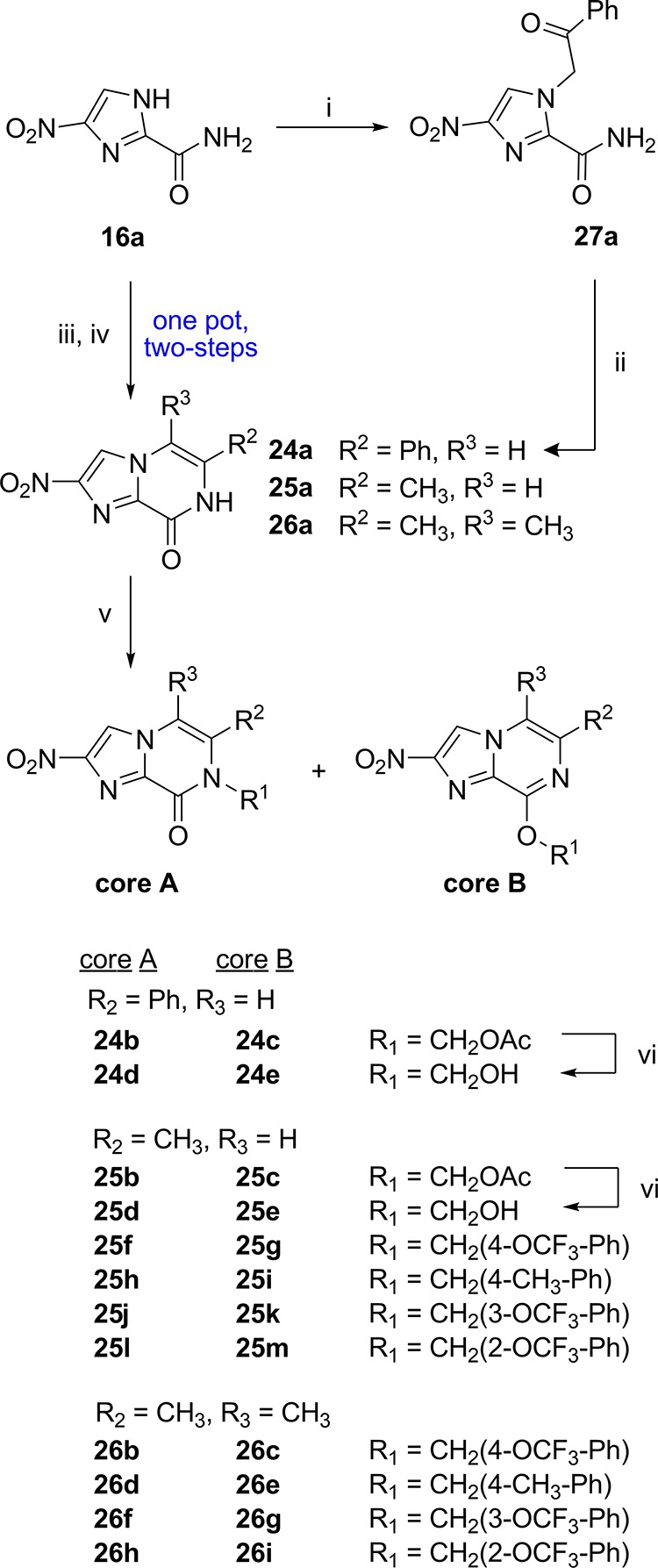
(i) 2-Bromoacetophenone, K2CO3, DMF, rt, 60%; (ii) 2 M HCl, 1,4-dioxane, μW 120 °C, 84%; (iii) 25a & 26a: α-halo ketone, K2CO3, DMF, rt; (iv) 25a & 26a: 2 M HCl, 1,4-dioxane, rt, 40–67% over two steps; (v) K2CO3 or Cs2CO3, DMF, rt—μW 80 °C, 41–90% yield inclusive of both isomers; (vi) K2CO3, MeOH, rt, 10–83% yield. Compound 25k was detected but not isolated.
The synthesis of the nitroimidazopyrazinone 24a (R2 = Ph) was initially performed as in Scheme 3 in a similar manner as described earlier. Intermediate 16a was alkylated with 2-bromoacetophenone at room temperature with K2CO3 as a base catalyst to yield 27a, which was then purified by silica chromatography. The condensation–dehydration cyclization of 27a was achieved with 2 M aq HCl and 1,4-dioxane under microwave irradiation to form the phenyl substituted nitroimidazopyrazinone core 24a. To synthesize the nitroimidazopyrazinone cores with R2 = CH3 (25a) and R2 = R3 = CH3 (26a), a more concise synthesis was designed that involved a two-step, one-pot tandem alkylation–dehydration reaction (Scheme 3). For 25a and 26a, this procedure worked well. The conditions were mild as both steps of the reaction (alkylation and condensation–dehydration) proceeded at room temperature and the products were obtained in high purity after filtration and washes (H2O and MeOH) of the precipitate (67% yield over two steps). The one pot, two-step methodology was also tested for 24a (R2 = phenyl), although conversion and yields were reduced [40% isolated yield with product of 57% purity: Abs % UV 254 nm liquid chromatography–mass spectrometry (LCMS)] due to impurities that formed in both steps because of the different reactivity of the alkylation and condensation–dehydration steps. In addition, product 24a was slightly soluble in the methanol used in the wash step to remove more nonpolar impurities. An intermediate work-up procedure or optimization of the initial alkylation conditions could be explored to improve this method for 24a.
Alkylation of nitroimidazopyrazinones 24a, 25a, and 26a under basic conditions provided both the N- and O-alkylated constitutional isomers that were readily separated by chromatography. Alkylation conditions that used heating in the presence of Cs2CO3 generally favored the O-alkylated nitroimidazopyrazine product. The method employed here was sufficient to provide both possible isomers for biological evaluation, but further optimization of the reaction conditions could be explored in the future to alter the ratio of N- to O-alkylated product.38 Again, the acetate groups were removed using K2CO3/MeOH to give the nitroimidazopyrazin-ones/-es 24d–e and 25d–e.
Results and Discussion
Antiparasitic and Antimicrobial Activity of Pretomanid
Both pretomanid 6 and its enantiomer 8 were found to be ∼2-fold more active than metronidazole 1 against G. lamblia (Table 1). Both enantiomers also had activity against E. histolytica, with 8 active at comparable levels to 1, and 6 ∼2-fold less active than 1. Compound 6 had moderate activity against C. difficile (MIC = 4 μg/mL), which was 8-fold less potent than metronidazole 1 (MIC = 0.5 μg/mL) (Table 1). These results expand the spectrum of action of 6, which was previously described to inhibit M. tuberculosis, Leishmania spp., and Trypanosoma spp.16,26 The enantiomers 6/8 have been reported to have more significant differences in activities against L. donovani, trypanosomes, and M. tuberculosis.16 For example, 8 was ∼5-fold more active than 6 against L. donovani in both the promastigote and amastigote (intracellular macrophage) assays.16 While 6 had weak activity against T. cruzi and T. b. brucei in the parasite life stages relevant to mammalian infection, 8 had slightly enhanced activity, similar to the results against L. donovani.16 In contrast, 8 was inactive against M. tuberculosis.39 Compound 8 has previously been shown to bind to the M. tuberculosis Ddn enzyme involved in reductive activation of 6, but it could not be turned over by the enzyme.39 These differences in selectivity of the enantiomers 6 and 8 likely indicate differences in the mode of action, respective targets, or uptake of compound by the respective organisms. Nonetheless, the results suggest that 6 or newer derivatives may also be repurposed toward enteric parasites. More potent compounds could also potentially be identified in the future by screening a library of pretomanid analogues from the TB alliance/DNDi collections, given that more than 1000 analogues have been prepared.19
Table 1. Activity of Pretomanid (6) Against Enteric Pathogens.
| antiparasitic IC50 (μM) (pIC50 ± SE) |
antibacterial MIC (μg/mL) | ||
|---|---|---|---|
| compound | G. lamblia | E. histolytica | C. difficile |
| metronidazole (1) | 7.2 (5.1 ± 0.02) | 4.3 (5.4 ± 0.02) | 0.5 |
| pretomanid (6) | 3.0 (5.5 ± 0.02) | 9.3 (5.0 ± 0.03) | 4 |
| (R)-PA-824 (8) | 3.2 (5.5 ± 0.08) | 5.3 (5.3 ± 0.08) | N.D. |
SAR of Bicyclic Nitroimidazoles with Variation at R1, R2 = R3 = H
Given the broad activity of pretomanid 6, bicyclic nitroimidazoles 17a–t, 18i, and 18m were tested against a wide panel of organisms. These organisms included M. tuberculosis grown under aerobic (normoxic) and nonreplicating (hypoxic) conditions, G. lamblia, E. histolytica, L. donovani (intracellular amastigote assay), T. b. brucei, and C. difficile. To gain a clear understanding of the spectra of action, compounds were also screened against representative ESKAPE bacterial pathogens S. aureus (ATCC 43300), E. coli (ATCC 25922), K. pneumoniae (ATCC 700603), A. baumannii (ATCC 19606), and P. aeruginosa (ATCC 27853) and fungal pathogens, C. albicans (ATCC 90028) and C. neoformans (H99 type strain, ATCC 208821) at the Community for Open Antimicrobial Drug Discovery (CO-ADD).40 The cytotoxicity against mammalian liver HepG2 and kidney HEK293 cell lines was also evaluated.
Gratifyingly, several compounds showed potent activity against M. tuberculosis, G. lamblia, and T. b. brucei with the SAR described in Table 2. The N- and O-alkylated isomers had varying profiles, with the nitroimidazopyrazinones 17a–t consistently lacking activity against L. donovani, E. histolytica, or C. difficile. However, nitroimidazopyrazine 18m was distinguished by its activity against E. histolytica (IC50 = 7.8 μM, Supporting Information, Table S5) and also G. lamblia (IC50 = 5.2 μM), despite no activity against M. tuberculosis compared to the nitroimidazopyrazinone derivative 17m. The screen against ESKAPE and fungal pathogens revealed that these compounds were selective for M. tuberculosis, G. lamblia, and T. b. brucei as they were not active against the other organisms (MIC >32 μg/mL) (Supporting Information, Table S5). Importantly, the majority of the compounds were not cytotoxic at the highest concentration tested (CC50 > 100 μM) against mammalian liver HepG2 and kidney HEK293 cell lines (Supporting Information, Table S5). However, compounds 17b (R1 = CH2Ph) and 17t (R1 = cyclohexyl) were moderately cytotoxic against mammalian liver and kidney cell lines (CC50 = 98–123 μM) compared to compounds 17a and 17c–s (CC50 > 100 μM). Nitroimidazopyrazine derivatives 18i and 18m were also not cytotoxic against mammalian cell lines (CC50 > 100 μM). This activity profile suggests a specific mechanism of action in M. tuberculosis, G. lamblia, and T. b. brucei rather than a general nonspecific toxic effect.
Table 2. Activity of Nitroimidazopyrazinones and Nitroimidazopyrazines with Variation at R1, R2 = R3 = H.
| compound |
molecular weight (g/mol) | antibacterial
MIC (μg/mL) |
antiparasitic IC50 (μM) (pIC50 ± SE)d |
||||
|---|---|---|---|---|---|---|---|
| no. | core | R1 | M. tuberculosisa normoxia | M. tuberculosisa hypoxia | G. lambliab | T. b. bruceic | |
| metronidazole 1 | 171.2 | >32 | >32 | 7.2 (5.1 ± 0.02) | >40 (<4.4) | ||
| pretomanid 6 | 359.3 | 0.25–0.5 | 1 | 3.0 (5.5 ± 0.02) | 19 (4.7 ± 2.4) | ||
| 17a | A | H | 180.1 | >32 | >32 | >25 (<4.6) | 97% I @ 40 μM |
| 17b | A | CH2Ph | 270.2 | 0.5–1 | 4–8 | 5.0 (5.3 ± 0.05) | 73% I @ 40 μM |
| 17c | A | CH2(4-OCF3-Ph) | 354.2 | 0.5 | 1–4 | 3.5 (5.5 ± 0.01) | 1.4 (5.9 ± 0.08) |
| 17d | A | CH2(4-CH3-Ph) | 284.3 | 0.125 | 1–4 | 3.4 (5.5 ± 0.03) | >40 (<4.4) |
| 17e | A | CH2(4-F-Ph) | 288.2 | 1 | 2 | 6.4 (5.2 ± 0.07) | 2.9 (5.5 ± 0.41) |
| 17f | A | CH2(3-OCF3-Ph) | 354.2 | 0.125 | 0.5–2 | 1.7 (5.8 ± 0.03) | 38% I @ 16 μM |
| 17g | A | CH2(3-CH3-Ph) | 284.3 | 0.06 | 70–90% I @ 0.125–8 μg/mL | 9.0 (5.0 ± 0.03) | 5.4 (5.3 ± 0.23) |
| 17h | A | CH2(3-CF3-Ph) | 338.2 | 0.06 | 70–90% I @ 0.06–16 μg/mL | 7.1 (5.1 ± 0.03) | 5.3 (5.3 ± 0.18) |
| 17i | A | CH2(2-OCF3-Ph) | 354.2 | 1 | 8 | 8.2 (5.1 ± 0.03) | 6.0 (5.2 ± 1.5) |
| 17j | A | CH2(2,4-F-Ph) | 306.2 | 16–32 | >32 | >50 (<4.3) | >40 (<4.4) |
| 17k | A | CH2(2-pyridyl) | 271.2 | >32 | >32 | 14 (4.8 ± 0.05) | 9.7 (5.0 ± 0.18) |
| 17l | A | CHMe(4-F-Ph) | 302.3 | 0.5 | 4–6.3 | 2.1 (5.7 ± 0.03) | 94% I @ 40 μM |
| 17m | A | CH2CH2(4-OCF3-Ph) | 368.3 | 8* (70% I) | >32 | 1.6 (5.8 ± 0.04) | 44% I @ 40 μM |
| 17n | A | CH2CH2(4-CH3-Ph) | 298.3 | 4* (78% I) | >32 | 3.2 (5.5 ± 0.04) | >40 (<4.4) |
| 17o | A | CH2CH2OAc | 266.2 | >32 | >32 | 65 (4.2 ± 0.03) | 6.5 (5.2 ± 0.22) |
| 17p | A | CH2CH2OH | 224.2 | >32 | >32 | >50 (<4.3) | 87% I @ 40 μM |
| 17q | A | CH2CH2-morpholine | 293.3 | >32 | >32 | ∼50 (∼4.3) | 2.9 (5.5 ± 0.14) |
| 17r | A | CH2CO-morpholine | 307.3 | >32 | >32 | >50 (<4.3) | 4.1 (5.4 ± 6.8) |
| 17s | A | CH2CONH2 | 237.2 | >32 | >32 | >50 (<4.3) | >40 (<4.4) |
| 17t | A | cyclohexyl | 262.3 | >32 | >32 | 5.2 (5.3 ± 0.03) | 1.4 (5.9 ± 0.13) |
| 18i | B | CH2(2-OCF3-Ph) | 354.2 | >32 | >32 | 12 (4.9 ± 0.05) | >40 (<4.4) |
| 18m | B | CH2CH2(4-OCF3-Ph) | 368.3 | >32 | >32 | 5.2 (5.3 ± 0.03) | N.D |
H37Rv, M. tuberculosis-normoxia primary screen at 32 μg/mL n = 3, M. tuberculosis-hypoxia primary screen at 32 μg/mL n = 1–3, MIC-normoxia/hypoxia of active compounds n = 3–6. Isoniazid control M. tuberculosis-normoxia MIC = 0.04 μg/mL, M. tuberculosis-hypoxia MIC >5 μg/mL.
WB.
Pentamidine control IC50 = 0.002 μM (pIC50 = 8.7), diminazine aceturate IC50 = 0.062 μM (pIC50 = 7.2), puromycin IC50 = 0.05 μM (pIC50 = 7.3), n = 2.
The percentage inhibition (% I) at the highest concentration tested is reported for compounds that were not sufficiently active to determine an IC50. *Increasing concentrations of compound did not inhibit growth further.
Nitroimidazopyrazinones 17b–i with a monosubstituted benzyl group at R1 displayed potent activity against M. tuberculosis (MICnormoxia = 0.06–1 μg/mL) at levels up to 2-fold better than pretomanid 6 (MICnormoxia = 0.25–0.5 μg/mL). There was a slight preference for substitution at the 3′ over the 2′ and 4′ positions of the phenyl group (cf. 17f–g with 17i and 17c–d). However, there was otherwise little electronic effect; both electron donating (17g, R1 = CH2(3-CH3-Ph)) and withdrawing substituents [17h, R1 = CH2(3-CF3-Ph)] had equal activity (MICnormoxia = 0.06 μg/mL). Poor solubility of 17j in the compound stock solutions was suspected to have contributed to the unexpected weak activity of this disubstituted-fluorine derivative against M. tuberculosis and the other organisms. Replacement of the phenyl group (17b) with a pyridyl moiety (17k) was unfavorable and resulted in a loss of activity against M. tuberculosis (17k MICnormoxia >32 μg/mL). A methyl substituent at the benzylic position (17l) was well tolerated (MICnormoxia = 0.5 μg/mL), but extension of the aromatic linkage with an ethyl bridge in the phenethyl derivatives (17m–n) resulted in a 16–32-fold loss of activity against M. tuberculosis, with bacteriostatic activity of MICnormoxia = 4–8 μg/mL compared to respective benzyl derivatives 17c–d with MICnormoxia = 0.125–0.5 μg/mL. None of the derivatives with polar groups at R1 (17o–s) had activity against M. tuberculosis (MICnormoxia >32 μg/mL). Lastly, compound 17t with an aliphatic cyclohexyl group at R1 was also inactive (MICnormoxia >32 μg/mL), indicating a preference for an aromatic group rather than a nonspecific lipophilic group.
In general, compounds with activity against M. tuberculosis grown in normoxic conditions were also active against nonreplicating M. tuberculosis under hypoxic conditions, although generally 2–16-fold less active. By comparison, isoniazid, a compound that inhibits cell wall synthesis in aerobically respiring M. tuberculosis (MIC = 0.04 μg/mL) did not inhibit growth in the hypoxic conditions (MIC >5 μg/mL). Metronidazole 1 was inactive (MIC >32 μg/mL) in this hypoxic-recovery assay, indicating that the medium was not sufficiently anaerobic to show activity of 1 against M. tuberculosis.
The SARs observed for G. lamblia were similar to M. tuberculosis, and several compounds (17c–d, 17f, and 17l–n) were found to have improved activity (up to 4-fold) relative to metronidazole 1 (IC50 = 1.6 to 3.5 μM cf. 1 IC50 = 7.2 μM). Interestingly, an expanded range of the derivatives with lipophilic R1 groups had potent activity against G. lamblia. For example, 17m (R1 = CH2CH2(4-OCF3-Ph)) and 17n (R1 = CH2CH2(4-Me-Ph)) had similar activity to the benzyl-substituted analogues 17c–d against G. lamblia (IC50 = 1.6–3.2 μM vs 3.4–3.5 μM, respectively). In addition, the cyclohexyl derivative 17t was potent against G. lamblia at equivalent levels to the metronidazole 1 control (17tG. lamblia IC50 = 5.2 μM).
There were also similar trends in the SAR observed against T. b. brucei, although with a number of differences compared to M. tuberculosis and G. lamblia. For example, the activity of the benzyl series 17b–i against T. b. brucei was sensitive to the particular nature of the benzyl substitution. For example, 17c [R1 = CH2(4-OCF3-Ph)] and 17e [R1 = CH2(4-F-Ph)] were active against T. b. brucei (IC50 = 1.4 and 2.9 μM, respectively), but compounds 17b (R1 = CH2Ph) and 17f [R1 = CH2(3-OCF3-Ph)] had reduced activity at the highest concentrations tested (40 and 16 μM, respectively). Another difference observed for T. b. brucei was an increased tolerance for polar substituents at R1. For example, compounds with polar ethyl acetate (17o) or morpholino groups (17q–r) displayed moderate activity against T. b. brucei (IC50 = 2.9–6.5 μM), whereas against M. tuberculosis and G. lamblia, these examples were essentially devoid of activity. The unsubstituted nitroimidazopyrazinone compound 17a with H at R1 displayed some activity against T. b. brucei (97% inhibition at 40 μM) but was insufficiently active at 20 μM to determine an IC50 value. The cyclohexyl derivative 17t also had good activity against T. b. brucei (IC50 = 1.4 μM), similarly to G. lamblia. These examples demonstrate that it is possible to identify compounds with selective antiparasitic activity, although all compounds with M. tuberculosis activity tended to display some activity toward parasites. The selectivity observed for the particular organisms may be due to differences in the compound activation or uptake of the compounds and is an area of ongoing investigation.
Comparison of Bicyclic Antimicrobial Activity to Monocyclic 4- and 4(5)-Nitroimidazole Carboxamide Analogues
Given the activity of the bicyclic derivatives against M. tuberculosis, we screened a number of previously reported28 4-nitroimidazole carboxamides 28b–k and 4(5)-nitroimidazole carboxamides 16a, 16c, 16e, and 16f, along with intermediates 16b and 16d prepared for this study, against M. tuberculosis under normoxic and hypoxic growth conditions. This would allow us to compare the SAR between the monocyclic and bicyclic series. Note that the 2-nitro group of the imidazopyrazinones occupies the equivalent position of the nitro group in the 4-nitroimidazole series (Figure 3). Interestingly, none of the 4- or 4(5)-monocyclic nitroimidazoles tested were active against M. tuberculosis grown aerobically at 32 μg/mL (Supporting Information, Table S6). Under hypoxic growth conditions, weak activity was observed for 28j and 16c (60–75% inhibition at 32 μg/mL), but this also correlated with increased cytotoxicity against the mammalian kidney cell line for these compounds (Supporting Information, Table S6). Therefore, the rigid, bicyclic nature of 17b–f appears necessary for antitubercular activity, especially against actively dividing M. tuberculosis cells, but is not essential for activity against other parasites. A comparison of the activity of the 4- and 4(5)-nitroimidazole carboxamide series with that of the bicyclic derivatives is summarized in Figure 3.
Figure 3.

4-Nitroimidazoles 28b–k and 4(5)-nitroimidazole carboxamide 16b–f, compared to nitroimidazopyrazinones 17b–f.
Evaluation of Des-nitro Derivatives for Biological Activity
Two des-nitro derivatives 20b–c were prepared as negative control compounds to confirm that the proposed mode of action relies on reduction of the nitro group. The des-nitro imidazopyrazinone derivatives 20b–c were tested for activity and indeed were found to be inactive against M. tuberculosis, G. lamblia, T. b. brucei, and E. histolytica (complete profiling results are detailed in the Supporting Information, Table S7). This result is consistent with the inactivity of a des-nitro pretomanid analogue 9 against M. tuberculosis under both aerobic and anaerobic growth (MIC >160 and >500 μM, respectively)41 and supports a critical role for the nitro group in the mechanism of action of nitroimidazopyrazinones.
SAR of Bicyclic Nitroimidazoles with Variation at R2 = Ph or CH3, R3 = H or CH3
Compounds 24a–e, 25a–m, and 26a–i with R2 = Ph or CH3 and R3 = H or CH3 were evaluated for antiparasitic and antimicrobial in the same manner as described for 17a–t, 18i, and 18m (Table 3). Both the R2 and R3 sites were found to influence activity against different pathogens to varying degrees. Interestingly, the modifications of R2 and R3 on the nitroimidazopyrazinone series were found to be particularly beneficial for activity against T. b. brucei. A number of nitroimidazopyrazinones were active against T. b. brucei between 0.2 and 0.9 μM (24a–b, 25b, 25f, 25h, 26b, 26d, 26f, and 26h). While some of the pyrazine matched pairs generally lost activity, several compounds (24c, 25m, 26e, 26g, and 26i) maintained activity in the 1.2–2.2 μM range. Furthermore, when R1 = CH2CH2OAc, compound 24b (R2 = Ph) and 25b (R2 = CH3) showed 7.6–10-fold improvement compared to 17o (R2 = H). This benefit was also clear for the derivatives with benzyl substitutions at R1. For example, compound 17f (R1 = CH2(3-OCF3-Ph), R2 = H, R3 = H) showed incomplete inhibition at 16 μM, while the matched pairs 25j (R2 = CH3, R3 = H) and 26f (R2 = CH3, R3 = CH3) inhibited T. b. brucei at 1.1 and 0.40 μM, respectively. This last example also demonstrates a slight preference for methyl group substitutions at both R2 and R3 on the pyrazinone ring, as this resulted in activity that was consistently about twofold improved.
Table 3. SAR Exploring the Differences between the R2 and R3 Groups for the Imidazopyrazinone (Core A) vs the Imidazopyrazine (Core B).
| compound |
molecular weight (g/mol) | antibacterial MIC (μg/mL) |
antiparasitic IC50 (μM) (pIC50 ± SE)e |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|
| no. | core | R1 | R2 | R3 | M. tuberculosisa normoxia | M. tuberculosisa hypoxia | G. lambliab | E. histolyticac | T. b bruceid | |
| metronidazole 1 | 171.2 | >32 | >32 | 7.2 (5.1 ± 0.02) | 4.3 (5.4 ± 0.02) | >40 (<4.4) | ||||
| pretomanid 6 | 359.3 | 0.25–0.5 | 1 | 3.0 (5.5 ± 0.02) | 9.3 (5.0 ± 0.03) | 19 (4.7 ± 2.4) | ||||
| 24a | A | H | Ph | H | 256.2 | >32 | >32 | 10 (5.0 ± 0.03) | >25 (<4.6) | 0.22 (6.7 ± 0.071) |
| 24b | A | CH2CH2OAc | Ph | H | 342.3 | >32 | >32 | 11 (5.0 ± 0.05) | 34 (4.5 ± 0.1) | 0.86 (6.1 ± 0.085) |
| 24c | B | CH2CH2OAc | Ph | H | 342.3 | >32 | >32 | 1.9 (5.7 ± 0.05) | >50 (<4.3) | 1.5 (5.8 ± 0.085) |
| 24d | A | CH2CH2OH | Ph | H | 300.3 | >32 | >32 | 38 (4.4 ± 0.03) | >50 (<4.3) | N.D. |
| 24e | B | CH2CH2OH | Ph | H | 300.3 | >32 | >32 | 5.2 (5.3 ± 0.05) | >50 (<4.3) | 1.1 (6.0 ± 0.12) |
| 25a | A | H | CH3 | H | 194.2 | >32 | >32 | ∼50 (∼4.3) | >50 (<4.3) | 3.6 (5.4 ± 0.20) |
| 25b | A | CH2CH2OAc | CH3 | H | 280.2 | >32 | >32 | ∼50 (∼4.3) | 41% I @ 50 μM | 0.65 (6.2 ± 0.021) |
| 25c | B | CH2CH2OAc | CH3 | H | 280.2 | >32 | >32 | 21 (4.7 ± 0.06) | 42% I @ 50 μM | >40 (<4.4) |
| 25d | A | CH2CH2OH | CH3 | H | 238.2 | >32 | >32 | >50 (<4.3) | >50 (<4.3) | 4.6 (5.3 ± 0.092) |
| 25e | B | CH2CH2OH | CH3 | H | 238.2 | >32 | >32 | 80% I @ 50 μM | >50 (<4.3) | 56% I @ 40 μM |
| 25f | A | CH2(4-OCF3-Ph) | CH3 | H | 368.3 | >32 | >32 | 5.8 (5.2 ± 0.04) | >50 (<4.3) | 0.41 (5.4 ± 0.057) |
| 25g | B | CH2(4-OCF3-Ph) | CH3 | H | 368.3 | >32 | >32 | 7.4 (5.1 ± 0.03) | 9.5 (5 ± 0.07) | >40 (<4.4) |
| 25h | A | CH2(4-CH3-Ph) | CH3 | H | 298.3 | >32 | >32 | 4.4 (5.4 ± 0.03) | 10 (5.0 ± 1.0) | 0.56 (6.3 ± 0.028) |
| 25i | B | CH2(4-CH3-Ph) | CH3 | H | 298.3 | N.D. | N.D. | 5.7 (5.2 ± 0.03) | 30 (4.5 ± 0.1) | N.D. |
| 25j | A | CH2(3-OCF3-Ph) | CH3 | H | 368.3 | >32 | >32 | 2.8 (5.6 ± 0.01) | 16 (4.8 ± 0.1) | 1.1 (5.9 ± 0.021) |
| 25l | A | CH2(2-OCF3-Ph) | CH3 | H | 368.3 | >32 | >32 | 3.3 (5.5 ± 0.01) | 9.1 (5 ± 0.08) | 2.1 (5.7 ± 0.29) |
| 25m | B | CH2(2-OCF3-Ph) | CH3 | H | 368.3 | >32 | >32 | 5.7 (5.2 ± 0.02) | >50 (<4.3) | 1.2 (5.9 ± 0.13) |
| 26a | A | H | CH3 | CH3 | 208.2 | >32 | >32 | ∼50 (∼4.3) | >50 (<4.3) | 2.6 (5.6 ± 0.17) |
| 26b | A | CH2(4-OCF3-Ph) | CH3 | CH3 | 382.3 | 1 | 4 | 4.2 (5.4 ± 0.04) | 10 (5 ± 0.1) | 0.24 (5.7 ± 0.0071) |
| 26c | B | CH2(4-OCF3-Ph) | CH3 | CH3 | 382.3 | >32 | >32 | 8.6 (5.1 ± 0.04) | 17 (4.8 ± 0.09) | 10 (5.0 ± 0.20) |
| 26d | A | CH2(4-CH3-Ph) | CH3 | CH3 | 312.3 | 2 | 4 | 5.2 (5.3 ± 0.02) | 33 (4.5 ± 0.04) | 0.25 (6.6 ± 0.0071) |
| 26e | B | CH2(4-CH3-Ph) | CH3 | CH3 | 312.3 | >32 | >32 | 5.8 (5.2 ± 0.02) | >50 (<4.3) | 2.2 (5.6 ± 0.31) |
| 26f | A | CH2(3-OCF3-Ph) | CH3 | CH3 | 382.3 | 1 | 16–32 | 1.7 (5.8 ± 0.01) | 15 (4.8 ± 0.03) | 0.40 (6.4 ± 0.0071) |
| 26g | B | CH2(3-OCF3-Ph) | CH3 | CH3 | 382.3 | >32 | >32 | 3.9 (5.4 ± 0.01) | 27 (4.6 ± 0.02) | 1.4 (5.9 ± 0.24) |
| 26h | A | CH2(2-OCF3-Ph) | CH3 | CH3 | 382.3 | >32 | >32 | 5.3 (5.3 ± 0.03) | 18 (4.7 ± 0.02) | 0.78 (6.1 ± 0.090) |
| 26i | B | CH2(2-OCF3-Ph) | CH3 | CH3 | 382.3 | >32 | >32 | >50 (<4.3) | >50 (<4.3) | 1.9 (5.7 ± 0.27) |
H37Rv, M. tuberculosis-normoxia primary screen at 32 μg/mL n = 3, M. tuberculosis-hypoxia primary screen at 32 μg/mL n = 1–3, MIC-normoxia/hypoxia of active compounds n = 3–6. Isoniazid control M. tuberculosis-normoxia MIC = 0.04 μg/mL, M. tuberculosis-hypoxia MIC >5 μg/mL.
WB.
HM1:IMSS.
Pentamidine control IC50 = 0.002 μM (pIC50 = 8.7), diminazine aceturate IC50 = 0.062 μM (pIC50 = 7.2), puromycin IC50 = 0.05 μM (pIC50 = 7.3), n = 2.
The percentage inhibition (% I) at the highest concentration tested is reported for compounds that were not sufficiently active to determine an IC50.
The methyl and phenyl groups explored at R2 and R3 did not result in any overall improvement or loss of activity against G. lamblia, as the activity was generally in the same range. As observed for nitroimidazopyrazine 18m, the expanded series of nitroimidazopyrazine derivatives also had activity against G. lamblia. When R2 = Ph, R3 = H, the pyrazine derivatives 24c and 24e performed better than the pyrazinone analogues 24b and 24d against G. lamblia. However, similar activity was observed when R2 = CH3, R3 = H (e.g., matched pair 25h and 25i) and R2 = CH3, R3 = CH3 (e.g., matched pair 26d and 26e), although the pyrazine was slightly worse by twofold for a few derivatives.
Activity against E. histolytica was also improved for the nitroimidazopyrazine series when R2 = CH3 and R3 = CH3. Both nitroimidazopyrazinones (e.g., 26b and 26f) and nitroimidazopyrazines (e.g., 26c and 26g) were active, with a slight preference for the nitroimidazopyrazinone core. Despite this improvement, none of the derivatives had activity comparable to metronidazole 1, with the active derivatives having only moderate to weak activity (10–34 μM). This suggests distinct requirements for activity in E. histolytica compared to T. b. brucei and G. lamblia. Nonetheless, it is possible that further modification of the scaffold may lead to identification of an optimal “sweet spot” for activity against E. histolytica.
Modifications explored at R2 and R3 were mostly detrimental for activity against M. tuberculosis. Activity against M. tuberculosis activity was abolished when R2 = CH3 or Ph and R3 = H. Consistent with the previous SAR study, all of the nitroimidazopyrazines were found to be inactive (MIC > 32 μg/mL). Nonetheless, activity was retained for three nitroimidazopyrazinone examples (26b, 26d, and 26f) when R2 = R3 = CH3 and R1 was a benzyl group. Compounds 26b, 26d, and 26f also displayed activity against M. tuberculosis under hypoxic growth conditions and against G. lamblia, E. histolytica, and T. b. brucei. These results indicate that it is possible to identify compounds with broad spectrum activity against both M. tuberculosis and parasites.
Continuing the trend previously observed for compounds 17a–t, 18i, and 18m, none of the additional nitroimidazopyrazin-ones/-es synthesized had appreciable activity against ESKAPE bacteria or fungal pathogens (MIC ≥ 32 μg/mL) (Supporting Information, Table S8). Both the nitroimidazopyrazinone and nitroimidazopyrazine series were also not cytotoxic against mammalian liver and kidney cell lines (CC50 > 100 μM, 24d > 75 μM due to compound limitations), except for 26f that displayed moderate cytotoxicity in both cell lines (CC50 = 26–80 μM) (Supporting Information, Table S8).
To summarize, the SAR of the R2, R3 sites was determined for both the nitroimidazopyrazinone and nitroimidazopyrazine series. Several compounds with potent activity against M. tuberculosis, G. lamblia, and T. b. brucei were identified. Further work confirming the target/mode of action in M. tuberculosis, G. lamblia, and T. b. brucei could aid in explaining the differences in activity. Additional studies to measure the reduction potential may provide insight into the differences in the activity between the nitroimidazopyrazinones and nitroimidazopyrazines.
Microsomal Stability, Plasma Stability, and PPB
A set of 10 compounds was selected for initial assessment of drug-like properties (microsomal stability, plasma stability, and PPB) based on their potency and structural diversity (Table 4). It was also envisioned that these studies would provide clarity as to whether there was a clear benefit to either the nitroimidazopyrazinone or nitroimidazopyrazine series from the perspective of ADMET properties. Nitroimidazopyrazinones were stable in human liver microsomes (HLM) after 2 h of incubation, regardless of the structural differences in R1, R2, and R3. However, 18i, 25g, and 26c from the nitroimidazopyrazine series only showed moderate stability in HLM, independent of differences at R1, R2, and R3, with 44–69% of the compound remaining after 2 h (Table 4).
Table 4. In Vitro Physicochemical Properties for Representative Compoundsa.
| microsomal
stability (% remaining at 2 h) |
plasma stability (% remaining at 2 h) |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| compound | core | R1 | R2 | R3 | human | mouse (CD-1) | human | mouse (CD-1) | PPB (%) |
| pretomanid 6 | 97 ± 5.0 | 92 ± 2.7 | 96 ± 6.9 | 96 ± 1.9 | 97 ± 1.1 | ||||
| 17c | A | CH2(4-OCF3-Ph) | H | H | >99 | >99 | >99 | >99 | 98 ± 0.4 |
| 17d | A | CH2(4-CH3-Ph) | H | H | 97 ± 1.1 | 96 ± 6.0 | >99 | >99 | N.D |
| 17e | A | CH2(4-F-Ph) | H | H | 99 | >99 | 98 ± 1.4 | >99 | 82 ± 0.3 |
| 17f | A | CH2(3-OCF3-Ph) | H | H | >99 | >99 | >99 | 97 ± 2.2 | 99 ± 0.2 |
| 17i | A | CH2(2-OCF3-Ph) | H | H | >99 | >99 | N.D | N.D | N.D |
| 18i | B | CH2(2-OCF3-Ph) | H | H | 51 ± 18 | <0.5 | N.D | N.D | N.D |
| 25f | A | CH2(4-OCF3-Ph) | CH3 | H | >99 | >99 | 92 ± 4.1 | 99 ± 4.8 | 98 ± 0.4 |
| 25g | B | CH2(4-OCF3-Ph) | CH3 | H | 69 ± 4.3 | 7.9 ± 4.0 | 94 ± 4.7 | >99 | >99 |
| 26b | A | CH2(4-OCF3-Ph) | CH3 | CH3 | 92 ± 1.1 | 3.6 ± 0.1 | 97 ± 1.4 | >99 | 99 ± 0.1 |
| 26c | B | CH2(4-OCF3-Ph) | CH3 | CH3 | 44 ± 0.98 | <0.5 | >99 | >99 | N.D |
Values are presented as mean of three replicates ±SD. N.D represents not determined. Microsome stability verapamil control = 2% (MLM), 9% (HLM) remaining at 30 min; plasma stability eucatropine control = 28% (mouse), 21% (human) remaining at 2 h; and PPB sulfamethoxazole control = 68% bound.
To further understand how the metabolic stability could impact in vivo efficacy studies, metabolic stability was evaluated in CD-1 mouse liver microsomes (MLM) because the CD-1 mouse strain is intended to be used for future in vivo efficacy studies. As for HLM, the majority of the nitroimidazopyrazinones 17c–f, 17i, and 25f displayed excellent stabilities toward MLM. However, one of the imidazopyrazinone analogues, 26b (R2 = R3 = CH3) showed significant degradation after 2 h, with only 3.6% of intact compound remaining (compared to 92% in HLM) (Table 4). Comparison of 25f with 26b, which differ by the presence of a hydrogen or methyl group at R3, respectively, indicated that the methyl group at R3 was responsible for the high intrinsic clearance of 26b in MLM. Nitroimidazopyrazines, 18i, 25g, and 26c were also not stable in MLM (<0.5–8% remaining), suggesting a metabolic liability of this series.
Compound stability in both human and mouse (CD-1) plasma was assessed for compounds 17c–f, 25f–g, and 26b–c (Table 4). All of the examples had >90% of compound remaining after 2 h at 37 °C, similar to pretomanid 6. Therefore, both the nitroimidazopyrazinone and nitroimidazopyrazine series were stable in human and mouse plasma, despite the instability of nitroimidazopyrazines observed in liver microsomes.
PPB affects compound bioavailability and tissue distribution in vivo. Therefore, to understand PPB for the nitroimidazopyrazinone and nitroimidazopyrazine series, selected potent compounds (17c, 17e–f, 25f–g, and 26b) were evaluated for PPB using the ultrafiltration method (Table 4). Most of the tested compounds, except 17e (82% bound), had high PPB at >95%, which might limit the concentration of free compounds at the site of infection. However, as many approved and clinical drugs, including 6, tend to have high PPB, this parameter is not recommended to be optimized in early drug design.42
Caco-2 Intestinal Epithelium Permeability
An in vitro Caco-2 monolayer bidirectional assay was used to evaluate intestinal epithelium permeability and to predict the oral absorption of the nitroimidazopyrazinone series.43 Three derivatives (17c, 17f, and 26b) were selected based on potency against M. tuberculosis, G. lamblia, and T. b. brucei and to permit direct comparison of R2 = R3 = H (17c) versus R2 = R3 = CH3 (26b). Pleasingly, compounds 17c, 17f, and 26b were highly permeable (Table 5). The apparent permeability coefficient (Papp) apical to basal (A to B) had values of >20 × 10–6 cm/s, similar to pretomanid 6 and propranolol, with the latter used as a positive control for high permeability. No metabolism of the compounds by Caco-2 cells was observed. This was evident from the high recovery of tested nitroimidazopyrazinones obtained in both directions, similar to the control propranolol (A to B measured). Nitroimidazopyrazinones 17c, 17f, and 26b and 6 also showed a low efflux ratio of <2 (cf. digoxin efflux ratio >360 as it is a substrate for P-gp). Therefore, the tested bicyclic nitroimidazoles do not appear to be substrates of efflux transporters. These results suggest that 17c, 17f, and 26b have desirable therapeutic properties and may display good oral absorption properties in vivo.
Table 5. Caco-2 Permeability of Selected Analoguesa.
|
Papp (10–6 cm/s) |
mean recovery (%) |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| compound | core | R1 | R2 | R3 | A to B | B to A | A to B | A to B | efflux ratio |
| pretomanid 6* | 29 | 22 | 81 | 101 | 0.74 | ||||
| fenoterol | 0.27 | N.D | 92 | N.D | N.D | ||||
| propranolol | 21 | N.D | 70 | N.D | N.D | ||||
| digoxin | <0.020 | 8.4 | <72 | 88 | >360 | ||||
| 17c | A | CH2(4-OCF3-Ph) | H | H | 25 | 22 | 83 | 97 | 0.88 |
| 17f | A | CH2(3-OCF3-Ph) | H | H | 24 | 24 | 88 | 99 | 1.0 |
| 26b | A | CH2(4-OCF3-Ph) | CH3 | CH3 | 21 | 20 | 83 | 97 | 0.95 |
N.D represents not determined. * Comparable results with the literature.22
Kinetic Solubility
Compound solubility impacts in vivo absorption, efficacy, and dosing and is thus an important parameter to optimize drug development. For example, the recently approved TB drug, delamanid has limited solubility which requires twice daily dosing,44 and new bicyclic analogues with improved solubility would be of great interest. According to the generic criteria for hits and leads in infectious diseases identified by Japanese Global Health Innovative Technology (GHIT) Fund and its key partners, a lead should have acceptable physicochemical properties, with solubility at least >10 μM in phosphate-buffered saline (PBS) pH 7.4.45
The kinetic solubility of 48 compounds from the nitroimidazopyrazin-one/-e libraries in water and PBS buffer (pH 7.4) was determined using LC–UV (Figure 4, for full data, see Supporting Information, Table S9). The solubility of the derivatives varied over a wide range reflective of the different properties that the R1 side chain can impart. As anticipated, polar groups at R1 such as morpholine (17r: CH2CO-morpholine and 17q: CH2CH2-morpholine), amide (17s: CH2CONH2), and alcohol groups (17p: CH2CH2OH) had >30-fold (Δlog S ≈ 1.6 units) better solubility than benzyl derivatives (17b–j). The nitroimidazopyrazinones had equivalent or improved solubility relative to the nitroimidazopyrazine series, and this is also reflected in their lower log P values. Encouragingly, when the solubility versus activity was compared, a number of compounds with antitrypanosomal activity (T. b. brucei IC50 ≤ 10 μM) had good solubility at >100 μM (equivalent to log S > −4), including two potential hits 24b and 25b with IC50 < 1 μM. The most potent compound 24a achieved moderate aqueous solubility (27 μM in water; 17 μM in PBS), whereas 26b and 26d with comparable activity were poorly soluble (<10 μM, or log S < −5 in water and PBS) (Figure 4). These results demonstrate the potential to achieve desirable solubility and activity profile by modifying the R1 group of the bicyclic nitroimidazoles.
Figure 4.

Comparison of activity vs aqueous solubility data. Several compounds, for example, 25b active against T. b. brucei (in blue) had good solubility (>100 μM or log S > −4). Most of the potent compounds, for example, 17h, against the other organisms displayed poor solubility (log S ≤ −5). Dashed lines represent the minimum and maximum range of solubility determined experimentally.
However, compounds with potent activity against M. tuberculosis, G. lamblia, and E. histolytica generally had poor solubility. Most of the potent compounds against M. tuberculosis showed poor solubility with <10 μM in both water and PBS, which might require complicated formulations for in vivo efficacy. This indicates the importance of lipophilicity at the R1 side chain, which is potentially required for penetration of these molecules through the mycobacterial cell walls. Although changing R1 to CH2-pyridinyl decreased activity against M. tuberculosis, this derivative (17k) had significantly improved water solubility (176 μM in water; 184 μM in PBS) and decreased lipophilicity (Δlog P −0.94 units). Exploring other heterocyclic analogues may provide the desired balance of potency and solubility, which has been a successful strategy in the literature to identify analogues of pretomanid 6 with improved solubility.46,47
Conclusions
In this study, pretomanid 6 was shown to have potent activity against enteric pathogens including G. lamblia, E. histolytica, and C. difficile. These results expand the current understanding of the spectrum of action of 6 from Mycobacterium and Leishmania to include anaerobic protozoan parasites and an anaerobic Gram-positive bacterium. The in vitro activity of 6 against G. lamblia and E. histolytica at equivalent levels to metronidazole 1 suggests that this class of compounds and other subclasses of bicyclic nitroimidazoles could be a beneficial avenue to explore for the development of new nitroimidazole-based therapeutics against these pathogens.
The activity of 6 against enteric pathogens inspired the synthesis and biological evaluation of two new bicyclic scaffolds; nitroimidazopyrazin-one/-es derived from the monocyclic 4(5)-nitroimidazole carboxamide framework previously reported. A modular synthetic approach enabled the exploration of bicyclic imidazopyrazinone derivatives with different substitutions at R1, R2, and R3, along with the isomeric O-alkylated nitroimidazopyrazine core scaffold. Through these studies, compounds with potent activity against M. tuberculosis, G. lamblia, and T. b. brucei were identified. Several compounds also displayed moderate activity against E. histolytica. To our knowledge, this is the first example of a nitroimidazopyrazinone heterocyclic core with potent antitubercular activity against M. tuberculosis under both normoxic and hypoxic growth conditions as well as promising antiparasitic activity against G. lamblia and T. b. brucei. In addition, the bicyclic core was essential for antitubercular activity.
This study highlights the potential to prepare new subclasses of bicyclic nitroimidazoles with varied ring systems to gain different selectivity profiles toward M. tuberculosis and a range of parasitic organisms. All of the compounds with M. tuberculosis activity also displayed activity toward at least some of the parasites tested. However, we showed that the activity profile of the series could be tuned toward parasites away from M. tuberculosis, which may be relevant for treating parasitic diseases without inducing resistance in M. tuberculosis in co-exposed patients. For example, nitroimidazopyrazinones and nitroimidazopyrazines, particularly 24c and 24e (R2 = Ph) and 25h, 25j, and 25l–m (R2 = CH3), were active against G. lamblia (IC50 = 1.9–5.7 μM) and T. b. brucei (IC50 = 0.56–2.1 μM) but not M. tuberculosis (MIC >32 μg/mL).
In general, these new subclasses displayed desirable therapeutic properties. Low cytotoxicity against mammalian cell lines was observed generally (CC50 > 100 μM) for both series. In addition, many of the potent derivatives were stable in human and mouse liver microsomes, although the nitroimidazopyrazine series was metabolized, particularly in MLM, and when R2 and R3 were methyl substituents. Selected derivatives also gave similar results to 6 in plasma stability, PPB, and Caco-2 intestinal permeability assays. Like other early stage antitubercular leads including bicyclic nitroimidazoles, solubility was identified as an important parameter for future optimization. While a number of active compounds against T. b. brucei had moderate solubility, the most potent antitubercular compounds had poor aqueous kinetic solubility (<10 μM). Optimization of the R1, R2, and R3 sites, particularly the R1 site, to arrive at more potent compounds, while maintaining desirable drug-like properties and improving the solubility will be explored in future studies.
This study lays the foundation for future work focused on determining and optimizing the efficacy of selected derivatives in in vivo models of M. tuberculosis, G. lamblia, and T. b. brucei and understanding their mode of action in these pathogens. These studies are expected to provide further insight into nitroimidazole activation and guide the development of bicyclic nitroimidazoles with therapeutic potential against both M. tuberculosis and a wide range of parasitic organisms.
Experimental Section
General Information
Reagents and anhydrous solvents were used as received. Reactions requiring anhydrous conditions were performed under an inert atmosphere of nitrogen. Reactions were monitored by thin-layer chromatography (TLC) or LCMS. Analytical TLC was performed on Merck TLC aluminum sheets pre-coated with Silica Gel 60 F-254 and compounds were visualized using a UV254 lamp and potassium permanganate stain. Melting points were determined using a Gallenkamp melting point apparatus and are corrected to a standard curve of the measured and literature melting points of vanillin, acetyl salicylic acid, 3-phenoxybenzoic acid, and caffeine standards. Analytical LCMS was performed on a Shimadzu LCMS using 0.05% formic acid in water as solvent A and 0.05% formic acid in acetonitrile as solvent B. Standard conditions unless otherwise specified: column Zorbax Eclipse XDB-Phenyl, 3.0 × 100 mm, 3.5 μ. Alternative column: Waters Atlantis T3, 3.0 × 100 mm, 3 μ. For detection, PDA UV, evaporative light scattering detector (ELSD), and electrospray MS were used. Compounds were purified by MPLC (Biotage Isolera or Grace Reveleris X2 chromatography systems) or by HPLC (Agilent Preparative HPLC 1260 Infinity Series). Commercially available cartridges were used for MPLC chromatography (Biotage SNAP cartridge HP-Silica 10, 25 or 50 g, Reveleris 4 g or 12 g silica (40 μm) cartridge or Reveleris C18 reversed-phase 12 g cartridge). Column for HPLC: Agilent XDB Phenyl 5 μm, 30 × 100 mm. All products tested for biological activity were obtained in >95% purity as determined by HPLC using UV at 254 nm, ESIMS, and ELSD detection. NMR data were collected and calibrated in DMSO-d6 or CDCl3 with 0.05% TMS at 298 K on a Varian Unity 400 MHz or Bruker AVANCE-600 MHz spectrometer. Where appropriate, 1H-coupling constants were examined using resolution enhancement with MestReNova software. High-resolution MS (HRMS) was performed on a Bruker micrOTOF mass spectrometer using (+)-ESI calibrated to HCOONa. For compounds purified by reverse phase chromatography, the exact concentration of the compounds for assay was determined by the quantitative NMR integration “PULCON” experiment.48 These settings were used for all PULCON experiments: relaxation delay of 30 s, 8 scans, 2 dummy scans, 90° pulse and temperature at 298 K. The mass calculated from PULCON experiments was used to calculate the reaction percentage yields.
General Procedure A: Alkylation of Imidazole and Imidazopyrazinones
To a stirred solution of imidazole/imidazopyrazinone (1 equiv) in anh. DMF (15–30 vol) was added K2CO3 or Cs2CO3 (3 equiv) followed by alkyl or benzyl halide (1.2–1.5 equiv). The reaction was stirred at rt or heated in a microwave reactor at 80–180 °C for 15 min and monitored by LCMS. If necessary, an additional portion of alkylating agent (0.5–1.5 equiv) was added and the reaction was heated at μW 120–180 °C for a further 15 min to consume the imidazole starting material. The work-up procedure A is as follows: the reaction was poured into H2O and extracted with EtOAc. The combined organic layers were washed with brine, dried over MgSO4, and filtered. Volatiles were removed in vacuo to give the crude product. The work-up procedure B is as follows: the reaction was poured into water and the precipitate was collected by filtration, washed with water, and dried in vacuo.
General Procedure B: Alkylation of Imidazopyrazinones
To a stirred suspension of imidazopyrazinone (1 equiv) in toluene (20 vol) was added Ag2CO3 (1.2 equiv) followed by benzyl halide (2 equiv). The reaction was heated at 80 °C overnight and monitored by LCMS. The general aqueous work up is as follows: the reaction was poured into H2O and extracted with EtOAc. The combined organic layers were washed with brine, dried over MgSO4, and filtered. Volatiles were removed in vacuo to give the crude product.
General Procedure C: Ring Closure to Synthesize Imidazopyrazinones
To a stirred solution of 4-nitroimidazole (1 equiv) in 1,4-dioxane (10 vol) was added 2 M aq HCl (10 vol). The reaction was heated at 120 °C for 30 min in the microwave. The work-up procedure A is as follows: the crystalline solid was collected by vacuum filtration. The precipitate was washed with water and dried in vacuo to give a solid. The work-up procedure B is as follows: after heating the reaction, the volatiles were evaporated in vacuo to yield a crude material that was purified by recrystallization or chromatography.
General Procedure D: Deprotection of the Acetate-Protecting Group
Anh. K2CO3 (1.5 equiv) was added to a stirred suspension of ethyl acetate imidazopyrazinone (1 equiv) in MeOH (20 vol). After 1 h, volatiles were removed under a stream of N2. The solid was suspended with H2O, filtered, washed with water, and dried under vacuum. The work-up procedure B is as follows: the reaction was acidified with a 5% solution of trifluoroacetic acid (TFA) in MeOH. The suspension was then concentrated onto C18 silica gel and purified by MPLC.
General Procedure E: Two Step, One-Pot Synthesis of Imidazopyrazinones
The alkylating agent (1.2–1.5 equiv) was added slowly to a stirred suspension of 13a (1 equiv), anh. K2CO3 (3 equiv) in anh. DMF (15 vol). The reaction was stirred at rt until complete (10 min to 5 h), and 2 M aq HCl (15 vol) was then added slowly to control effervescence (CO2). The reaction was then stirred at rt (overnight to 4 days) or for 22a heated with microwave irradiation until the cyclization was complete. The solid precipitate was collected by vacuum filtration, washed with water and MeOH, and then dried in vacuo.
N-Benzyl-5-nitro-1H-imidazole-2-carboxamide; 16b
The title compound was prepared according to the procedure reported by Jarrad et al.;28 amine: benzylamine (300 μL, 2.74 mmol). The crude material was purified over silica gel by MPLC (Biotage 20–100% EtOAc in pet. spirits gradient) to yield a colorless solid (399 mg, 71%). LCMS: Rt = 3.13 min, 99 A % @ 254 nm, [M – H]− = 245.0. 1H NMR (600 MHz, DMSO-d6): δ 14.31 (s, 1H), 9.45 (t, J = 6.4 Hz, 1H), 8.46 (s, 1H), 7.31 (d, J = 4.4 Hz, 4H), 7.27–7.20 (m, 1H), 4.43 (d, J = 6.4 Hz, 2H). 13C NMR (150 MHz, DMSO-d6): δ 157.2, 146.8, 139.7, 139.1, 128.3, 127.4, 126.9, 121.6, 42.3. HRMS (ESI) m/z: calcd for C11H10N4NaO3 [M + Na]+, 269.0645; found, 269.0639.
N-(4-Methylbenzyl)-5-nitro-1H-imidazole-2-carboxamide; 16d
It was prepared according to the procedure reported by Jarrad et al.;28 amine: 4-methylbenzylamine (155 μL, 1.37 mmol). The crude material was purified over silica gel by MPLC (Grace Reveleris X2, 20–100% EtOAc in pet. spirits gradient) to obtain a yellow solid (201 mg, 68%). LCMS: Rt = 3.27 min, 99 A % @ 254 nm, [M – H]− = 259.0. 1H NMR (600 MHz, DMSO-d6): δ 14.30 (s, 1H), 9.37 (t, J = 6.4 Hz, 1H), 8.43 (s, 1H), 7.20 (d, J = 8.2 Hz, 2H), 7.11 (d, J = 7.8 Hz, 2H), 4.37 (d, J = 6.4 Hz, 2H), 2.26 (s, 3H). 13C NMR (150 MHz, DMSO-d6): δ 157.2, 146.8, 140.0, 136.1, 135.9, 128.8, 127.4, 121.8, 42.0, 20.7. HRMS (ESI) m/z: calcd for C12H12N4NaO3 [M + Na]+, 283.0802; found, 283.0794.
2-Nitroimidazo[1,2-a]pyrazin-8(7H)-one; 17a
To a stirred suspension of 19a (551 mg, 2.02 mmol) in H2O (11 mL) was added 5% aq HCl (1.44 mL, 1 equiv). The reaction was refluxed for 4.5 h. The solvent was removed in vacuo. The crude product was purified by recrystallization (slurry equilibration with hot MeOH) to give fine off-white needles (241 mg, 66%). mp = 350 °C (decomposed). LCMS: Rt = 1.49 min, 99 A % @ 254 nm, [M + H]+ = 180.8. 1H NMR (600 MHz, DMSO-d6): δ 11.58 (s, 1H), 8.81 (s, 1H), 7.50 (d, J = 5.6 Hz, 1H), 7.07 (d, J = 5.7 Hz, 1H). 13C NMR (150 MHz, DMSO-d6): δ 153.2, 147.6, 135.5, 119.8, 116.6, 107.1. The position of the nitro group was confirmed by HMBC and X-ray crystallography. X-ray diffraction data were collected on Oxford Diffraction Gemini Ultra dual source (Mo and Cu) CCD Diffractometer with Cu radiation (λ = 1.54184 Å), T = 190(2) K. Additional crystallographic data and HMBC data are available in the Supporting Information, Figure S1 and Tables S1–S3. HRMS (ESI) m/z: calcd for C12H8N8NaO6 [2M + Na]+, 383.0459; found, 383.0454.
7-Benzyl-2-nitroimidazo[1,2-a]pyrazin-8(7H)-one; 17b
Compound 19b (150 mg, 0.414 mmol) was reacted according to general procedure C, work-up procedure B. The crude material was purified by recrystallization (hot slurry from DCM/MeOH) to yield a tan solid (88 mg, 78%). mp = 298–299 °C (decomposed). LCMS: Rt = 3.17 min, 99 A % @ 254 nm, [M + H]+ = 271.0. 1H NMR (600 MHz, DMSO-d6): δ 8.81 (s, 1H), 7.60 (d, J = 5.9 Hz, 1H), 7.42 (d, J = 5.9 Hz, 1H), 7.37–7.34 (m, 4H), 7.32–7.24 (m, 1H), 5.13 (s, 2H). 13C NMR (150 MHz, DMSO-d6): δ 152.8, 148.0, 136.5, 135.2, 128.7, 127.8, 127.7, 123.6, 116.7, 107.4, 50.2. HRMS (ESI) m/z: calcd for C13H10N4NaO3 [M + Na]+, 293.0645; found, 293.0640.
2-Nitro-7-(4-(trifluoromethoxy)benzyl)imidazo[1,2-a]pyrazin-8(7H)-one; 17c
Compound 19c (300 mg, 0.672 mmol) was reacted according to general procedure C, work-up procedure A, to yield a lemon yellow crystalline solid (207 mg, 87%). mp = 264–266 °C (decomposed). LCMS: Rt = 3.47 min, 99 A % @ 254 nm, [M + H]+ = 355.0. 1H NMR (600 MHz, DMSO-d6): δ 8.82 (s, 1H), 7.62 (d, J = 5.9 Hz, 1H), 7.52–7.47 (m, 2H), 7.46 (d, J = 5.9 Hz, 1H), 7.39–7.33 (m, 2H), 5.16 (s, 2H).·13C NMR (150 MHz, DMSO-d6): δ 152.8, 148.0, 147.7, 136.0, 135.1, 129.7, 123.5, 121.2, 120.0 (q, J = 257.6 Hz), 116.6, 107.5, 49.6. HRMS (ESI) m/z: calcd for C14H9F3N4NaO4 [M + Na]+, 377.0468; found, 377.0467.
7-(4-Methylbenzyl)-2-nitroimidazo[1,2-a]pyrazin-8(7H)-one; 17d
Compound 19d (100 mg, 0.266 mmol) was reacted according to general procedure C, work-up procedure B. The crude material was purified by recrystallization (slurry equilibration with hot DCM/MeOH) to yield a tan solid (53 mg, 70%). mp = 311–312 °C (decomposed). LCMS: Rt = 3.30 min, 99 A % @ 254 nm, [M + H]+ = 285.1. 1H NMR (600 MHz, DMSO-d6): δ 8.80 (s, 1H), 7.58 (d, J = 5.9 Hz, 1H), 7.39 (d, J = 5.9 Hz, 1H), 7.27–7.22 (m, 2H), 7.18–7.13 (m, 2H), 5.07 (s, 2H), 2.27 (s, 3H). 13C NMR (151 MHz, DMSO-d6): δ 152.7, 148.0, 137.1, 135.1, 133.5, 129.2, 127.8, 123.5, 116.6, 107.4, 49.9, 20.7. HRMS (ESI) m/z: calcd for C14H13N4O3 [M + H]+, 285.0982; found, 285.0973.
7-(4-Fluorobenzyl)-2-nitroimidazo[1,2-a]pyrazin-8(7H)-one; 17e
Compound 19e (384 mg, 1.01 mmol) was reacted according to the general procedure C, work-up procedure A, to yield a yellow crystalline solid (249 mg, 86%). mp = 297 °C (decomposed). LCMS: Rt = 3.23 min, 98 A % @ 254 nm, [M + H]+ = 289.0. 1H NMR (600 MHz, DMSO-d6): δ 8.81 (s, 1H), 7.60 (d, J = 5.9 Hz, 1H), 7.44 (d, J = 5.9 Hz, 1H), 7.44–7.39 (m, 2H), 7.23–7.15 (m, 2H), 5.11 (s, 2H). 13C NMR (150 MHz, DMSO-d6): δ 161.7 (d, J = 243.9 Hz), 152.8, 148.0, 135.1, 132.7, 130.0 (d, J = 8.6 Hz), 123.4, 116.6, 115.4 (d, J = 20.1 Hz), 107.4, 49.5. HRMS (ESI) m/z: calcd for C13H9FN4NaO3 [M + Na]+, 311.0551; found, 311.0551.
2-Nitro-7-(3-(trifluoromethoxy)benzyl)imidazo[1,2-a]pyrazin-8(7H)-one; 17f
Compound 19f (100 mg, 0.224 mmol) was reacted according to general procedure C, work-up procedure B. The crude material was purified by recrystallization (DCM/EtOH) with hot filtration to obtain a colorless pearlescent solid (33 mg, 42%). mp = 230–231 °C (decomposed). LCMS: Rt = 3.45 min, 99 A % @ 254 nm, [M + H]+ = 355.0. 1H NMR (600 MHz, DMSO-d6): δ 8.82 (s, 1H), 7.61 (d, J = 5.8 Hz, 1H), 7.49 (t, J = 7.9 Hz, 1H), 7.45 (d, J = 5.9 Hz, 1H), 7.41–7.38 (m, 1H), 7.37 (ddd, J = 7.5, 1.6, 0.7 Hz, 1H), 7.33–7.28 (m, 1H), 5.17 (s, 2H). 13C NMR (150 MHz, DMSO-d6): δ 152.9, 148.5, 148.0, 139.3, 135.2, 130.7, 126.7, 123.5, 120.3, 120.2, 120.1 (q, J = 257.2 Hz), 116.7, 107.6, 49.8. HRMS (ESI) m/z: calcd for C14H9F3N4NaO4 [M + Na]+, 377.0468; found, 377.0471.
7-(3-Methylbenzyl)-2-nitroimidazo[1,2-a]pyrazin-8(7H)-one; 17g
Compound 17a (70 mg, 0.389 mmol) was reacted according to general procedure B. The crude material contained imidazopyrazinone (major) and imidazopyrazine (minor) isomers in a ratio of 2:1 as detected by LC-MS (UV 254 nm). The crude was partially purified over silica gel by MPLC (Biotage Isolera, 0–6% DCM/MeOH), then purified over C18-reversed phase silica (Grace Reveleris X2, A: H2O + 0.1% TFA, B: ACN + 0.1% TFA, 0–100% B) to yield imidazopyrazinone 17g as white powder (11 mg, 7%) and imidazopyrazine 18g as white powder (9 mg, 7%). Major isomer imidazopyrazinone 17g: LCMS: Rt = 3.25 min, 99 A % @ 254 nm, [M + H]+ = 285.1. 1H NMR (600 MHz, DMSO-d6): δ 8.81 (s, 1H), 7.59 (d, J = 5.9 Hz, 1H), 7.40 (d, J = 5.9 Hz, 1H), 7.24 (t, J = 7.6 Hz, 1H), 7.18–7.09 (m, 3H), 5.09 (s, 2H), 2.28 (s 3H). 13C NMR (150 MHz, DMSO-d6): δ 152.7, 148.0, 137.9, 136.4, 135.1, 128.5, 128.4, 128.2, 124.8, 123.5, 116.6, 107.3, 50.1, 20.9. HRMS (ESI) m/z: calcd for C14H12N4O3Na [M + Na]+, 307.0802; found, 307.0800. Characterization data of 18g are listed below following compounds 17a–t.
2-Nitro-7-(3-(trifluoromethyl)benzyl)imidazo[1,2-a]pyrazin-8(7H)-one; 17h
Compound 17a (70 mg, 0.389 mmol) was reacted with Cs2CO3 and 3-(trifluoromethyl)benzyl bromide (1.2 equiv) according to general procedure A at μW 100 °C (15 min), work-up B. The crude material containing imidazopyrazinone (major) and imidazopyrazine (minor) isomers was detected in a ratio of 10.1:1 by LC–MS (UV 254 nm). The crude material was purified over silica gel by MPLC (Biotage Isolera, 0–5% DCM/MeOH) to give final products imidazopyrazinone 17h as white powder (11 mg, 7%) and imidazopyrazine 18h as white powder (9 mg, 7%). Major isomer imidazopyrazinone 17h (cream powder, 72 mg, 55%): LCMS: Rt = 2.95 min, 99 A % @ 254 nm, [M + H]+ = 339.1; 1H NMR (600 MHz, DMSO-d6): δ 8.82 (s, 1H), 7.77 (d, J = 1.7 Hz, 1H), 7.67 (dd, J = 12.3, 7.4 Hz, 2H), 7.63–7.58 (m, 2H), 7.48 (d, J = 5.9 Hz, 1H), 5.22 (s, 2H); 13C NMR (150 MHz, DMSO-d6): δ 152.9, 148.0, 137.9, 135.2, 131.9, 129.7, 129.3 (q, J = 31.8 Hz), 124.5, 124.5, 124.1 (q, J = 272.2 Hz), 123.5, 116.7, 107.6, 49.9. HRMS (ESI) m/z: calcd for C14H9F3N4O3Na [M + Na]+, 361.0519; found, 361.0517. Characterization data of 18h are listed below following compounds 17a–t.
2-Nitro-7-(2-(trifluoromethoxy)benzyl)imidazo[1,2-a]pyrazin-8(7H)-one; 17i
Compound 17a (70 mg, 0.389 mmol) was reacted according to general procedure B. The crude material contained imidazopyrazinone (major) and imidazopyrazine (minor) isomers in a ratio of 1.9:1 as detected by LC–MS (UV 254 nm). The crude material was purified over C18-reversed phase silica (Grace Reveleris X2, A: H2O + 0.1% TFA, B: ACN +0.1% TFA, 30–80% B) to give the final product: Major isomer imidazopyrazinone 17i (cream powder, 20 mg, 5%): LCMS: Rt = 2.95 min, 98 A % @ 254 nm, [M + H]+ = 355.1; 1H NMR (600 MHz, DMSO-d6): δ 8.84 (s, 1H), 7.63 (d, J = 5.9 Hz, 1H), 7.47 (ddd, J = 8.2, 7.1, 1.8 Hz, 1H), 7.42 (dt, J = 8.2, 1.6 Hz, 1H), 7.39–7.27 (m, 3H), 5.21 (s, 2H); 13C NMR (150 MHz, DMSO-d6): δ 152.7, 148.0, 146.2, 135.1, 129.7, 129.5, 128.5, 127.6, 123.6, 120.5, 120.2 (q, J = 257.1 Hz), 116.7, 107.5, 45.7. HRMS (ESI) m/z: calcd for C14H9F3N4O4Na [M + Na]+, 377.0468; found, 377.0469. Mixed fractions were combined and purified over silica gel by MPLC (Biotage Isolera, 0–6% DCM/MeOH) to yield minor isomer imidazopyrazine 18i (white powder, 11 mg, 3%). Characterization data of 18i are listed below following compounds 17a–t.
7-(2,4-Difluorobenzyl)-2-nitroimidazo[1,2-a]pyrazin-8(7H)-one; 17j
Compound 17a (50 mg, 0.278 mmol) was reacted with Cs2CO3 and 2,4-difluorobenzyl bromide (1.2 equiv) according to general procedure A at rt for 1 h, with work-up procedure B. The crude product was purified by recrystallization (slurry equilibration with hot DCM/EtOH) to yield the final product imidazopyrazinone 17j as a white powder (64 mg, 75%). LCMS: Rt = 2.79 min, 99 A % @ 254 nm, [M + H]+ = 307.1; 1H NMR (600 MHz, DMSO-d6): δ 8.82 (s, 1H), 7.61 (d, J = 5.9 Hz, 1H), 7.43 (td, J = 8.7, 6.5 Hz, 1H), 7.37 (d, J = 5.9 Hz, 1H), 7.30 (ddd, 10.7, 9.3, 2.6 Hz, 1H), 7.10–7.06 (m, 1H), 5.14 (s, 2H); 13C NMR (150 MHz, DMSO-d6): δ 161.9 (dd, J = 246.9, 12.2 Hz), 160.3 (dd, J = 247.9, 12.2 Hz), 152.7, 148.0, 135.0, 131.5 (dd, J = 10.3, 5.6 Hz), 123.6, 119.5 (dd, J = 15.2, 3.9 Hz), 116.7, 111.6 (dd, J = 21.4, 3.2 Hz), 107.4, 104.0 (t, J = 25.7 Hz), 44.6 (d, J = 3.2 Hz). HRMS (ESI) m/z: calcd for C13H8F2N4O3Na [M + Na]+, 329.0457; found, 329.0449.
2-Nitro-7-(pyridin-2-ylmethyl)imidazo[1,2-a]pyrazin-8(7H)-one, 1TFA; 17k
Compound 19k (150 mg, 0.413 mmol) was reacted according to general procedure C, work-up procedure B. The crude material was purified over C18 silica gel by MPLC (Grace Reveleris X2, A: H2O + 0.1% TFA, B: ACN +0.1% TFA, 5–30% B) to yield a colorless powder (88 mg, 56%). LCMS: Rt = 2.61 min, 99 A % @ 254 nm, [M + H]+ = 272.0. 1H NMR (600 MHz, DMSO-d6): δ 8.85 (s, 1H), 8.53 (dt, J = 4.8, 1.4, 1H), 7.86 (td, J = 7.7, 1.8 Hz, 1H), 7.63 (d, J = 5.9 Hz, 1H), 7.44 (dd, J = 10.1, 6.9 Hz, 2H), 7.40–7.34 (m, 1H), 5.26 (s, 2H). 13C NMR (150 MHz, DMSO-d6): δ 155.0, 152.8, 148.6, 148.0, 137.8, 135.1, 124.6, 123.1, 122.1, 116.6, 107.1, 51.8. HRMS (ESI) m/z: calcd for C12H9N5O3 [M + H]+, 272.0778; found, 272.0782.
7-(1-(4-Fluorophenyl)ethyl)-2-nitroimidazo[1,2-a]pyrazin-8(7H)-one; 17l
Compound 19l (100 mg, 0.254 mmol) was reacted according to general procedure C, work-up procedure B. The crude material was purified by recrystallization (DCM/EtOH) with hot filtration to yield a colorless pearlescent solid (34 mg, 45%). mp = 275–277 °C (decomposed). LCMS: Rt = 3.31 min, 99 A % @ 254 nm, [M + H]+ = 303.0. 1H NMR (600 MHz, DMSO-d6): δ 8.77 (s, 1H), 7.57 (d, J = 6.1 Hz, 1H), 7.47–7.41 (m, 2H), 7.24–7.19 (m, 3H), 6.15 (q, J = 7.1 Hz, 1H), 1.71 (d, J = 7.2 Hz, 3H). 13C NMR (150 MHz, DMSO-d6): δ 161.6 (d, J = 244.4 Hz), 152.6, 148.0, 136.2 (d, J = 2.90 Hz), 134.9, 129.3 (d, J = 8.49 Hz), 119.8, 116.4, 115.5 (d, J = 21.3 Hz), 107.8, 51.6, 18.4. HRMS (ESI) m/z: calcd for C14H11FN4NaO3 [M + Na]+, 325.0707; found, 325.0718.
2-Nitro-7-(4-(trifluoromethoxy)phenethyl)imidazo[1,2-a]pyrazin-8(7H)-one; 17m
Imidazopyrazinone 17a (75 mg, 0.42 mmol), K2CO3, and 1-(2-bromoethyl)-4-(trifluoromethoxy)benzene (1.2 equiv) were reacted according to general procedure A (μW 80 °C, 30 min). The crude material contained imidazopyrazinone (major) and imidazopyrazine (minor) isomers in a 10:1 ratio (1H NMR integration). The major imidazopyrazinone isomer 17m was obtained by purification of the crude residue over C18 silica gel (Grace Reveleris X2, A: H2O + 0.1% TFA, B: ACN + 0.1% TFA, 40–100% B) to yield a cream solid (33 mg, 22%). Purification of mixed fractions over silica gel by MPLC (Grace Reveleris X2, 0–7% MeOH in DCM gradient) yielded an additional portion of imidazopyrazinone 17m as a cream solid (34 mg, 22%) and the imidazopyrazine isomer 18m as a colorless solid (6 mg, 4%). Major isomer imidazopyrazinone 17m: LCMS: Rt = 3.51 min, 99 A % @ 254 nm, 95 A % @ 200 nm, [M + H]+ = 369.0. 1H NMR (600 MHz, DMSO-d6): δ 8.79 (s, 1H), 7.54 (d, J = 5.8 Hz, 1H), 7.41–7.35 (m, 2H), 7.29 (d, J = 8.0 Hz, 2H), 7.27 (d, J = 5.9 Hz, 1H), 4.13 (t, J = 7.5 Hz, 2H), 3.00 (t, J = 7.5 Hz, 2H). 13C NMR (150 MHz, DMSO-d6): δ 152.6, 148.0, 147.0, 137.4, 135.0, 130.7, 123.6, 121.1, 120.1 (q, J = 257.1 Hz), 116.5, 106.8, 48.6, 33.3. HRMS (ESI) m/z: calcd for C15H12F3N4O4 [M + H]+, 369.0805; found, 369.0814. Characterization data of 18m are listed below following compounds 17a–t.
7-(4-Methylphenethyl)-2-nitroimidazo[1,2-a]pyrazin-8(7H)-one; 17n
Compound 19n (100 mg, 0.273 mmol) was reacted according to general procedure C, work-up procedure B. The crude material was purified by recrystallization (DCM/acetone) with hot filtration to yield a pearlescent tan solid (45 mg, 59%). mp = 262–264 °C. LCMS: Rt = 3.38 min, 99 A % @ 254 nm, [M + H]+ = 299.1. 1H NMR (600 MHz, DMSO-d6): δ 8.78 (s, 1H), 7.52 (d, J = 5.8 Hz, 1H), 7.26 (d, J = 5.9 Hz, 1H), 7.12 (d, J = 7.8 Hz, 2H), 7.09 (d, J = 7.8 Hz, 2H), 4.10 (dd, J = 8.4, 6.7 Hz, 2H), 2.92 (t, J = 7.5 Hz, 2H), 2.25 (s, 3H). 13C NMR (150 MHz, DMSO-d6): δ 152.6, 148.0, 135.5, 135.0, 134.7, 129.1, 128.7, 123.7, 116.4, 106.7, 49.0, 33.7, 20.7. HRMS (ESI) m/z: calcd for C15H15N4O3 [M + H]+, 299.1139; found, 299.1132.
2-(2-Nitro-8-oxoimidazo[1,2-a]pyrazin-7(8H)-yl)ethyl Acetate; 17o
Imidazopyrazinone 17a (70 mg, 0.39 mmol), K2CO3, and 2-bromoethylacetate (1.5 equiv) were reacted according to general procedure A (μW 80 °C, 30 min). The imidazopyrazinone:imidazopyrazine isomers were detected in a 10:1 ratio by LCMS (UV 254 nm). The reaction was poured into H2O (20 mL), and the precipitate is filtered to yield a cream solid (72 mg, 69%). LCMS: Rt = 2.18 min, 99 A % @ 254 nm, [M + H]+ = 267.0. 1H NMR (600 MHz, DMSO-d6): δ 8.81 (s, 1H), 7.58 (d, J = 5.9 Hz, 1H), 7.35 (d, J = 5.9 Hz, 1H), 4.33–4.27 (m, 2H), 4.19–4.14 (m, 2H), 1.97 (s, 3H). 13C NMR (150 MHz, DMSO-d6): δ 170.2, 152.8, 147.9, 134.9, 124.0, 116.5, 106.8, 61.3, 46.7, 20.6. HRMS (ESI) m/z: calcd for C10H10N4NaO5 [M + Na]+, 289.0543; found, 289.0545.
7-(2-Hydroxyethyl)-2-nitroimidazo[1,2-a]pyrazin-8(7H)-one; 17p
Compound 17o (45 mg, 0.17 mmol) was reacted according to general procedure D. The product was purified over C18 silica gel by MPLC (Grace Reveleris X2, A: H2O + 0.1% TFA, B: ACN + 0.1% TFA, 5–22% B) to yield a pale lemon powder (18 mg, 45%). LCMS: Rt = 1.79 min, 99 A % @ 254 nm, [M + H]+ = 225.0. 1H NMR (600 MHz, DMSO-d6): δ 8.81 (s, 1H), 7.55 (d, J = 5.9 Hz, 1H), 7.29 (d, J = 5.8 Hz, 1H), 4.90 (t, J = 5.7 Hz, 1H), 3.96 (t, J = 5.5 Hz, 2H), 3.66 (q, J = 5.6 Hz, 2H). 13C NMR (150 MHz, DMSO-d6): δ 152.8, 147.9, 135.1, 124.7, 116.3, 106.2, 58.4, 50.3. HRMS (ESI) m/z: calcd for C8H8N4NaO4 [M + Na]+, 247.0438; found, 247.0435.
4-(2-(2-Nitro-8-oxoimidazo[1,2-a]pyrazin-7(8H)-yl)ethyl)morpholin-4-ium 2,2,2-Trifluoroacetate; 17q
Imidazopyrazinone 17a (75 mg, 0.42 mmol), 4-(2-chloroethyl)morpholine hydrochloride (1.5 equiv), potassium iodide (0.06 equiv), K2CO3 (3 equiv), and anh. DMF (20 vol) were reacted according to general procedure A (μW 120 °C, 15 min). To achieve conversion of the imidazopyrazinone starting material, further portions of potassium iodide (0.09 equiv), 4-(2-chloroethyl)morpholine hydrochloride (1.5 equiv) and anh. K2CO3 (1.5 equiv) were added and the reaction was heated in the microwave for a further 30 min at 120 °C and then for 15 min at 150 °C. The reaction was then evaporated to dryness to give crude material containing imidazopyrazinone (major) and imidazopyrazine (minor) isomers in a 13:1 ratio by LCMS (UV 254 nm). The crude solid was purified over C18 silica gel by MPLC (Grace Reveleris X2, A: H2O + 0.1% TFA, B: ACN +0.1% TFA, 5–15% B) to yield imidazopyrazinone 17q as a sticky solid (143 mg, 84%) and imidazopyrazine 18q as a red residue (6 mg, 5%). Major isomer imidazopyrazinone 17q: LCMS: Rt = 1.58 min, 99 A % @ 254 nm, [M + H]+ = 294.1. 1H NMR (600 MHz, DMSO-d6): δ 9.94 (br s, 1H), 8.86 (s, 1H), 7.67 (d, J = 5.9 Hz, 1H), 7.37 (d, J = 5.9 Hz, 1H), 4.31 (t, J = 6.2 Hz, 2H), 4.12–3.89 (m, 2H), 3.85–3.32 (m, 4H), 3.29–2.95 (m, 2H), 2.92–2.60 (m, 2H). 13C NMR (150 MHz, DMSO-d6): δ 153.6, 148.0, 135.0, 123.2, 116.6, 107.7, 63.3, 54.3, 52.4, 51.5, 51.0, 41.4. HRMS (ESI) m/z: calcd for C12H16N5O4 [M + H]+, 294.1197; found, 294.1197. Characterization data of 18q are listed below following compounds 17a–t.
7-(2-Morpholino-2-oxoethyl)-2-nitroimidazo[1,2-a]pyrazin-8(7H)-one; 17r
Imidazopyrazinone 17a (75 mg, 0.42 mmol), K2CO3, and 4-(chloroacetyl)morpholine (1.2 equiv) were reacted according to general procedure A (rt, 2.5 h). The reaction volatiles were removed in vacuo. The imidazopyrazinone isomer was detected in a 99:1 ratio by LCMS (UV 254 nm). A solid precipitated from the ACN: (H2O + 0.1% TFA) mixture and was filtered. The solid was then washed with water (500 μL) and ACN (500 μL) and volatiles were removed in vacuo to yield a tan pearlescent solid (80 mg, 63%). LCMS: Rt = 2.44 min, 99 A % @ 254 nm, [M + H]+ = 308.1. 1H NMR (600 MHz, DMSO-d6): δ 8.85 (s, 1H), 7.59 (d, J = 5.9 Hz, 1H), 7.24 (d, J = 5.9 Hz, 1H), 4.89 (s, 2H), 3.66 (t, J = 4.8 Hz, 2H), 3.59 (t, J = 4.9 Hz, 2H), 3.54 (t, J = 4.9 Hz, 2H), 3.45 (t, J = 4.9 Hz, 2H), 3.33 (s, 1H). 13C NMR (150 MHz, DMSO-d6): δ 164.8, 152.8, 148.0, 134.8, 124.7, 116.8, 106.6, 66.0, 65.9, 48.7, 44.6, 41.9. HRMS (ESI) m/z: calcd for C12H13N5NaO5 [M + Na]+, 330.0809; found, 330.0819.
2-(2-Nitro-8-oxoimidazo[1,2-a]pyrazin-7(8H)-yl)acetamide; 17s
Imidazopyrazinone 17a (75 mg, 0.42 mmol), K2CO3, and 2-bromoacetamide (1.2 equiv) were reacted according to general procedure A at rt for 1.5 h. The imidazopyrazinone:imidazopyrazine isomers were detected in a 32:1 ratio by LCMS (UV 254 nm). The reaction was then filtered, and the precipitate was washed with water (4 × 250 μL) and MeOH (2 × 250 μL). The crude material was purified over C18 silica gel by MPLC (Grace Reveleris X2, A: H2O + 0.1% TFA, B: ACN +0.1% TFA, 5–25% B) to yield a tan solid (5 mg, 4%). LCMS: Rt = 1.75 min, 99 A % @ 254 nm, [M + H]+ = 238.0. 1H NMR (600 MHz, DMSO-d6): δ 8.84 (s, 1H), 7.66 (br s, 1H), 7.57 (d, J = 5.9 Hz, 1H), 7.31 (br s, 1H), 7.30 (d, J = 5.8 Hz, 1H), 6.57 (s, 0.3H), 4.52 (s, 2H). A broad singlet that was exchangeable with water was observed at 6.57 ppm integrating for 0.3H. This has previously been observed for this class of compounds and is proposed to be due to protonation of the imidazopyrazinone core. 13C NMR (150 MHz, DMSO-d6): δ 168.2, 152.9, 148.0, 135.1, 124.8, 116.6, 106.6, 49.9. HRMS (ESI) m/z: calcd for C8H7N5NaO4 [M + Na]+, 260.0390; found, 260.0392.
7-Cyclohexyl-2-nitroimidazo[1,2-a]pyrazin-8(7H)-one; 17t
Compound 19t (79 mg, 0.22 mmol) was reacted according to general procedure C, work-up procedure B. The crude material was purified by recrystallization (DCM/EtOH) to yield a cream-colored crystalline solid (42 mg, 68%). mp = 275–277 °C (decomposed). LCMS: Rt = 3.20 min, 99 A % @ 254 nm, [M + H]+ = 263.1. 1H NMR (600 MHz, DMSO-d6): δ 8.76 (s, 1H), 7.58 (d, J = 6.0 Hz, 1H), 7.43 (d, J = 6.1 Hz, 1H), 4.66 (tt, J = 12.1, 3.8 Hz, 1H), 1.83 (d, J = 13.6 Hz, 2H), 1.74 (d, J = 12.0 Hz, 2H), 1.64 (qd, J = 12.3, 3.6 Hz, 3H), 1.41 (qt, J = 12.5, 3.4 Hz, 2H), 1.20 (qt, J = 12.5, 4.0 Hz, 1H). 13C NMR (150 MHz, DMSO-d6): δ 152.3, 148.0, 135.0, 119.8, 116.2, 107.1, 53.3, 30.8, 25.3, 24.7. HRMS (ESI) m/z: calcd for C12H14N4NaO3 [M + Na]+, 285.0958; found, 285.0953.
8-((3-Methylbenzyl)oxy)-2-nitroimidazo[1,2-a]pyrazine; 18g
Minor isomer imidazopyrazine 18g was isolated as described in the preparation of 17g. LCMS: Rt = 3.58 min, 99 A % @ 254 nm, [M + H]+ = 285.1; 1H NMR (600 MHz, DMSO-d6): δ 9.02 (t, J = 1.8 Hz, 1H), 8.25 (dt, J = 3.8, 1.7 Hz, 1H), 7.65 (dt, J = 4.0, 1.8 Hz, 1H), 7.36–7.28 (m, 3H), 7.20 (dd, J = 6.8, 3.5 Hz, 1H), 5.55–5.51 (m, 2H), 3.42 (s, 1H), 2.34 (d, J = 2.1 Hz, 3H). Impurities were detected by 1H NMR and this compound was not subjected to biological evaluation.
2-Nitro-8-((3-(trifluoromethyl)benzyl)oxy)imidazo[1,2-a]pyrazine; 18h
Minor isomer imidazopyrazine 18h was isolated as described in the preparation of 17h. LCMS: Rt = 3.22 min, 99 A % @ 254 nm, [M + H]+ = 339.1; 1H NMR (600 MHz, CDCl3): δ 8.31 (s, 1H), 7.81–7.76 (m, 2H), 7.76–7.72 (m, 1H), 7.63–7.58 (m, 2H), 7.54–7.48 (m, 1H), 5.67 (s, 2H). Impurities were detected by 1H NMR, and this compound was not subjected to biological evaluation.
2-Nitro-8-((2-(trifluoromethoxy)benzyl)oxy)imidazo[1,2-a]pyrazine; 18i
Minor isomer imidazopyrazine 18i was isolated as described in the preparation of 17i. LCMS: Rt = 3.25 min, 99 A % @ 254 nm, [M + H]+ = 355.1; 1H NMR (600 MHz, DMSO-d6): δ 9.04 (s, 1H), 8.27 (d, J = 4.7 Hz, 1H), 7.76–7.72 (m, 1H), 7.66 (d, J = 4.7 Hz, 1H), 7.60–7.53 (m, 1H), 7.51–7.44 (m, 2H), 5.64 (s, 2H); 13C NMR (150 MHz, DMSO-d6): δ 153.8, 148.2, 146.7, 131.2, 130.6, 130.1, 128.3, 128.0, 127.7, 120.7, 120.2 (q, J = 258.3 Hz), 116.5, 115.0, 62.7.δ HRMS (ESI) m/z: calcd for C14H9F3N4O4Na [M + Na]+, 377.0468; found, 377.0470.
2-Nitro-8-(4-(trifluoromethoxy)phenethoxy)imidazo[1,2-a]pyrazine; 18m
Minor isomer imidazopyrazine 18m was isolated as described in the preparation of 17m. LCMS: Rt = 3.76 min, 99 A % @ 254 nm, [M + H]+ = 369.1. 1H NMR (600 MHz, DMSO-d6): δ 9.01 (s, 1H), 8.22 (d, J = 4.7 Hz, 1H), 7.62 (d, J = 4.7 Hz, 1H), 7.52–7.46 (m, 2H), 7.34–7.29 (m, 2H), 4.73 (t, J = 6.7 Hz, 2H), 3.20 (t, J = 6.7 Hz, 2H). 13C NMR (150 MHz, DMSO-d6): δ 154.3, 148.2, 147.0, 137.7, 130.8, 128.2, 121.1, 120.1 (q, J = 256.2 Hz), 116.0, 114.9, 67.1, 33.5. HRMS (ESI) m/z: calcd for C15H12F3N4O4 [M + H]+, 369.0805; found, 369.0799.
4-(2-((2-Nitroimidazo[1,2-a]pyrazin-8-yl)oxy)ethyl)morpholin-4-ium 2,2,2-Trifluoroacetate; 18q
Minor isomer imidazopyrazine 18q was isolated as described in the preparation of 17q. LCMS: Rt = 2.31 min, 97 A % @ 254 nm, [M + H]+ = 294.1. 1H NMR (600 MHz, DMSO-d6): δ 9.08 (s, 1H), 8.31 (d, J = 4.7 Hz, 1H), 7.67 (d, J = 4.7 Hz, 1H), 4.85 (br s, 2H), 4.27–3.02 (m, 10H). Impurities were detected in the 1H NMR analysis, and therefore, the compound was not subjected to biological analysis.
1-(2,2-Diethoxyethyl)-4-nitro-1H-imidazole-2-carboxamide; 19a
Imidazole-2-carboxamide 16a (650 mg, 3.38 mmol) was reacted according to general procedure A with K2CO3, bromoacetaldehyde diethyl acetal (2 × 1.5 equiv) at μW 180 °C (2 × 15 min). The crude material was triturated with pet. spirits (3 × 3 mL) and dried in vacuo to yield an orange solid (710 mg, 77%). LCMS: Rt = 3.04 min, 88 A % @ 254 nm, [M + H – EtOH]+ = 227.0. 1H NMR (400 MHz, CDCl3): δ 7.93 (s, 1H), 7.93 (br s, 1H), 5.62 (br s, 1H), 4.73 (dd, J = 5.2, 4.5 Hz, 1H), 4.64 (d, J = 5.0 Hz, 2H), 3.75 (dq, J = 9.4, 7.1 Hz, 2H), 3.53 (dq, J = 9.4, 7.0 Hz, 2H), 1.19 (t, J = 7.0 Hz, 6H). 13C NMR (150 MHz, CDCl3): δ 159.5, 145.3, 136.2, 125.4, 100.5, 64.2, 51.8, 15.2. HRMS (ESI) m/z: calcd for C10H16N4NaO5 [M + Na]+, 295.1013; found, 295.1010.
N-Benzyl-1-(2,2-diethoxyethyl)-4-nitro-1H-imidazole-2-carboxamide; 19b
Imidazole-2-carboxamide 16b (150 mg, 0.609 mmol) was reacted according to general procedure A with K2CO3 and bromoacetaldehyde diethyl acetal (1.5 equiv, second portion = 0.5 equiv) at μW 150 °C (30 min, followed by 15 min) to yield a waxy yellow solid (219 mg, 99%). LCMS: Rt = 3.72 min, 97 A % @ 254 nm, [M + H – EtOH]+ = 317.1. 1H NMR (400 MHz, CDCl3): δ 7.91 (s, 1H), 7.68 (t, J = 4.5 Hz, 1H), 7.40–7.27 (m, 5H), 4.75 (dd, J = 5.3, 4.5 Hz, 1H), 4.68 (d, J = 4.8 Hz, 2H), 4.57 (d, J = 6.1 Hz, 2H), 3.75 (dq, J = 9.0, 7.3 Hz, 2H), 3.54 (dq, J = 9.4, 6.9 Hz, 2H), 1.19 (t, J = 7.1 Hz, 6H). 13C NMR (150 MHz, CDCl3): δ 157.5, 145.2, 136.9, 136.8, 128.8, 128.0, 127.9, 125.2, 100.6, 64.2, 51.6, 43.4, 15.2. HRMS (ESI) m/z: calcd for C17H22N4NaO5 [M + Na]+, 385.1482; found, 385.1480.
1-(2,2-Diethoxyethyl)-4-nitro-N-(4-(trifluoromethoxy)benzyl)-1H-imidazole-2-carboxamide; 19c
Imidazole-2-carboxamide 16c (125 mg, 0.332 mmol) was reacted according to general procedure A with K2CO3 and bromoacetaldehyde diethyl acetal (2 × 1.5 equiv) at μW 180 °C (2 × 15 min). The crude residue was purified over silica gel by MPLC (Biotage, 12–100% EtOAc in pet. spirits gradient) to yield a pale yellow waxy solid (112 mg, 76%). LCMS: Rt = 3.40 min, 99 A % @ 254 nm, [M – H]− = 445.0. 1H NMR (600 MHz, CDCl3): δ 7.92 (s, 1H), 7.75 (br t, 1H), 7.39–7.33 (m, 2H), 7.20 (m, 2H), 4.74 (dd, J = 5.1, 4.5 Hz, 1H), 4.67 (d, J = 4.8 Hz, 2H), 4.57 (d, J = 6.2 Hz, 2H), 3.75 (dq, J = 9.4, 7.0 Hz, 2H), 3.53 (dq, J = 9.4, 7.0 Hz, 2H), 1.18 (t, J = 7.0 Hz, 6H). 13C NMR (150 MHz, CDCl3): δ 157.8, 148.9, 145.4, 136.8, 136.0, 129.5, 125.4, 121.5, 120.6 (q, J = 256.5 Hz), 100.7, 64.3, 51.7, 42.7, 15.3. HRMS (ESI) m/z: calcd for C18H21F3N4NaO6 [M + Na]+, 469.1305; found, 469.1316.
1-(2,2-Diethoxyethyl)-N-(4-methylbenzyl)-4-nitro-1H-imidazole-2-carboxamide; 19d
Imidazole-2-carboxamide 16d (75 mg, 0.29 mmol) was reacted according to general procedure A with K2CO3 and bromoacetaldehyde diethyl acetal (2 × 1.5 equiv) at μW 150 °C (2 × 30 min) to yield a yellow oil which was used without further purification (116 mg with 43 mol % residual DMF by 1H NMR, 98%). LCMS: Rt = 3.80 min, 97 A % @ 254 nm, [M + H – EtOH]+ = 331.1. 1H NMR (600 MHz, CDCl3): δ 7.92 (s, 1H), 7.67 (d, J = 5.7 Hz, 1H), 7.25–7.19 (m, 2H), 7.19–7.14 (m, 2H), 4.76 (t, J = 4.9 Hz, 1H), 4.68 (d, J = 4.9 Hz, 2H), 4.54 (d, J = 6.1 Hz, 2H), 3.77 (dq, J = 9.4, 7.0 Hz, 2H), 3.55 (dq, J = 9.4, 7.0 Hz, 2H), 2.35 (s, 3H), 1.19 (t, J = 7.0 Hz, 5H). 13C NMR (151 MHz, CDCl3): δ 157.5, 145.2, 137.6, 136.9, 133.9, 129.5, 128.0, 125.2, 100.6, 64.2, 51.6, 43.2, 21.1, 15.2. HRMS (ESI) m/z: calcd for C18H24N4NaO5 [M + Na]+, 399.1639; found, 399.1643.
1-(2,2-Diethoxyethyl)-N-(4-fluorobenzyl)-4-nitro-1H-imidazole-2-carboxamide; 19e
Imidazole-2-carboxamide 16e (311 mg, 1.18 mmol) was reacted according to general procedure A with K2CO3 and bromoacetaldehyde diethyl acetal (2 × 1.5 equiv) at μW 180 °C (2 × 15 min). The crude residue was purified over silica gel by MPLC (Biotage, 10–60% EtOAc in pet. spirits gradient) to yield a pale yellow oil (411 mg, 92%). LCMS: Rt = 3.76 min, 99 A % @ 254 nm, [M + H – EtOH]+ = 335.0. 1H NMR (400 MHz, CDCl3): δ 7.91 (s, 1H), 7.70 (br s, 1H), 7.35–7.27 (m, 2H), 7.09–6.98 (m, 2H), 4.74 (dd, J = 5.2, 4.4 Hz, 1H), 4.67 (d, J = 4.8 Hz, 2H), 4.60–4.50 (m, 2H), 3.75 (dq, J = 9.4, 7.0 Hz, 2H), 3.54 (dq, J = 9.4, 7.0 Hz, 2H), 1.18 (t, J = 7.0 Hz, 6H). 13C NMR (150 MHz, CDCl3): δ 162.4 (d, J = 245.19 Hz), 157.6, 145.3, 136.8, 132.8 (d, J = 3.8 Hz), 129.7 (d, J = 8.1 Hz), 125.2, 115.7 (d, J = 21.6 Hz), 100.6, 64.2, 51.6, 42.7, 15.2. HRMS (ESI) m/z: calcd for C17H21F1N4NaO5 [M + Na]+, 403.1388; found, 403.1389.
1-(2,2-Diethoxyethyl)-4-nitro-N-(3-(trifluoromethoxy)benzyl)-1H-imidazole-2-carboxamide; 19f
Imidazole-2-carboxamide 16f (75 mg, 0.23 mmol) was reacted according to general procedure A with K2CO3 and bromoacetaldehyde diethyl acetal (2 × 1.5 equiv) at μW 150 °C (2 × 30 min) to yield an orange solid which was used without further purification (108 mg, quant.). LCMS: Rt = 3.88 min, 95 A % @ 254 nm, [M + H – EtOH]+ = 401.1. 1H NMR (600 MHz, CDCl3): δ 7.94 (s, 1H), 7.81 (t, J = 6.1 Hz, 1H), 7.39 (t, J = 7.9 Hz, 1H), 7.30–7.24 (m, 1H), 7.21–7.14 (m, 2H), 4.75 (dd, J = 5.2, 4.5 Hz, 1H), 4.69 (d, J = 4.8 Hz, 2H), 4.60 (d, J = 6.3 Hz, 2H), 3.76 (dq, J = 9.4, 7.1 Hz, 2H), 3.54 (dq, J = 9.4, 7.0 Hz, 2H), 1.19 (t, J = 7.0 Hz, 6H). 13C NMR (150 MHz, CDCl3): δ 157.7, 149.5, 145.2, 139.5, 136.6, 130.2, 126.1, 125.3, 120.4 (q, J = 260.9 Hz), 120.4, 120.2, 100.5, 64.2, 51.6, 42.7, 15.1. HRMS (ESI) m/z: calcd for C18H21F3N4NaO6 [M + Na]+, 469.1305; found, 469.1306.
1-(2,2-Diethoxyethyl)-4-nitro-N-(pyridin-2-ylmethyl)-1H-imidazole-2-carboxamide; 19k
Imidazole-2-carboxamide 16k (150 mg, 0.607 mmol) was reacted according to general procedure A with K2CO3 and bromoacetaldehyde diethyl acetal (1.5 equiv, second portion = 0.5 equiv) at μW 150 °C (30 min, followed by 15 min) to yield a red oil which was used without further purification (153 mg, 69%). LCMS: Rt = 3.13 min, 97 A % @ 254 nm, [M + H – EtOH]+ = 318.1. 1H NMR (600 MHz, DMSO-d6): δ 9.41 (t, J = 6.1 Hz, 1H), 8.52 (s, 1H), 8.51 (ddd, J = 4.7, 1.7, 1.0 Hz, 1H), 7.76 (td, J = 7.7, 1.8 Hz, 1H), 7.32 (d, J = 8.3 Hz, 1H), 7.27 (ddd, J = 7.6, 4.9, 1.1 Hz, 1H), 4.79 (t, J = 5.2 Hz, 1H), 4.60 (d, J = 5.2 Hz, 2H), 4.53 (d, J = 6.1 Hz, 2H), 3.61 (dq, J = 9.7, 7.0 Hz, 2H), 3.41 (dq, J = 9.7, 7.0 Hz, 2H), 1.02 (t, J = 7.0 Hz, 6H). 13C NMR (150 MHz, DMSO-d6): δ 157.9, 157.7, 148.9, 144.4, 137.7, 136.7, 126.4, 122.2, 120.9, 100.0, 62.8, 50.3, 44.1, 15.1. HRMS (ESI) m/z: calcd for C16H22N5O5 [M + H]+, 364.1615; found, 364.1622.
1-(2,2-Diethoxyethyl)-N-(1-(4-fluorophenyl)ethyl)-4-nitro-1H-imidazole-2-carboxamide; 19l
Imidazole-2-carboxamide 16l (75 mg, 0.27 mmol) was reacted according to general procedure A with K2CO3 and bromoacetaldehyde diethyl acetal (1.5 equiv, second portion = 1 equiv) at μW 150 °C (2 × 30 min) to yield a yellow oil (113 mg, quant) which was used without further purification. LCMS: Rt = 3.80 min, 97 A % @ 254 nm, [M + H – EtOH]+ = 349.1. 1H NMR (600 MHz, CDCl3): δ 7.92 (s, 1H), 7.60 (d, J = 8.2 Hz, 1H), 7.37–7.31 (m, 2H), 7.08–7.01 (m, 2H), 5.20 (p, J = 7.2 Hz, 1H), 4.74–4.66 (m, 2H), 4.65–4.56 (m, 1H), 3.77–3.69 (m, 2H), 3.52 (dq, J = 9.4, 7.0 Hz, 2H), 1.60 (d, J = 7.0 Hz, 3H), 1.17 (dt, J = 12.1, 7.0 Hz, 6H). 13C NMR (150 MHz, CDCl3): δ 162.1 (d, J = 245.3 Hz), 156.8, 145.2, 138.1 (d, J = 3.0 Hz), 136.8, 127.8 (d, J = 8.2 Hz), 125.3, 115.6 (d, J = 21.8 Hz), 100.5, 64.1, 51.6, 48.5, 21.9, 15.1. HRMS (ESI) m/z: calcd for C18H23FN4NaO5 [M + Na]+, 417.1545; found, 417.1537.
1-(2,2-Diethoxyethyl)-N-(4-methylphenethyl)-4-nitro-1H-imidazole-2-carboxamide; 19n
Imidazole-2-carboxamide 16n (75 mg, 0.27 mmol) was reacted according to general procedure A with K2CO3 and bromoacetaldehyde diethyl acetal (2 × 1.5 equiv) at μW 150 °C (2 × 30 min) to yield a yellow oil which was used without further purification (112 mg, quant.). LCMS: Rt = 3.85 min, 98 A % @ 254 nm, [M + H – EtOH]+ = 345.1. 1H NMR (600 MHz, CDCl3): δ 7.90 (s, 1H), 7.46 (t, J = 6.2 Hz, 1H), 7.19–7.03 (m, 4H), 4.72 (t, J = 4.9 Hz, 1H), 4.64 (d, J = 4.9 Hz, 2H), 3.76 (dq, J = 9.4, 7.0 Hz, 2H), 3.64 (dt, J = 7.7, 6.5 Hz, 2H), 3.53 (dq, J = 9.4, 7.0 Hz, 2H), 2.91–2.85 (m, 2H), 2.34 (s, 3H), 1.19 (t, J = 7.0 Hz, 6H). 13C NMR (150 MHz, CDCl3): δ 157.6, 145.1, 136.9, 136.2, 135.1, 129.4, 128.6, 125.1, 100.7, 64.2, 51.6, 40.8, 35.3, 21.0, 15.2. HRMS (ESI) m/z: calcd for C19H26N4NaO5 [M + Na]+, 413.1795; found, 413.1797.
N-Cyclohexyl-1-(2,2-diethoxyethyl)-4-nitro-1H-imidazole-2-carboxamide; 19t
Imidazole-2-carboxamide 16t (75 mg, 0.31 mmol) was reacted according to general procedure A with K2CO3 and bromoacetaldehyde diethyl acetal (1.5 equiv, second portion = 1 equiv) at μW 150 °C (2 × 30 min) to yield an orange solid (107 mg, 96%) which was used without further purification. LCMS: Rt = 3.78 min, 97 A % @ 254 nm, [M + H]+ = 309.1. 1H NMR (600 MHz, CDCl3): δ 7.90 (s, 1H), 7.24 (d, J = 8.5 Hz, 1H), 4.75 (t, J = 5.0 Hz, 1H), 4.65 (d, J = 5.0 Hz, 2H), 3.88 (tdt, J = 11.9, 8.2, 3.9 Hz, 1H), 3.76 (dq, J = 9.3, 7.1 Hz, 2H), 3.54 (dq, J = 9.3, 7.0 Hz, 2H), 2.02–1.96 (m, 2H), 1.83–1.77 (m, 2H), 1.72–1.62 (m, 1H), 1.44–1.35 (m, 2H), 1.35–1.25 (m, 3H), 1.19 (t, J = 7.0 Hz, 6H). 13C NMR (150 MHz, CDCl3): δ 156.8, 145.1, 137.2, 125.1, 100.7, 64.2, 51.7, 48.8, 32.9, 25.3, 25.0, 15.2. HRMS (ESI) m/z: calcd for C16H26N4NaO5 [M + Na]+, 377.1795; found, 377.1794.
Imidazo[1,2-a]pyrazin-8(7H)-one; 20a
To a stirred suspension of imidazole 23a (168 mg, 0.741 mmol) in H2O (20 vol) was added 5% w/v HCl (530 μL, 1 equiv). The solution was heated at reflux for 2 h, cooled to rt, and concentrated in vacuo to dryness. The solid was purified by recrystallization (slurry equilibration with hot MeOH) to yield beige crystals (50 mg, 50%). mp = 307–308 °C (decomposed). LCMS (Waters Atlantis): Rt = 1.75 min, 99 A % @ 254 nm, [M + H]+ = 136.1 1H NMR (400 MHz, DMSO-d6): δ 8.20 (d, J = 1.7 Hz, 1H), 8.01 (d, J = 1.7 Hz, 1H), 7.75 (d, J = 5.6 Hz, 1H), 7.23 (t, J = 5.6 Hz, 1H). 13C NMR (150 MHz, DMSO-d6): δ 151.3, 135.1, 126.3, 119.7, 118.3, 107.2. HRMS calcd for C12H10N6NaO2 [2M + Na]+, 293.0757; found, 293.0764. The proton spectrum was consistent with the literature.35
7-Benzylimidazo[1,2-a]pyrazin-8(7H)-one; 20b
To a stirred suspension of compound 23b (120 mg, 0.378 mmol) in H2O (20 vol.) was added 5% aq HCl (270 μL, 1 equiv). The reaction was heated at 80 °C for 3 days before further addition of 5% aq HCl (135 μL, 0.5 equiv). The reaction mixture was then washed with EtOAc (3 × 5 mL followed by 3 × 10 mL). The aqueous layer was then evaporated to dryness. The crude material was purified over C18 silica gel by MPLC (Grace Reveleris X2. A: H2O + 0.1% TFA, B: ACN +0.1% TFA, 10–30% B) to yield a colorless solid (47 mg, 39%). LCMS: Rt = 2.14 min, 98 A % @ 254 nm, [M + H]+ = 226.0. 1H NMR (600 MHz, DMSO-d6): δ 7.93 (d, J = 1.2 Hz, 1H), 7.69–7.65 (m, 2H), 7.40–7.27 (m, 6H), 5.14 (s, 2H). 13C NMR (151 MHz, DMSO-d6): δ 152.1, 136.7, 136.0, 130.3, 128.5, 127.6, 127.6, 121.6, 117.8, 107.4, 49.8. HRMS (ESI) m/z: calcd for C13H11N3NaO [M + Na]+, 248.0794; found, 249.0799.
7-(4-Fluorobenzyl)imidazo[1,2-a]pyrazin-8(7H)-one; 20c
Imidazopyrazinone 20a (100 mg, 0.740 mmol), K2CO3, and 4-fluorobenzyl bromide (1.2 equiv) were reacted according to general procedure A at rt for 45 min. The imidazopyrazinone isomer was detected as the preferred isomer (24:1 ratio) by HPLC (UV 254 nm). The crude material was purified by recrystallization (EtOH) to yield a colorless solid (34 mg, 19%). mp = 168–169 °C. LCMS: Rt = 2.94 min, 99 A % @ 254 nm, [M + H]+ = 244.1. 1H NMR (400 MHz, DMSO-d6): δ 7.84–7.78 (m, 1H), 7.61 (d, J = 5.8 Hz, 1H), 7.52–7.46 (m, 1H), 7.45–7.36 (m, 2H), 7.24 (d, J = 5.9 Hz, 1H), 7.22–7.12 (m, 2H), 5.09 (s, 2H). 13C NMR (150 MHz, DMSO-d6): δ 161.6 (d, J = 243.2 Hz), 153.0, 137.0, 133.4 (d, J = 3.0 Hz), 132.7, 129.9 (d, J = 8.5 Hz), 120.4, 117.6, 115.4 (d, J = 21.7 Hz), 107.5, 49.1. HRMS (ESI) m/z: calcd for C13H11FN3O [M + H]+, 244.0881; found, 244.0884.
N-(Benzyl)-1H-imidazole-2-carboxamide; 22b
1H-Imidazole-2-carboxylic acid 21a (1.08 g, 9.60 mmol) was refluxed in SOCl2 (10 mL) under N2 for 22 h. Volatiles were removed in vacuo, and residual SOCl2 was removed by co-evaporation with toluene to give the crude acid chloride intermediate. Benzylamine (478 μL, 4.38 mmol) was added drop wise to acid chloride (476 mg, 3.65 mmol) suspended in anh. THF (13 mL) under an atmosphere of N2. The light brown precipitate was filtered and washed with THF to yield a tan solid (685 mg, 79%) that was used without further purification. LCMS: Rt = 2.68 min, 99 A % @ 254 nm, [M + H]+ = 202.1. 1H NMR (600 MHz, CDCl3): δ 8.97 (t, J = 5.7 Hz, 1H), 7.49–7.04 (m, 7H), 4.43 (d, J = 6.2 Hz, 2H). 13C NMR (150 MHz, CDCl3): δ 158.3, 140.9, 139.5, 134.0, 128.1, 127.1, 126.6, 119.6 (br), 41.8.
1-(2,2-Diethoxyethyl)-1H-imidazole-2-carboxamide; 23a
Imidazole-2-carboxamide 22a (650 mg, 5.85 mmol) was reacted according to general procedure A with K2CO3, bromoacetaldehyde diethyl acetal (2 × 1.5 equiv) at μW 120 °C (2 × 15 min) to yield a yellow solid that was used without further purification (1.08 g, 81%). LCMS: Rt = 2.65 min, 99 A % @ 254 nm, [M + H – EtOH]+ = 182.1. 1H NMR (400 MHz, CDCl3): δ 7.23 (br s, 1H), 7.12 (d, J = 1.1 Hz, 1H), 7.01 (d, J = 1.1 Hz, 1H), 5.33 (br s, 1H), 4.84–4.60 (m, 1H), 4.53 (d, J = 5.3 Hz, 2H), 3.73 (dq, J = 9.4, 7.0 Hz, 2H), 3.48 (dq, J = 9.4, 7.0 Hz, 2H), 1.16 (t, J = 7.0 Hz, 6H). 13C NMR (150 MHz, CDCl3): δ 161.1, 137.8, 127.6, 126.7, 101.8, 64.0, 51.0, 15.2. NMR data was consistent with literature.35
N-Benzyl-1-(2,2-diethoxyethyl)-1H-imidazole-2-carboxamide; 23b
Imidazole-2-carboxamide 22b (201 mg, 0.846 mmol) was reacted according to general procedure A with K2CO3, bromoacetaldehyde diethyl acetal (1 equiv, second portion = 0.2 equiv) at μW 180 °C (2 × 15 min). The crude product was obtained as a yellow oil (246 mg, 92%) which was used without further purification. LCMS: Rt = 2.37 min, 89 A % @ 254 nm, [M + H]+ = 318.2. 1H NMR (600 MHz, CDCl3): δ 7.79 (t, J = 6.3 Hz, 1H), 7.40–7.34 (m, 4H), 7.34–7.30 (m, 1H), 7.14 (d, J = 1.0 Hz, 1H), 7.01 (d, J = 1.0 Hz, 1H), 4.79 (t, J = 5.5 Hz, 1H), 4.61 (d, J = 6.3 Hz, 2H), 4.60 (d, J = 5.5 Hz, 2H), 3.77 (dq, J = 9.4, 7.0 Hz, 2H), 3.52 (dq, J = 9.4, 7.0 Hz, 2H), 1.20 (t, J = 7.0 Hz, 6H). 13C NMR (100 MHz, CDCl3): δ 159.0, 138.5, 137.9, 128.7, 127.7, 127.5, 127.4, 126.3, 101.9, 64.0, 50.8, 43.1, 15.2. HRMS (ESI) m/z: calcd for C34H46N6NaO6 [2M + Na]+, 657.3371; found, 657.3392.
2-Nitro-6-phenylimidazo[1,2-a]pyrazin-8(7H)-one; 24a
Compound 27a (230 mg, 0.839 mmol) was reacted according to general procedure C to yield a crystalline yellow solid (181 mg, 84%). mp = 342 °C (decomposed). LCMS: Rt = 3.05 min, 99 A % @ 254 nm, [M + H]+ = 257.0. 1H NMR (600 MHz, DMSO-d6): δ 11.97 (s, 1H), 8.79–8.73 (s, 1H), 7.83 (s, 1H), 7.71–7.64 (m, 2H), 7.57–7.48 (m, 3H). 13C NMR (150 MHz, DMSO-d6): δ 153.6, 147.9, 134.6, 132.1, 130.7, 129.9, 129.0, 126.8, 116.7, 104.6. HRMS (ESI) m/z: calcd for C12H8N4NaO3 [M + Na]+, 279.0489; found, 279.0482.
2-Nitro-6-phenylimidazo[1,2-a]pyrazin-8(7H)-one; 24a
Imidazole-2-carboxamide 13a (450 mg, 2.34 mmol) was reacted according to general procedure E with 2-bromoacetophenone (1.2 equiv) in anh. DMF (20 vol). HCl (2 M aq, 20 vol) was then added slowly and the reaction was then heated (μW 120 °C for 1 h, 150 °C for 15 min), and the product was isolated to yield a brown solid (242 mg, 40%). LCMS: Rt = 3.02 min, 98 A % @ 254 nm, [M + H]+ = 257.0. 1H NMR (600 MHz, DMSO-d6): δ 11.99 (s, 1H), 8.77 (s, 1H), 7.83 (s, 1H), 7.69–7.67 (m, 2H), 7.55–7.50 (m, 3H). The proton spectrum is consistent with the preparation that used 4-nitroimidazole 21a as the starting material.
2-((2-Nitro-6-phenylimidazo[1,2-a]pyrazin-8-yl)oxy)ethyl Acetate; 24b and 2-(2-Nitro-8-oxo-6-phenylimidazo[1,2-a]pyrazin-7(8H)-yl)ethyl Acetate; 24c
Imidazopyrazinone 24a (280 mg, 1.09 mmol) was reacted according to general procedure A with K2CO3 and 2-bromoethylacetate (2 × 1.5 equiv) with heating (2 × μW 80 °C, 30 min). The crude material contained imidazopyrazinone (minor) and imidazopyrazine (major) isomers in a 1:5.7 ratio as determined by NMR. The crude material was purified over C18 silica gel by MPLC (Grace Reveleris, A: H2O + 0.1% TFA, B: ACN +0.1% TFA, 20–100% B) to yield imidazopyrazine as a yellow solid (231 mg, 62%) and imidazopyrazinone as a yellow solid (19 mg, 5%). Minor isomer imidazopyrazinone 24b: LCMS: Rt = 2.59 min, 99 A % @ 254 nm, [M + H]+ = 343.0. 1H NMR (600 MHz, DMSO-d6): δ 8.74 (s, 1H), 7.60–7.52 (m, 5H), 7.50 (s, 1H), 4.10–4.02 (m, 4H), 1.89 (s, 3H). 13C NMR (150 MHz, DMSO-d6): δ 170.0, 153.3, 148.1, 134.6, 134.1, 131.3, 129.9, 129.8, 128.8, 116.1, 106.8, 60.7, 43.4, 20.6. HRMS (ESI) m/z: calcd for C16H14N4Na1O5 [M + Na]+, 365.0856; found, 365.0858. Major isomer imidazopyrazine 24c: LCMS: Rt = 2.93 min, 99 A % @ 254 nm, [M + H]+ = 343.0. 1H NMR (600 MHz, DMSO-d6): δ 8.95 (s, 1H), 8.86 (s, 1H), 8.03–7.98 (m, 2H), 7.57–7.51 (m, 2H), 7.49–7.43 (m, 1H), 4.89–4.84 (m, 2H), 4.53–4.48 (m, 2H), 2.05 (s, 3H). 13C NMR (150 MHz, DMSO-d6): δ 170.3, 153.0, 148.4, 136.8, 135.0, 129.5, 129.2, 128.9, 125.7, 115.1, 111.9, 65.0, 62.0, 20.7. HRMS (ESI) m/z: calcd for C16H14N4Na1O5 [M + Na]+, 365.0856; found, 365.0861.
7-(2-Hydroxyethyl)-2-nitro-6-phenylimidazo[1,2-a]pyrazin-8(7H)-one; 24d
Compound 24b (19 mg, 55 μmol) was reacted according to general procedure D. After 25 min, volatiles were removed under a stream of N2. The residue was diluted with H2O (2 mL) and then extracted with EtOAc (3 × 2 mL), followed by DCM (3 × 2 mL). The organic layer was further diluted with EtOAc (14 mL), washed with brine (20 mL), dried with anh. MgSO4, and filtered. Volatiles were removed in vacuo to give a yellow residue which was purified over silica gel by MPLC (Grace Reveleris X2, 4–15% MeOH in DCM) then over C18 silica gel by MPLC (Grace Reveleris, A: H2O + 0.1% TFA, B: ACN +0.1% TFA, 15–40% B) to yield a yellow-brown solid (3.8 mg, 17%). LCMS: Rt = 2.52 min, 99 A % @ 254 nm, [M + H]+ = 301.1. 1H NMR (600 MHz, DMSO-d6): δ 8.73 (s, 1H), 7.59–7.51 (m, 5H), 7.46 (s, 1H), 4.77 (s, 1H), 3.85 (t, J = 6.3 Hz, 2H), 3.43 (br t, J = 6.7 Hz, 2H). 13C NMR (150 MHz, DMSO-d6): δ 153.2, 148.0, 135.1, 134.4, 131.7, 129.9, 129.7, 128.6, 115.9, 106.5, 57.5, 46.6. HRMS (ESI) m/z: calcd for C14H12N4Na1O4 [M + Na]+, 323.0751; found, 323.0759.
2-((2-Nitro-6-phenylimidazo[1,2-a]pyrazin-8-yl)oxy)ethan-1-ol; 24e
Compound 24c (146 mg, 0.427 mmol) was reacted according to general procedure D, work-up procedure B. The crude material was purified by MPLC (Grace Reveleris X2, A: H2O + 0.1% TFA, B: ACN +0.1% TFA, 30–100% B) to yield a bright yellow solid (13 mg, 10%). LCMS: Rt = 3.36 min, 98 A % @ 254 nm, [M + H]+ = 301.0. 1H NMR (600 MHz, DMSO-d6): δ 8.94 (s, 1H), 8.83 (s, 1H), 8.04–7.98 (m, 2H), 7.57–7.51 (m, 2H), 7.49–7.43 (m, 1H), 4.69–4.64 (m, 2H), 3.91–3.86 (m, 2H). 13C NMR (150 MHz, DMSO-d6): δ 153.4, 148.3, 137.0, 135.1, 129.6, 129.1, 128.9, 125.7, 115.0, 111.6, 68.7, 58.9. HRMS (ESI) m/z: calcd for C14H12N4NaO4 [M + Na]+, 323.0751; found, 323.0756.
6-Methyl-2-nitroimidazo[1,2-a]pyrazin-8(7H)-one; 25a
Imidazole-2-carboxamide 16a (1.50 g, 7.79 mmol) was reacted according to general procedure E with chloroacetone (1.3 equiv) to yield a yellow solid (1.01 g, 67%). LCMS: Rt = 2.35 min, 99 A % @ 254 nm, [M + H]+ = 195.0. 1H NMR (600 MHz, DMSO-d6): δ 11.66 (s, 1H), 8.76 (s, 1H), 7.34–7.29 (m, 1H), 2.12 (d, J = 1.2 Hz, 3H). 13C NMR (150 MHz, DMSO-d6): δ 153.4, 147.7, 134.4, 129.5, 116.2, 103.9, 15.7. HRMS (ESI) m/z: calcd for C14H12N8NaO6 [2M + Na]+, 411.0772; found, 411.0782.
2-(6-Methyl-2-nitro-8-oxoimidazo[1,2-a]pyrazin-7(8H)-yl)ethyl Acetate; 25b and 2-((6-Methyl-2-nitroimidazo[1,2-a]pyrazin-8-yl)oxy)ethyl; 25c
Compound 25a (150 mg, 0.773 mmol) was reacted according to general procedure A with K2CO3 and 2-bromoethyl acetate (2 × 2 equiv) with heating (2 × μW 80 °C, 30 min). The crude material contained imidazopyrazinone (major) and imidazopyrazine (minor) isomers in a 2.4:1 ratio as determined by NMR. The crude solid was purified over silica gel by MPLC (Grace Reveleris X2, 0–10% MeOH in DCM gradient) to yield imidazopyrazinone as a yellow solid (140 mg, 48%) and imidazopyrazine as a yellow solid (57 mg, 21%). Major isomer imidazopyrazinone 25b: LCMS: Rt = 2.80 min, 99 A % @ 254 nm, [M + H]+ = 281.1. 1H NMR (600 MHz, DMSO-d6): δ 8.76 (s, 1H), 7.45 (q, J = 1.2 Hz, 1H), 4.27 (t, J = 5.6 Hz, 2H), 4.21 (t, J = 5.5 Hz, 2H), 2.35 (d, J = 1.2 Hz, 3H), 1.98 (s, 3H). 13C NMR (150 MHz, DMSO-d6): δ 170.2, 153.4, 147.9, 133.8, 131.2, 115.7, 104.8, 61.1, 42.4, 20.6, 16.8. HRMS (ESI) m/z: calcd for C11H13N4O5 [M + H]+, 281.0880; found, 281.0878. Minor isomer imidazopyrazine 25c: LCMS: Rt = 3.23 min, 99 A % @ 254 nm, [M + H]+ = 281.1. 1H NMR (600 MHz, DMSO-d6): δ 8.96 (s, 1H), 8.06 (q, J = 1.1 Hz, 1H), 4.70–4.65 (m, 2H), 4.46–4.41 (m, 2H), 2.33 (d, J = 1.1 Hz, 3H), 2.05 (s, 3H). 13C NMR (151 MHz, DMSO): δ 170.4, 152.8, 148.1, 136.9, 129.0, 114.4, 112.4, 64.8, 62.0, 20.7, 20.4. HRMS (ESI) m/z: calcd for C11H13N4O5 [M + H]+, 281.0880; found, 281.0890.
7-(2-Hydroxyethyl)-6-methyl-2-nitroimidazo[1,2-a]pyrazin-8(7H)-one; 25d
Imidazopyrazinone 25b (50 mg, 0.18 mmol) was reacted according to general procedure D, work-up procedure B. The crude material was purified over C18 silica gel by MPLC (Grace Reveleris, A: H2O + 0.1% TFA, B: ACN +0.1% TFA, 5–25% B) to yield a yellow solid (35 mg, 83%). LCMS: Rt = 2.42 min, 99 A % @ 254 nm, [M + H]+ = 239.0. 1H NMR (600 MHz, DMSO-d6): δ 8.75 (s, 1H), 7.42 (q, J = 1.2 Hz, 1H), 4.95 (t, J = 5.8 Hz, 1H), 4.02 (t, J = 5.7 Hz, 2H), 3.64 (q, J = 5.7 Hz, 2H), 2.36 (d, J = 1.2 Hz, 3H). 13C NMR (150 MHz, DMSO-d6): δ 153.3, 147.9, 134.0, 132.0, 115.5, 104.3, 58.1, 45.9, 17.3. HRMS (ESI) m/z: calcd for C18H20N8NaO8 [2M + Na]+, 499.1296; found, 499.1296.
2-((6-Methyl-2-nitroimidazo[1,2-a]pyrazin-8-yl)oxy)ethan-1-ol; 25e
Imidazopyrazine 25c (20 mg, 71 μmol) was reacted according to general procedure D, work-up procedure B. The crude material was purified over C18 silica gel by MPLC (Grace Reveleris, A: H2O + 0.1% TFA, B: ACN +0.1% TFA, 10–30% B) to yield a yellow solid product (11 mg, 67%). LCMS: Rt = 2.78 min, 99 A % @ 254 nm, [M + H]+ = 239.0. 1H NMR (600 MHz, DMSO-d6): δ 8.94 (s, 1H), 8.03 (q, J = 1.1 Hz, 1H), 5.00 (t, J = 5.5 Hz, 1H), 4.51–4.46 (m, 2H), 3.84–3.78 (m, 2H), 2.32 (d, J = 1.1 Hz, 3H). 13C NMR (150 MHz, DMSO-d6): δ 153.3, 148.1, 137.1, 129.2, 114.3, 112.0, 68.6, 58.9, 20.5. HRMS (ESI) m/z: calcd for C18H20N8NaO8 [2M + Na]+, 499.1296; found, 499.1315.
6-Methyl-2-nitro-7-(4-(trifluoromethoxy)benzyl)imidazo[1,2-a]pyrazin-8(7H)-one; 25f and 6-Methyl-2-nitro-8-((4-(trifluoromethoxy)benzyl)oxy)imidazo[1,2-a]pyrazine; 25g
Compound 25a (150 mg, 0.773 mmol), K2CO3, and 1-(bromomethyl)4-(trifluoromethoxy)benzene (1.2 equiv) were reacted according to general procedure A at rt for 3 h, work-up B. The crude material contained imidazopyrazinone (major) and imidazopyrazine (minor) isomers in a 7.2:1 ratio by NMR. The crude solid was purified over silica gel by MPLC (Grace Reveleris X2, 0–7% MeOH in DCM gradient) to yield imidazopyrazinone as an off-white solid (197 mg, 69%) and imidazopyrazine as a colorless solid (27 mg, 9%). Major isomer imidazopyrazinone 25f: LCMS: Rt = 3.52 min, 99 A % @ 254 nm, [M + H]+ = 369.0. 1H NMR (600 MHz, DMSO-d6): δ 8.81 (s, 1H), 7.50 (q, J = 1.2 Hz, 1H), 7.40–7.36 (m, 2H), 7.36–7.32 (m, 2H), 5.31 (s, 2H), 2.21 (d, J = 1.2 Hz, 3H). 13C NMR (150 MHz, DMSO-d6): δ 153.7, 148.0, 147.4, 136.1, 134.0, 131.0, 128.3, 121.4, 120.1 (q, J = 256.4 Hz), 115.9, 105.2, 45.3, 16.6. HRMS (ESI) m/z: calcd for C15H12F3N4O4 [M + H]+, 369.0805; found, 369.0803. Minor isomer imidazopyrazine 25g: LCMS: Rt = 3.86 min, 99 A % @ 254 nm, [M + H]+ = 369.1. 1H NMR (600 MHz, DMSO-d6): δ 8.96 (s, 1H), 8.08 (q, J = 1.1 Hz, 1H), 7.72–7.66 (m, 2H), 7.46–7.41 (m, 2H), 5.58 (s, 2H), 2.36 (d, J = 1.1 Hz, 3H). 13C NMR (151 MHz, DMSO): δ 152.7, 148.2, 148.2, 137.0, 135.3, 130.6, 129.1, 121.1, 120.1 (q, J = 257.1 Hz), 114.4, 112.5, 67.2, 20.4. HRMS (ESI): m/z: calcd for C15H12F3N4NaO4 [M + Na]+, 391.0625; found, 391.0629.
6-Methyl-7-(4-methylbenzyl)-2-nitroimidazo[1,2-a]pyrazin-8(7H)-one; 25h and 6-Methyl-8-((4-methylbenzyl)oxy)-2-nitroimidazo[1,2-a]pyrazine; 25i
Compound 25a (150 mg, 0.773 mmol) was reacted according to general procedure method A with Cs2CO3 and 4-methylbenzyl bromide (1.2 equiv) with heating (μW 80 °C for 10 min and then 90 °C at 10 min) followed by rt overnight, work up B. The crude material contained imidazopyrazinone (major) and imidazopyrazine (minor) isomers in a ratio of 1.8:1 by LC-MS (UV 254 nm). The crude product was purified over silica gel by MPLC (Biotage Isolera, 0–4% DCM/MeOH) to give imidazopyrazinone as light yellow powder (150 mg, 65%) and imidazopyrazine as a light orange solid (21 mg, 9%). Major isomer imidazopyrazinone 25h: LCMS: Rt = 2.92 min, 99 A % @ 254 nm, [M + H]+ = 299.1. 1H NMR (600 MHz, DMSO-d6): δ 8.79 (s, 1H), 7.48 (d, J = 1.3 Hz, 1H), 7.17–7.08 (m, 4H), 5.24 (s, 2H), 2.27 (s, 3H), 2.20 (d, J = 1.3 Hz, 3H); 13C NMR (150 MHz, DMSO-d6): δ 153.6, 148.0, 136.4, 133.9, 133.5, 131.1, 129.2, 126.1, 115.8, 105.0, 45.5, 20.6, 16.5. HRMS (ESI) m/z: calcd for C15H14N4O3Na [M + Na]+: 321.0958, found 321.0957. Minor isomer imidazopyrazine 25i: LCMS: Rt = 3.26 min, 99 A % @ 254 nm, [M + H]+ = 299.1; 1H NMR (600 MHz, DMSO-d6): δ 8.94 (d, J = 0.9 Hz, 1H), 8.06 (t, J = 1.1 Hz, 1H), 7.43 (d, J = 7.8 Hz, 2H), 7.23 (d, J = 7.7 Hz, 2H), 5.49 (s, 2H), 2.36 (d, J = 1.1 Hz, 3H), 2.32 (s, 3H); 13C NMR (150 MHz, DMSO-d6): δ 152.9, 148.1, 137.7, 137.0, 132.7, 129.1, 129.0, 128.7, 114.3, 112.3, 68.1, 20.8, 20.4. HRMS (ESI) m/z: calcd for C15H14N4O3Na [M + Na]+: 321.0958, found 321.0958.
6-Methyl-2-nitro-7-(3-(trifluoromethoxy)benzyl)imidazo[1,2-a]pyrazin-8(7H)-one; 25j
Compound 25a (120 mg, 0.618 mmol), Cs2CO3, and 1-(bromomethyl)-3-(trifluoromethoxy)benzene (1.2 equiv) were reacted according to general procedure A with heating (μW 80 °C) for 10 min, work-up B. The crude material contained imidazopyrazinone (major) and imidazopyrazine (minor) isomers in a ratio of 4.3:1 by LC–MS (UV 254 nm). The crude product was purified over silica gel by MPLC (Biotage Isolera, 2–4% DCM/MeOH) to give imidazopyrazinone as white powder (122 mg, 54%) and imidazopyrazine as a white solid. Major isomer imidazopyrazinone 25j: LCMS: Rt = 3.49 min, 99 A % @ 254 nm, [M + H]+ = 368.8. 1H NMR (600 MHz, DMSO-d6): δ 8.80 (s, 1H), 7.51–7.45 (m, 2H), 7.32–7.23 (m, 3H), 5.33 (s, 2H), 2.20 (d, J = 1.3 Hz, 3H). 13C NMR (150 MHz, DMSO-d6): δ 153.7, 148.6, 147.9, 139.5, 134.0, 130.9, 130.7, 125.2, 120.0 (q, J = 256.9 Hz), 119.6, 119.1, 115.9, 105.2, 45.4, 16.6. HRMS (ESI) m/z: calcd for C15H11F3N4O4Na [M + Na]+, 391.0624; found, 391.0625. Minor isomer imidazopyrazine 25k: LCMS: Rt = 3.79 min, 65 A % @ 254 nm, [M + H]+ = 368.8; impurity detected at Rt = 4.03 min, 35 A % @ 254 nm, [M + H]+ = 737.2. This compound was not subjected to biological evaluation.
6-Methyl-2-nitro-7-(2-(trifluoromethoxy)benzyl)imidazo[1,2-a]pyrazin-8(7H)-one; 25l and 6-Methyl-2-nitro-8-((2-(trifluoromethoxy)benzyl)oxy)imidazo[1,2-a]pyrazine; 25m
Compound 25a (120 mg, 0.618 mmol), Cs2CO3, and 2-(trifluoromethoxy)benzyl bromide (1.2 equiv) were reacted according to general procedure A with heating (μW 80 °C) for 10 min, work-up B. The crude material contained imidazopyrazinone (major) and imidazopyrazine (minor) isomers in a ratio of 1.3:1 by LC–MS (UV 254 nm). The crude product was purified over silica gel by MPLC (Biotage Isolera, 0–4% DCM/MeOH) to yield imidazopyrazinone as white powder (76 mg, 33%) and imidazopyrazine as cream powder (cream powder, 18 mg, 8%). Major isomer imidazopyrazinone 25l: LCMS: Rt = 3.48 min, 99 A % @ 254 nm, [M + H]+ = 369.0; 1H NMR (600 MHz, DMSO-d6): δ 8.83 (s, 1H), 7.54 (d, J = 1.3 Hz, 1H), 7.49–7.42 (m, 2H), 7.36–7.30 (m, 1H), 7.11 (dt, J = 7.8, 1.0 Hz, 1H), 5.30 (s, 2H), 2.18 (d, J = 1.2 Hz, 3H). 13C NMR (150 MHz, DMSO-d6): δ 153.6, 148.0, 145.6, 133.9, 130.8, 129.2, 128.7, 127.9, 127.1, 120.8, 120.2 (q, J = 257.4 Hz), 116.0, 105.4, 41.3, 16.2. HRMS (ESI) m/z: calcd for C15H11F3N4O4Na [M + Na]+, 391.0625; found, 391.0613. Minor isomer imidazopyrazine 25m: LCMS: Rt = 3.78 min, 99 A % @ 254 nm, [M + H]+ = 369.0; 1H NMR (600 MHz, DMSO-d6): δ 8.96 (s, 1H), 8.09 (d, J = 1.3 Hz, 1H), 7.75 (dd, J = 7.6, 1.7 Hz, 1H), 7.57 (td, J = 7.9, 1.8 Hz, 1H), 7.51–7.44 (m, 2H), 5.63 (s, 2H), 2.36 (d, J = 1.1 Hz, 3H); 13C NMR (150 MHz, DMSO-d6): δ 152.5, 148.2, 146.8, 136.9, 131.4, 130.6, 128.9, 128.3, 127.7, 120.6, 120.1 (q, J = 256.4 Hz), 114.4, 111.6, 62.5, 20.3. HRMS (ESI) m/z: calcd for C15H11F3N4O4Na [M + Na]+, 391.0625; found, 391.0621.
5,6-Dimethyl-2-nitroimidazo[1,2-a]pyrazin-8(7H)-one; 26a
Imidazole-2-carboxamide 16a (750 mg, 3.89 mmol) was reacted according to general procedure E with 3-bromo-2-butanone (1.5 equiv) to yield a yellow solid (477 mg, 67%). LCMS: Rt = 2.57 min, 99 A % @ 254 nm, [M + H]+ = 209.1. 1H NMR (600 MHz, DMSO-d6): δ 11.55 (s, 1H), 8.82 (s, 1H), 2.32 (q, J = 1.0 Hz, 3H), 2.16 (q, J = 1.0 Hz, 3H). 13C NMR (150 MHz, DMSO-d6): δ 152.8, 147.9, 134.6, 124.6, 115.2, 111.0, 14.7, 12.5. HRMS (ESI) m/z: calcd for C16H16N8NaO6 [2M + Na]+, 439.1085; found, 439.1090.
6-Methyl-2-nitro-7-(4-(trifluoromethoxy)benzyl)imidazo[1,2-a]pyrazin-8(7H)-one; 26b and 5,6-Dimethyl-2-nitro-8-((4-(trifluoromethoxy)benzyl)oxy)imidazo[1,2-a]pyrazine; 26c
Compound 26a (150 mg, 0.721 mmol) was reacted according to general procedure A with K2CO3 and 1-(bromomethyl)4-(trifluoromethoxy)benzene (1.2 equiv) at rt overnight. The ratio of 26b/26c in the crude material was 3:1 as determined by LCMS. The crude solid was purified over silica gel by MPLC (Grace Reveleris X2, 0–7% MeOH in DCM gradient) to yield 26b as a yellow solid (178 mg, 69%) and 25c as a yellow solid (57 mg, 21%). Major isomer imidazopyrazinone 26b: LCMS: Rt = 3.61 min, 99 A % @ 254 nm, [M + H]+ = 383.1. 1H NMR (600 MHz, DMSO-d6): δ 8.93 (s, 1H), 7.38–7.31 (m, 4H), 5.37 (s, 2H), 2.43 (q, J = 1.0 Hz, 3H), 2.23 (q, J = 1.0 Hz, 3H). 13C NMR (150 MHz, DMSO-d6): δ 153.2, 148.1, 147.4, 136.3, 133.9, 128.2, 126.3, 121.4, 120.1 (q, J = 256.1 Hz), 115.3, 112.5, 45.7, 14.9, 13.9. HRMS (ESI) m/z: calcd for C16H14F3N4O4 [M + H]+, 383.0962; found, 383.0958. Minor isomer imidazopyrazine 26c: LCMS: Rt = 3.94 min, 99 A % @ 254 nm, [M + H]+ = 383.0. 1H NMR (600 MHz, DMSO-d6): δ 9.03 (s, 1H), 7.70–7.65 (m, 2H), 7.45–7.40 (m, 2H), 5.56 (s, 2H), 2.54 (q, J = 0.9 Hz, 3H), 2.40 (q, J = 0.9 Hz, 3H).·13C NMR (150 MHz, DMSO-d6): δ 150.8, 148.5, 148.1, 135.6, 132.8, 130.5, 129.4, 121.1, 120.6, 120.1 (q, J = 255.5 Hz), 113.2, 66.8, 19.6, 13.7. HRMS (ESI) m/z: calcd for C16H14F3N4O4 [M + H]+, 383.0962; found, 383.0961.
5,6-Dimethyl-7-(4-methylbenzyl)-2-nitroimidazo[1,2-a]pyrazin-8(7H)-one; 26d and 5,6-Dimethyl-8-((4-methylbenzyl)oxy)-2-nitroimidazo[1,2-a]pyrazine; 26e
Compound 24a (65 mg, 0.312 mmol) was reacted according to general procedure A with Cs2CO3 and 4-methylbenzyl bromide (1.2 equiv) with heating (μW 80 °C, 20 min then μW 90 °C, 10 min) followed by stirring overnight at rt, work up B. The ratio of 26d:26e in the crude material was 1.5:1 by LC–MS (UV 254 nm). The crude was purified over silica gel by MPLC (Biotage Isolera, 0–4% DCM/MeOH) to yield 26d as a yellow powder (39 mg, 40%) and 26e as a light yellow powder (18 mg, 19%). Major isomer imidazopyrazinone 26d: LCMS: Rt = 3.45 min, 99 A % @ 254 nm, [M + H]+ = 313.1. 1H NMR (600 MHz, DMSO-d6): δ 8.90 (s, 1H), 7.14 (d, J = 7.9 Hz, 2H), 7.08 (d, J = 8.1 Hz, 2H), 5.30 (s, 2H), 2.42 (d, J = 1.0 Hz, 3H), 2.27 (s, 3H), 2.22 (d, J = 1.0 Hz, 3H). 13C NMR (150 MHz, DMSO-d6): δ 153.2, 148.1, 136.4, 133.9, 133.7, 129.3, 126.5, 126.1, 115.2, 112.3, 46.0, 20.6, 14.8, 13.8. HRMS (ESI) m/z: calcd for C16H16N4O3Na [M + Na]+, 335.1115; found, 335.1116. Minor isomer imidazopyrazine 26e: Rt = 3.81 min, 99 A % @ 254 nm, [M + H]+ = 313.1. 1H NMR (600 MHz, DMSO-d6): δ 9.01 (s, 1H), 7.44–7.40 (m, 2H), 7.25–7.20 (m, 2H), 5.48 (s, 2H), 2.55–2.52 (m, 3H), 2.40 (d, J = 1.0 Hz, 3H), 2.32 (s, 3H). 13C NMR (150 MHz, DMSO-d6): δ 151.0, 148.4, 137.5, 132.9, 132.8, 129.4, 128.9, 128.7, 120.3, 113.1, 67.7, 20.8, 19.6, 13.6. HRMS (ESI) m/z: calcd for C16H16N4O3Na [M + Na]+, 335.1115; found, 335.1115.
5,6-Dimethyl-2-nitro-7-(3-(trifluoromethoxy)benzyl)imidazo[1,2-a]pyrazin-8(7H)-one; 26f and 5,6-Dimethyl-2-nitro-8-((3-(trifluoromethoxy)benzyl)oxy)imidazo[1,2-a]pyrazine; 26g
Compound 26a (120 mg, 0.576 mmol) was reacted according to general procedure A with Cs2CO3 and 1-(bromomethyl)-3-(trifluoromethoxy)benzene (1.2 equiv) with heating (μW 100 °C) for 10 min. The ratio of 26f/26g in the crude material was 1.2:1 by LC-MS (UV 254 nm). The crude material was purified over silica gel by MPLC (Biotage Isolera, 0–4% dichloromethane/methanol) to yield 26f as a light brown solid (131 mg, 60%) and 26g as a yellow gel (66 mg, 30%). Major isomer imidazopyrazinone 26f: LCMS: Rt = 3.56 min, 99 A % @ 254 nm, [M + H]+ = 383.1. 1H NMR (600 MHz, DMSO-d6): δ 8.93 (s, 1H), 7.51–7.43 (m, 1H), 7.29 (s, 1H), 7.28–7.21 (m, 2H), 5.39 (s, 2H), 2.43 (d, J = 1.1 Hz, 3H), 2.24–2.20 (m, 3H). 13C NMR (150 MHz, DMSO-d6): δ 153.3, 148.6, 148.1, 139.7, 133.9, 130.7, 126.2, 125.1, 120.0 (q, J = 256.2 Hz), 119.6, 119.0, 115.2, 112.4, 45.8, 14.9, 13.9. HRMS (ESI) m/z: calcd for C32H26F6N8O8Na [2M + Na]+, 787.1678; found, 787.1670. Minor isomer imidazopyrazine 26g: LCMS: Rt = 3.88 min, 99 A % @ 254 nm, [M + H]+ = 383.1. 1H NMR (600 MHz, DMSO-d6): δ 9.03 (s, 1H), 7.61–7.51 (m, 3H), 7.40–7.35 (m, 1H), 5.58 (s, 2H), 2.54 (s, 3H), 2.39 (s, 3H). 13C NMR (150 MHz, DMSO-d6): δ 150.7, 148.5, 148.3, 138.8, 132.7, 130.5, 129.4, 127.5, 120.9, 120.9, 120.7, 120.0 (q, J = 256.2 Hz), 113.2, 66.8, 19.6, 13.7. HRMS (ESI) m/z: calcd for C16H13F3N4O4Na [M + Na]+, 405.0797; found, 405.0781.
5,6-Dimethyl-2-nitro-7-(2-(trifluoromethoxy)benzyl)imidazo[1,2-a]pyrazin-8(7H)-one; 26h and 5,6-Dimethyl-2-nitro-8-((2-(trifluoromethoxy)benzyl)oxy)imidazo[1,2-a]pyrazine; 26i
Compound 26a (120 mg, 0.576 mmol) was reacted according to general procedure A with Cs2CO3 and 2-(trifluoromethoxy)benzyl bromide (1.2 equiv) with heating (μW 100 °C) for 10 min. The ratio of 26h/26i in the crude material was 1:2.1 by LC–MS (UV 254 nm). The crude material was purified over silica gel by MPLC (Biotage Isolera, 0–4% DCM/MeOH) to yield 26h as a light yellow powder (46 mg, 21%) and 26i as a light yellow powder (97 mg, 44%). Minor isomer imidazopyrazinone 26h: LCMS: Rt = 3.15 min, 99 A % @ 254 nm, [M + H]+ = 383.1. 1H NMR (600 MHz, DMSO-d6): δ 8.95 (s, 1H), 7.48–7.41 (m, 2H), 7.35–7.29 (m, 1H), 7.06 (dt, J = 8.0, 1.0 Hz, 1H), 5.36 (s, 2H), 2.45 (d, J = 1.0 Hz, 3H), 2.19 (d, J = 1.0 Hz, 3H). 13C NMR (150 MHz, DMSO-d6): δ 153.1, 148.1, 145.6, 133.8, 129.2, 128.9, 127.9, 127.1, 126.1, 120.9, 120.3 (q, J = 256.8 Hz), 115.3, 112.6, 41.7, 14.5, 13.9. HRMS (ESI) m/z: calcd for C16H13N4O4F3Na [M + Na]+, 405.0781; found, 405.0796. Major isomer imidazopyrazine 26i: LCMS: Rt = 3.44 min, 99 A % @ 254 nm, [M + H]+ = 383.1. 1H NMR (600 MHz, DMSO-d6): δ 9.03 (s, 1H), 7.74 (dd, J = 7.6, 1.8 Hz, 1H), 7.55 (td, J = 7.8, 1.8 Hz, 1H), 7.50–7.43 (m, 2H), 5.61 (s, 2H), 2.54 (d, J = 0.9 Hz, 3H), 2.39 (d, J = 1.0 Hz, 3H). 13C NMR (150 MHz, DMSO-d6): δ 150.6, 148.5, 146.8, 132.7, 131.3, 130.5, 129.3, 128.5, 127.7, 120.8, 120.6, 120.2 (q, J = 256.8 Hz), 113.2, 62.2, 19.6, 13.6. HRMS (ESI) m/z: calcd for C16H13N4O4F3Na [M + Na]+, 405.0781; found, 405.0793.
4-Nitro-1-(2-oxo-2-phenylethyl)-1H-imidazole-2-carboxamide; 27a
Compound 16a (1.20 g, 5.53 mmol), K2CO3, and 2-bromoacetophenone (1.1 equiv) were reacted according to general procedure A at rt for 1 h. The crude was purified over silica gel by MPLC (Grace Reveleris X2 15–100% EtOAc in pet. spirits gradient) to yield an orange oily-solid (907 mg, 60%). LCMS: Rt = 3.15 min, 98 A % @ 254 nm, [M + H]+ = 274.9. 1H NMR (600 MHz, DMSO-d6): δ 8.52 (s, 1H), 8.24 (s, 1H), 8.08–8.02 (m, 2H), 7.78–7.72 (m, 2H), 7.66–7.59 (m, 2H), 6.13 (s, 2H). Minor impurities in 1H NMR. 13C NMR (150 MHz, DMSO-d6): δ 191.9, 159.3, 144.5, 137.7, 134.3, 134.0, 129.1, 128.0, 126.4, 55.7. HRMS (ESI) m/z: calcd for C12H10N4NaO4 [M + Na]+, 297.0594; found, 297.0592.
Minimum Inhibition Concentration (MIC) Assay
All compounds were tested for activity against C. difficile bacteria (630, ATCC BAA-1382) and representative ESKAPE pathogens S. aureus (MRSA, ATCC 43300), E. coli (FDA control strain, ATCC 25922), K. pneumoniae (MDR, ATCC 700603), A. baumannii (type strain, ATCC 19606), and P. aeruginosa (quality control strain, ATCC 27853) and fungi C. albicans (CLSI reference, ATCC 90028) and C. neoformans (H99 type strain, ATCC 208821) using a standard broth microdilution assay essentially as previously described.49 MICs for each strain were determined as the lowest concentration without visible growth. Variance between replicates was typically within one twofold dilution. Median MICs are reported with a range given when the median MIC was between two tested concentrations.
M. tuberculosis H37Rv Minimum Inhibition Assays
The potency of the inhibitors was measured by a resazurin reduction microplate assay as previously described50,51 with some alterations. M. tuberculosis (H37Rv) was grown in Middlebrook 7H9 broth medium supplemented with ADC (Difco Laboratories), 0.5% glycerol, and 0.02% tyloxapol. Freshly seeded cultures were grown at 37 °C for approximately 14 days to mid-exponential phase (OD600 0.4–0.8) for use in the inhibition assays. Assay plates (96-well microtiter) were prepared with the compound serially diluted in 100 μL of 7H9S media (7H9 with 10% ADC, 0.5% glycerol, 0.05% Tween-80 and 1% tryptone). Mid-exponential phase culture of M. tuberculosis (OD600 0.4–0.8) was diluted to OD600 0.001 in 7H9S media, and 100 μL (representing ∼2 × 104 cfu/mL) was added to each well. For normoxic conditions, the plates were incubated for 5 days at 37 °C in a humidified incubator prior to the addition of 30 μL of a 0.02% resazurin solution and 12.5 μL of 20% Tween-80 to each well. After 24 h incubation (37 °C), sample fluorescence was measured on a Fluorostar Omega fluorescent plate reader (BMG) with an excitation wavelength of 530 nm and emission read at 590 nm. Percent fluorescence relative to the positive control wells (H37Rv without compound) minus the negative control wells (without H37Rv) was plotted for the determination of the MIC (≤90% reduction in growth). The assays were performed in replicate on independent occasions (n = 3–6). For hypoxic assays the same method was used except assay plates were incubated for 5 days at 0.1% oxygen, and after addition of the resazurin solution, the fluorescence was measured after a prolonged incubation time of 48 h.
Antiparasitic Assay: G. lamblia and E. histolytica
Compounds were screened for antiparasitic activity in a 96-well plate using an ATP-bioluminescence based assay for cell growth and survival as previously described.49 Briefly, trophozoites of G. lamblia (WB line52) and E. histolytica (HM1:IMSS) were axenically maintained in TYI-S-33 medium supplemented with penicillin (100 U/mL) and streptomycin (100 μg/mL).53 All experiments were performed using trophozoites harvested during the logarithmic phase of growth. Twofold serial dilutions were prepared from compound stocks (10 mM in DMSO) yielding a concentration range of 78 μM–10 mM. From this dilution plate, 0.5 μL from each well was transferred into 96-well microtitre plates followed by addition of 99.5 μL of trophozoites (5000 parasites) to yield a final 8-point concentration range spanning 0.39–50 μM. Assay plates were incubated for 24–48 h at 37 °C in the GasPak EZ Anaerobe Gas Generating Pouch Systems (VWR, West Chester, PA) to maintain anaerobic conditions throughout the incubation period. Viable cell numbers were determined in triplicate using the CellTiter-Glo Luminescent Cell Viability Assay.54
Antiparasitic Assay: T. b. brucei
Compounds were screened for antitrypanosomal activity against T. b. brucei (strain 427, BS427) using an established 384-well resazurin viability assay, as previously described.55−57 Briefly, 55 μL of HMI-9 media supplemented with 10% FCS containing 1200 cells/mL of logarithmic phase T. b. brucei bloodstream parasites were added to a 384-well microtiter plate (BD Biosciences, USA) and incubated for 24 h at 37 °C/5% CO2. Serial compound concentrations were prepared in 100% DMSO and diluted 1:21 in Dulbecco’s modified Eagles medium. Five microliters of these dilutions was subsequently added to assay plates to give final compound concentrations ranging from a top final assay concentration of 40 or 20 μM, to 4 × 10–3 μM or 2 × 10–3 μM, respectively. Plates were incubated for 48 h at 37 °C/5% CO2. Ten microlitres of 0.49 mM resazurin (Sigma-Aldrich, USA) in HMI-9 media + 10% FCS was added to assay plates and incubated for a further 2 h at 37 °C/5% CO2 followed by 22 h at room temperature. Final assay concentrations of puromycin (5 μM) and 0.4% DMSO were used as in-plate controls for all experiments. Assay plates were read at 535 nm excitation/590 nm emission on an Envision multiplate reader (PerkinElmer, USA). Nonlinear sigmoidal dose response curves with no constraints were plotted, and IC50 values were calculated in GraphPad Prism 6 (La Jolla, California, USA). The IC50 value was determined for compounds that exhibited a plateau of inhibition (above 90% inhibition at the top two concentrations) and were calculated from two independent experiments.
Antiparasitic Assay: L. donovani Intracellular Amastigote
Compounds were screened for antileishmanial activity in an established DD8 intracellular (THP-1) amastigote assay as previously described.57 Briefly, seven day old L. donovani MHOM/IN/80/DD8 (ATCC50212) culture containing metacyclic promastigotes was added to the 384-well assay plates containing transformed THP-1 cells (ATCC TIB202) at 72 h after the initial cell seeding with a multiplicity of infection of 1:5 (ratio host cells/parasites). Assay plates were incubated 24 h at 37 °C/5% CO2. Noninternalized parasites were removed by washing six times in PBS before the addition of 45 μL RMPI (10% FBS and 25 ng/mL phorbol 12-myristate 13-acetate). Five microliters of compounds was added to wells to give final assay concentrations ranging from 80 to 4 × 10–3 μM. Final assay concentrations of 2 μM of amphotericin B and 0.4% DMSO were used as in-plate controls for all experiments. Plates were incubated for 96 h at 37 °C/5% CO2, fixed with 4% paraformaldehyde, and stained with SYBR green and CellMask Deep Red plasma membrane dye. Images were acquired on an Opera high-content imaging system (PerkinElmer). Healthy host (THP-1) cells were identified based on fluorescent staining with CellMask Deep Red to characterize the cytoplasm in which the parasite resides and SYBR green to identify both host and parasite nuclei. An infected cell was defined as a host cell containing >3 parasites within the cytoplasm boundary. Nonlinear sigmoidal dose response curves with no constraints were plotted, and IC50 values were calculated in GraphPad Prism 6 (La Jolla, California, USA). The IC50 value was determined for compounds that exhibited a plateau of inhibition (above 90% inhibition at the top two concentrations) and were calculated from two independent experiments.
Mammalian Cytotoxicity Assay
Human HEK293 and HepG2 cells were seeded at 3000 and 5000 cells per well in 384-well plates, respectively. Cells were cultured in Dulbecco’s modified Eagle’s medium with 10% FBS for 24 h at 37 °C, 5% CO2. A dilution series of compounds was added, with the highest concentration of 100 μM. The final concentration of DMSO in culture media was 0.5%, which showed no effect on cell growth. After 24 h incubation with the compounds, 5 μM resazurin was added into each well and incubated at 37 °C for 2 h. As a negative control, 1% Triton X-100 was added into the culture media to lyse all of the cells. The fluorescence intensity was read using POLARstar Omega with excitation/emission 560/590 nm. Data were analyzed with GraphPad Prism 6 software (La Jolla, California USA) to calculate CC50 values.
Microsome Stability
Metabolic stability was determined using pooled human (HMMC-PL, Lot# PL050B–B, Thermo Fisher Scientific USA) and mouse (CD-1) (MSMCPL, Lot#MS033D, Thermo Fisher) liver microsomes. The test compound (3 μM, final DMSO concentration 0.2%) and liver microsomes (1 mg/mL) were mixed in 100 mM potassium phosphate buffer, pH 7.4 preincubated at 37 °C (500 μL volume). The reaction was initiated by addition of NADPH solution (cofactor) in 0.1 M potassium phosphate buffer at a final NADPH concentration of 1 mM. The reaction was incubated in a shaking incubator at 37 °C, 150 rpm. Aliquots (90 μL) from the reaction mixture were withdrawn (t = 0, 10, 30, 60, and 120 min) and quenched by adding ice-cold precipitating solution (270 μL) comprising 0.5 μM carbutamide internal standard in acetonitrile/methanol/formic acid (1:1:0.001 v/v/v). Reaction samples were incubated at 4 °C for 30 min and centrifuged at 14 000g for 8 min, and the clear supernatant was analyzed by LC/MS/MS. The percentage of the compound remaining at different times was calculated by comparing the peak area ratio of the parent compound (compound peak area/internal standard peak area) at the start of incubation (t = 0 min sample). All samples were tested in triplicate except for the control samples (without NADPH), matrix blank, and verapamil standard (time points = 0, 10 and 30 min). LC/MS/MS parameters are detailed in the Supporting Information Table S4.
Plasma Stability
Plasma stability studies were performed using human (HMPLNAHP, Lot#BRH1324758, BioReclamationIVT) and mouse (CD-1) plasma (MSEPLNAHP, Lot#MSE261215, BioReclamationIVT) at five different time points. A solution of plasma and PBS, pH 7.4 (50:50; v/v) were preheated at 37 °C for 30 min (240 μL volume). The reaction was initiated by the addition of the test compounds (3 μM, final DMSO concentration 1%), and the reaction was incubated in a shaking incubator at 37 °C, 150 rpm. Aliquots (40 μL) from the reaction mixture were withdrawn and processed as described for microsome stability assay. All samples were tested in triplicate, and eucatropine was used as a positive control.
Plasma Protein Binding
PPB was performed using an Ultrafiltration method.58,59 Fresh frozen human plasma was pooled from O-positive (product number 2799882) and O-negative (product number 5398256) blood from the R & D division of the Australian Red Cross Blood Services (Brisbane). Test compounds (5 μM) were incubated in 100% human plasma at 37 °C for 30 min (1 mL volume). For unfiltered samples, an aliquot (50 μL) was removed, diluted with PBS (50 μL), and quenched with ice-cold precipitating solution comprising 0.5 μM carbutamide MS internal standard in acetonitrile/methanol/formic acid (1:1:0.001). Samples were incubated at 4 °C for 30 min and then centrifuged at 14 000g for 8 min before the clear supernatant was transferred to a vial for LC/MS/MS analysis. For filtered samples, the plasma sample (250 μL) was filtered using Amicon Ultra-0.5 Centrifugal Filter Devices 30K NMWL at 14 000g for 7 min, and then an aliquot (50 μL) was processed as described for unfiltered samples. The fraction of unbound compounds was calculated by determining the concentration of the filtered sample and the concentration of the unfiltered sample. All samples were tested in triplicate with sulfamethoxazole as a control.
Caco-2 Permeability Assay
This study was conducted by WuXi AppTec Co. Ltd. (Shanghai). Caco-2 cells from ATCC were seeded onto polystyrene membranes in 96-well insert plate at 1 × 105 cells/cm2 until the formation of the confluent cell monolayer at 21st–28th day. Transport buffer used was Hanks’ balanced salt solution with 10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid, pH 7.4. Compounds (2 μM) were tested bidirectionally in duplicate. The plate was incubated for 2 h at 37 °C and 5% CO2 at saturated humidity without shaking. All samples were then mixed with acetonitrile containing internal standard and centrifuged at 4000 rpm for 20 min and the clear supernatant was diluted 1:1 with water prior to LC/MS/MS analysis. Test compounds and controls (fenoterol, propranolol, and digoxin) in starting solution, donor solution, and receiver solution were quantified using a peak area ratio of analyte/internal standard. To confirm the Caco-2 cell monolayer integrity after the transport assay, lucifer yellow rejection assay was performed. The apparent permeability coefficient Papp (cm/s) was calculated using the equation: Papp = (dCr/dt) × Vr/(A × C0) where dCr/dt is the cumulative concentration of the compound in the receiver chamber as a function of time (μM/s); Vr is the solution volume in the receiver chamber (0.075 mL on the apical side, 0.25 mL on the basolateral side); A is the surface area for the transport, that is, 0.0804 cm2 for the area of the monolayer; C0 is the initial concentration in the donor chamber (μM). The efflux ratio was calculated using the equation: efflux ratio = Papp (BA)/Papp (AB). Percent recovery was calculated using the equation: % recovery = 100 × [(Vr × Cr) + (Vd × Cd)]/(Vd × C0) where Vd is the volume in the donor chambers (0.075 mL on the apical side, 0.25 mL on the basolateral side); Cd and Cr are the final concentrations of the transport compound in donor and receiver chambers, respectively.
Solubility Determination
Stock compound solution (20 mM in DMSO) was aliquoted into water and PBS, pH 7.4 and 0.1 M HCl (pH 1) to a final concentration of 200 μM, 1% DMSO. After 24 h of incubation in a shaking incubator at room temperature, 130 rpm, solutions were filtered using centrifuge filter tubes (Corning Costar Spin-X centrifuge tube filters, CLS8169) at 8000 rpm for 1 min. The filtrates were further diluted with acetonitrile (1:1, v/v) prior to analysis using LC–UV as detailed in the general Experimental Section. The solubility was determined based on the peak area at UV absorbance 254 nm, with reference to the standard calibration curve prepared from 20 mM DMSO stock. Compounds and standards (caffeine and pretomanid) were prepared in duplicate, and each sample was analyzed in duplicate by LC–UV.
Acknowledgments
We thank Johnny Huang, Geraldine Kaeslin, and Janet Reid for assisting with the cytotoxicity assays and the Community for Open Antimicrobial Drug Discovery40 for performing MIC assays against the ESKAPE bacteria and fungal pathogens. We thank Alan Fairlamb and Stephen Patterson of the University of Dundee for providing the R-enantiomer of PA-824. The antimicrobial screening performed by CO-ADD was funded by the Wellcome Trust (UK) and The University of Queensland (Australia). AMJ was supported by an Australian Postgraduate Award, IMB Research Scholarship and QLD Government Smart Futures PhD scholarship and CWA by a Research Training Program scholarship. AD was supported by the National Institutes of Health (grant no.1KL2TR001444). MLS and AJ were supported by ARC grant LP140100560 and NHMRC Project grant APP1067728 awarded to VMA. MAB was supported by Wellcome Trust Strategic Award 104797/Z/14/Z and NHMRC Development Grant APP113719. MAC was supported by an NHMRC Principal Research Fellowship APP1059354.
Glossary
Abbreviations
- TB
tuberculosis
- MIC
minimal inhibitory concentration
- SAR
structure activity relationships
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem.8b01578.
Author Present Address
# Novasep (Dynamit Nobel Explosivstoff und Systemtechnik GmbH), Leverkusen, Germany.
Author Present Address
¶ Department of Chemical Biology, Helmholtz Centre for Infection Research, Inhoffenstrasse 7, 38124, Braunschweig, Germany.
Author Contributions
⊥ A.M.J. and C.W.A. contributed equally.
The authors declare the following competing financial interest(s): MAC currently holds a fractional Professorial Research Fellow appointment at the University of Queensland with his remaining time as CEO of Inflazome Ltd. a company headquartered in Dublin, Ireland that is developing drugs to address clinical unmet needs in inflammatory disease by targeting the inflammasome.
Supplementary Material
References
- Ang C. W.; Jarrad A. M.; Cooper M. A.; Blaskovich M. A. T. Nitroimidazoles: Molecular Fireworks That Combat a Broad Spectrum of Infectious Diseases. J. Med. Chem. 2017, 60, 7636–7657. 10.1021/acs.jmedchem.7b00143. [DOI] [PubMed] [Google Scholar]
- Schwebke J. R.; Morgan F. G.; Koltun W.; Nyirjesy P. A Phase-3, Double-Blind, Placebo-Controlled Study of the Effectiveness and Safety of Single Oral Doses of Secnidazole 2 g for the Treatment of Women with Bacterial Vaginosis. Am. J. Obstet. Gynecol. 2017, 217, 678.e1–678.e9. 10.1016/j.ajog.2017.08.017. [DOI] [PubMed] [Google Scholar]
- FDA Approves Symbiomix Therapeutics’ Solosec (Secnidazole) Oral Granules for the Treatment of Bacterial Vaginosis in Adult Women. https://symbiomix.com/fda-approves-symbiomix-therapeutics-solosec-secnidazole-oral-granules-treatment-bacterial-vaginosis-adult-women/ (accessed Sep 1, 2018).
- Traynor K. Benznidazole Approved for Chagas Disease in Children. Am. J. Health-Syst. Pharm. 2017, 74, 1519. 10.2146/news170065. [DOI] [PubMed] [Google Scholar]
- Simarro P. P.; Cecchi G.; Franco J. R.; Paone M.; Diarra A.; Ruiz-Postigo J. A.; Fèvre E. M.; Mattioli R. C.; Jannin J. G. Estimating and Mapping the Population at Risk of Sleeping Sickness. PLoS Neglected Trop. Dis. 2012, 6, e1859. 10.1371/journal.pntd.0001859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiberi S.; du Plessis N.; Walzl G.; Vjecha M. J.; Rao M.; Ntoumi F.; Mfinanga S.; Kapata N.; Mwaba P.; McHugh T. D.; Ippolito G.; Migliori G. B.; Maeurer M. J.; Zumla A. Tuberculosis: Progress and Advances in Development of New Drugs, Treatment Regimens, and Host-Directed Therapies. Lancet Infect. Dis. 2018, 18, e183–e198. 10.1016/s1473-3099(18)30110-5. [DOI] [PubMed] [Google Scholar]
- Geneva: World Health Organisation . Global Tuberculosis Report 2017, 2017; pp 1–262.
- Matsumoto M.; Hashizume H.; Tomishige T.; Kawasaki M.; Tsubouchi H.; Sasaki H.; Shimokawa Y.; Komatsu M. OPC-67683, a Nitro-dihydro-imidazooxazole Derivative with Promising Action Against Tuberculosis In Vitro and in Mice. PLoS Med. 2006, 3, e466. 10.1371/journal.pmed.0030466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki H.; Haraguchi Y.; Itotani M.; Kuroda H.; Hashizume H.; Tomishige T.; Kawasaki M.; Matsumoto M.; Komatsu M.; Tsubouchi H. Synthesis and Antituberculosis Activity of a Novel Series of Optically Active 6-Nitro-2,3-dihydroimidazo[2,1-b]oxazoles. J. Med. Chem. 2006, 49, 7854–7860. 10.1021/jm060957y. [DOI] [PubMed] [Google Scholar]
- Nagarajan K.; Shankar R. G.; Rajappa S.; Shenoy S.; Costa-pereira R. Nitroimidazoles XXI 2,3-Dihydro-6-nitroimidazo [2,1-b] Oxazoles with Antitubercular Activity. Eur. J. Med. Chem. 1989, 24, 631–633. 10.1016/0223-5234(89)90034-2. [DOI] [Google Scholar]
- Ashtekar D. R.; Costa-Perira R.; Nagrajan K.; Vishvanathan N.; Bhatt A. D.; Rittel W. In Vitro and In Vivo Activities of the Nitroimidazole CGI 17341 Against Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 1993, 37, 183–186. 10.1128/aac.37.2.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upton A. M.; Cho S.; Yang T. J.; Kim Y.; Wang Y.; Lu Y.; Wang B.; Xu J.; Mdluli K.; Ma Z.; Franzblau S. G. In Vitro and In Vivo Activities of the Nitroimidazole TBA-354 Against Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2015, 59, 136–144. 10.1128/aac.03823-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manjunatha U.; Boshoff H. I. M.; Barry C. E. The Mechanism of Action of PA-824: Novel Insights From Transcriptional Profiling. Commun. Integr. Biol. 2014, 2, 215–218. 10.4161/cib.2.3.7926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh R.; Manjunatha U.; Boshoff H. I. M.; Ha Y. H.; Niyomrattanakit P.; Ledwidge R.; Dowd C. S.; Lee I. Y.; Kim P.; Zhang L.; Kang S.; Keller T. H.; Jiricek J.; Barry C. E. PA-824 Kills Nonreplicating Mycobacterium tuberculosis by Intracellular NO Release. Science 2008, 322, 1392–1395. 10.1126/science.1164571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wayne L. G.; Hayes L. G. An In Vitro Model for Sequential Study of Shiftdown of Mycobacterium tuberculosis Through Two Stages of Nonreplicating Persistence. Infect. Immun. 1996, 64, 2062–2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson S.; Wyllie S.; Stojanovski L.; Perry M. R.; Simeons F. R. C.; Norval S.; Osuna-Cabello M.; De Rycker M.; Read K. D.; Fairlamb A. H. The R Enantiomer of the Antitubercular Drug PA-824 as a Potential Oral Treatment for Visceral Leishmaniasis. Antimicrob. Agents Chemother. 2013, 57, 4699–4706. 10.1128/aac.00722-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyllie S.; Roberts A. J.; Norval S.; Patterson S.; Foth B. J.; Berriman M.; Read K. D.; Fairlamb A. H. Activation of Bicyclic Nitro-Drugs by a Novel Nitroreductase (NTR2) in Leishmania. PLoS Pathog. 2016, 12, e1005971. 10.1371/journal.ppat.1005971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyllie S.; Patterson S.; Stojanovski L.; Simeons F. R. C.; Norval S.; Kime R.; Read K. D.; Fairlamb A. H. The Anti-Trypanosome Drug Fexinidazole Shows Potential for Treating Visceral Leishmaniasis. Sci. Transl. Med. 2012, 4, 119re1. 10.1126/scitranslmed.3003326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson A. M.; Marshall A. J.; Maes L.; Yarlett N.; Bacchi C. J.; Gaukel E.; Wring S. A.; Launay D.; Braillard S.; Chatelain E.; Mowbray C. E.; Denny W. A. Assessment of a Pretomanid Analogue Library for African Trypanosomiasis: Hit-to-Lead Studies on 6-Substituted 2-Nitro-6,7-dihydro-5H-imidazo[2,1-b][1,3]thiazine 8-oxides. Bioorg. Med. Chem. Lett. 2018, 28, 207–213. 10.1016/j.bmcl.2017.10.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson A. M.; Bonnet M.; Lee H. H.; Franzblau S. G.; Wan B.; Wong G. S.; Cooper C. B.; Denny W. A. Antitubercular Nitroimidazoles Revisited: Synthesis and Activity of the Authentic 3-Nitro Isomer of Pretomanid. ACS Med. Chem. Lett. 2017, 8, 1275–1280. 10.1021/acsmedchemlett.7b00356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim P.; Kang S.; Boshoff H. I.; Jiricek J.; Collins M.; Singh R.; Manjunatha U. H.; Niyomrattanakit P.; Zhang L.; Goodwin M.; Dick T.; Keller T. H.; Dowd C. S.; Barry C. E. Structure–Activity Relationships of Antitubercular Nitroimidazoles. 2. Determinants of Aerobic Activity and Quantitative Structure–Activity Relationships. J. Med. Chem. 2009, 52, 1329–1344. 10.1021/jm801374t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakshminarayana S. B.; Boshoff H. I. M.; Cherian J.; Ravindran S.; Goh A.; Jiricek J.; Nanjundappa M.; Nayyar A.; Gurumurthy M.; Singh R.; Dick T.; Blasco F.; Barry C. E.; Ho P. C.; Manjunatha U. H. Pharmacokinetics-Pharmacodynamics Analysis of Bicyclic 4-Nitroimidazole Analogs in a Murine Model of Tuberculosis. PLoS One 2014, 9, e105222. 10.1371/journal.pone.0105222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson A. M.; Blaser A.; Anderson R. F.; Shinde S. S.; Franzblau S. G.; Ma Z.; Denny W. A.; Palmer B. D. Synthesis, Reduction Potentials, and Antitubercular Activity of Ring A/B Analogues of the Bioreductive Drug (6S)-2-Nitro-6-{[4-(trifluoromethoxy)benzyl]oxy}-6,7-dihydro-5H-imidazo[2,1-b][1,3]-oxazine (PA-824). J. Med. Chem. 2009, 52, 637–645. 10.1021/jm801087e. [DOI] [PubMed] [Google Scholar]
- Drugs for Neglected Diseases initiative . Nitroimidazoles (VL)—DNDi. https://www.dndi.org/diseases-projects/portfolio/nitroimidazole/ (accessed Sep 1, 2018).
- Thompson A. M.; O’Connor P. D.; Marshall A. J.; Yardley V.; Maes L.; Gupta S.; Launay D.; Braillard S.; Chatelain E.; Franzblau S. G.; Wan B.; Wang Y.; Ma Z.; Cooper C. B.; Denny W. A. 7-Substituted 2-Nitro-5,6-dihydroimidazo[2,1-b][1,3]oxazines: Novel Antitubercular Agents Lead to a New Preclinical Candidate for Visceral Leishmaniasis. J. Med. Chem. 2017, 60, 4212–4233. 10.1021/acs.jmedchem.7b00034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson A. M.; O’Connor P. D.; Marshall A. J.; Blaser A.; Yardley V.; Maes L.; Gupta S.; Launay D.; Braillard S.; Chatelain E.; Wan B.; Franzblau S. G.; Ma Z.; Cooper C. B.; Denny W. A. Development of (6R)-2-Nitro-6-[4-(trifluoromethoxy)phenoxy]-6,7-dihydro-5H-imidazo[2,1-b][1,3]oxazine (DNDI-8219): A New Lead for Visceral Leishmaniasis. J. Med. Chem. 2018, 61, 2329–2352. 10.1021/acs.jmedchem.7b01581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson A. M.; Blaser A.; Palmer B. D.; Anderson R. F.; Shinde S. S.; Launay D.; Chatelain E.; Maes L.; Franzblau S. G.; Wan B.; Wang Y.; Ma Z.; Denny W. A. 6-Nitro-2,3-dihydroimidazo-[2,1-b][1,3]thiazoles: Facile Synthesis and Comparative Appraisal Against Tuberculosis and Neglected Tropical Diseases. Bioorg. Med. Chem. Lett. 2017, 27, 2583–2589. 10.1016/j.bmcl.2017.03.069. [DOI] [PubMed] [Google Scholar]
- Jarrad A. M.; Debnath A.; Miyamoto Y.; Hansford K. A.; Pelingon R.; Butler M. S.; Bains T.; Karoli T.; Blaskovich M. A. T.; Eckmann L.; Cooper M. A. Nitroimidazole Carboxamides as Antiparasitic Agents Targeting Giardia lamblia, Entamoeba histolytica and Trichomonas vaginalis. Eur. J. Med. Chem. 2016, 120, 353–362. 10.1016/j.ejmech.2016.04.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodacre S. C.; Hallett D. J.; Carling R. W.; Castro J. L.; Reynolds D. S.; Pike A.; Wafford K. A.; Newman R.; Atack J. R.; Street L. J. Imidazo[1,2-a]pyrazin-8-ones, Imidazo[1,2-d][1,2,4]-triazin-8-ones and Imidazo[2,1-f][1,2,4]triazin-8-ones as α2/α3 Subtype Selective GABAA Agonists for the Treatment of Anxiety. Bioorg. Med. Chem. Lett. 2006, 16, 1582–1585. 10.1016/j.bmcl.2005.12.027. [DOI] [PubMed] [Google Scholar]
- Mignani S.; Aloup J.-C.; Barreau M.; Blanchard J. C.; Böhme G. A.; Boireau A.; Damour D.; Debono M.-W.; Dubroeucq M.-C.; Genevois-Borella A.; Imperato A.; Jimonet P.; Pratt J.; Randle J. C. R.; Reibaud M.; Ribeill Y.; Stutzmann J.-M. Synthesis and Pharmacological Properties of 5H, 10H-imidazo[1,2-a]indeno[1,2-e]pyrazine-4-one, A New Competitive AMPA/KA Receptor Antagonist. Drug Dev. Res. 1999, 48, 121–129. 10.1002/(sici)1098-2299(199911)48:3<121::aid-ddr4>3.0.co;2-e. [DOI] [Google Scholar]
- Stutzmann J.-M.; Bohme G. A.; Boireau A.; Damour D.; Debono M. W.; Genevois-Borella A.; Imperato A.; Jimonet P.; Pratt J.; Randle J. C. R.; Ribeill Y.; Vuilhorgne M.; Mignani S. 4,10-Dihydro-4-oxo-4H-imidazo[1,2-a]indeno[1,2-e]pyrazin-2-carboxylic Acid Derivatives. Bioorg. Med. Chem. Lett. 2000, 10, 1133–1137. 10.1016/s0960-894x(00)00181-5. [DOI] [PubMed] [Google Scholar]
- Plouvier B. M. C.; Fedida D.; Beatch G. N.; Chou D. T. H.; Yifru A. S.; Jung G.. Imidazo Compounds and Uses Thereof. WO 2005/034837, April 21, 2005.
- Couturier C.; Silve S.; Morales R.; Pessegue B.; Llopart S.; Nair A.; Bauer A.; Scheiper B.; Pöverlein C.; Ganzhorn A.; Lagrange S.; Bacqué E. Nanomolar Inhibitors of Mycobacterium tuberculosis Glutamine Synthetase 1: Synthesis, Biological Evaluation and X-Ray Crystallographic Studies. Bioorg. Med. Chem. Lett. 2015, 25, 1455–1459. 10.1016/j.bmcl.2015.02.035. [DOI] [PubMed] [Google Scholar]
- Rao A. K. S. B.; Rao C. G.; Singh B. B. Regioselective Alkylation of 4(5)-Nitro-1H-imidazoles in Acidic Media: Study of Temperature Effects. J. Chem. Soc., Perkin Trans. 1 1994, 2399–2402. 10.1039/p19940002399. [DOI] [Google Scholar]
- Prévot I.; Leumann C. J. Evaluation of Novel Third-Strand Bases for the Recognition of a C·G Base Pair in the Parallel DNA Triple-Helical Binding Motif. Helv. Chim. Acta 2002, 85, 502–515. 10.1002/1522-2675(200202)85:2<502::aid-hlca502>3.0.co;2-l. [DOI] [Google Scholar]
- Plettenburg O.; Hofmeister A.; Kadereit D.; Brendel J.; Loehn M.. Cyclohexylamin Isoquinolone Derivatives as Rho-Kinase Inhibitors. WO 2007012422A1, Feb 1, 2007.
- Van der Eycken E.; Singh B.; Cavalluzzo C.; De Maeyer M.; Debyser Z.; Parmar V. Microwave-Assisted Silver(I)-Mediated Selective O-Alkylation of Aromatic Imidate Systems. Synthesis 2009, 16, 2725–2728. 10.1055/s-0029-1216878. [DOI] [Google Scholar]
- Semple G.; Andersson B.-M.; Chhajlani V.; Georgsson J.; Johansson M. J.; Rosenquist Å.; Swanson L. Synthesis and Biological Activity of Kappa Opioid Receptor Agonists. Part 2: Preparation of 3-Aryl-2-pyridone Analogues Generated by Solution- and Solid-Phase Parallel Synthesis Methods. Bioorg. Med. Chem. Lett. 2003, 13, 1141–1145. 10.1016/s0960-894x(03)00033-7. [DOI] [PubMed] [Google Scholar]
- Gurumurthy M.; Mukherjee T.; Dowd C. S.; Singh R.; Niyomrattanakit P.; Tay J. A.; Nayyar A.; Lee Y. S.; Cherian J.; Boshoff H. I.; Dick T.; Barry C. E. III; Manjunatha U. H. Substrate Specificity of the Deazaflavin-Dependent Nitroreductase from Mycobacterium tuberculosis Responsible for the Bioreductive Activation of Bicyclic Nitroimidazoles. FEBS J. 2011, 279, 113–125. 10.1111/j.1742-4658.2011.08404.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaskovich M. A. T.; Zuegg J.; Elliott A. G.; Cooper M. A. Helping Chemists Discover New Antibiotics. ACS Infect. Dis. 2015, 1, 285–287. 10.1021/acsinfecdis.5b00044. [DOI] [PubMed] [Google Scholar]
- Kim P.; Zhang L.; Manjunatha U. H.; Singh R.; Patel S.; Jiricek J.; Keller T. H.; Boshoff H. I.; Barry C. E. III; Dowd C. S. Structure–Activity Relationships of Antitubercular Nitroimidazoles. 1. Structural Features Associated with Aerobic and Anaerobic Activities of 4- and 5-Nitroimidazoles. J. Med. Chem. 2009, 52, 1317–1328. 10.1021/jm801246z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X.; Wright M.; Hop C. E. C. A. Rational Use of Plasma Protein and Tissue Binding Data in Drug Design. J. Med. Chem. 2014, 57, 8238–8248. 10.1021/jm5007935. [DOI] [PubMed] [Google Scholar]
- De Angelis I.; Turco L. Caco-2 Cells as a Model for Intestinal Absorption. Curr. Protoc. Toxicol. 2011, 47, 20.6.1–20.6.15. 10.1002/0471140856.tx2006s47. [DOI] [PubMed] [Google Scholar]
- Xavier A.; Lakshmanan M. Delamanid: A New Armor in Combating Drug-Resistant Tuberculosis. J. Pharmacol. Pharmacother. 2014, 5, 222–224. 10.4103/0976-500x.136121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsuno K.; Burrows J. N.; Duncan K.; van Huijsduijnen R. H.; Kaneko T.; Kita K.; Mowbray C. E.; Schmatz D.; Warner P.; Slingsby B. T. Hit and Lead Criteria in Drug Discovery for Infectious Diseases of the Developing World. Nat. Rev. Drug Discovery 2015, 14, 751–758. 10.1038/nrd4683. [DOI] [PubMed] [Google Scholar]
- Blaser A.; Palmer B. D.; Sutherland H. S.; Kmentova I.; Franzblau S. G.; Wan B.; Wang Y.; Ma Z.; Thompson A. M.; Denny W. A. Structure-Activity Relationships for Amide-, Carbamate-, And Urea-Linked Analogues of the Tuberculosis Drug (6S)-2-Nitro-6-{[4-(trifluoromethoxy)benzyl]oxy}-6,7-dihydro-5H-imidazo-[2,1-b][1,3]oxazine (PA-824). J. Med. Chem. 2011, 55, 312–326. 10.1021/jm2012276. [DOI] [PubMed] [Google Scholar]
- Sutherland H. S.; Blaser A.; Kmentova I.; Franzblau S. G.; Wan B.; Wang Y.; Ma Z.; Palmer B. D.; Denny W. A.; Thompson A. M. Synthesis and Structure–activity Relationships of Antitubercular 2-Nitroimidazooxazines Bearing Heterocyclic Side Chains. J. Med. Chem. 2010, 53, 855–866. 10.1021/jm901378u. [DOI] [PubMed] [Google Scholar]
- Dreier L.; Wider G. Concentration Measurements by PULCON Using X-Filtered or 2D NMR Spectra. Magn. Reson. Chem. 2006, 44, S206–S212. 10.1002/mrc.1838. [DOI] [PubMed] [Google Scholar]
- Jarrad A. M.; Karoli T.; Debnath A.; Tay C. Y.; Huang J. X.; Kaeslin G.; Elliott A. G.; Miyamoto Y.; Ramu S.; Kavanagh A. M.; Zuegg J.; Eckmann L.; Blaskovich M. A. T.; Cooper M. A. Metronidazole-Triazole Conjugates: Activity Against Clostridium difficile and Parasites. Eur. J. Med. Chem. 2015, 101, 96–102. 10.1016/j.ejmech.2015.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran A. T.; West N. P.; Britton W. J.; Payne R. J. Elucidation of Mycobacterium tuberculosis Type II Dehydroquinase Inhibitors using a Fragment Elaboration Strategy. ChemMedChem 2012, 7, 1031–1043. 10.1002/cmdc.201100606. [DOI] [PubMed] [Google Scholar]
- Taneja N. K.; Tyagi J. S. Resazurin Reduction Assays for Screening of Anti-Tubercular Compounds Against Dormant and Actively Growing Mycobacterium tuberculosis, Mycobacterium bovis BCG and Mycobacterium smegmatis. J. Antimicrob. Chemother. 2007, 60, 288–293. 10.1093/jac/dkm207. [DOI] [PubMed] [Google Scholar]
- Townson S. M.; Laqua H.; Upcroft P.; Boreham P. F. L.; Upcroft J. A. Induction of Metronidazole and Furazolidone Resistance in Giardia. Trans. R. Soc. Trop. Med. Hyg. 1992, 86, 521–522. 10.1016/0035-9203(92)90095-t. [DOI] [PubMed] [Google Scholar]
- Diamond L. S.; Harlow D. R.; Cunnick C. C. A New Medium for the Axenic Cultivation of Entamoeba histolytica and Other Entamoeba. Trans. R. Soc. Trop. Med. Hyg. 1978, 72, 431–432. 10.1016/0035-9203(78)90144-x. [DOI] [PubMed] [Google Scholar]
- Debnath A.; Parsonage D.; Andrade R. M.; He C.; Cobo E. R.; Hirata K.; Chen S.; García-Rivera G.; Orozco E.; Martínez M. B.; Gunatilleke S. S.; Barrios A. M.; Arkin M. R.; Poole L. B.; McKerrow J. H.; Reed S. L. A High-Throughput Drug Screen for Entamoeba histolytica Identifies a New Lead and Target. Nat. Med. 2012, 18, 956–960. 10.1038/nm.2758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sykes M. L.; Avery V. M. Development of an Alamar Blue Viability Assay in 384-Well Format for High Throughput Whole Cell Screening of Trypanosoma brucei brucei Bloodstream Form Strain 427. Am. J. Trop. Med. Hyg. 2009, 81, 665–674. 10.4269/ajtmh.2009.09-0015. [DOI] [PubMed] [Google Scholar]
- Sykes M. L.; Baell J. B.; Kaiser M.; Chatelain E.; Moawad S. R.; Ganame D.; Ioset J.-R.; Avery V. M. Identification of Compounds with Anti-Proliferative Activity Against Trypanosoma brucei brucei Strain 427 by a Whole Cell Viability Based HTS Campaign. PLoS Neglected Trop. Dis. 2012, 6, e1896. 10.1371/journal.pntd.0001896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffy S.; Sykes M. L.; Jones A. J.; Shelper T. B.; Simpson M.; Lang R.; Poulsen S.-A.; Sleebs B. E.; Avery V. M.. Screening the Medicines for Malaria Venture Pathogen Box Across Multiple Pathogens Reclassifies Starting Points for Open-Source Drug Discovery. Antimicrob. Agents Chemother. 2017, 61 ( (9), ). 10.1128/aac.00379-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F.; Xue J.; Shao J.; Jia L. Compilation of 222 Drugs’ Plasma Protein Binding Data and Guidance for Study Designs. Drug Discovery Today 2012, 17, 475–485. 10.1016/j.drudis.2011.12.018. [DOI] [PubMed] [Google Scholar]
- Di L.; Kerns E. H.. Plasma Protein Binding Methods. Drug-like Properties: Concepts, Structure Design and Methods; Academic Press: San Diego, CA, 2008; pp 372–377. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.