

Abstract
Employing live cells as therapeutics is a direction of future drug discovery. An easy and robust method to modify the surfaces of cells directly to incorporate novel functionalities is highly desirable. However, genetic methods for cell-surface engineering are laborious and limited by low efficiency for primary cell modification. Here we report a chemoenzymatic approach that exploits a fucosyltransferase to transfer bio-macromolecules, such as an IgG antibody (MW∼ 150 KD), to the glycocalyx on the surfaces of live cells when the antibody is conjugated to the enzyme’s natural donor substrate GDP-Fucose. Requiring no genetic modification, this method is fast and biocompatible with little interference to cells’ endogenous functions. We applied this method to construct two antibody–cell conjugates (ACCs) using both cell lines and primary cells, and the modified cells exhibited specific tumor targeting and resistance to inhibitory signals produced by tumor cells, respectively. Remarkably, Herceptin-NK-92MI conjugates, a natural killer cell line modified with Herceptin, exhibit enhanced activities to induce the lysis of HER2+ cancer cells both ex vivo and in a human tumor xenograft model. Given the unprecedented substrate tolerance of the fucosyltransferase, this chemoenzymatic method offers a general approach to engineer cells as research tools and for therapeutic applications.
Short abstract
Here we report a single-step approach to construct the antibody−cell conjugate. The modified cells exhibited novel functions of specific tumor targeting or resistance to inhibitory signals.
Molecules presented on the cell surface determine how cells interact with their partners and their environment. Methods for engineering the cell-surface landscape are instrumental for the study of cell–cell communications and the downstream signaling. Such methods also have brought breakthroughs to therapeutic intervention.1 The most remarkable example is Kymriah, a chimeric antigen receptor T-cell (CAR-T) therapy that was approved recently as the first cell-based gene therapy in the United States for the treatment of patients with B-cell precursor acute lymphoblastic leukemia.2,3
The major technical challenge in cell engineering is to confer new properties to the manipulated cells with little interference with the cells’ endogenous functions. As the most common and robust cell-engineering approach, genetic engineering is limited by technical complications and safety concerns (Figure 1A), such as the inconsistent reproducibility of viral transduction efficiency of primary cells, heterogeneous expression levels, and the potential for endogenous gene disruption.4−6 Therefore, engineering cell surfaces from “outside” using chemical biology tools has emerged as a complementary and generally applicable approach.7,8 Preeminent examples include metabolic oligosaccharide engineering (MOE) developed by Bertozzi et al. (Figure 1A) and sortagging, the transpeptidation reaction catalyzed by bacterial sortases, among others.7,9−11 MOE requires a two-step procedure, combining metabolic labeling with bioorthogonal chemistry to endow cell-surface glycans with new functions. Sortagging involves only a single-step treatment. However, cells without genetic modification only have a few thousand naturally exposed glycine residues that can be functionalized by this approach.12 Therefore, direct functionalization of the cell surface in a noninvasive and highly efficient way is still difficult to achieve.7,11 Cell engineering would benefit from a single-step method that efficiently modifies native substrates on the surface of cells to incorporate novel functionalities.
Figure 1.

One-step fucosylation-based strategy for cell-surface engineering: (A) Two representative cell-surface engineering approaches. Metabolic engineering is used to install a reaction handle (X) onto the surface of the cell, which can react with a complementary handle (Y) on a molecule of interest. Genetic engineering allows the direct expression of functional molecules or the installation of reaction handles (X) on the surfaces of cells. (B) This work reports on an enzymatic glycoengineering approach capable of transferring a variety of functional molecules to the surfaces of cells in one step. The reaction between LacNAc/sialyl LacNAc and GDP-Fucose derivatives on the surfaces of cells is enabled by H. pylori α1,3FucT that tolerates modifications as large as a whole IgG conjugated at the C6 position of fucose. (C) One-pot protocol for the synthesis of GF-Al and GF-Az derivatives. The new functional group (Z) conjugated to fucose includes bioorthogonal handles (tetrazine, Tz), biophysical probes (biotin, Cy3), and biomaterials (ssDNA).
In situ glycan editing via glycosylation enzymes is a single-step approach to modify glycocalyx on the cell surface. The most notable example of its application is ex vivo fucosylation of mesenchymal stem cells and regulatory T cells using GDP-Fucose (GF) and recombinant human α(1,3)-fucosyltransferase (FucT) VI to convert cell-surface α2,3 sialyl LacNAc (Neu5NAcα2,3Galβ1,4GlcNAc) residues into sialyl Lewis X.13,14 This procedure, currently undergoing several clinical trials, improves adhesion, homing, and engraftment of adoptively transferred cells. However, enzymatic glycoengineering on the cell surface has not been widely used in therapeutic interventions.7 A major limitation is that current enzymatic transferable substrates are confined to small, synthetic molecules (MW < 5000),15−17 while biopolymers (e.g., monoclonal antibodies, mAbs) that have high therapeutic value are not accessible.
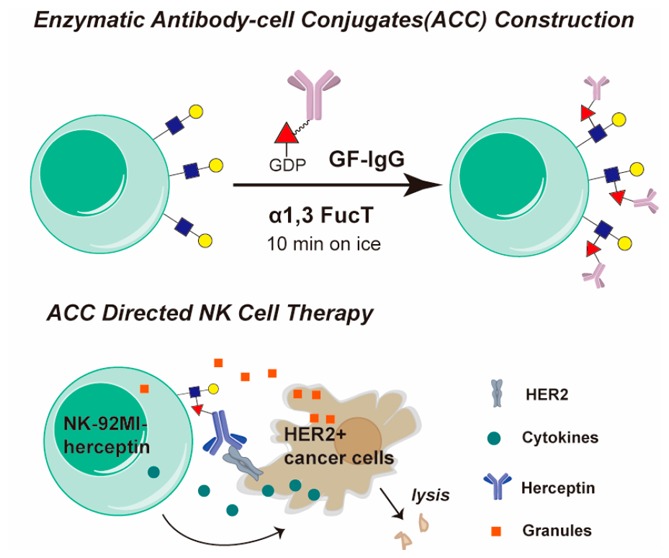
Here, we report the discovery of the remarkable substrate tolerance of Helicobacter pylori 26695 α1,3FucT. This enzyme enables quantitative transfer of a full-length IgG antibody conjugated to the GDP-Fucose donor to LacNAc and α2,3 sialyl LacNAc, common building blocks of glycocalyx, on the cell surface of live cells within a few minutes (Figure 1B). A one-pot protocol that couples the synthesis of an unnatural GDP-Fucose derivative to the subsequent transfer of the derivative was developed and made this engineering approach practical and cost-effective. Using this technique, we constructed two types of antibody–cell conjugates (ACCs) using a natural killer cell line (NK-92MI) and primary CD8+ OT-1 T cells. We demonstrated, for the first time, the application of this technique to boost the activities of modified immune cells, including specific tumor targeting and resistance to inhibitory signals produced by tumor cells.
Results and Discussion
One-Pot Protocol for Preparing and Transferring GDP-Fucose Derivatives
To develop the enzyme-based glycan modification as a general method for cell-surface engineering, a practical and scalable approach for the preparation and transfer of nucleotide sugar donors equipped with new functional groups is required.18 We discovered that GDP-l-6-ethynylfucose (GF-Al) or GDP-l-6-azidofucose (GF-Az) produced in situ can be coupled directly with a wide variety of probes using the ligand accelerated copper(I)-catalyzed alkyne–azide cycloaddition (CuAAC)19−21 (Figure 1C). These probes include biotin, a fluorescent probe Cy3, a bioorthogonal reaction handle tetrazine (Tz), and a dye-labeled (fluorescein amidite, FAM), single-strand DNA (ssDNA) (Supporting Information, Figure S1). All reactions attained near quantitative yields (>90%), and the crude products were rendered biocompatible for direct transfer by α1,3 FucT onto the cell surface after quenching the reaction with the FDA-approved copper chelator bathocuproine sulfonate (BCS) (Supporting Information, Figure S2). Compared to the conventional two-step labeling protocol,16 i.e., enzymatic transfer followed by cell-surface click chemistry, the one-step enzymatic labeling using the substrate of one-pot product was significantly more efficient and biocompatible (Supporting Information, Figure S3). In addition, we found that the enzymatic transfer of the one-pot Tz derivative made from GF-Az was more efficient than that made from GF-Al (Supporting Information, Figure S2D).
Sortagging is probably the best known enzymatic covalent ligation reaction without the need for genetic manipulation of the target cell population.12,22 We directly compared the efficiency of the FucT-mediated cell-surface modification with that catalyzed by sortase (SrtA 5M) using biotin-conjugated substrates. At the optimal substrate concentration (500 μM biotin-LPETG), negligible transpeptidation reaction took place within 2 h in the presence of 1 μM sortase (Supporting Information, Figure S4A). By contrast, 0.6 μM FucT afforded robust cell-surface labeling within 2 min in the presence of 50 μM GDP-Fucose–biotin (Supporting Information, Figure S4B). Even at 20 μM enzyme concentration, it took 120 min for sortagging to reach signal saturation.22 Moreover, when 0.6 μM FucT and 20 μM sortase were used for cell-surface modification, respectively, the labeling intensity of the FucT-catalyzed process was found to be at least 80 times higher than that of the sortase-catalyzed process (Supporting Information, Figure S4A).
Enzymatic Transfer of Full-Length IgG Molecules to the Cell Surface Using FucT
The remarkable efficiency of fucosylation to transfer small-molecule probes with diverse structures to the cell surface combined with the previous known plasticity of mammalian sialyltransferases for protein PEGylation suggests the possibility that FucT may tolerate even larger molecules conjugated to the fucose C6 position.16,23 To assess this possibility, we conjugated GDP-Fucose with monoclonal antibodies (mAbs, full-length IgG), the fastest growing class of protein drugs. The bioorthogonal handle trans-cyclooctene (TCO) with a PEG linker was installed onto mAbs or their isotype controls via standard amine-coupling procedures.24 Subsequently, mAbs bearing TCO moieties were reacted with GF-Az-Tz via the highly efficient inverse electron-demand Diels–Alder reaction (IEDDA)25−27 to generate GDP-Fucose-conjugated IgG molecules (GF-IgG) (Figure 2A). GDP-Fucose-modified antibodies were characterized by MALDI-TOF MS and were found to exhibit similar antigen-binding capacities compared to their parent antibodies (Supporting Information, Figure S5).
Figure 2.

Enzymatic transfer of IgG to the surfaces of Lec2 CHO cells: (A) The synthesis of a GDP-Fucose-conjugated IgG (GF-IgG). (B) Workflow of the FucT-catalyzed transfer of GF-IgG to the surface of Lec2 CHO cells. (C) Flow cytometry analysis of Lec2 cells treated with the enzyme FucT, the substrate GF-rIgG, or both. (D) Titration of GF-rIgG, concentrations ranging from 0.005 to 0.2 mg/mL in the reaction buffer; each reaction used 60 mU FucT and proceeded at room temperature for 30 min; mean ± SD (error bars), representative graph from three independent experiments. (E) Time course of enzymatic transfer of GF-rIgG to Lec2 cells on ice; reaction at 37 °C was used as the maximum labeling control. (F) Fluorescent gel imaging for detecting and quantifying rIgG (Alexa Fluor 647-labeled) molecules conjugated on the Lec2 cell surface. (G) Confocal microscopy images of Lec2 cells treated with or without FucT when incubated with Alexa Fluor 647-labeled GF-rIgG; nuclei were stained with Hoechst 33342. Scale bar: 2 μm. (H) Flow cytometry analysis of Lec2 cells simultaneously labeled with rIgG and mIgG. (I) Lec8 CHO cells without LacNAc expression were compared with Lec2 cells in the enzymatic IgG transfer as a negative control; mean ± SD (error bars), representative graph from three independent experiments. α1,3FucT is abbreviated as FT in all figures.
The one-pot product of GDP-Fucose-conjugated rat IgG (GF-rIgG) was then incubated with Lec2 CHO cells that express abundant terminal LacNAc units in the presence of FucT (Figure 2B). Remarkably, the signal of rIgG conjugated onto the cell surface was detectable after a 2-min incubation with FucT (60 mU) and GF-rIgG (0.1 mg/mL) (Figure 2C). The labeling efficiency was concentration-dependent (GF-rIgG), which reached saturation at 0.1 mg/mL during a 30-min reaction course (Figure 2D). Notably, the conjugation reaction was completed in 10 min even on ice (Figure 2E). At the saturated condition, approximately 2.5 × 105 rIgG molecules were introduced to the cell surface (Figure 2F, and Supporting Information, Figure S6A). The viability of the rIgG-labeled cells was similar to that of unlabeled cells, which confirmed the biocompatibility of this one-pot procedure (Supporting Information, Figure S7). Furthermore, confocal microscopy analysis verified that most of the labeled rIgGs were located on the cell membrane (Figure 2G). It is worth noting that multiple functionalities can be introduced to the cell-surface simultaneously, e.g., two antibodies (GF-rIgG and GF-mIgG—GDP-Fucose-modified mouse IgG) (Figure 2H, and Supporting Information, Figure S8). Such results are difficult to achieve via genetic approaches, especially when three or four antibodies need to be installed.
To confirm that cell-surface LacNAc and sialyl LacNAc are still the conjugation sites of this enzyme-mediated IgG transfer, Lec8 cells, a mutant CHO cell line that do not express galactose and sialic acid and accordingly without LacNAc, were used as a negative control. As expected, only background fluorescence was displayed by Lec8 cells after the enzymatic reaction (Figure 2I). To further validate this observation, we performed a competition experiment in which the known acceptor substrate, free LacNAc or α2,3 sialyl LacNAc,16,28 was mixed together with NK-92 cells to compete for the FucT-mediated GF-IgG transfer. Not surprisingly, a dose-dependent inhibition was observed in each case with LacNAc showing more pronounced blocking of the GF-IgG transfer than α2,3 sialyl LacNAc (Supporting Information, Figure S9A,B). Likewise, we observed that the transfer of GF-IgG to the cell surface could be blocked by the natural donor substrate GDP-Fucose as well (Supporting Information, Figure S9A,C,D). Significantly, ACCs can also be constructed using other human fucosyltransferases, such as FucT 6 and 9 (Supporting Information, Figure S10A), and sialyltransferases (Supporting Information, Figure S10B) despite with much lower efficiency compared with H. pylori 26695 α1,3FucT.
To demonstrate that this approach can be applied to modify other cell types, primary human cells, e.g., T cells, were subjected to the FucT-mediated conjugation; robust cell labeling with IgGs was achieved within 15 min (Supporting Information, Figures S11 and S6B). We confirmed that the bioconjugation of IgG molecules to the cell surface had no short-term interference with the expression of cell-surface markers (Supporting Information, Figure S12). The half-life of IgG molecules conjugated to the human T cell surface is approximately 24 h, and the conjugation had no effect on the proliferation of the modified cells (Supporting Information, Figure S11C,D).
Taken together, we confirmed that the transfer of GF-IgG to LacNAc on the cell surface via FucT is a highly efficient one-step approach to construct ACC. With this powerful method in hand, we explored its application to construct ACCs using various immune cells for boosting the efficacy of cell-based therapies.
Herceptin-NK-92MI Conjugates Enable Specific Killing of HER2+ Tumor Cells in a Murine Model
Specific targeting is key for the success of cell-based cancer immunotherapy. In innate immunity human natural killer (NK) cells play crucial roles in the rejection of tumors and virally infected cells.29 NK-92, a constantly active and nonimmunogenic natural killer (NK) cell line, is being developed in bulk quantities as an “off-the-shelf therapeutic” for adoptive NK-based cancer immunotherapy in clinical evaluations.29,30 However, NK-92 cells do not express Fc receptors for antibody-dependent cell-mediated cytotoxicity (ADCC), a mechanism for specific cell lysis, which significantly limits their clinical applications.29 We speculate that modifying NK-92 cells with antibodies against specific tumor antigens via the chemoenzymatic conjugation may confer NK-92 cells with specific targeting capability (Figure 3A). We chose NK-92MI cells, an IL-2-independent variant of the NK-92 cell line, as the candidate for bioconjugation with Herceptin because it expresses a high level of LacNAc to be modified by GDP-Fucose-conjugated human IgG (GF-hIgG) (Supporting Information, Figure S13A). Herceptin, also known as Trastuzumab, is a FDA approved antibody to treat human epidermal growth factor receptor 2-positive (HER2+) breast cancer. We calculated that approximately 3 × 105 Herceptin molecules were conjugated to the surface of a NK-92MI cell, which equaled ∼7.5 ng Herceptin/105 cells (Supporting Information, Figure S6C). Herceptin conjugated to the surface of NK-92MI cells maintains exclusive binding to the HER2 antigen (Figure 3B, and Supporting Information, Figure S13B,C), and its cell-surface half-life is approximately 20 h (Supporting Information, Figure S13D). As revealed by flow cytometry analysis and fluorescent microscopy imaging NK-92MI cells conjugated with Herceptin formed clusters with BT474—a HER2+ breast cancer cell—in a coculture assay, whereas no cluster formation was observed between unmodified NK-92MI cells and BT474 (Figure 3C,D). Moreover, NK-92MI cells modified with Herceptin induced the lysis of BT474 cells at least 7 times more efficiently than unmodified NK-92MI cells (Figure 3E). Neither isotype control hIgG labeling nor cotreatment with excess, free Herceptin (1 μg/105 NK cells) could enhance the killing activity of NK-92MI on BT474, indicating that covalent conjugation of Herceptin to the surface of NK-92MI cells is required (Figure 3E). Importantly, the cell-lysis efficiency of the modified NK-92MI cells also was dependent on the loading of Herceptin on the cell surface, which reached the plateau when 0.1 mg/mL of GF-Herceptin was used for enzymatic transfer (Figure 3F, Supporting Information, Figure S14). The enhanced killing effect of Herceptin-NK92-MI conjugates later was confirmed on other HER2+ cancer cells, including SKBR3 and MDA-MB-435/HER2+, but not on HER2 negative (HER2−) cancer cells, such as MDA-MB-435 and MDA-MB-468 (Figure 3G). In addition, the total secretion of granzyme B was elevated only when Herceptin-NK-92MI conjugates were mixed with BT474 (Supporting Information, Figure S13E), strongly suggesting that the interaction between Herceptin and HER2 is key to enhance NK-92MI cell activation. Similar to other cell-mediated cytotoxicity, higher effector-to-target cell ratios also show better killing, but only in the Herceptin-labeled NK-92MI group, which reaches saturation at E/T 5:1 (Figure 3H). As mentioned above, another distinct advantage of enzymatic cell engineering is that several antibodies can be conjugated onto the surface of a cell at the same time. As proof-of-concept, we conjugated NK-92MI cells with both Herceptin and an anti-EGFR antibody (Supporting Information, Figure S15), and the dually modified cells exhibited better killing efficiency on SKOV3 cells (HER2+EGFR+) than that induced by the single-mAb-modified counterparts (Figure 3I).
Figure 3.

Construction of Herceptin-NK-92MI conjugates for targeting HER2+ cancer cells: (A) Herceptin-NK-92MI conjugates specifically bind to HER2+ cancer cells and exhibit enhanced killing activities due to proximity effects. (B) Analysis of HER2 binding of Hereceptin-NK-92MI conjugates. Mean ± SD (error bars). Flow cytometry analysis (C) and fluorescent microscopy images (D) of specific binding between Herceptin-NK-92MI conjugates and BT474 (HER2+); NK-92MI cells were stained with CellTracker Orange (red), and BT474 cells were stained with CellTracker Green (green). The merged channels of fluorescence and phase contrast are shown; the green fields represent clusters of BT474 cells. Scale bar: 50 μm. (E) LDH release assay for quantifying induced lysis of BT474 cells by NK-92MI cells; Herceptin-NK-92MI conjugates were compared with parental NK-92MI with or without additionally added free Herceptin (5 μg/mL). hIgG-NK-92MI conjugates were used as a negative control. Mean ± SD (error bars), representative graph from three independent experiments. (F) Killing activity of Herceptin-NK-92MI conjugates constructed at different GF-Herceptin concentrations. Mean ± SD (error bars). (G) Comparisons of NK-92MI and Herceptin-NK-92MI conjugates in killing different cancer cell lines with or without HER2 expression. Mean ± SD (error bars), representative graph from three independent experiments. (H) Comparisons of NK-92MI and Herceptin-NK-92MI conjugates in killing BT474 at different effector-to-target cell ratios. Mean ± SD (error bars). (I) Herceptin and α-EGFR dual-labeled NK-92MI cells were compared with Herceptin-NK-92MI conjugates and α-EGFR-NK-92MI conjugates in killing HER2+EGFR+ SKOV3 cancer cells. Mean ± SD (error bars). (J) Comparison of nonirradiated and irradiated (6 Gy) NK-92MI cells in killing BT474 cells. (K) In vivo antitumor activity of Herceptin-NK-92MI conjugates. NSG mice were injected intravenously with 0.5 million MDA-MB-435/HER2+/F-luc cells. Then, the animals were treated once by IV injection of 3 million NK-92MI or Herceptin-NK-92MI cells on day 1 after the injection of the tumor cells. The control mice received HBSS. Six days after the tumor challenge, the mice were injected IP with d-luciferin and imaged by IVIS system. The sizes of the tumors of the mice and mean values ± SD are shown; n = 10. Representative images also are shown. In all figures, ns, P > 0.05; *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001; one-way ANOVA followed by Tukey’s multiple comparisons test, two-way ANOVA followed by Sidak’s multiple comparisons test.
The promising results of enhanced ex vivo killing ability of Herceptin-NK92-MI conjugates led us to test their efficacy in vivo. Since NK-92 is developed from a patient with lymphoma, as a safety measure, it is usually irradiated prior to clinical use to prevent permanent engraftment in the human body. As expected, irradiation of 6 Gy prevented the proliferation of NK-92MI cells (Supporting Information, Figure S16A). We found that γ-irradiated NK-92MI cells maintained their cytotoxicity (Figure 3J), and the half-life of Herceptin conjugated to the irradiated NK-92MI was slightly longer than that of the nonirradiated cells (Supporting Information, Figure S16B). To evaluate the in vivo efficacy of Herceptin-NK-92MI conjugates, we chose an experimental lung metastasis model in which NSG mice received intravenous (IV) injections of MDA-MB-435/HER2+/F-luc cells (stably transduced with firefly luciferase). One day after being inoculated with the tumor cells, the mice were treated by IV injections of irradiated parental NK-92MI cells or Herceptin-NK-92MI conjugates while the nontreated group was injected with Hank’s balanced salt solution (HBSS). Six days after tumor inoculations, the volumes of the tumors in the lungs were determined by longitudinal, noninvasive bioluminescence imaging. While treatment with parental NK-92MI cells only moderately reduced the formation of tumors in the lungs (∼48% less than the HBSS group), Herceptin-labeled NK-92MI cells exhibited significantly enhanced in vivo tumor killing activity (∼83% less than the HBSS group) (Figure 3K). To assess if Herceptin-NK92-MI conjugates were effective to treat established tumors, NSG mice were injected with luciferase-bearing MDA-MB-435/HER2+ cells intravenously. Three days later, the growth of tumor cells in the lung region was revealed by a 3fold increase in bioluminescence (Supporting Information, Figure S17). The mice were then treated by IV injections of irradiated parental NK-92MI cells or Herceptin-NK-92MI conjugates on day 3, day 9, and day 17. On day 23, bioluminescence imaging showed that tumor growth was significantly suppressed by Herceptin-labeled NK-92MI cells compared to unmodified NK-92MI cells (Supporting Information, Figure S17).
Anti-PD-L1 (α-PD-L1) Conjugated on the Surfaces of CD8+ T Cells Could Block the PD-1–PD-L1 Pathway and Enhance the Proliferation of T Cells ex Vivo
Even after infiltrating the tumor bed, cytotoxic functions of effector cells may be dampened by factors produced in the microenvironment of the tumor.31 As another application of our new technique, we sought to determine whether CD8+ T cells modified by cell-surface mAb conjugation could counteract such inhibitory signals to maintain their activities. The interaction between programmed death 1 (PD-1) receptor, found on T cells, and PD-Ligand (PD-L) expressed by tumor cells plays a major role in inhibiting the cytotoxicity of T cells32 (Figure 4A). We hypothesize that the installation of α-PD-L1 on the surfaces of T cells may block the PD-1–PD-L1 interaction in situ to enhance the activation of the T cells and thus enforcing tumor cell lysis (Figure 4B).
Figure 4.

Enzymatic transfer of α-PD-L1 to OT-1 T cells for enhanced T-cell activation and specific killing: (A) Schematic illustration of the interaction between OT-1 T cells and B16-OVA melanoma cells. MHC molecules on B16-OVA present OVA antigen to OVA-specific TCR on OT-1 T cells to induce activation, while PD-L1 on B16-OVA interact with PD-1 on OT-1 T cells to inhibit the T-cell effector function. (B) Schematic illustration of the blockade of PD-1–PD-L1 pathway via α-PD-L1 conjugated on the surfaces of OT-1 T cells. The in situ blockade could enhance T-cell activation and the killing of cancer cells. (C) Analysis of PD-L1 antigen binding on OT-1 T cells for different treatments. Mean ± SD (error bars). (D) Analysis of PD-L1 antigen binding of OT-1 T cells modified with various concentrations of GF-α-PD-L1. Mean ± SD (error bars). (E) Quantifying α-PD-L1-OT-1 T cell conjugates mediated killing of B16-OVA cells. Mean ± SD (error bars), representative graph from three independent experiments. OT-1 T cells conjugated with rIgG and P14 T cells conjugated with α-PD-L1 were shown as negative control. (F) Comparison of OT-1 T cells and α-PD-L1-OT-1 T cell conjugates in killing B16-OVA at different effector-to-target cell ratios. Mean ± SD (error bars). (G) Killing activities of OT-1 T cells and α-PD-L1-OT-1 T cell conjugates constructed at different GF-α-PD-L1 concentrations. Mean ± SD (error bars). (H) Comparison of α-PD-L1-OT-1 T cell conjugates with OT-1 T cells with or without additionally added free α-PD-L1 (5 μg/mL) in killing B16-OVA (3 h incubation). (I) IFN-γ secretion analysis of modified or unmodified OT-1 T cells in a B16-OVA cell co-culture assay. P14 T cells conjugated with α-PD-L1 were used as a negative control. Mean ± SD (error bars), representative graph from three independent experiments. (J) Microscopy images of OT-1 T cell-induced killing of B16-OVA with or without α-PD-L1 labeling. The blue arrow indicates fewer cancer cells, and the purple arrow indicates larger clusters of T cells. Scale bar: 50 μm. (K) Analysis of OT-1 T cell proliferation via CFSE dilution in a B16-OVA cell co-culture assay. Mean ± SD (error bars). In all figures, ns, P > 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001; one-way ANOVA followed by Tukey’s multiple comparisons test, two-way ANOVA followed by Sidak’s multiple comparisons test.
OT-1 transgenic mice were chosen as the model system whose CD8+ T cells express a T cell receptor (TCR) specific for the SIINFEKL peptide (OVA257–264) of ovalbumin presented on major histocompatibility complex I (MHC I) molecules. Splenocytes from the OT-1 mice were first stimulated with OVA257–264 peptide and expanded ex vivo in the presence of cytokines IL2 or IL7/IL15 (Supporting Information, Figure S18C). The expanded OT-1 T cells were confirmed to express high levels of LacNAc (Supporting Information, Figure S18B) and were subjected to the chemoenzymatic modification with GF-rIgG (Supporting Information, Figure S18C). Restimulation experiments confirmed that the modified T cells had a similar proliferation rate to that of the unmodified cells, suggesting that the conjugated IgG molecules do not block the interaction between TCR and the MHC I-complex (Supporting Information, Figure S18D).
Subsequently, CD8+ T cells from OT-1 or P14 mice that carry a transgenic TCR recognizing the gp33-41 epitope of lymphocytic choriomeningitis virus, were modified with GF-α-PD-L1 using chemoenzymatic glycan engineering (Supporting Information, Figure S19A,B). α-PD-L1 maintained its antigen-binding capacity upon cell-surface conjugation (Figure 4C, and Supporting Information, Figure S19C). As expected, the degrees of both cell-surface conjugation and antigen binding were dependent on the concentration of GF-α-PD-L1 (Figure 4D, and Supporting Information, Figure S19D). At the saturated condition, approximately 7 × 104 anti-PD-L1 molecules were conjugated to the OT-1 T cell surface (Supporting Information, Figure S6D). Then, the modified OT-1 CD8+ T cells were subjected to an ex vivo killing assay, in which a B6-derived melanoma cell line B16F10 expressing ovalbumin (B16-OVA) was used as model target cells. After incubation for 20 h, the OT-1 T cells conjugated with α-PD-L1 showed significantly enhanced lysis of B16-OVA cells (Figure 4E), and the enhanced killing effect was observed only when the effector-to-target cell ratio was above 1 (Figure 4F). By contrast, the specific lysis of B16-OVA induced by the rIgG-OT-1 T cell conjugates was much weaker, which is similar to that of the control, unmodified T cells (Figure 4E). CD8+ T cells of irrelevant specificity from P14 mice also were conjugated with α-PD-L1 as a negative control. Remarkably, this ACC only induced background killing (Figure 4E), suggesting that trace levels of α-PD-L1 conjugated to the surfaces of the cells could not mediate significant target cell lysis. Although the antigen-binding capacity reached the maximum when 0.1 mg/mL GF-α-PD-L1 was used in the enzymatic transfer reaction (Figure 4D), the optimal killing capacity was achieved at ∼0.05 mg/mL GF-α-PD-L1 (Figure 4G), suggesting that only half of the maximum cell-surface conjugation is required for efficient blocking of the PD-1–PD-L1 interaction. Moreover, using the same killing assay we observed that α-PD-L1-modified OT-1 cells exhibited significantly better lysis than a simple combination of OT-1 cells with free α-PD-L1 (Figure 4H). The antibodies conjugated to the T cells’ surface was approximately 1.8 ng/1 × 105 cells, which is much lower than the concentration of free antibodies used in the control experiment (500 ng/1 × 105 cells).
To further determine whether the cell-surface-conjugated α-PD-L1 could suppress PD-1–PD-L1 coinhibitory signaling, we measured the cytokine production of the modified OT-1 T cells when they were mixed with B16-OVA. Enhanced IFN-γ and TNF-α secretion was only observed in T cells conjugated with α-PD-L1, which exhibited dependency on the dose of conjugated α-PD-L1 (Figure 4I, and Supporting Information, Figure S20). Enhanced T-cell activation also was observed directly using microscopy due to the formation of larger clusters of T cells in the α-PD-L1-labeled group (Figure 4J, and Supporting Information, Figure S21A). Finally, we found that the cell-surface conjugated α-PD-L1 also promoted T cell proliferation as confirmed by a CFSE dilution assay when the modified OT-1 T cells were re-stimulated with B16-OVA (Figure 4K, and Supporting Information, Figure S21B).
Summary
The one-step FucT-based chemoenzymatic method developed here is a fast, simple, and cost-effective technique for cell engineering. Via this technique, both bio-macromolecules, including proteins and nucleic acids, and small-molecule probes, e.g., fluorescent and biophysical probes, can be introduced to the cell surface without genetic modification of the host cells. Targeting oligosaccharides—the most abundant biopolymers found on the cell surface—we have demonstrated that as many as 300 000 copies of functionalities can be incorporated.
Using this method, we successfully constructed two ACCs, which exhibited enhanced activities in two critical stages of anticancer immune responses, i.e., targeting and killing. Although mAbs conjugated on the cell surface are diluted due to internalization and cell proliferation (cell-surface half-life ∼8–24 h in our experiments), this method offers several advantages over genetic-based engineering, e.g., rapid and homogeneous modification, capability to install multiple mAbs simultaneously, and therefore serving as a nice complement or synergistic method to the permanent, genetic engineering approach that has found great success in making CAR constructs, including CAR-NKs based on NK-92 cells.33 Because γ-irradiation is employed as a potential safety measure for clinical application to prevent NK-92 cell replication while preserving their antitumor activities,33 there are likely no obvious advantages of using these CAR constructs than using ACCs disclosed here for NK-92 cell engineering. The fact that NK-92MI cells are currently undergoing clinical trials, and Herceptin is already a FDA-approved drug heralds the potential of Herceptin-NK-92MI conjugates for further development as a clinical candidate.
Given its broad substrate scope and simple procedure, we predict that the technique described here will provide new avenues for landscaping cell surface beyond the applications demonstrated above, such as controlling cell–cell interactions,34 regulating cell-surface channel activity,35 and realizing targeted drug delivery.36 In the new era of cell-based therapy, to endow living cells with new properties a simple and robust reaction like the one described here will be highly sought after.
Acknowledgments
This work was supported by the NIH (GM113046 and GM093282 to P.W.; GM103390 to K.W.M.). We thank Prof. Peter Schultz (TSRI, USA) for MDA-MB-435/HER2+/F-luc cells, Prof. John Teijaro (TSRI, USA) for P14 mice, and Prof. Philippe A. Gallay (TSRI, USA) for NSG mice.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscentsci.8b00552.
Additional experimental details and figures including chemical structures, characterizations, fluorescent SDS-PAGE gel analysis, flow cytometry analysis, TLC results, ESI-TOF MS, 1H NMR, 13C NMR, and 31P NMR (PDF)
Author Contributions
# J.L. and M.C. contributed equally to this work. P.W., J.L., and M.C. designed the experimental strategy and wrote the manuscript. J.L. and M.C. performed the experiments with the help of L.Z., Z.L., and M.A. P.W. and J.L. prepared the figures, and all authors edited the manuscript.
The authors declare the following competing financial interest(s): J.L., M.C., and P.W. are listed as inventors on a patent application (application number: PCT/US2018/016503) that discloses the enzymatic construction of antibody-cell conjugates.
Notes
Safety statement: no unexpected or unusually high safety hazards were encountered in this line of research.
Supplementary Material
References
- Abbina S.; Siren E. M. J.; Moon H.; Kizhakkedathu J. N. Surface Engineering for Cell-Based Therapies: Techniques for Manipulating Mammalian Cell Surfaces. ACS Biomater. Sci. Eng. 2018, 4, 3658. 10.1021/acsbiomaterials.7b00514. [DOI] [PubMed] [Google Scholar]
- Rosenberg S. A.; Restifo N. P. Adoptive Cell Transfer as Personalized Immunotherapy for Human. Science 2015, 348, 62–68. 10.1126/science.aaa4967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasad V. Tisagenlecleucel — the First Approved CAR-T-cell Therapy: Implications for Payers and Policy Makers. Nat. Rev. Clin. Oncol. 2017, 15, 11–12. 10.1038/nrclinonc.2017.156. [DOI] [PubMed] [Google Scholar]
- Cornetta K.; Pollok K. E.; Miller A. D. Transduction of Primary Hematopoietic Cells by Retroviral Vectors. Cold Spring Harbor Protocols 2008, 2008, 4884. 10.1101/pdb.prot4884. [DOI] [PubMed] [Google Scholar]
- Dupré L.; Trifari S.; Follenzi A.; Marangoni F.; Lain de Lera T.; Bernad A.; Martino S.; Tsuchiya S.; Bordignon C.; Naldini L.; Aiuti A.; Roncarolo M.-G. Lentiviral Vector-Mediated Gene Transfer in T Cells from Wiskott–Aldrich Syndrome Patients Leads to Functional Correction. Mol. Ther. 2004, 10, 903–915. 10.1016/j.ymthe.2004.08.008. [DOI] [PubMed] [Google Scholar]
- Shearer R. F.; Saunders D. N. Experimental Design for Stable Genetic Manipulation in Mammalian Cell Lines: Lentivirus and Alternatives. Genes Cells 2015, 20, 1–10. 10.1111/gtc.12183. [DOI] [PubMed] [Google Scholar]
- Stephan M. T.; Irvine D. J. Enhancing Cell Therapies from the Outside in: Cell Surface Engineering using Synthetic Nanomaterials. Nano Today 2011, 6, 309–325. 10.1016/j.nantod.2011.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swartz M. A.; Hirosue S.; Hubbell J. A. Engineering Approaches to Immunotherapy. Sci. Transl. Med. 2012, 4, 148rv9–148rv9. 10.1126/scitranslmed.3003763. [DOI] [PubMed] [Google Scholar]
- Griffin M. E.; Hsieh-Wilson L. C. Glycan Engineering for Cell and Developmental Biology. Cell Chem. Biol. 2016, 23, 108–121. 10.1016/j.chembiol.2015.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudak J. E.; Bertozzi C. R. Glycotherapy: New Advances Inspire a Reemergence of Glycans in Medicine. Chem. Biol. 2014, 21, 16–37. 10.1016/j.chembiol.2013.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi X.; Yin J.; Chen Guanbang A.; Liu C.-F. Chemical and Enzymatic Strategies for Bacterial and Mammalian Cell Surface Engineering. Chem. - Eur. J. 2018, 24, 8042–8050. 10.1002/chem.201705049. [DOI] [PubMed] [Google Scholar]
- Pishesha N.; Bilate A. M.; Wibowo M. C.; Huang N.-J.; Li Z.; Deshycka R.; Bousbaine D.; Li H.; Patterson H. C.; Dougan S. K.; Maruyama T.; Lodish H. F.; Ploegh H. L. Engineered Erythrocytes Covalently Linked to Antigenic Peptides Can Protect Against Autoimmune Disease. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, 3157–3162. 10.1073/pnas.1701746114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parmar S.; Liu X.; Najjar A.; Shah N.; Yang H.; Yvon E.; Rezvani K.; McNiece I.; Zweidler-McKay P.; Miller L.; Wolpe S.; Blazar B. R.; Shpall E. J. Ex vivo Fucosylation of Third-party Human Regulatory T Cells Enhances Anti–Graft-Versus-Host Disease Potency in Vivo. Blood 2015, 125, 1502–1506. 10.1182/blood-2014-10-603449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sackstein R.; Merzaban J. S.; Cain D. W.; Dagia N. M.; Spencer J. A.; Lin C. P.; Wohlgemuth R. Ex vivo Glycan Engineering of CD44 Programs Human Multipotent Mesenchymal Stromal Cell Trafficking to Bone. Nat. Med. 2008, 14, 181–187. 10.1038/nm1703. [DOI] [PubMed] [Google Scholar]
- Srivastava G.; Kaur K. J.; Hindsgaul O.; Palcic M. M. Enzymatic Transfer of a Preassembled Trisaccharide Antigen to Cell Surfaces Using a Fucosyltransferase. J. Biol. Chem. 1992, 267, 22356–22361. [PubMed] [Google Scholar]
- Zheng T.; Jiang H.; Gros M.; Soriano del Amo D.; Sundaram S.; Lauvau G.; Marlow F.; Liu Y.; Stanley P.; Wu P. Tracking N-Acetyllactosamine on Cell-Surface Glycans In Vivo. Angew. Chem. 2011, 123, 4199–4204. 10.1002/ange.201100265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capicciotti C. J.; Zong C.; Sheikh M. O.; Sun T.; Wells L.; Boons G.-J. Cell-Surface Glyco-Engineering by Exogenous Enzymatic Transfer Using a Bifunctional CMP-Neu5Ac Derivative. J. Am. Chem. Soc. 2017, 139, 13342–13348. 10.1021/jacs.7b05358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W.; Hu T.; Frantom P. A.; Zheng T.; Gerwe B.; del Amo D. S.; Garret S.; Seidel R. D.; Wu P. Chemoenzymatic Synthesis of GDP-l-fucose and the Lewis X Glycan Derivatives. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 16096–16101. 10.1073/pnas.0908248106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rostovtsev V. V.; Green L. G.; Fokin V. V.; Sharpless K.B. a Stepwise Huisgen Cycloaddition Process: Copper(I)-Catalyzed Regioselective “Ligation” of Azides and Terminal Alkynes. Angew. Chem., Int. Ed. 2002, 41, 2596–2599. . [DOI] [PubMed] [Google Scholar]
- Tornøe C. W.; Christensen C.; Meldal M. Peptidotriazoles on Solid Phase: [1,2,3]-Triazoles by Regiospecific Copper(I)-Catalyzed 1,3-Dipolar Cycloadditions of Terminal Alkynes to Azides. J. Org. Chem. 2002, 67, 3057–3064. 10.1021/jo011148j. [DOI] [PubMed] [Google Scholar]
- Besanceney-Webler C.; Jiang H.; Zheng T.; Feng L.; Soriano del Amo D.; Wang W.; Klivansky L. M.; Marlow F. L.; Liu Y.; Wu P. Increasing the Efficacy of Bioorthogonal Click Reactions for Bioconjugation: a Comparative Study. Angew. Chem., Int. Ed. 2011, 50, 8051–8056. 10.1002/anie.201101817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swee L. K.; Lourido S.; Bell G. W.; Ingram J. R.; Ploegh H. L. One-Step Enzymatic Modification of the Cell Surface Redirects Cellular Cytotoxicity and Parasite Tropism. ACS Chem. Biol. 2015, 10, 460–465. 10.1021/cb500462t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giorgi M. E.; Agusti R.; de Lederkremer R. M. Carbohydrate PEGylation, an Approach to Improve Pharmacological Potency. Beilstein J. Org. Chem. 2014, 10, 1433–1444. 10.3762/bjoc.10.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahim M. K.; Kota R.; Haun J. B. Enhancing Reactivity for Bioorthogonal Pretargeting by Unmasking Antibody-Conjugated trans-Cyclooctenes. Bioconjugate Chem. 2015, 26, 352–360. 10.1021/bc500605g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveira B. L.; Guo Z.; Bernardes G. J. L. Inverse Electron Demand Diels-Alder Reactions in Chemical Biology. Chem. Soc. Rev. 2017, 46, 4895–4950. 10.1039/C7CS00184C. [DOI] [PubMed] [Google Scholar]
- Blackman M. L.; Royzen M.; Fox J. M. Tetrazine Ligation: Fast Bioconjugation Based on Inverse-Electron-Demand Diels–Alder Reactivity. J. Am. Chem. Soc. 2008, 130, 13518–13519. 10.1021/ja8053805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selvaraj R.; Fox J. M. Trans-Cyclooctene—a Stable, Voracious Dienophile for Bioorthogonal Labeling. Curr. Opin. Chem. Biol. 2013, 17, 753–760. 10.1016/j.cbpa.2013.07.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soriano del Amo D.; Wang W.; Besanceney C.; Zheng T.; He Y.; Gerwe B.; Seidel R. D.; Wu P. Chemoenzymatic synthesis of the sialyl Lewis X glycan and its derivatives. Carbohydr. Res. 2010, 345, 1107–1113. 10.1016/j.carres.2010.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morvan M. G.; Lanier L. L. NK Cells and Cancer: You Can Teach Innate Cells New Tricks. Nat. Rev. Cancer 2016, 16, 7. 10.1038/nrc.2015.5. [DOI] [PubMed] [Google Scholar]
- Suck G.; Odendahl M.; Nowakowska P.; Seidl C.; Wels W. S.; Klingemann H. G.; Tonn T. NK-92: an ‘Off-the-Shelf Therapeutic’ for Adoptive Natural Killer Cell-based Cancer Immunotherapy. Cancer Immunol. Immunother. 2016, 65, 485–492. 10.1007/s00262-015-1761-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munn D. H.; Bronte V. Immune Suppressive Mechanisms in the Tumor Microenvironment. Curr. Opin. Immunol. 2016, 39, 1–6. 10.1016/j.coi.2015.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou W.; Wolchok J. D.; Chen L. PD-L1 (B7-H1) and PD-1 Pathway Blockade for Cancer Therapy: Mechanisms, Response Biomarkers, and Combinations. Sci. Transl. Med. 2016, 8, 328rv4–328rv4. 10.1126/scitranslmed.aad7118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schönfeld K.; Sahm C.; Zhang C.; Naundorf S.; Brendel C.; Odendahl M.; Nowakowska P.; Bönig H.; Köhl U.; Kloess S.; Köhler S.; Holtgreve-Grez H.; Jauch A.; Schmidt M.; Schubert R.; Kühlcke K.; Seifried E.; Klingemann H. G.; Rieger M. A.; Tonn T.; Grez M.; Wels W. S. Selective Inhibition of Tumor Growth by Clonal NK Cells Expressing an ErbB2/HER2-Specific Chimeric Antigen Receptor. Mol. Ther. 2015, 23, 330–338. 10.1038/mt.2014.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi P.; Ju E.; Yan Z.; Gao N.; Wang J.; Hou J.; Zhang Y.; Ren J.; Qu X. Spatiotemporal Control of Cell–Cell Reversible Interactions Using Molecular Engineering. Nat. Commun. 2016, 7, 13088. 10.1038/ncomms13088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ai X.; Lyu L.; Zhang Y.; Tang Y.; Mu J.; Liu F.; Zhou Y.; Zuo Z.; Liu G.; Xing B. Remote Regulation of Membrane Channel Activity by Site-Specific Localization of Lanthanide-Doped Upconversion Nanocrystals. Angew. Chem., Int. Ed. 2017, 56, 3031–3035. 10.1002/anie.201612142. [DOI] [PubMed] [Google Scholar]
- Stephan M. T.; Moon J. J.; Um S. H.; Bershteyn A.; Irvine D. J. Therapeutic Cell Engineering with Surface-conjugated Synthetic Nanoparticles. Nat. Med. 2010, 16, 1035–1041. 10.1038/nm.2198. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.