

Summary
Background
Haemopoietic progenitor cell (HPC) transplantation can cure sickle cell disease. Non-myeloablative conditioning typically results in donor-derived erythrocytes and stable mixed chimerism of recipient-derived and donor-derived leucocytes. Exposure to donor antigens from the HPC graft and new red cell antibodies induced by transfusion can lead to immunohaematological complications. We assessed the incidence of such complications among HPC transplant recipients with sickle cell disease.
Methods
The study population was all patients with sickle cell disease enrolled before March 31, 2015, in the three clinical trials of non-myeloablative HPC transplantation at the National Institutes of Health. We assessed formation of new red cell antibodies after transplantation and red cell incompatibility between donors and recipients.
Findings
61 patients were enrolled, 42 were HLA matched and 19 were haploidentical. Nine (15%) had immunohaematological complications. Before HPC transplantation, three patients had antibodies incompatible with their donors. After HPC transplantation, new red cell antibodies were seen in six patients (11 alloantibodies and two autoantibodies), among whom three developed antibodies incompatible with donor or recipient red cells and three developed compatible antibodies. The clinical course of complications was highly variable, from no severe effects attributable to antibodies, to sustained reticulocytopenia, to near-fatal haemolysis. We found no significant correlation between immunohaematological complications and graft failure, graft rejection, or death.
Interpretation
Clinical effects ranged from seemingly not clinically important to potentially fatal. In patients with sickle cell disease, donor and recipient red cell phenotypes should be carefully assessed before transplantation to minimise and manage the risk of immunohaematological complications.
Introduction
Haemopoietic progenitor cell (HPC) transplantation can cure patients with sickle cell disease. In the first case report in 1984, a child with sickle cell disease developed acute myeloid leukaemia, and HPC transplantation treated both diseases.1 In the 1990s, several different myeloablative HPC transplantation regimens involving matched related donors led to cure, but mortality approached 10%.2 In 2001, two reports described patients undergoing HPC transplantations who developed stable mixed chimerism of donor-derived and recipient-derived leucocytes after transplantation.3 The haemoglobin concentrations of recipients were generally normal and haemoglobin S expression was similar to that of donors, some of whom had sickle cell trait. Acute events and progression of organ damage ceased, and no recipient developed chronic graft-versus-host disease.3
Prompted by these findings, various non-myeloablative and reduced-intensity conditioning regimens were assessed in clinical trials.4–14 These approaches sought to lessen the risks of treatment-related mortality and toxic effects while establishing stable mixed chimerism. Early trials reported substantial graft rejection,5,6 morbidity,4 and mortality.4 In later trials involving related HPC donors, refined conditioning and immunomodulatory regimens led to immunosuppression of the recipient, induced tolerance towards donor-derived cells,15 and remission. Transplant-related mortality decreased to 1%, chronic graft-versus-host disease to 5%, and graft failure to 8%.7–13 In children with sickle cell disease and unrelated HPC donors, 62% develop chronic graft-versus-host disease, and graft failure occurs in 10%.14 Because of decreased toxicity, non-myeloablative regimens can be tolerated by adults who have sustained end-organ damage that renders them ineligible for standard myeloablative regimens.12
Stable mixed blood cell chimerism after non-myeloablative HPC transplantation carries the risk of immunohaematological complications. Donor and recipient leucocytes coexist alongside donor red cells.9,12,15 In particular, recipient plasma cells might persist even after most other cell populations have converted fully to donor cells.16 This unusual haematological and immunological milieu creates the potential for the recipient’s residual leucocytes to generate alloantibodies against the donor’s red-cell antigens, or vice versa, which can cause haemolysis in the immediate transplantation process and suppression of red cell production long term. Any red cell antibody, whether pre-existing or newly formed, might increase the risk of clinically relevant haemolysis and limit the supply of compatible blood.
Among patients without sickle cell disease who undergo non-myeloablative HPC transplantation, pure red cell aplasia and delayed production of donor red cells,17 increased transfusion requirements,18 delayed engraftment,18 graft rejection, and transplant-related mortality19 can occur in ABO-mismatched recipients. Non-ABO antibodies might also develop after transplantation, and can cause severe haemolysis.20–22 The effects of non-myeloablative HPC transplantation in patients with sickle cell disease, in whom red cell alloimmunisation ranges from 18% to 30%, have not, however, been substantially assessed.23 In one study of children with sickle cell disease undergoing myeloablative HPC transplantation, a median of seven red blood cell transfusions was needed, whereas fewer transfusions were needed in those receiving phenotype-matched units, and no new antibodies formed during the process.24 In a case series of children with sickle cell disease undergoing myeloablative HPC transplantation despite red cell incompatibility between donors and recipients, chimerism affected the transition to donor type red cell transfusions, but showed no severe haemolytic events.21 We assessed the immunohaematological complications and clinical outcomes of 61 patients in the three clinical trials of non-myeloablative HPC transplantation in patients with sickle cell disease at the National Institutes of Health (NIH).9,12,25
Materials and methods
Patients
Inclusion criteria for this study were enrolment in the NIH trials NCT00061568,9,12 NCT02105766, or NCT0097769125 before March 31, 2015, at least one haemoglobin S gene, and scheduled HPC transplantation. The trials were single-centre, single-arm, phase 1 and 2 studies of HPC transplantation in patients with haemoglobinopathies. NCT00061568 and NCT02105766 were designed to assess transplantation from HLA-matched related donors, and NCT00977691 transplantation from HLA-haploidentical related donors. All donor-recipient pairs were identical or minor incompatible for ABO antigens. The institutional review board of the National Heart, Lung, and Blood Institute approved the three study protocols, and informed consent was obtained from all participants. Clinical data were collected up to March 31, 2016, from the electronic health records maintained at NIH.
Haemopoietic progenitor cell transplantation
The transplantation regimens have been described previously.9,12,25 Briefly, patients underwent red cell exchange to achieve a haemoglobin S fraction of 30% or less, followed by non-myeloablative conditioning with alemtuzumab and total body irradiation (300 cGy9,12 or 400 cGy25). The HPC grafts typically contained at least 10 × 106 peripheral blood CD34-positive cells per kg of recipient weight. After transplantation, patients with haploidentical grafts received cyclophosphamide with dose escalation. Sirolimus was used for immunosuppression, but could be tapered off after 1 year in patients who had received HLA-matched transplants from siblings if donor T-cell chimerism was greater than 50% in the absence of graft-versus- host disease.
Antibody identification was done before or at enrolment, and the referring hospitals were contacted to obtain the antibody histories of the patients. Transfusions were used to maintain concentrations of haemoglobin at 90–100 g/L and platelet concentrations greater than 50 × 109 cells per pL whenever possible. AH patients received leucocyte-reduced and irradiated red cell and apheresis platelet products. Red cell transfusions were matched to donors and recipients, with antigen-negative units provided when one or both individuals were phenotypically negative. At a minimum, the D, C, E, c, e, and K antigens were matched. Patients who had previously developed an alloantibody received red cell products that were negative for the corresponding antigen and matched for the D, C, E, c, e, K, Jka, Jkb, Fya, Fyb, S, and s antigens.
Immunohaematology
Routine blood group type, antibody screening, antibody identification, and direct antiglobulin tests were done with haemagglutination in gel matrix (ID-Micro Typing System, Ortho Clinical Diagnostics, Raritan, NJ, USA). Other serological platforms were used as needed for antibody identification. A reported alloantibody of unknown specificity that had been undetectable at enrolment in patient 170–32, who had antibodies against C, E, and S antigens, was excluded from analysis. We defined immunohaematological complications as the formation of new red cell antibodies after HPC transplantation, the presence of a red cell antibody and its cognate antigen in a donor-recipient pair, or both. HLA typing was outside the scope of this study.
Molecular immunohaematology
Red cell genotyping was done with commercial platforms and kits (PreciseType, BioArray RHCE, and RHD BeadChip, Immucor, Norcross, GA, USA). Some RHCE and RHD haplotypes were determined by nucleotide sequencing, as described previously.25 DNA samples from two donors were derived from recipients’ myeloid cells after transplantation, in which the donor represented 95% or greater of the DNA, as confirmed by chimerism studies.
Statistics
We used two-sided Fisher’s exact, Mann-Whitney U, and Wilcoxon W tests, to assess differences. We took p<0·05 to be significant. All analyses were done with R Statistical Programming Language (version 3.3.1).
Results
Of the 65 patients enrolled in the three NIH studies, one died before transplantation and three were treated for β thalassaemia but had no haemoglobin S genes. We therefore assessed 61 patients with sickle cell disease who underwent non-myeloablative allogeneic HPC transplantation from relatives (table 1). Of the 57 patients who were still alive at the time of writing (40 [95%] of 42 with identical HLA matches and 17 [89%] of 19 with haploidential HLA matches), all had maintained follow-up, 54 by attending appointments in person and three by email or telephone contact. The median follow-up was 4·3 years (IQR 1·8–5·8). 47 (77%) of 61 patients had functional grafts. Five of these patients developed new red cell antibodies after transplantation (table 2). In the other 14 patients, durable HPC engraftment failed (appendix p 1) and only one developed new red cell antibodies after transplantation (table 2).
Table 1:
Characteristics of patients at the time of HPC transplantation
| Identical HLA match (n=42) |
Haploidentical HLA match (n=19) |
Total (n=61) | |
|---|---|---|---|
| Haemoglobinopathy | |||
| SS | 37 (88%) | 17 (89%) | 54 (89%) |
| SC | 2 (5%) | 1 (5%) | 3 (5%) |
| S-β thalassaemia | 3 (7%) | 1 (5%) | 4 (7%) |
| Sex | |||
| Men | 24 (57%) | 10 (53%) | 34 (56%) |
| Women | 18 (43%) | 9 (47%) | 27 (44%) |
| Age (years) | |||
| Mean (SD) | 31·5 (10·1) | 34·9 (9·9) | 32·5 (10·1) |
| Median (IQR) | 29·0 (23·6·37·2) | 36·3 (27·1·39·8) | 31·2 (24·4·37·8) |
| HPC transplantation | |||
| HPC engraftment failed | 0 | 4 (21%) | 4 (7%) |
| Secondary graft rejection* | 5 (12%) | 5 (26%) | 10 (16%) |
| Graft maintained long term† | 37 (88%) | 10 (53%) | 47 (77%) |
| Follow-up (days) | |||
| Mean (SD) | 1865 (988) | 1150 (566) | 1643 (907) |
| Median (IQR) | 1764 (565·1991) | 1021 (499·1341) | 1575 (516·1831) |
| RBC transfusion | |||
| Before enrolment | |||
| Unknown | 5 (12%) | 0 | 5 (8%) |
| 1·10 units | 6 (14%) | 1 (5%) | 7 (11%) |
| 11·50 units | 16 (38%) | 5 (26%) | 21 (34%) |
| >50 units | 15 (36%) | 13 (68%) | 28 (46%) |
| After enrolment (units) | |||
| Mean (SD) | 17 (15) | 27 (18) | 20 (17) |
| Median (IQR) | 12 (10·18) | 20 (16·29) | 15 (10·22) |
HPC=haemopoietic progenitor cells. RBC=red blood cell.
All secondary graft rejections occurred between days 50 and 187, except one on day 215 (patient 225·01).
Includes three patients who showed signs of graft rejection (patients 170·02, 225·07, and 225·11) and received repeat HPC transplantation boosts from their original donors.
Table 2:
New red cell antibodies seen after enrolment
| Antibody specificity | Time to detection after transplantation (days) |
Cognate antigen | Donor chimerism* | Presumed antibody source† |
||||
|---|---|---|---|---|---|---|---|---|
| Antigen | Clinically relevant |
Recipient | Donor | CD3 (%) | Myeloid (%) |
|||
| Patient 170·20 | V | Yes | 61 | Negative | Positive | 15 | 100 | Recipient |
| Patient 170·28 | Knops‡, Jsa, D | No, yes, yes | 0, 7, 25 | NT, negative, positive | NT, negative, negative | NT | NT | Recipient |
| NT | NT | Recipient | ||||||
| 2 | 100 | Donor | ||||||
| Patient 170·29 | McCa | No | 21 | NT | NT | 7 | 76 | Donor |
| Patient 170·31 | E | Yes | 13 | Negative | Positive | 10 | 50 | Recipient |
| Patient 170·39 | Rh‡, Leb, autoantibody§ | Yes, no, possible | 1, 8, 99 | Negative, negative, NA | Negative, negative, NA | NT | NT | Recipient |
| NT | NT | Recipient | ||||||
| 5 | 100 | NA | ||||||
| Patient 225·01|| | Autoantibody§, C, K, Jsa | Possible, yes, yes, yes | 334, ~365, ~365, ~365 | NA, negative, negative, negative | NA, negative, negative, negative | 0 | 0 | NA |
| 0 | 0 | Recipient | ||||||
| 0 | 0 | Recipient | ||||||
| 0 | 0 | Recipient | ||||||
NT=not tested. NA=not applicable.
Before or within 1 day of antibody detection.
Based on phenotype and timing of antibody development.
Antibody against an antigen in the system but exact antigen not determined.
Warm-reactive to a high-prevalence antigen of the donor and recipient.
After graft loss at day 214, the patient received transfusions at an outside institution, and a change in prophylactic antigen matching might have contributed to antibody formation.
22 (36%) of 61 patients had red cell alloantibodies detectable at enrolment or noted by history (appendix p 2). No patients had only autoantibodies, but eight had red cell autoantibodies and alloantibodies. The 22 patients carried 58 alloantibodies, 34 (59%) of which had not been detected at enrolment (appendix p 3). Only four alloantibodies seen in three patients at enrolment were incompatible with donor antigens, and only one donor had a red cell alloantibody.
Among the new red cell antibodies seen in six (10%) patients after enrolment, 11 were alloantibodies and two were autoantibodies (table 2). Three patients developed incompatible antibodies, in two of whom the donor antigens were incompatible and in one the recipient antigen was incompatible. All new antibodies were detected at or after transplantation, and seven (64%) formed within 30 days of transplantation (table 2, figure 1). Event-free survival at 1 year was 90% (68–100). The median time to the event first antibody development was 17·0 days (IQR 4·0-51·0). Patient 225–01 lost the graft at day 214 and formed new alloantibodies at 1 year, but was included in the intention-to-treat analysis.
Figure 1:
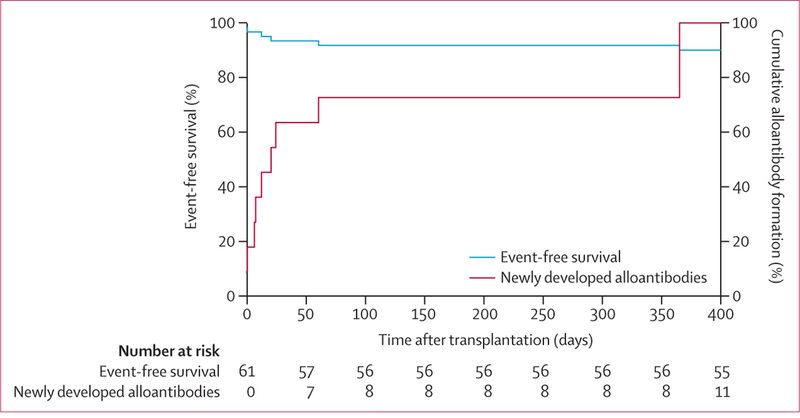
Alloantibody formation
The occurrence of new alloantibodies was not significantly associated with alloantibodies detected or reported at enrolment, a positive direct antiglobulin test before transplantation, autoantibodies detected at enrolment, detectable (rather than historical) alloantibodies at enrolment, number of transfusions before enrolment, sex of the recipient, sex of the donor, diagnosis, or ABO blood group (appendix p 4). The occurrence of new autoantibodies correlated significantly with development of new alloantibodies (p=0·0082, appendix p 4).
Patients required a mean of 20·0 (SD 16·7) red blood cell units after enrolment (table 1), of which a mean of 12.3 (15.3) were transfused after transplantation. The three patients with antibodies incompatible with donor red cells at enrolment received more red cell transfusions and depended on transfusion for longer time periods than other patients (figure 2, appendix p 5). The effect of new alloantibodies (table 2) in decreasing the number of available red cell units from regular blood donors differed widely between the six affected patients (table 3). In one patient (170–39) no compatible allogeneic red cell units were available, and haemoglobin concentration fell to 24 g/L despite aggressive therapy.
Figure 2: Transfusion support and clinical outcomes in engrafted patients by alloantibody status.
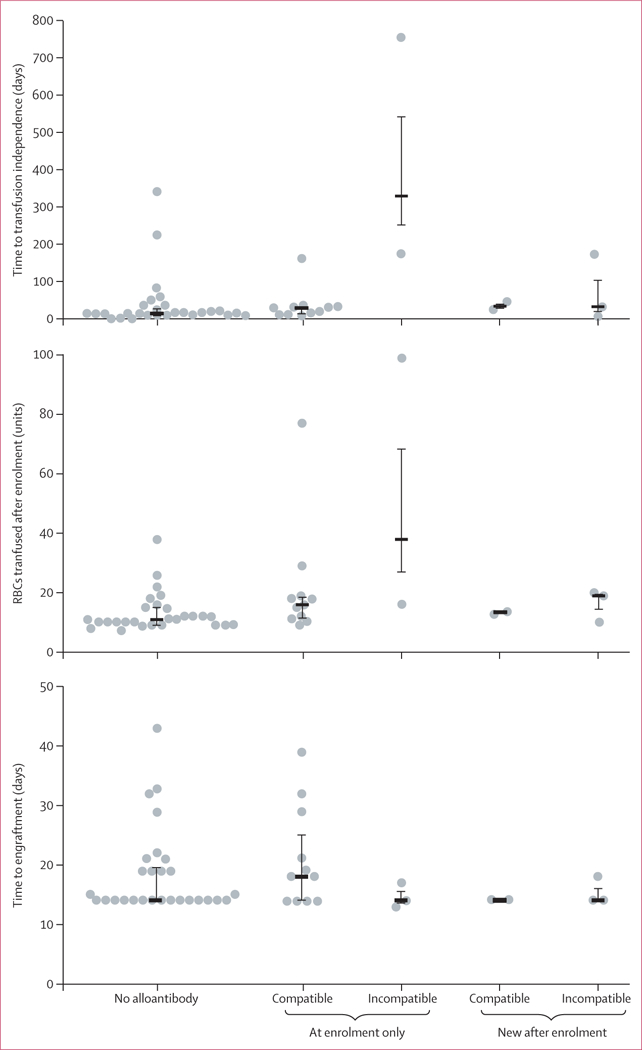
The 47 patients who maintained their grafts long term were divided into five groups: those with no alloantibodies (n=28); those with alloantibodies at enrolment that were either compatible (n=11) or incompatible (n=3) with their donors; and those who developed new alloantibodies after enrolment that were either compatible (n=2) or incompatible (n=3) with donor and recipient. The number of RBCs transfused after enrolment (A), time to transfusion independence (B), and time to engraftment (C) were compared across the five subgroups. Each circle represents one patient, horizontal bars show the median, and vertical bars indicate the first and third quartiles. For detailed data and statistics see the appendix (p 5). RBCs=red blood cells.
Table 3:
Reduction of available blood units caused by new alloantibodies during the clinical course
| Red cell alloantibodies | Probability of finding compatible blood donor units, by ethnicity (%)* |
Reduction of available units in the least affected donor population (%) |
|||||
|---|---|---|---|---|---|---|---|
| Before transplantation | After transplantation | Before transplantation |
After transplantation |
||||
| White | Black | White | Black | ||||
| Patient 170·20 | E, K | V | 65% | 76% | 65% | 54% | 0 in white |
| Patient 170·28 | S, C, E, V, K, Lea†, Fya | Jsa, D, Knops† | 6% | 29% | 1% | 2% | 83% in white |
| Patient 170·29 | None | McCa† | 100% | 100% | 100% | 100% | 0 any donor |
| Patient 170·31 | None | E | 100% | 100% | 71% | 78% | 22% in black |
| Patient 170·39 | Fya | Rh, Leb† | 32% | 87% | ~0 | ~0 | 100% any donor |
| Patient 225·01 | None | C, K, Jsa | 100% | 100% | 29% | 57% | 43% in black |
Calculations are based on published antigen frequencies, including only clinically relevant antigens; ABO compatibility and prophylactic matching for other antigens further restricts the number of available units.
Not judged to be clinically relevant.
Among the nine patients with immunohaematological complications, related severe clinical events occurred in five (56%). Sustained reticulocytopenia was seen in three patients (figure 3), two of whom had antibodies at enrolment that were incompatible with the donor, and one of whom developed a new antibody incompatible with the donor. All three patients needed transfusion support during their erythropoietic nadir (appendix pp 6–7). Acute haemolysis with severe anaemia occurred in two other patients who developed new antibodies and required complicated transfusion support (appendix p 7). Patient 170·39, who had an RHD-RHCE (C)ceS type 1 haplotype25 and RHD*weak D type 4.2.2-RHCE*ceAR haplotype, developed an alloantibody against a high- prevalence Rh antigen 1 day after transplantation (table 2), which was followed by hyperhaemolysis and a near fatal decrease in haemoglobin concentration to 24 g/L. This patient’s haemoglobin concentration at enrolment had been 66 g/L and remained at or above 110 g/L for the duration of follow-up of 1·5 years.
Figure 3: Reticulocyte concentrations in patients with sustained reticulocytopenia.
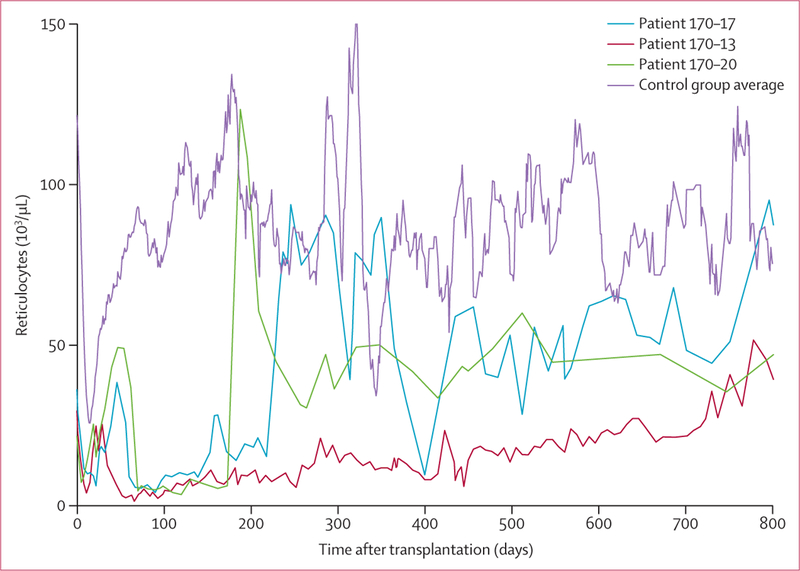
41 patients with haemopoietic progenitor cell engraftment and no red cell incompatibility experienced low reticulocyte concentrations around 10 days after transplantation that generally returned to normal concentrations by day 50. Three patients developed sustained reticulocytopenia, of whom two had pre-existing antibodies and one developed a new antibody incompatible with donors. Reticulocyte concentrations were >3 SD below control (14-day moving average of reticulocyte counts in 41 patients with red cell incompatability and sustained engraftment) at day 90. Reticulocytes recovered in two of these patients after >180 days, and in one after >2 years.
Seven of the nine patients with immunohaematological complications were alive at the time of writing and had been followed up for 1·5–8·0 years (appendix p 6). Patient 170–13 died 4·5 years after transplantation from a lower gastrointestinal haemorrhage, and patient 225–01 lost the graft on day 214 and died 5 years after transplantation from sepsis. Among the 52 patients without immunohaematological complications, 50 were alive at the time of writing (table 4), and had follow-up of 1·0-11·5 years (median 4·4 years, IQR 1·9-6·1). Patient 170–27 lost the graft on day 106 and died 10 months after transplantation from intracranial haemorrhage related to moyamoya disease. Patient 225–05 lost the graft on day 128 and died 6 months after transplantation from an infectious complication. Neither HPC graft failure or loss (p=1·0) nor death (p=0·10) correlated significantly with immunohaematological complications (table 4).
Table 4:
Survival and outcomes after haemopoietic progenitor cell transplantation, by alloantibody status, at March 31, 2016
| Without immunohaematological complications |
With immunohaematological complications |
Total | ||||||
|---|---|---|---|---|---|---|---|---|
| No alloantibody | Compatible alloantibody at enrolment only |
Subtotal | Incompatible alloantibody at enrolment only |
New alloantibody detected | Subtotal | |||
| Compatible | Incompatible | |||||||
| Graft recipients | ||||||||
| Total | 36 | 16 | 52 | 3 | 3 | 3 | 9 | 61 |
| Alive | 35 | 15 | 50 | 2 | 2 | 3 | 7 | 57 |
| Dead | 1 | 1 | 2 | 1 | 1 | 0 | 2 | 4 |
| Survival (95% CI) | 97% (68·100; p=NA) | 94% (52·100; p=NA) | 96% (71·100; reference) | 67% (8·100; p=NA) | 67% (8·100; p=NA) | 100% (20·100; p=NA) | 78% (31·100; p=0·10) | 93% (71·100; p=NA) |
| Engraftment failed | 1 | 3 | 4 | 0 | 0 | 0 | 0 | 4 |
| Secondary graft rejection | 7 | 2 | 9 | 0 | 1 | 0 | 1 | 10 |
| Graft maintained long term (95% CI) | 78% (52·100; p=NA) | 69% (26·100; p=NA) | 75% (53·100; reference) | 100% (21·100; p=NA) | 67% (8·100; p=NA) | 100% (21·100; p=NA) | 89% (38·100; p=1·0) | 77% (57·100; p=NA) |
| Sustained engraftment | ||||||||
| Number of patients | 28 | 11 | 39 | 3 | 2 | 3 | 8 | 47 |
| Mean (SD) haemoglobin concentration (g/L) | ||||||||
| At enrolment* | 86 (16) | 81 (12) | 85 (15) | 90 (6) | 84 (4) | 76 (32) | 83 (19) | 84 (15) |
| After transfusion independence† | 121 (21; | 112 | 118 | 109 | 112 | 104 | 108 | 117 |
| reference) | (18; p=0·16) | (20; p=NA) | (15; p=0·26) | (10; p=0·36) | (4; p=0·11) | (10; p=NA) | (1·9; p=0·13) | |
| Haemoglobin AA donors | ||||||||
| Number of patients | 10 | 6 | 16 | 1 | 0 | 1 | 2 | 18 |
| Means (SD) haemoglobin S expression (%) | ||||||||
| At enrolment* | 53·2 (30·6) | 53·5 (28·0) | 53·3 (28·7) | 83·4 (0) | NA | 75·4 (0) | 79·4 (5·7) | 56·2 (28·3) |
| After transfusion independence† | 0 | 0 | 0 | 0 | NA | 0 | 0 | 0 |
| Haemoglobin S trait donors | ||||||||
| Number of patients | 18 | 5 | 23 | 2 | 2 | 2 | 6 | 29 |
| Mean (SD) haemoglobin S expression (%) | ||||||||
| At enrolment* | 56·3 (23·0) | 58·5 (19·8) | 56·8 (21·9) | 50·5 (8·2) | 57·1 (21·8) | 84·3 (9·8) | 64·0 (19·6) | 58·3 (21·3) |
| After transfusion independence† | 38·1 (6·8; reference) | 35·8 (4·1; p=0·53) | 37·6 (6·3; p=NA) | 35·0 (9·1; p=0·90) | 35·1 (1·9; p=0·31) | 41·6 (0·7; p=0·78) | 37·2 (5·4; p=NA) | 37·5 (6·0; p=0·25) |
NA=not applicable.
Many patients had been recently transfused.
Determined ≥3 months after the last red blood cell transfusion.
Haemoglobin concentration and haemoglobin S expression after transfusion independence were excellent in all 47 patients in whom grafts were maintained long term (table 4). We found no significant differences between the eight patients with and the 39 patients without immunohaematological complications or between the subgroups of these patients (table 4).
Discussion
Among 61 patients with sickle cell disease who underwent non-myeloablative HPC transplantation, immunohaematological complications were seen in nine (15%), and the outcomes ranged from seemingly not clinically relevant to potentially fatal. At enrolment, three patients had known red cell alloantibodies that were incompatible with their donors’ antigens, and after transplantation, two of these patients experienced sustained reticulocytopenia for at least 180 days. New red cell alloantibodies were observed in six (10%) patients, of which three were compatible and three were incompatible with donor or recipient antigens. We did not identify any clinical or laboratory parameters predictive of which patients would form new red cell antibodies, suggesting that all patients are at risk.
The consequences of incompatible alloantibodies seemed to be unrelated to the blood group system involved. For example, patient 170–13 had an antibody against Jka antigen that was incompatible with the donor antigens, and developed sustained reticulocytopenia (2 years), whereas patient 170–09 had an antibody against Jkb antigen that was incompatible with the donor antigens and was associated with no clinically relevant outcomes. In a study of haemopoietic stem-cell transplantation in adults with sickle cell disease,13 two recipients of major ABO-incompatible HPC grafts did not develop complications, although ABO antibodies are clinically relevant.
The presence of mixed chimerism after transplantation raises the question of whether the donor or recipient immune system generated the antibody response. Although recipient response was more common in this study (table 2), we did see some donor-derived antibodies. Patient 170–28 developed a donor-derived antibody against D antigen, which is consistent with previous reports that in cases of D mismatch, antibodies are most commonly seen when the donor is D negative and the recipient D positive.22 Additionally, transplantation raises the question of whether the source of sensitisation is transfused, donor, or recipient red blood cells. Patient 170–39 had arguably the most dangerous complications, including severe anaemia and the complete lack of available allogeneic donor units (table 3). The Rh antibody was probably a response to transfused red blood cells. Thus, the clinical course could not have been anticipated, and having the identical red cell genotype to the HPC donor contributed to survival.
Because immunohaematological complications have unpredictable onset and effects, prevention is crucial. The allogeneic red blood cell units and the HPC graft are both sources of sensitising red cells. To prevent alloimmun- isation from red blood cell units, they must be phenotyped and matched to the recipient, which requires access to a large inventory of extensively typed red cell units. HPC donor antigens should be assessed and considered during the donor selection process. Serological phenotyping might be inadequate in both settings. Red cell alloantibodies can form despite matching for Rh and Kell antigens, including in patients who seem to express the corresponding antigen.26,27 These outcomes are thought to be related to the high prevalence of variant RHD and RHCE alleles in individuals with sickle cell disease.27 We therefore recommend careful assessment of compatibility between the recipient and all potential HPC donors by red cell genotyping before transplantation. Additionally, the risks of using an incompatible donor and the feasibility of providing long-term transfusion support should be considered.
There is no medical therapy to prevent antibody formation and no method to predict which patients are at risk. For patients with existing antibodies, desensitisation regimens are available that might be useful. The availability of antigen-negative red cell units determines whether patients with existing or new alloantibodies can safely receive transfusions. The number of donors who must be screened to find a compatible red cell unit increases with increasing rarity of blood type (table 3). High-throughput red cell genotyping methods have been developed that enable rapid and cost-efficient screening of tens of thousands of donors for millions of antigen data.28 When fully implemented to encompass all donors and provide comprehensive blood group typing, including RHD and RHCE genes, genotypically matched red cell units might become available for all patients, either to manage alloantibodies or to support prophylactic matching.
Red cell incompatibility or development of new red cell alloantibodies did not correlate with graft failure or secondary graft rejection (table 4). HPCs and leucocytes, specifically T cells, rather than red cells, mediate these processes and do not express most red cell antigens. Although red cell alloimmunisation does not present a barrier to HPC engraftment and maintenance of a functional graft, it can cause severe complications, such as anaemia, haemolysis, and sustained reticulocytopenia. Nevertheless, standard care also carries a risk oftransfusion and subsequent alloimmunisation, with 18–30% of patients with sickle cell disease developing red blood cell antibodies.23 In this study, 10% of patients showed new alloimmunisation, which is comparatively low but ocurred over a short time period. With involvement by transfusion medicine specialists to prevent and manage these complications, however, HPC transplantation might be preferable to a lifetime of transfusion.
The number of immunohaematological complications reported in this study reflects a specific conditioning regimen that used the lymphocyte-depleting agent alemtuzumab. When given early (around 20 days before transplantation), alemtuzumab leads to recipient immunoablation and prevents graft rejection, and when given closer to transplantation (around 7 days before), it depletes donor T cells and prevents graft-versus-host disease.29 In the patients we assessed, alemtuzumab given from 7 to 3 days before transplantation prevented graft-versus-host disease and might have lessened the risk of immunohaematological complications. If myeloablative conditioning were used, severe complications would be unlikely in donor-recipient pairs with red blood cell incompatibility.21 The toxic effects, though, cannot be tolerated by most adults with sickle cell disease. Additionally, distinct HLA haplotypes are thought to affect the immune response to blood group antigens.30 Although outside the scope of this study, future research could investigate whether this mechanism alters the development of antibodies, and would allow stratification of patients for myeloablative and non-myeloablative HPC transplantation.
Our results were obtained in a highly selected population of patients for whom transplantation was judged to be feasible and safe. Several patients who initially passed screening for the three NIH clinical trials were not enrolled because of known alloimmunisation or transfusion-related complications. The frequency of immunohaematological complications would probably have been greater if these patients had also undergone HPC transplantation. Studying cohorts of patients with sickle cell disease who have been assessed for HPC transplantation, including those ineligible for immunohaematological reasons, could document the progress needed to serve all patients with sickle cell disease. Additionally, our findings might be applicable to other transplantation scenarios, especially as HPC transplantation in sickle cell disease is changing rapidly.9,12,25 For example, autologous transplantation with gene therapy is being explored as an alternative to related-donor transplantation. In this setting, there is no allogeneic HPC donor and, therefore, no risk of alloimmunisation. Nevertheless, exposure to allogeneic red cell units would still be important to the transplantation course and could still lead to immunohaematological complications like those we describe.
HPC transplantation is increasingly being used to cure patients with sickle cell disease. Immunohaematological complications occurred in nine (15%) of 61 of our patients, some of which were life threatening. Whether related HPC donors or autologous gene therapy is used, treatment is most often be supported by red cell transfusion. Advanced typing of red cells in patients, HPC donors, and allogeneic blood donors at the molecular level is technically feasible. Transfusion medicine specialists can advise on donor selection and transfusion support for red cell antigen matching. Implementation of red cell genotyping should further improve care of patients and lessen risks, and might prove the conclusive way to prevent immuno- haematological complications.
Supplementary Material
Research in context
Evidence before this study
Special matching of transfused red cell units is needed for patients with sickle cell disease because they are more likely than the general population to produce red cell antibodies and are prone to transfusion reactions, such as delayed haemolysis and hyperhaemolysis. Haemopoietic progenitor cell (HPC) transplantation can cure sickle cell disease. Various regimens are being investigated, but all require transfusion. We searched PubMed for clinical trials of HPC transplantation in sickle cell disease, including those describing non-myeloablative conditioning regimens, published in English up to Nov 30, 2015. We used the search terms “sickle cell”, “hematopoietic progenitor cell transplantation”, and “antibodies” (and synonyms). Besides a small case series, we found no trials that systematically analysed complications related to red cell antibodies.
Added value of this study
We assessed the effects of existing and newly formed red cell antibodies on clinical outcomes in all patients with sickle cell disease prospectively enrolled since 2003 in the three clinical trials of non-myeloablative HPC transplantation at the National Institutes of Health. Immunohaematological complications did not affect HPC engraftment or overall survival, but ranged from mild and not clinically relevant to serious and potentially fatal. No subsets of patients were particularly prone to developing complications, and we conclude that all patients are at risk.
Implications of all the available evidence
Understanding the risks of developing immunohaematological complications and possible associated outcomes will improve the safety of non-myeloablative HPC transplantation in patients with sickle cell disease. Careful assessment of the red cell antigen match and planning of transfusion support before HPC transplantation should help to minimise serious immunohaematological complications.
Acknowledgments
We thank the patients and family members who enrolled in National Heart, Lung, and Blood Institute (NHLBI) protocols 03-H-0170, 09-H-0225, and 14-H-0077 We thank Donna Chauvet, Wynona Coles, Mary Elizabeth Link, Monica Schmitt, and Tiffani Taylor at NHLBI for research coordination and clinical care, Sherry Sheldon, Sharon Adams, Nadine Dowling, Rebecca Perry, and Hallie Lee-Stroka and National Institutes of Health (NIH) Clinical Center for laboratory testing and transfusion support, David Stiles at NIH Clinical Center for assistance with statistics and graphic design, and Judith Welsh at NIH Library for assistance with the literature search. This work was supported by the Intramural Research Program (project ID Z99 CL999999) of the NIH Clinical Center. The views expressed do not necessarily represent the view of the NIH, the Department of Health and Human Services, or the US Federal Government.
Funding Intramural Research Program and National Institutes of Health.
Role of the funding source
The funder of the study had no role in the study design, data collection, data analysis, data interpretation, or the writing of the report. The corresponding author had full access to all the data in the study and had final responsibility for the decision to submit for publication
Footnotes
Declarations of interest
We declare no competing interests.
Contributor Information
Elizabeth S Allen, Department of Transfusion Medicine, NIH Clinical Center, National Institutes of Health, Bethesda, MD, USA.
Kshitij Srivastava, Department of Transfusion Medicine, NIH Clinical Center, National Institutes of Health, Bethesda, MD, USA.
Matthew M Hsieh, Molecular and Clinical Hematology Branch, National Heart, Lung, and Blood Institute, National Institutes of Health, Bethesda, MD, USA.
Courtney D Fitzhugh, Sickle Cell Branch, National Heart, Lung, and Blood Institute, National Institutes of Health, Bethesda, MD, USA.
Harvey G Klein, Department of Transfusion Medicine, NIH Clinical Center, National Institutes of Health, Bethesda, MD, USA.
John F Tisdale, Molecular and Clinical Hematology Branch, National Heart, Lung, and Blood Institute, National Institutes of Health, Bethesda, MD, USA.
Prof Willy A Flegel, Department of Transfusion Medicine, NIH Clinical Center, National Institutes of Health, Bethesda, MD, USA.
References
- 1.Johnson FL, Look AT, Gockerman J, Ruggiero MR, Dalla-Pozza L, Billings FT 3rd. Bone-marrow transplantation in a patient with sickle-cell anemia. N Engl J Med 1984; 311: 780–83. [DOI] [PubMed] [Google Scholar]
- 2.Vermylen C, Cornu G, Ferster A, et al. Haematopoietic stem cell transplantation for sickle cell anaemia: the first 50 patients transplanted in Belgium. Bone Marrow Transplant 1998; 22: 1–6. [DOI] [PubMed] [Google Scholar]
- 3.Walters MC, Patience M, Leisenring W, et al. Stable mixed hematopoietic chimerism after bone marrow transplantation for sickle cell anemia. Biol Blood Marrow Transplant 2001; 7: 665–73. [DOI] [PubMed] [Google Scholar]
- 4.van Besien K, Bartholomew A, Stock W, et al. Fludarabine-based conditioning for allogeneic transplantation in adults with sickle cell disease. Bone Marrow Transplant 2000; 26: 445^−9. [DOI] [PubMed] [Google Scholar]
- 5.Iannone R, Casella JF, Fuchs EJ, et al. Results of minimally toxic nonmyeloablative transplantation in patients with sickle cell anemia and beta-thalassemia. Biol Blood Marrow Transplant 2003; 9: 519–28. [DOI] [PubMed] [Google Scholar]
- 6.Horan JT, Liesveld JL, Fenton P, Blumberg N, Walters MC. Hematopoietic stem cell transplantation for multiply transfused patients with sickle cell disease and thalassemia after low-dose total body irradiation, fludarabine, and rabbit anti-thymocyte globulin. Bone Marrow Transplant 2005; 35: 171–77 [DOI] [PubMed] [Google Scholar]
- 7.Horwitz ME, Spasojevic I, Morris A, et al. Fludarabine-based nonmyeloablative stem cell transplantation for sickle cell disease with and without renal failure: clinical outcome and pharmacokinetics. Biol Blood Marrow Transplant 2007; 13: 1422–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Krishnamurti L, Kharbanda S, Biernacki MA, et al. Stable long-term donor engraftment following reduced-intensity hematopoietic cell transplantation for sickle cell disease. Biol Blood Marrow Transplant 2008; 14: 1270–78. [DOI] [PubMed] [Google Scholar]
- 9.Hsieh MM, Kang EM, Fitzhugh CD, et al. Allogeneic hematopoietic stem-cell transplantation for sickle cell disease. N Engl J Med 2009; 361: 2309–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Matthes-Martin S, Lawitschka A, Fritsch G, et al. Stem cell transplantation after reduced-intensity conditioning for sickle cell disease. Eur J Haematol 2013; 90: 308–12. [DOI] [PubMed] [Google Scholar]
- 11.Bhatia M, Jin Z, Baker C, et al. Reduced toxicity, myeloablative conditioning with BU, fludarabine, alemtuzumab and SCT from sibling donors in children with sickle cell disease. Bone Marrow Transplant 2014; 49: 913–20. [DOI] [PubMed] [Google Scholar]
- 12.Hsieh MM, Fitzhugh CD, Weitzel RP, et al. Nonmyeloablative HLA-matched sibling allogeneic hematopoietic stem cell transplantation for severe sickle cell phenotype. JAMA 2014; 312: 48–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Saraf SL, Oh AL, Patel PR, et al. Nonmyeloablative stem cell transplantation with alemtuzumab/low-dose irradiation to cure and improve the quality of life of adults with sickle cell disease. Biol Blood Marrow Transplant 2016; 22: 441–48. [DOI] [PubMed] [Google Scholar]
- 14.Shenoy S, Eapen M, Panepinto JA, et al. A trial of unrelated donor marrow transplantation for children with severe sickle cell disease. Blood 2016; 128: 2561–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hsieh MM, Fitzhugh CD, Tisdale JF. Allogeneic hematopoietic stem cell transplantation for sickle cell disease: the time is now. Blood 2011; 118: 1197–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Griffith LM, McCoy JP Jr, Bolan CD, et al. Persistence of recipient plasma cells and anti-donor isohaemagglutinins in patients with delayed donor erythropoiesis after major ABO incompatible non-myeloablative haematopoietic cell transplantation. Br J Haematol 2005; 128: 668–75. [DOI] [PubMed] [Google Scholar]
- 17.Bolan CD, Leitman SF, Griffith LM, et al. Delayed donor red cell chimerism and pure red cell aplasia following major ABO-incompatible nonmyeloablative hematopoietic stem cell transplantation. Blood 2001; 98: 1687–94. [DOI] [PubMed] [Google Scholar]
- 18.Canals C, Muniz-Diaz E, Martinez C, et al. Impact of ABO incompatibility on allogeneic peripheral blood progenitor cell transplantation after reduced intensity conditioning. Transfusion 2004; 44: 1603–11. [DOI] [PubMed] [Google Scholar]
- 19.Worel N, Kalhs P, Keil F, et al. ABO mismatch increases transplant-related morbidity and mortality in patients given nonmyeloablative allogeneic HPC transplantation. Transfusion 2003; 43: 1153–61. [DOI] [PubMed] [Google Scholar]
- 20.Franchini M, Gandini G, Aprili G. Non-ABO red blood cell alloantibodies following allogeneic hematopoietic stem cell transplantation. Bone Marrow Transplant 2004; 33: 1169–72. [DOI] [PubMed] [Google Scholar]
- 21.Senzel L, Boulad F, Wuest D, Reid ME. Transfusion policy: when to stop the use of extremely rare blood for an allogeneic hematopoietic progenitor cell transplant recipient with a history of red cell alloimmunization. Transfusion 2007; 47: 781–87 [DOI] [PubMed] [Google Scholar]
- 22.Cid J, Lozano M, Klein HG, Flegel WA. Matching for the D antigen in haematopoietic progenitor cell transplantation: definition and clinical outcomes. Blood Transfus 2014; 12: 301–06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Klein HG, Anstee DJ. Mollison’s blood transfusion in clinical medicine, 12th edn Hoboken, NJ: Wiley-Blackwell, 2014. [Google Scholar]
- 24.McPherson ME, Anderson AR, Haight AE, et al. Transfusion management of sickle cell patients during bone marrow transplantation with matched sibling donor. Transfusion 2009; 49: 1977–86. [DOI] [PubMed] [Google Scholar]
- 25.Fitzhugh CD, Hsieh MM, Taylor T, et al. Cyclophosphamide improves engraftment in patients with SCD and severe organ damage who undergo haploidentical PBSCT. Blood Adv 2017; 1: 652–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fasano RM, Monaco A, Meier ER, et al. RH genotyping in a sickle cell disease patient contributing to hematopoietic stem cell transplantation donor selection and management. Blood 2010; 116: 2836–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chou ST, Jackson T, Vege S, Smith-Whitley K, Friedman DF, Westhoff CM. High prevalence of red blood cell alloimmunization in sickle cell disease despite transfusion from Rh-matched minority donors. Blood 2013; 122: 1062–71. [DOI] [PubMed] [Google Scholar]
- 28.Flegel WA, Gottschall JL, Denomme GA. Integration of red cell genotyping into the blood supply chain: a population-based study. Lancet Haematol 2015; 2: e282–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.King AA, Kamani N, Bunin N, et al. Successful matched sibling donor marrow transplantation following reduced intensity conditioning in children with hemoglobinopathies. Am J Hematol 2015; 90: 1093–98. [DOI] [PubMed] [Google Scholar]
- 30.Tatari-Calderone Z, Gordish-Dressman H, Fasano R, et al. Protective effect of HLA-DQB1 alleles against alloimmunization in patients with sickle cell disease. Hum Immunol 2016; 77: 35–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.