

Abstract
Systemic administration of antibiotics can cause severe side-effects such as liver and kidney toxicity, destruction of healthy gut bacteria, as well as multidrug resistance. Here, we present a bio-orthogonal chemistry-based strategy toward local prodrug concentration and activation. The strategy is based on the inverse electron-demand Diels–Alder chemistry between trans-cyclooctene and tetrazine and involves a biomaterial that can concentrate and activate multiple doses of systemic antibiotic therapy prodrugs at a local site. We demonstrate that a biomaterial, consisting of alginate hydrogel modified with tetrazine, is efficient at activating multiple doses of prodrugs of vancomycin and daptomycin in vitro as well as in vivo. These results support a drug delivery process that is independent of endogenous environmental markers. This approach is expected to improve therapeutic efficacy with decreased side-effects of antibiotics against bacterial infections. The platform has a wide scope of possible applications such as wound healing, and cancer and immunotherapy.
Short abstract
A biomaterial, consisting of alginate hydrogel modified with tetrazine, is efficient at local activation of multiple doses of prodrugs of vancomycin and daptomycin using bio-orthogonal chemistry.
Introduction
Bacterial infections are a critical health threat of our time.1,2 The standard approach to treat bacterial infections is through systemic administration of antibiotics. The choice of the antibiotic for a given infection is based on the minimum inhibitory concentration (MIC) profile of the targeted bacteria.3 The MIC is defined as the minimum concentration of an antibiotic that will inhibit bacterial growth in vitro during an overnight incubation.4 Historically, the MIC correlates with a specific dosing regimen (magnitude and frequency). Ideally, a supratherapeutic dose would be used to ensure complete elimination of the infection and to prevent emergence of antibiotic-resistant bacterial strains. However, high doses of systemic antibiotic therapy cause adverse side-effects including musculoskeletal, hepatic, and renal toxicity.5−7 There is a growing need for better antibiotics. We believe that improved drug delivery methods that supply high doses of antibiotics specifically to the area of infection would fulfill this need.
An example that underscores the aforementioned challenges is the eradication of implant-associated infections (IAIs).8 IAIs due to bacterial biofilms formed on the surface of soft tissues and medical devices are more resistant to conventional antibiotics than planktonic bacteria. Staphylococcus aureus is one of the most frequent germs causing biofilm-associated infections on the medical indwelling devices such as contact lenses, catheters, endotracheal tubes, mechanical heart valves, pacemakers, and prosthetic implants.9−12 Among S. aureus isolates the methicillin-resistant (MRSA) strains remain the most frequent multidrug-resistant hospital-associated infection. Microorganisms like S. aureus adhere to an extracellular polymeric matrix and develop into organized, complex communities with structural and functional heterogeneity, known as biofilms. Depletion of metabolic substances and accumulation of waste products causes biofilm bacteria to enter into a slow-growing state, rendering them more resistant to antibiotic drugs than their planktonic counterparts.13 Moreover, the spread of antibiotic-resistant strains, including MRSA isolates, further reduces therapeutic options. A drug delivery method capable of supplying multiple high doses of antibiotics directly to the location of the biofilm infected implant would greatly improve the efficacy of IAI treatment.
It has recently been shown that biocompatible polymers, strategically implanted in the vicinity of the infected part of the body, can serve as local depots of large doses of medication.14 Currently, the TYRX envelope15 is the only clinically approved implantable mesh that is impregnated with rifampin and minocyclin. Biocompatible hydrogels have also been explored to achieve local release of antibiotics.16−18 Hydrogels are a particularly important class of modern drug delivery materials due to their physical similarity to the physiological environment.19 They can be engineered to resemble the native interstitial matrix. The biocompatible material has a high water content (70–99%), and the physical properties, such as stiffness, porosity, and shape, can be tuned for different medical applications.20−22 The reported hydrogel systems release drugs either by spontaneous diffusion or through controlled or uncontrolled hydrogel degradation. These systems have a key limitation—they cannot be reloaded. The course of treatment is predetermined at the time of hydrogel implantation. Additional invasive procedures are necessary to alter the course of treatment or to provide additional medication. Moreover, the systems are usually extremely specific to the therapeutic being delivered.
A number of approaches have emerged in recent years to address this problem. In 2014, we presented our first step toward the construction of an implantable biomaterial based on click chemistry for targeting small molecules and showed that we could enhance the delivery of a suitable radiolabeled molecules by an order of magnitude to an implanted polysaccharide.23 Mooney and co-workers designed a hydrogel drug-reloading system based on nucleic acid sequence complementarity.24 In the described construct, small molecule drugs were modified with DNA strands that were captured by the complementary strands attached to the implanted hydrogel. Chen et al. reported self-healing hydrogel enabling reloading via diffusive chemical transport across the gel–gel interface.25 Hsieh and co-workers described a reloadable system involving hydrogel suspended anti-PEG immunoglobulin M (IgM) antibodies that were capable of catching pegylated proteins.26 None of these systems, however, have been successfully applied in vivo to tackle bacterial infections.
Herein, we report a reloadable hydrogel system that is capable of activating antibiotic prodrugs at the site of bacterial infection. It is based on the previously reported local drug activation approach that combines the temporal flexibility of systemic drug delivery and the spatial control of injectable biomaterials and could enhance the localization of small molecule therapeutics.27 Our previous report demonstrated that the approach is capable of local activation of therapeutically meaningful quantities of a chemotherapeutic agent. The approach has been applied to treat soft-tissue sarcoma xenografts in mice and showed superior efficacy and significantly lower side-effects than conventional doxorubicin standard of care treatment. In this report, we demonstrate that the local drug activation approach is indeed reloadable, and its versatility can be utilized to tackle planktonic and biofilm infections.
To illustrate our approach, we chose two FDA-approved antibiotics, vancomycin and daptomycin that have widespread clinical significance in the treatment of S. aureus infections, including MRSA strains, and whose versatility is limited by harmful side-effects. Vancomycin is a tricyclic glycopeptide antibiotic that is indicated to fight severe infections caused by Gram-positive bacteria. It has a well-understood mechanism of action involving inhibition of bacterial cell wall biosynthesis. Vancomycin is often used as a drug of “last resort” against microorganisms that proved resistant against other antimicrobial agents.28 Vancomycin is also used in the treatment of planktonic bacteria in the surrounding of the device-related infections.29,30 Wider indication of vancomycin is limited due to its adverse effects like hypotension and tachycardia, phlebitis, nephrotoxicity, and ototoxicity.31 Meanwhile, daptomycin is a cyclic lipopeptide antibiotic. Its activity is dependent on the presence of Ca2+ that facilitates oligomerization and insertion into bacterial membranes. Oligomers of daptomycin are thought to form toxic pores inside of bacterial membranes causing membrane depolarization. Daptomycin has a narrow therapeutic window. It is approved at a dose of 4 mg/kg for the treatment of complicated skin and soft-tissue infection and at a dose of 6 mg/kg for S. aureus bloodstream infection.32 Daptomycin has recently been reported to be effective against biofilms and therefore is a promising option for IAI treatment.30 However, during phase 1 clinical trials, higher doses of daptomycin such as 8 mg/kg led to unacceptable adverse effects involving the musculoskeletal system with accompanying increases in creatine phosphokinase levels.33 Moreover, its applications to joint infections have been limited due to limited biodistribution and the aforementioned side-effect profile.34 In this work we will show that systemically administered prodrugs of vancomycin and daptomycin can be converted into active antibiotics at the site of bacterial infection using bio-orthogonal chemistry.
Results and Discussion
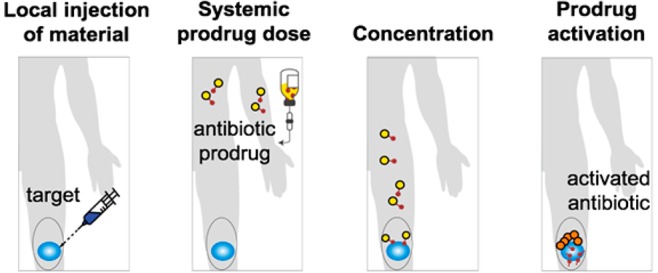
Our local prodrug activation approach is based on the inverse electron-demand Diels–Alder (IEDDA) chemistry between trans-cyclooctene (TCO) and tetrazine (Tz). During the past decade, this chemistry has become increasingly popular for in vivo bioconjugation due to fast reaction kinetics and inertness of the two bio-orthogonal groups to endogenous biomolecules and bioprocesses.35,36 IEDDA chemistry has been shown to be virtually nontoxic and highly effective under physiological conditions.37 As illustrated in Figure 1, the strategy starts with the injection of a biocompatible tetrazine-modified alginate gel (TAG) at the site of an infection. A prodrug, synthesized by covalent modification of a clinically tested antibiotic with a releasable TCO moiety, is injected intravenously (Figure 1B). When the prodrug reaches the site of the hydrogel, the bio-orthogonal agents (TCO and Tz) react with each other through the IEDDA chemistry, concentrating the therapeutic payload at the site of an infection (Figure 1C).27,38 Finally, the resulting intermediate isomerizes spontaneously releasing the active antibiotic from the hydrogel to perform its therapeutic function locally (Figure 1D). The multivalency of Tz on the hydrogel’s surface allows catching of multiple doses of systemically administered TCO-modified compounds.
Figure 1.

A bio-orthogonal chemistry-based strategy for concentration and activation of systemically administered antibiotic prodrugs. (A) Tz-modified alginate gel (TAG) is injected into the infected area. (B) An antibiotic, covalently modified with a TCO (prodrug), is given to the patient. (C) When the prodrug and the TAG come in contact, the IEDDA reaction enhances the amount of antibiotic present near the infected site. (D) The resulting cycloaddition product spontaneously isomerizes, releasing an equivalent of carbon dioxide and most importantly the active antibiotic.
To evaluate the strategy, TAG was prepared by modifying an ultrapure medium viscosity (>200 mPa s) sodium alginate, containing a minimum of 60% guluronate monomer units, with the Tz (Figure S1). Based on 1H NMR analysis, TAG contained about 400 nmol of Tz per mg of the material (∼8% of alginate monomers modified). The extent of Tz loading reflects the maximum amount of prodrugs that can theoretically be activated. To determine if Tz incorporation altered the flow properties of alginate, the viscosities of 2.5% w/v aqueous solutions of TAG and unmodified gel (UG) were measured. In general, aqueous polysaccharide solutions are known to behave as non-Newtonian fluids, such that their dynamic viscosities decrease in response to an increase in the applied shear rate. Consistently, both TAG and UG displayed such a shear-thinning or pseudoplastic behavior (Figure S2). Furthermore, zeta (ζ) potential, a surrogate measurement for surface charge, at 1 mg/mL dilution in distilled water at pH 7.0 was found to be −78.6 ± 5.2 mV for TAG and −75.6 ± 5.7 mV for UG. These values suggest that the low extent of modification with Tz did not affect the inherent surface charge of the alginate gel. Additionally, the high magnitude of ζ (>±30 mV) of these gels indicates that they are stable in water and do not aggregate, as consistent with the literature.
The prodrugs of vancomycin and daptomycin, shown in Scheme 1, were synthesized by covalent modification of the parent antibiotics with the releasable TCO moiety. The detailed syntheses are illustrated in Figures S9 and S16. Vancomycin was modified at the N-terminus which is involved in binding the C-terminal portion of bacterial cell wall precursor peptides ending in the sequence −Lys–d-Ala–d-Ala. This binding sequesters substrates necessary for construction of the bacterial cell wall, eventually causing cell death. Extensive studies by Williams have shown that acylation of the N-terminus of vancomycin causes a 17-fold decrease in antibiotic activity.39 Daptomycin was converted into a prodrug by modifying the most reactive, ornithine residue (Orn6). The precise role of this residue in daptomycin’s mechanism of action is not fully understood. However, Marahiel reported that a prodrug formed by acylation of the ornithine residue is 4 times less active.40 Due to its well-known reactivity and documented significance, Orn6 was chosen as the site of attachment of the releasable TCO group.
Scheme 1. Prodrugs of Vancomycin and Daptomycin Modified with the Releasable TCO Group.

In Vitro Prodrug Activation
TAG can efficiently activate multiple doses of prodrugs of vancomycin and daptomycin, TCO–Vanco and TCO–Dapto, under simulated physiological conditions. When 25 mg of TAG and 5 nmol of either prodrug were mixed in phosphate buffered saline for 2 h at room temperature, over 99% of the compounds detected in the supernatant were activated vancomycin and daptomycin as shown by LC-MS and HPLC analyses (Figure 2A). Subsequent measurements over a 24 h period detected only the release of the corresponding antibiotic. Calibration of the acquired spectra revealed that TCO–Vanco and TCO–Dapto are rapidly captured by TAG and that over 80% of the activated antibiotics are released within 24 h (Figure 2B). To test the ability of TAG to be reloaded, the hydrogel was treated with second and third doses of 5 nmol of TCO–Vanco and TCO–Dapto 24 and 48 h after the first dose. As illustrated in Figure 2, TAG is capable of efficiently “catching” multiple doses of prodrugs and subsequently releasing their corresponding activated antibiotics. The kinetics of drug release after the second and the third doses followed essentially the same trend as those seen after the first dose.
Figure 2.

In vitro activation of TCO–Vanco and TCO–Dapto by TAG. (A) HPLC analysis of in vitro activation of TCO–Vanco. A 25 mg portion of TAG was treated with the 5 nmol doses of TCO–Vanco at 0, 24, and 48 h. (B) Cumulative release of vancomycin after mixing TAG with the three doses of TCO–Vanco. (C) LC-MS analysis of in vitro activation of TCO–Dapto. A 25 mg portion of TAG was treated with the 5 nmol doses of TCO–Dapto at 0, 24, and 48 h. (D) Cumulative release of daptomycin after mixing TAG with the three doses of TCO–Dapto.
Microcalorimetric Analysis of Inhibition of Growth of Planktonic Bacteria
Isothermal microcalorimetry experiments using two laboratory strains of S. aureus (MSSA and MRSA) confirmed efficient in vitro activation of the antibiotic prodrugs by TAG. The experiments were based on the known principle that metabolically active (live and reproducing) bacteria produce heat that can be measured using an isothermal calorimeter.41 Heat generated by MSSA and MRSA bacteria when treated with different concentrations of TCO–Vanco or TCO–Dapto in the presence of either TAG or UG was plotted as a function of time (Figures S4–S7). Analogous experiments using the parent antibiotics were carried out as a positive control (Figure S9). An in-depth data analysis is described in Tables S1 and S2. A threshold of 4 μW for total heat production was selected as a marker of bacterial growth, and the time required for bacteria heat to reach that threshold was measured as “time to detection” (TTD). For example, bacteria without any antibiotic treatment were the fastest to reach the heat threshold and hence had a low TTD (Figure 3). Conversely, higher TTD values indicate a delay in heat production due to bacterial susceptibility to antibiotics. Inability to reach the 4 μW threshold within 24 h corresponds to 99% inhibition of bacterial growth. The TTD was assigned to be nondetectable (ND), and the minimum antibiotic dosage required to achieve 99% inhibition of bacterial growth was considered as the minimum heat inhibitory concentration (MHIC), the term which is analogous to MIC.
Figure 3.

TTD values corresponding to MRSA and MSSA bacteria treated with different concentrations of TCO–Vanco and TCO–Dapto in the presence of TAG or UG. (A) TTD values corresponding to MSSA bacteria treated with different concentrations of TCO–Vanco in the presence of TAG or UG. (B) TTD values corresponding to MRSA bacteria treated with different concentrations of TCO–Vanco in the presence of TAG or UG. (C) TTD values corresponding to MSSA bacteria treated with different concentrations of TCO–Dapto in the presence of TAG or UG. (D) TTD values corresponding to MRSA bacteria treated with different concentrations of TCO–Dapto in the presence of TAG or UG. Statistics was done with two-way ANOVA followed by multiple comparisons with Sidak’s post-test. An overall p-value less than 0.05 was accepted as significant. Adjusted p-values are indicated for individual comparisons as •• p < 0.05, ** p < 0.005, # p < 0.001, ○ p < 0.0001. Error bars represent standard error over mean (SEM) of n = 3–6. The minimum dosage at which TTD became nondetectable (ND) is marked as the MHIC. Blue and red correspond to MHIC of prodrug with TAG and UG, respectively.
Figure 3 shows TTD values corresponding to MRSA and MSSA bacteria treated with different concentrations of TCO–Vanco and TCO–Dapto in the presence of TAG or UG (negative control). In all cases, the untreated bacteria was able to reach the heat production threshold within 5 h. Increasing prodrug dosage consistently resulted in a delay in heat production and higher TTDs.
Figure 3 illustrates that bacterial treatment in the presence of TAG resulted in significantly higher TTD values, relative to UG. For example, MRSA treated with 2 μg/mL TCO–Vanco and TAG showed TTD of 15 h, while the same treatment with UG had a TTD of 9 h (Figure 3B). The difference was even more dramatic when MRSA was treated with 0.5 μg/mL TCO–Dapto. TTD of 23 h was observed with TAG, while the same treatment with UG had a TTD of 4 h (Figure 3D). Analogous trends were observed with MSSA treated with TCO–Vanco and TCO–Dapto. The MHIC values derived from these data are listed in Table 1. The MHIC values indicate that the antibiotic prodrugs have higher antibiotic activity when activated in the presence of TAG.
Table 1. Minimum Heat Inhibitory Concentration (MHIC) Measured from the Total Heat Produced by Bacteria at 24 h.
| MHIC
for MSSA [μg/mL] |
activity improvement [%] | ||
|---|---|---|---|
| TAG | UG | (UG – TAG)/UG | |
| TCO–Vanco | 3 | 4 | 25% |
| TCO–Dapto | 1 | 3.5 | 71% |
| MHIC
for MRSA [μg/mL] |
activity improvement [%] | ||
|---|---|---|---|
| TAG | UG | (UG – TAG)/UG | |
| TCO–Vanco | 2.5 | 3 | 16% |
| TCO–Dapto | 1 | 3 | 67% |
Microcalorimetric Analysis of Inhibition of Growth of Biofilm Bacteria
Among S. aureus strains, MRSA remains the most prevalent microorganism associated with hospital infections.42 We tested the ability of our prodrug activation system to eradicate MRSA biofilms grown on porous glass beads. First the beads were immersed in either TAG or UG and then incubated with different concentrations of daptomycin or TCO–Dapto. TCO–Vanco and vancomycin were not tested as the antibiotic has limited efficacy against S. aureus biofilms.43 After antibiotic treatment, the beads were sonicated, and the sonication fluid was plated on agar plates. The colony-forming units (CFUs) were counted after 24 h. The results are shown in Figure 4 as CFU/mL. The lowest antibiotic concentration that inhibited colony formation on the agar plate was defined as the minimum biofilm eradication concentration (MBEC).44 TCO–Dapto is significantly more effective at eliminating MRSA biofilms in the presence of TAG. MBEC was achieved at 64 μg/mL for TCO–Dapto with the tetrazine-modified gel, which is comparable to the standard daptomycin treatment. Meanwhile in the presence of UG, the TCO–Dapto was unable to eradicate biofilms even at concentrations of 256 μg/mL.
Figure 4.

Treatment of MRSA biofilm with variable concentrations of daptomycin and TCO–Dapto in the presence of TAG and UG. The data show colony-forming unit (CFU)/mL as a function of antibiotic concentration (μg/mL). Statistical analysis was done with one-way ANOVA (*p < 0.0001), and error bars indicate standard error measurement of n = 4.
Elimination of S. aureus Infection in Mice
The hypothesis that TAG is reloadable and capable of concentrating and activating antibiotic prodrugs at a location of choice was first tested using a fluorescent model system. TAG or UG (negative control) were implanted at the dorsum of mice, while TCO–TAMRA was administered systemically. Figure S8 shows that TAG was able to enhance the local delivery of the fluorescent payload over days, after multiple doses. The areas injected with UG or without a gel injection (negative controls) did not have an equivalent enhancement of fluorescence, confirming that the bio-orthogonal reaction is the reason for the enhancement. Moreover, the fact that the fluorescence decreased to background levels in all cases confirmed that the cargo was not trapped indefinitely but rather was concentrated and activated (released from the material).
To study the in vivo behavior of TAG, we synthesized a gel to contain Cyanine 5.5 dye (0.732 nmol/mg) in addition to Tz. We found that TAG injected in the thigh muscle remains at the site of the injection for at least 3 months (Figure 5). Fluorescence was observed throughout the study, and representative images of mice immediately postinjection, on days 7 and 90, are shown (Figure 5A). The fluorescence signal observed on day 7 showed a gradual decrease over time up to 58% on day 90 (Figure 5B). Postnecropsy relative fluorescence biodistribution showed Cy5.5 signal in the lymph nodes and liver, indicating that TAG is likely cleared from the body, with a preference for lymphatic and hepatic routes over renal clearance (Figure 5C). Histopathological analysis of the gel site showed the presence of infiltrating macrophages without any associated fibrosis or cell death, indicating a mild inflammatory response only (Figure 5D). Together, these results suggest that TAG undergoes gradual biodegradation, persists at the injection site for at least 90 days, and does not induce a fibrotic inflammatory response. These findings suggest that TAG is suitable for extended treatment durations.
Figure 5.

In vivo behavior of TAG after injection. TAG covalently modified with Cy 5.5 was injected in the thigh muscle of a mouse (100 μL of 2.5% w/w solution). (A) The animal was imaged at different time points confirming the location of the hydrogel. (B) Percent of gel remaining determined by fluorescence intensity of gel injection site relative to day 7 postinjection value, error bars represent SEM of n = 3, paired one-way ANOVA, Dunnett’s post-test **p < 0.005, *p < 0.05. (C) Fluorescence distribution in organs. (D) Histopathological analysis of the gel site. Unpaired t test, error bars represent SEM of n = 3.
The total therapeutic benefit of the platform comes from two components. On one hand we have the activation component. This can be determined by the difference of activity (MHIC) of the prodrug with TAG or UG as shown in Table 1. On the other hand, it is challenging to determine the benefits of the concentration step in vitro as it is hard to model the amount of prodrug that would be concentrated at the area labeled with TAG. In this study we used an in vivo efficacy study using a bioluminescent MRSA strain45 and TCO–Vanco, a prodrug that has a limited benefit from the activation factor as determined by the difference between the MHIC against MRSA when used with TAG and UG of only 16% (Table 1). Fluorescently labeled TAG and UG were synthesized to contain ∼0.5% Cy5 loading (w/w), in addition to tetrazine in the case of TAG. Bioluminescent MRSA was injected along with the Cy5-modified hydrogels into the thighs of neutropenic mice. Mice were subsequently administered a systemic dose of TCO–Vanco. Figure 6A shows that the bioluminescent bacteria was completely eradicated within 24 h of receiving TCO–Vanco. Analogous treatments involving UG or saline in combination with TCO–Vanco were unsuccessful at clearing the infection. Upon acquiring the in vivo images, mice were sacrificed, and bacterial load of the harvested thigh tissue was assessed in terms of colony-forming units (CFU) obtained per gram of the harvested tissue. As illustrated in Figure 6B, treatment with TAG and TCO–Vanco resulted in a 800-fold reduction of bacterial load relative to UG and TCO–Vanco. The difference of almost 3 orders of magnitude in eliminating bacteria surpasses the 16% improvement by the activation that was observed in vitro (Table 1). These results suggest that TAG is very efficient at concentrating prodrugs from the plasma circulation at the desired location.
Figure 6.
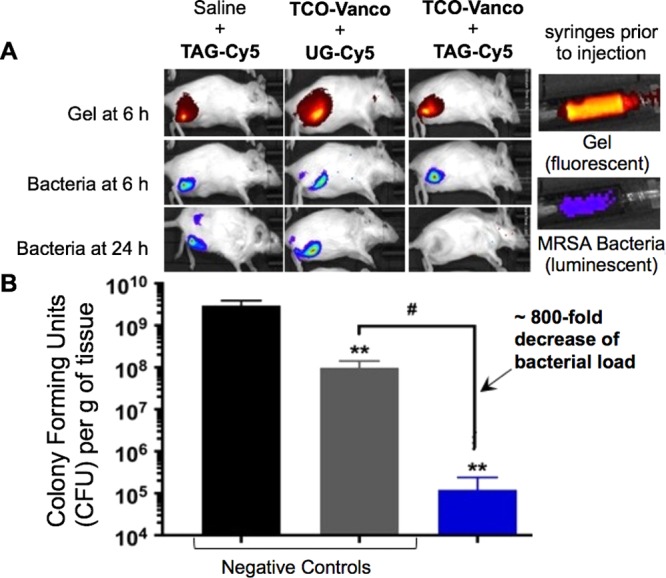
TAG + TCO–Vanco eliminates bacterial infection in 24 h. (A) Fluorescence images of mice injected with Cy5-labeled (red) modified or unmodified gels and MRSA bacteria (blue) at 6 and 24 h. (B) Bacterial loading of the harvested tissue from the respective mice. Statistical analysis was done with unpaired Welch’s t test, and error bars indicate standard deviation of means of n = 3. ** p < 0.005 compared to saline, # p < 0.005 compared to unmodified alginate-Cy5 control.
This is consistent with the unexpected results seen in our previous work with chemotherapeutic prodrugs, where Tz-mediated activation of 10 daily prodrug doses led to a sustained remission of tumors.27 Future studies will evaluate directly the pharmacokinetics of these antibiotic prodrugs as well as other prodrugs to quantify the effect directly.
Conclusion
Herein we have described a hydrogel-based reloadable platform that utilizes bio-orthogonal IEDDA chemistry to achieve local activation of systemically administered antibiotic prodrugs. The local drug activation platform was shown to be capable of activating multiple doses of prodrugs of vancomycin and daptomycin in vitro. Isothermal microcalorimetry experiments showed that the prodrugs activated by TAG were significantly more potent at inhibiting bacterial growth than the corresponding negative controls using unmodified hydrogel. In vivo experiments showed that treatment of TAG and TCO–Vanco is more effective than UG and TCO–Vanco, in eradicating luminescent MRSA infections.
A number of elements of the local drug activation platform require further optimization. First, the prodrug of daptomycin requires structural modification, as TCO–Dapto was found to have poor aqueous solubility, which limited its in vivo testing. We plan to report our efforts in this area shortly. In addition, we envision that other prodrug modifications would additionally attenuate the antibacterial activity of the resulting prodrug, as daptomycin’s mode of action heavily relies on its hydrophobicity for incorporation into bacterial membranes.46,47 This will be advantageous, as it will allow administration of higher systemic dosages of prodrugs toward local activation in the area of interest. These strategies will undoubtedly be pursued in subsequent studies.
Acknowledgments
The authors would like to thank the National Science Foundation and the National Institutes of Health for their generous support of this work. In particular, this work was funded in part by the National Science Foundation Grant 1664577 to M.R. and a phase II SBIR Grant 1660258 to J.M.M.O. This work was also funded by the National Institutes of Health Grant 1R21CA228997-01 to M.R. and a phase I Grant 1R43GM119864-01A1 to J.M.M.O. The authors would also like to thank the staff of the Life Sciences Research Building at the University at Albany for their help in acquiring the experimental data.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscentsci.8b00344.
Synthesis and characterization of TAG and the prodrugs of vancomycin and daptomycin; description of microcalorimetry and kinetic experiments; and detailed analysis of microcalorimetry data (PDF)
The authors declare the following competing financial interest(s): Jose M. Mejia Oneto is the founder of Shasqi, Inc. Nathan A. Yee and Sangeetha Srinivasan are employed by Shasqi, Inc. Its core technology is based on the research described herein.
Notes
Safety statement: the MRSA and MSSA bacteria described in this work belong to biosafety Level 2, as defined by the U.S. Public Health Services Guidelines. Handling of these pathogens requires Level 2 biosafety-compliant facilities, as increased concentration of bacteria poses significant risk of exposure.
Supplementary Material
References
- Carlet J.; Collignon P.; Goldmann D.; Goossens H.; Gyssens I. C.; Harbarth S.; Jarlier V.; Levy S. B.; N’Doye B.; Pittet D.; Richtmann R.; Seto W. H.; van der Meer J. W.; Voss A. Society’s failure to protect a precious resource: antibiotics. Lancet 2011, 378 (9788), 369–371. 10.1016/S0140-6736(11)60401-7. [DOI] [PubMed] [Google Scholar]
- Martinez J. L. Antibiotics and antibiotic resistance genes in natural environments. Science 2008, 321 (5887), 365–367. 10.1126/science.1159483. [DOI] [PubMed] [Google Scholar]
- O’Neill A. J.; Chopra I. Preclinical evaluation of novel antibacterial agents by microbiological and molecular techniques. Expert Opin. Invest. Drugs 2004, 13 (8), 1045–63. 10.1517/13543784.13.8.1045. [DOI] [PubMed] [Google Scholar]
- Andrews J. M. Determination of minimum inhibitory concentrations. J. Antimicrob. Chemother. 2001, 48 (S1), 5–16. 10.1093/jac/48.suppl_1.5. [DOI] [PubMed] [Google Scholar]
- Brooks B. D.; Brooks A. E. Therapeutic strategies to combat antibiotic resistance. Adv. Drug Delivery Rev. 2014, 78, 14–27. 10.1016/j.addr.2014.10.027. [DOI] [PubMed] [Google Scholar]
- Wu P.; Grainger D. W. Drug/device combinations for local drug therapies and infection prophylaxis. Biomaterials 2006, 27 (11), 2450–2467. 10.1016/j.biomaterials.2005.11.031. [DOI] [PubMed] [Google Scholar]
- Brooks B.; Brooks A.; Grainger D. W.. Antimicrobial technologies in preclinical and clinical medical devices. In Biomaterials associated infection: immunological aspects and antimicrobial strategies; Springer: NY, 2012; pp 307–354. [Google Scholar]
- Zimmerli W.; Trampuz A.; Ochsner P. E. Prosthetic-joint infections. N. Engl. J. Med. 2004, 351 (16), 1645–1654. 10.1056/NEJMra040181. [DOI] [PubMed] [Google Scholar]
- Donlan R. M. Biofilms and device-associated infections. Emerging Infect. Dis. 2001, 7 (2), 277–281. 10.3201/eid0702.010226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto M. Staphylococcal biofilms. Microbiol. Spectrum 2018, 6 (4), 1–17. 10.1128/microbiolspec.GPP3-0023-2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trampuz A.; Zimmerli W. Diagnosis and treatment of infections associated with fracture-fixation devices. Injury 2006, 37 (2), S59–S66. 10.1016/j.injury.2006.04.010. [DOI] [PubMed] [Google Scholar]
- Trampuz A.; Zimmerli W. Diagnosis and treatment of implant-associated septic arthritis and osteomyelitis. Curr. Infect. Dis. Rep. 2008, 10 (5), 394–403. 10.1007/s11908-008-0064-1. [DOI] [PubMed] [Google Scholar]
- Trampuz A.; Zimmerli W. Prosthetic joint infections: update in diagnosis and treatment. Swiss Med. Wkly. 2005, 135 (17–18), 243–251. 10.4414/smw.2005.10934. [DOI] [PubMed] [Google Scholar]
- Kearney C. J.; Mooney D. J. Macroscale delivery systems for molecular and cellular payloads. Nat. Mater. 2013, 12 (11), 1004–1017. 10.1038/nmat3758. [DOI] [PubMed] [Google Scholar]
- Hansen L. K.; Berg K.; Johnson D.; Sanders M.; Citron M. Efficacy of local rifampin/minocycline delivery (AIGIS(RX)(R)) to eliminate biofilm formation on implanted pacing devices in a rabbit model. Int. J. Artif. Organs 2010, 33 (9), 627–635. 10.1177/039139881003300908. [DOI] [PubMed] [Google Scholar]
- Romano C. L.; Scarponi S.; Gallazzi E.; Romano D.; Drago L. Antibacterial coating of implants in orthopaedics and trauma: a classification proposal in an evolving panorama. J. Orthop. Surg. Res. 2015, 10, 157. 10.1186/s13018-015-0294-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drago L.; De Vecchi E.; Bortolin M.; Toscano M.; Mattina R.; Romano C. L. Antimicrobial activity and resistance selection of different bioglass S53P4 formulations against multidrug resistant strains. Future Microbiol. 2015, 10 (8), 1293–1299. 10.2217/FMB.15.57. [DOI] [PubMed] [Google Scholar]
- Li W.; Dong K.; Ren J.; Qu X. A beta-lactamase-imprinted responsive hydrogel for the treatment of antibiotic-resistant bacteria. Angew. Chem., Int. Ed. 2016, 55 (28), 8049–8053. 10.1002/anie.201600205. [DOI] [PubMed] [Google Scholar]
- Li J.; Mooney D. J. Designing hydrogels for controlled drug delivery. Nat. Rev, Mater. 2016, 1, 16071. 10.1038/natrevmats.2016.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvert P. Hydrogels for soft machines. Adv. Mater. 2009, 21 (7), 743–756. 10.1002/adma.200800534. [DOI] [Google Scholar]
- Li J.; Illeperuma W. R. K.; Suo Z.; Vlassak J. J. Hybrid hydrogels with extremely high stiffness and toughness. ACS Macro Lett. 2014, 3 (6), 520–523. 10.1021/mz5002355. [DOI] [PubMed] [Google Scholar]
- Bodugoz-Senturk H.; Macias C. E.; Kung J. H.; Muratoglu O. K. Poly(vinyl alcohol)–acrylamide hydrogels as load-bearing cartilage substitute. Biomaterials 2009, 30 (4), 589–596. 10.1016/j.biomaterials.2008.10.010. [DOI] [PubMed] [Google Scholar]
- Mejia Oneto J. M.; Gupta M.; Leach J. K.; Lee M.; Sutcliffe J. L. Implantable biomaterial based on click chemistry for targeting small molecules. Acta Biomater. 2014, 10 (12), 5099–5105. 10.1016/j.actbio.2014.08.019. [DOI] [PubMed] [Google Scholar]
- Brudno Y.; Silva E. A.; Kearney C. J.; Lewin S. A.; Miller A.; Martinick K. D.; Aizenberg M.; Mooney D. J. Refilling drug delivery depots through the blood. Proc. Natl. Acad. Sci. U. S. A. 2014, 111 (35), 12722–12727. 10.1073/pnas.1413027111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J.; Li S.; Zhang Y.; Wang W.; Zhang X.; Zhao Y.; Wang Y.; Bi H. A Reloadable self-healing hydrogel enabling diffusive transport of C-dots across gel-gel interface for scavenging reactive oxygen species. Adv. Healthcare Mater. 2017, 6, 1700746. 10.1002/adhm.201700746. [DOI] [PubMed] [Google Scholar]
- Wu J. P.; Cheng B.; Roffler S. R.; Lundy D. J.; Yen C. Y.; Chen P.; Lai J. J.; Pun S. H.; Stayton P. S.; Hsieh P. C. Reloadable multidrug capturing delivery system for targeted ischemic disease treatment. Sci. Transl. Med. 2016, 8 (365), 365ra160. 10.1126/scitranslmed.aah6228. [DOI] [PubMed] [Google Scholar]
- Mejia Oneto J. M.; Khan I.; Seebald L.; Royzen M. In vivo bioorthogonal chemistry enables local hydrogel and systemic pro-drug to treat soft tissue sarcoma. ACS Cent. Sci. 2016, 2 (7), 476–482. 10.1021/acscentsci.6b00150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okano A.; Isley N. A.; Boger D. L. Total syntheses of vancomycin-related glycopeptide antibiotics and key analogues. Chem. Rev. 2017, 117 (18), 11952–11993. 10.1021/acs.chemrev.6b00820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith K.; Perez A.; Ramage G.; Gemmell C. G.; Lang S. Comparison of biofilm-associated cell survival following in vitro exposure of meticillin-resistant Staphylococcus aureus biofilms to the antibiotics clindamycin, daptomycin, linezolid, tigecycline and vancomycin. Int. J. Antimicrob. Agents 2009, 33 (4), 374–378. 10.1016/j.ijantimicag.2008.08.029. [DOI] [PubMed] [Google Scholar]
- Corona Perez-Cardona P. S.; Barro Ojeda V.; Rodriguez Pardo D.; Pigrau Serrallach C.; Guerra Farfan E.; Amat Mateu C.; Flores Sanchez X. Clinical experience with daptomycin for the treatment of patients with knee and hip periprosthetic joint infections. J. Antimicrob. Chemother. 2012, 67 (7), 1749–1754. 10.1093/jac/dks119. [DOI] [PubMed] [Google Scholar]
- Bruniera F. R.; Ferreira F. M.; Saviolli L. R. M.; Bacci M. R.; Feder D.; da Luz Goncalves Pedreira M.; Sorgini Peterlini M. A.; Azzalis L. A.; Campos Junqueira V. B.; Fonseca F. L. A. The use of vancomycin with its therapeutic and adverse effects: a review. Eur. Rev. Med. Pharmacol. Sci. 2015, 19 (4), 694–700. [PubMed] [Google Scholar]
- Bassetti M.; Nicco E.; Ginocchio F.; Ansaldi F.; de Florentiis D.; Viscoli C. High-dose daptomycin in documented Staphylococcus aureus infections. Int. J. Antimicrob. Agents 2010, 36 (5), 459–461. 10.1016/j.ijantimicag.2010.07.011. [DOI] [PubMed] [Google Scholar]
- Eisenstein B. I.; Oleson F. B. Jr.; Baltz R. H. Daptomycin: from the mountain to the clinic, with essential help from Francis Tally, MD. Clin. Infect. Dis. 2010, 50 (s1), S10–S15. 10.1086/647938. [DOI] [PubMed] [Google Scholar]
- Byren I.; Rege S.; Campanaro E.; Yankelev S.; Anastasiou D.; Kuropatkin G.; Evans R. Randomized controlled trial of the safety and efficacy of Daptomycin versus standard-of-care therapy for management of patients with osteomyelitis associated with prosthetic devices undergoing two-stage revision arthroplasty. Antimicrob. Agents Chemother. 2012, 56 (11), 5626–5632. 10.1128/AAC.00038-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devaraj N. K.; Thurber G. M.; Keliher E. J.; Marinelli B.; Weissleder R. Reactive polymer enables efficient in vivo bioorthogonal chemistry. Proc. Natl. Acad. Sci. U. S. A. 2012, 109 (13), 4762–4767. 10.1073/pnas.1113466109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devaraj N. K.; Weissleder R. Biomedical applications of tetrazine cycloadditions. Acc. Chem. Res. 2011, 44 (9), 816–827. 10.1021/ar200037t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Versteegen R. M.; Ten Hoeve W.; Rossin R.; de Geus M. A. R.; Janssen H. M.; Robillard M. S. Click-to-release from trans-cyclooctenes: mechanistic insights and expansion of scope from established carbamate to remarkable ether cleavage. Angew. Chem., Int. Ed. 2018, 57 (33), 10494–10499. 10.1002/anie.201800402. [DOI] [PubMed] [Google Scholar]
- Rossin R.; van Duijnhoven S. M. J.; Ten Hoeve W.; Janssen H. M.; Kleijn L. H. J.; Hoeben F. J. M.; Versteegen R. M.; Robillard M. S. Triggered drug release from an antibody-drug conjugate using fast ″click-to-release″ chemistry in mice. Bioconjugate Chem. 2016, 27 (7), 1697–1706. 10.1021/acs.bioconjchem.6b00231. [DOI] [PubMed] [Google Scholar]
- Kannan R.; Harris C. M.; Harris T. M.; Waltho J. P.; Skelton N. J.; Williams D. H. Function of the amino sugar and N-terminal amino acid of the antibiotic vancomycin in its complexation with cell wall peptides. J. Am. Chem. Soc. 1988, 110 (9), 2946–2953. 10.1021/ja00217a042. [DOI] [Google Scholar]
- Grunewald J.; Sieber S. A.; Mahlert C.; Linne U.; Marahiel M. A. Synthesis and derivatization of daptomycin: a chemoenzymatic route to acidic lipopeptide antibiotics. J. Am. Chem. Soc. 2004, 126 (51), 17025–17031. 10.1021/ja045455t. [DOI] [PubMed] [Google Scholar]
- Braissant O.; Bonkat G.; Wirz D.; Bachmann A. Microbial growth and isothermal microcalorimetry: growth models and their application to microcalorimetric data. Thermochim. Acta 2013, 555 (SC), 64–71. 10.1016/j.tca.2012.12.005. [DOI] [Google Scholar]
- Lakhundi S.; Zhang K. Methicillin-resistant Staphylococcus aureus: molecular characterization, evolution, and epidemiology. Clin. Microbiol. Rev. 2018, 31 (4), 1–103. 10.1128/CMR.00020-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravn C.; Furustrand Tafin U.; Betrisey B.; Overgaard S.; Trampuz A. Reduced ability to detect surface-related biofilm bacteria after antibiotic exposure under in vitro conditions. Acta Orthop. 2016, 87 (6), 644–650. 10.1080/17453674.2016.1246795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez Moreno M.; Trampuz A.; Di Luca M. Synergistic antibiotic activity against planktonic and biofilm-embedded Streptococcus agalactiae, Streptococcus pyogenes and Streptococcus oralis. J. Antimicrob. Chemother. 2017, 72 (11), 3085–3092. 10.1093/jac/dkx265. [DOI] [PubMed] [Google Scholar]
- Plaut R. D.; Mocca C. P.; Prabhakara R.; Merkel T. J.; Stibitz S. Stably luminescent Staphylococcus aureus clinical strains for use in bioluminescent imaging. PLoS One 2013, 8 (3), e59232. 10.1371/journal.pone.0059232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senneville E.; Caillon J.; Calvet B.; Jehl F. Towards a definition of daptomycin optimal dose: Lessons learned from experimental and clinical data. Int. J. Antimicrob. Agents 2016, 47 (1), 12–19. 10.1016/j.ijantimicag.2015.11.005. [DOI] [PubMed] [Google Scholar]
- Baltz R. H. Daptomycin: mechanisms of action and resistance, and biosynthetic engineering. Curr. Opin. Chem. Biol. 2009, 13 (2), 144–151. 10.1016/j.cbpa.2009.02.031. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.