

Abstract
Foxp3+ regulatory T (TR) cells are phenotypically and functionally diverse, and broadly distributed in lymphoid and non-lymphoid tissues. However, the pathways guiding the differentiation of tissue-resident TR populations have not been well defined. By regulating E-protein function, Id3 controls the differentiation of CD8+ effector T cells and is essential for TR maintenance and function. We show that dynamic expression of Id3 helps define three distinct mouse TR populations, Id3+CD62LhiCD44lo central (c)TR, Id3+CD62LloCD44hi effector (e)TR and Id3- eTR. Adoptive transfer experiments and transcriptome analyses support a stepwise model of differentiation from Id3+ cTR to Id3+ eTR to Id3- eTR. Furthermore, Id3- eTR have high expression of functional inhibitory markers and a transcriptional signature of tissue-resident TR. Accordingly, Id3- eTR are highly enriched in non-lymphoid organs, but virtually absent from blood and lymph. Thus, we propose that tissue-resident TR develop in a multi-step process associated with Id3 downregulation.
Introduction
Several recent studies have highlighted the phenotypic and functional heterogeneity of regulatory T (TR) cells during both steady state and inflammation (1–4). We and others have shown that at steady state in lymphoid organs TR can be broadly divided by expression of CD44 and CD62L into distinct subsets which differ in their localization, dependence on IL-2, and extent of PI3K signaling (2, 5, 6). Moreover, CD44hiCD62Llo effector (e)TR display diverse expression of transcription factors and chemokine receptors that promote their migration to inflamed tissues and their response to different types of inflammatory signals (1, 7). Accordingly, TR found in nonlymphoid tissues have a distinct molecular profile that includes high expression of Gata3 and ST2 (the IL-33R), and are functionally equipped to suppress inflammation at barrier sites (8, 9). Although these data highlight the anatomical, functional and molecular diversity of TR, the pathways by which these TR populations differentiate have not been completely defined.
The inhibitors of DNA binding (Id) proteins have been extensively studied in lymphocyte development (10, 11). Studies of CD8+ effector T cells revealed that Id2 and Id3 are powerful transcriptional regulators of differentiation that are dynamically regulated during T cell activation and effector/memory T cell differentiation (12, 13). Through their regulation of E protein function, Id2 and Id3 help to control expression of genes essential for CD8+ effector cell differentiation and survival such as Tcf7, Tbx21, Bcl2 and Klrg1 (14, 15). Although less well studied, Id proteins have been shown to have essential roles in CD4+ T cell function. For instance, Id2 and Id3 are essential for TR maintenance and function, with TR lacking both Id2 and Id3 having impaired proliferation and survival (16). In TR, Id3 helps to stabilize Foxp3 through restriction of the E protein E47 and its downstream targets Spi-B and SOCS3 (17). However, Id3 expression is not uniform in TR, and distinct populations of Id3+ and Id3- have been identified (16, 18). In this study, we show that Id3 is dynamically regulated in TR, and that progressive loss of Id3 correlates with the stepwise differentiation of a highly-functional TR population localized primarily in non-lymphoid tissues.
Materials and Methods
Mice
C57BL/6, RAG1-deficient and Foxp3-mRFP mice were purchased from The Jackson Laboratory. Id3-GFP mice were a gift from Ananda Goldrath (UCSD, La Jolla, California) and have been previously described (12, 14). Mice were bred and housed under the approval of the Institutional Animal Care and Use Committee of the Benaroya Research Institute.
Cell Isolation
Unless noted below, single cell suspensions isolated from tissues using manual disruption. PEC isolated by injecting sterile PBS into peritoneal cavity of euthanized mice, gentle disruption to dislodge cells and collection of injected PBS. IEL and LPL were isolated from pooled large and small intestine as previous described (19). Lymphocytes further purified by resuspension in 44% Percoll™ (GE Healthcare) layered over 67% Percoll™ and spun at 2,800rpm for 20 mins. Lung and fat were finely minced, digested with 0.26U/mL Liberase TM (Roche) and 10U/mL DNAse (Sigma) for 1 hr at 37°C and filtered. For skin tissue, ears were processed as above with 0.14U/mL Liberase TM and 10U/mL DNAse. For lymph collection, mice were fed 20mL/kg ‘Half and Half’ by oral gavage and sacrificed 2–3 hrs later. Lymph collected from the cisterna chyli was directly stained for flow cytometry.
Flow cytometry
Single cells suspensions were stained with fixable Viability Dye eFluor 780 (eBiosciences) in PBS for 10 min at RT. Cells were stained with directly conjugated Abs in PBS with 0.5% BCS for 20 min at 4°C. Abs purchased from BioLegend: CD4 (RM4–5), TCRβ (H57–597), CD44 (IM7), CD62L (MEL-14), CD25 (PC61), ICOS (C398.4A), TIGIT (1G9), CTLA4 (UC10–4B9), GITR (DTA.1), CD69 (53–7.3). Abs purchased from eBiosciences: KLRG1 (2F1) and CD103 (2E7). Intracellular stains were performed using a FixPerm Kit (eBiosciences). Data acquired on an LSR II (BD Biosciences) and analyzed using FlowJo software (TreeStar).
In vitro assays
CD4+ T cells isolated from spleen and LN using CD4 microbeads (Miltenyi). 1×106 T cells cultured with platebound α-CD3 (2C11) and α-CD28 (37.51) from BioXcell at 1μg/mL each for 48 or 66 hrs. Inhibitors purchased and used as follows: ZSTK474 (1μM, Sigma), Rapamycin (10nM, Selleckchem), NFAT inhibitor (10μM, Tocoris), Mek inhibitor PD0325901 (100nM, Peprotech) and Erk inhibitor FR180204 (10μM, Tocoris). In vitro TR suppression assays were performed as previous described (20). Chemotaxis assay performed as previously described (21).
In vivo TR transfer
Sorted TR isolated from spleen and LN as described for RNA-seq. 100,000 sorted cells were injected retro-orbitally into RAG1-deficient hosts. Spleen, LN and blood of recipient mice collected two wks later and analyzed by flow cytometry.
Statistical Analysis
The p values were calculated by Prism software (GraphPad) using either an unpaired Student’s t test or one way ANOVA as indicated. Values less than 0.05 were considered significant.
RNA-seq
CD4+ T cells were isolated using CD4 microbreads (Miltenyi) from either peripheral LNs or spleens of 3 littermate Id3-GFP x Foxp3-mRFP mice. Cells were sorted based on viability, CD4, CD44, CD62L, Id3-GFP and Foxp3-mRFP expression on a FACs Aria II (BD Biosciences). 500 cells were sorted directly into SMART-Seq v4 Ultra Low Input RNA Kit (Takara) lysis buffer and protocol followed to produce cDNA. Library construction was performed using a modified protocol of the NexteraXT DNA sample preparation kit (Illumina). Dual-index, single-read sequencing of pooled libraries was run on a HiSeq2500 sequencer (Illumina) with 58-base reads and an average depth of 8.6Mio reads per library. Base-calling and demultiplexing were performed automatically on BaseSpace (Illumina) to generate FASTQ files.
Results/Discussion
Id3 is dynamically expressed in TR and regulated by TCR signaling
To examine Id3 expression in TR we generated Id3-GFP x Foxp3-mRFP double reporter mice. In agreement with previously published reports, most CD4+ T cells in spleen and lymph nodes (LNs) were Id3+ (16, 18). However, we noted a subset of Id3- cells in both Foxp3+ TR and Foxp3- conventional CD4+ T cell populations (Fig 1A). Within TR, the Id3- population fell exclusively within the CD44hi CD62Llo eTR compartment (5), whereas Id3+ TR were found in both the CD44lo CD62Lhi central (c)TR and eTR compartments (Fig 1B). Thus, in secondary lymphoid organs (SLOs) TR can be divided into three distinct subsets based on Id3, CD62L and CD44 expression, Id3+ cTR, Id3+ eTR or Id3- eTR. Within the Id3+ TR populations Id3+ cTR had higher Id3 expression than Id3+ eTR measured by GFP-MFI (Fig 1B), leading us to hypothesize that TR may downregulate Id3 as they transition during their activation and differentiation from cTR into eTR. Indeed, Id3 expression can be downregulated by TCR signaling (16), and we observed a loss of Id3 expression in the TR compartment correlating with the strength of TCR stimulation when cells were activated with different concentrations of platebound αCD3/28 (Supplemental Fig 1). Furthermore, inhibiting either the MAP-kinase/Erk or PI3-kinase/mTOR signaling pathways blocked Id3 downregulation in TR (Fig 1C), consistent with reports that eTR development requires TCR stimulation, mTOR signaling and the PI3-kinase-dependent inactivation of the transcription factor Foxo1 (5, 6, 22).
Figure 1: Id3 is dynamically expressed in TR and regulated by TCR signaling.
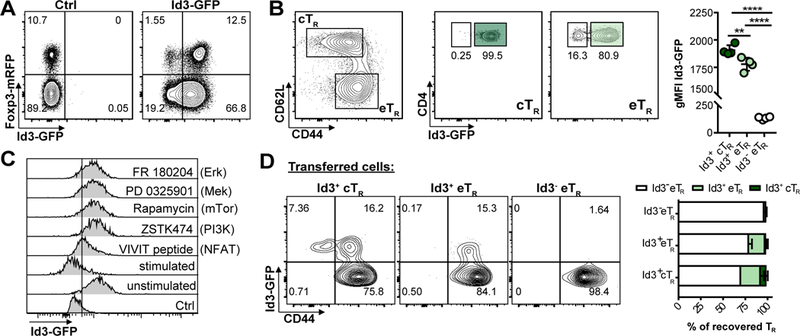
A) Representative flow cytometry plots of Id3-GFP and Foxp3-RFP expression by gated splenic TCRβ+ CD4+ T cells. B) Representative flow cytometry analysis of Id3-GFP expression by splenic CD44loCD62Lhi cTR and CD44hiCD62Llo eTR gated as indicated. (Bottom left) Graphical analysis of gMFI of Id3-GFP in each of the three gated TR populations. C) Representative flow cytometry plots of Id3-GFP expression by gated splenic TCRβ+ CD4+ Foxp3+ TR 66 hrs after stimulation of purified CD4+ T cells in the presence or absence of the idicated inhibitors, representative of 2 independent expts. D) Representative flow cytometry plots and graphical analysis of CD44 and Id3-GFP expression by gated TCRβ+CD4+Foxp3+ TR recovered from the LNs of RAG1-deficient mice 2 weeks after transfer of the indicated TR population, summary of 3 independent expts, 3–5 mice per group total. Significance determined by one way ANOVA with Tukey’s post-test for pairwise comparisons, *p <0.05, **p < 0.01, ****p < 0.001
To more precisely define the developmental relationship between these TR subsets we utilized an adaptive transfer model in which TR stimulation and expansion depends on TCR:MHCII interactions (23). For this we sorted Id3+ cTR, Id3+ eTR or Id3- eTR from spleen and LNs of reporter mice and transferred individual TR populations into RAG1-deficient animals, and evaluated the phenotype and expansion of transferred TR after 2 weeks. Importantly, we did not observe any difference in the extent of Foxp3 expression between the TR populations upon their recovery, which varied between ~30–80% in different experiments (not shown). Transferred Id3+ cTR gave rise to all three subsets, with some cells retaining Id3 and CD62L expression but the majority converting into Id3- eTR (Fig 1D). The bulk of Id3+ eTR downregulated Id3, with no cells regaining CD62L expression, whereas Id3- eTR did not give rise to either of the other populations, indicating that these cells are likely a terminal differentiated population. Thus, Id3- eTR appear to develop from Id3+ cTR in a stepwise manner during activation, with Id3+ eTR acting as an intermediate population.
Id3- eTR express inhibitory markers and are highly suppressive
To explore the phenotypic and functional differences between these three subsets of TR, we assessed expression of the TR-associated surface markers on each population of splenic TR. In agreement with previously published data, cTR and eTR showed distinct phenotypes, with eTR having lower expression of the high affinity IL-2 receptor component CD25, but higher levels of the activation and functional surface markers ICOS, KLRG1, TIGIT, GITR and CTLA4 (Fig 2A) (5, 6). Moreover, within the eTR compartment the Id3- TR had higher expression of activation and inhibitory molecules than their Id3+ TR counterparts, but had the lowest expression of CD25. As our lab previously described (5), elevated expression of ICOS and diminished expression of CD25 suggests that Id3- eTR are less dependent on IL-2 for their homeostatic maintenance, and instead likely rely on continued ICOS signaling. Additionally, increased expression of these TR functional molecules correlated with enhanced in vitro suppressive activity of Id3- eTR compared with either Id3+ cTR or Id3+ eTR (Fig 2B).
Figure 2: Id3- eTR express inhibitory markers and are highly suppressive.
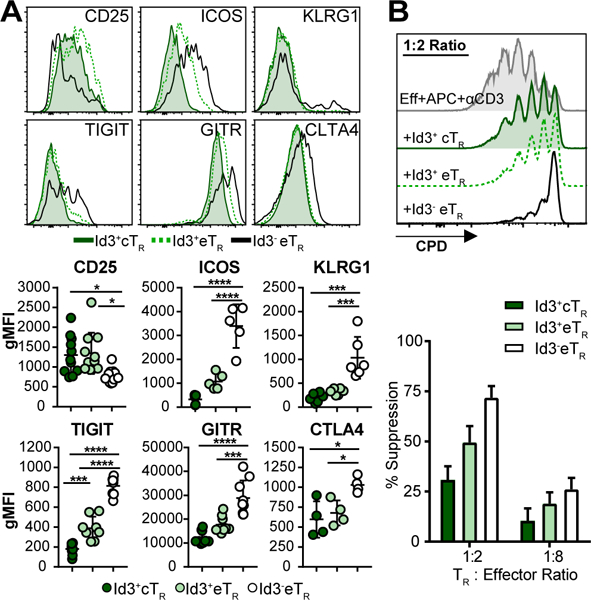
A) (Top) Representative flow cytometry histograms. (Bottom) Graphical analysis of expression of the indicated markers by gated splenic TR populations. B) (Top) Representative flow cytometry analysis of CPD dilution by CD4+Foxp3- effector T cells stimulated with or without the indicated TR populations. (Bottom) Graphical analysis of suppression by each of the indicated populations, n=3. Significance determined by one way ANOVA with Tukey’s post-test for pairwise comparisons, *p <0.05, **p < 0.01, ****p < 0.001
Transcriptional profiling highlights the stepwise differentiation of Id3- eTR
To identify and compare their unique transcriptional profiles, we performed RNA-seq on sorted Id3+ cTR, Id3+ eTR and Id3- eTR from spleen or LNs of Id3-GFP x Foxp3-mRFP reporter mice. Although there was little difference between LN and spleen samples, principle component analysis (PCA) showed that each of the three TR populations were transcriptionally distinct (Fig 3A), and accordingly we identified 1,672 significantly differentially expressed (DE) genes between the three TR populations (Fig 3B, Supplemental Table 1). The largest differences were found between Id3- eTR and Id3+ cTR with 1,471 DE genes, whereas only 474 genes differed between Id3+ and Id3- eTR (Fig 3B). Interestingly among the DE genes we observed a reciprocal increase in Id2 expression as TR lose Id3 (Fig 3C). This differential Id expression in TR is similar to what occurs in CD8+ T cells, which upregulate Id2 and downregulate Id3 while becoming activated and gaining effector function (12, 13). This suggests that as TR downregulate Id3, the closely related Id2 may take over some of its functions while also driving a unique E-protein-dependent signature promoting eTR development. Consistent with the stepwise differentiation model we propose for these populations, examination of the 300 most DE genes across the three TR subsets (based on highest F-value) showed that both up and downregulated genes were generally expressed in a gradient fashion, with expression in Id3+ eTR falling between that of Id3+ cTR and Id3- eTR (Fig 3D-E). Moreover, in accordance with our prior phenotypic analysis and their enhanced suppressive activity, Id3- eTR showed elevated expression of known TR function genes, including Il10, Ctla4, Pdcd1, Tnfrsf4, Lag3 and Ebi3 (Fig 3F). To further validate our RNA-seq results, we confirmed differential expression of several TR-associated genes by flow cytometry (Supplemental Fig 2). Gene Ontology (GO) term enrichment analysis of genes DE between Id3+ eTR and Id3- eTR identified specific molecular pathways altered between these closely related populations (Fig 3G). The top six enriched categories all related to cytokine and chemokine receptor signaling, with Id3- eTR expressing high levels of receptors indicating that they can tune their activity in response to key inflammatory cytokines such as IL-1, IL-18, IL-23, and IL-25 (Fig 3H). Moreover, among the DE chemokine receptors, Id3- eTR had the highest expression and subsequent responsiveness to chemokines that promote lymphocyte migration to inflamed tissues, such as Ccr4 and Cxcr3 (Fig 3H-I) (1) . Thus, Id3- eTR have a unique molecular profile indicative of their development from Id3+ TR precursors, their elevated suppressive function and altered migratory capacity.
Figure 3: Transcriptional profiling highlights the stepwise differentiation of Id3- eTR.
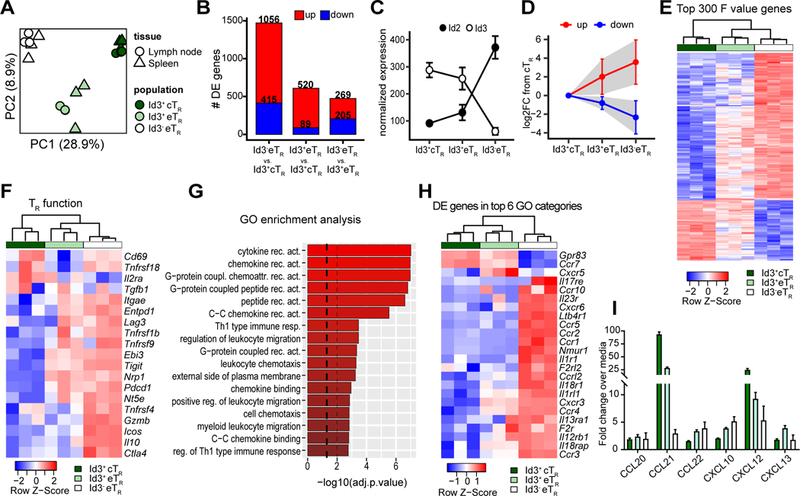
A) PCA of RNA-seq data from LN and splenic Id3+ cTR, Id3+ eTR and Id3- eTR populations sorted from three individual mice. B) Bar graphs showing the number of differentially expressed genes (adj.p.value < 0.05 and log2FC >1) for each of the indicated pairwise comparisons. C) Graphical analysis of normalized transcript reads for Id2 or Id3 from RNA-seq data. D) The 300 most differentially expressed genes (determined by F value) were split into the upregulated (red) and downregulated (blue) fractions based expression in Id3- eTR vs Id3+ cTR. Graph shows the mean log2FC compared to Id3+ cTR for both eTR populations. Error bars and shaded area represent 1×SD. E) Heatmap and hierarchical clustering of splenic RNA-seq samples based on the 300 most variably expressed genes. F) Heatmap and hierarchical clustering of splenic RNA-seq samples based on TR signature genes identified in reference (8). G) GO term enrichment analysis for DE genes of Id3- eTR vs. Id3+ eTR. Dashed lines represent adjusted p values of 0.05 and 0.01. H) Heatmap and hierarchical clustering of splenic RNA-seq samples based on DE genes found in the top 6 GO functional categories enriched in the comparison of Id3+ and Id3- eTR. I) Graphical analysis of chemotaxis assay.
Id3- TR are enriched and resident in non-lymphoid tissues
In contrast to their elevated expression of inflammatory chemokine receptors, Id3- eTR had the lowest expression of Ccr7 and S1pr1 which function together to promote TR recirculation through SLOs (24) (Fig 3H). Additionally, expression of CD103 and CD69, which together act to retain tissue-resident memory T(RM) cells in non-lymphoid sites, was strongly enriched in Id3- eTR, suggesting these cells may be tissue-resident (Fig 4A) (25, 26). Indeed, utilizing published gene signatures of CD8+ TRM or circulating memory T cells (27), we found that the CD8+ TRM signature gene set was enriched in Id3- eTR compared to either Id3+ cTR or Id3+ eTR, whereas the circulating memory gene set was enriched in the Id3+ TR populations (Fig 4B). Several groups have recently identified the ST2 (IL-33R)-Gata3 axis as a key determinant of TR residency and function in non-lymphoid tissues (8, 9, 28). Accordingly, Id3- eTR had the highest expression of all positively associated tissue TR genes such as Il1rl1 (ST2), Gata3, Areg, Irf4 and Rora, whereas Id3+ cTR had the lowest expression of these genes but high expression of negative regulators of tissue residence such as Tcf7, Klf2 and Lef1, and Id3+ eTR displayed intermediate expression for all of these genes (Fig 4C). Consistent with their TRM-like transcriptional signature, Id3- eTR were a minority of TR in LNs and spleen, but their frequency dramatically increased in nonlymphoid tissues, where they highly expressed the TRM surface markers CD103 and CD69 (Fig 4D-F). Indeed, tissues such as the fat and skin contained almost exclusively Id3- eTR. Of particular note Id3- eTR were rarest in the lymph and blood, indicating that Id3- eTR do not actively recirculate, but instead are retained as tissue-resident cells in non-lymphoid organs.
Figure 4: Id3- eTR are enriched and resident in non-lymphoid tissues.
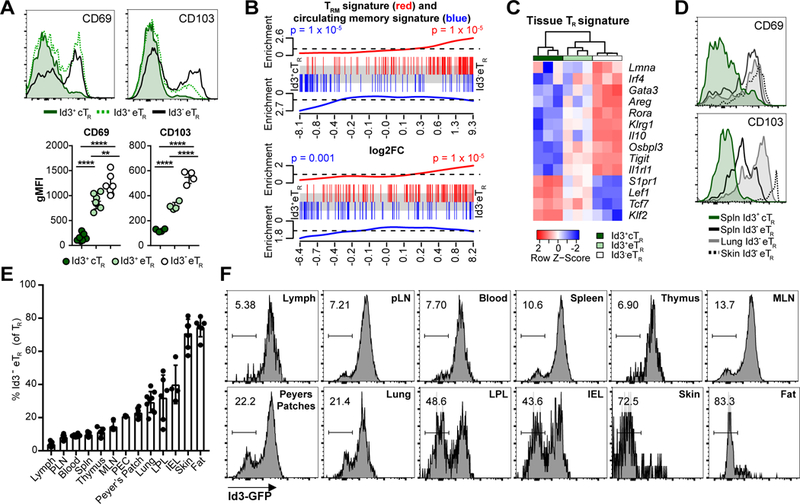
A) Representative flow cytometry histograms and graphical analysis of expression of the CD69 and CD103 by gated splenic TR populations. B) Enrichment of CD8+ TRM (red) or circulating T cell (blue) gene sets along ranked lists of the indicated pairwise comparisons. C) Heatmap and hierarchical clustering of splenic RNA-seq samples based on ST2+ tissue TR associated genes. D) Representative flow cytometry plots of CD103 and CD69 expression by gated TCRβ+ CD4+ Foxp3+ TR in the indicated tissues. E) Graphical analysis of Id3- eTR frequency among total TR in various tissues. F) Representative histograms of GFP expression from various tissues of Id3-GFP x Foxp3-mRFP mice, gated on TCRβ+ CD4+ Foxp3+ TR.
Despite significant interest in tissue-resident TR, the mechanisms regulating their differentiation and distribution are still poorly defined. Our data show that TR can be subdivided based on Id3 expression and known markers of cTR and eTR into distinct populations, and together our phenotypic, functional, transcriptional and transfer analyses strongly support a stepwise differentiation model in which TR downregulate Id3 as they progressively gain effector function, tissue homing capacity and residency in non-lymphoid organs. Similarly, Li and colleagues (28) recently proposed a stepwise model for the differentiation of TR in adipose tissue in which activation in the spleen allowed TR to migrate into the adipose tissue, where IL-33 signaling drove their terminal differentiation and functional specialization. High expression of co-stimulatory receptors such as ICOS and receptors for inflammatory cytokines would also allow Id3- eTR to respond to inflammatory signals that enhance Foxp3-mediated transcriptional repression and promote eTR differentiation and function (29), in part through activation of mTORC1 signaling (22). Although the direct role of Id3 downregulation in the functional differentiation of TR is still not established, our data highlight the complexity of tissue TR development, and identify novel molecular pathways that are modulated during their stepwise differentiation.
Supplementary Material
Acknowledgments
Acknowledgements:
We thank A. Goldrath for Id3-GFP mice. K. Arumuganathan and T. Nguyen for help with flow cytometry and cell sorting. V. Gersuk and the BRI Genomics Core for running RNA-seq samples. And members of the Campbell lab for helpful discussions.
Funding: This work was supported by grants to DJC from the National Institutes of Health (AI085130, AI124693). JMS was supported by NIH-NIAID T32 (AI106677, UW Immunology).
References
- 1.Campbell DJ 2015. Control of Regulatory T Cell Migration, Function, and Homeostasis. J Immunol 195: 2507–2513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.S. SK., Shivani S, Michael SJ, and D. J C. 2014. Regulatory T‐cell homeostasis: steady‐state maintenance and modulation during inflammation. Immunological Reviews 259: 40–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dominguez-Villar M, and Hafler DA 2018. Regulatory T cells in autoimmune disease. Nat Immunol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Josefowicz SZ, Lu LF, and Rudensky AY 2012. Regulatory T cells: mechanisms of differentiation and function. Annu Rev Immunol 30: 531–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Smigiel KS, Richards E, Srivastava S, Thomas KR, Dudda JC, Klonowski KD, and Campbell DJ 2014. CCR7 provides localized access to IL-2 and defines homeostatically distinct regulatory T cell subsets. J Exp Med 211: 121–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Luo CT, Liao W, Dadi S, Toure A, and Li MO 2016. Graded Foxo1 activity in Treg cells differentiates tumour immunity from spontaneous autoimmunity. Nature 529: 532–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gratz IK, and Campbell DJ 2014. Organ-specific and memory treg cells: specificity, development, function, and maintenance. Front Immunol 5: 333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Delacher M, Imbusch CD, Weichenhan D, Breiling A, Hotz-Wagenblatt A, Trager U, Hofer AC, Kagebein D, Wang Q, Frauhammer F, Mallm JP, Bauer K, Herrmann C, Lang PA, Brors B, Plass C, and Feuerer M 2017. Genome-wide DNA-methylation landscape defines specialization of regulatory T cells in tissues. Nat Immunol 18: 1160–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wohlfert EA, Grainger JR, Bouladoux N, Konkel JE, Oldenhove G, Ribeiro CH, Hall JA, Yagi R, Naik S, Bhairavabhotla R, Paul WE, Bosselut R, Wei G, Zhao K, Oukka M, Zhu J, and Belkaid Y 2011. GATA3 controls Foxp3(+) regulatory T cell fate during inflammation in mice. J Clin Invest 121: 4503–4515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kee BL 2009. E and ID proteins branch out. Nat Rev Immunol 9: 175–184. [DOI] [PubMed] [Google Scholar]
- 11.Quong MW, Romanow WJ, and Murre C 2002. E protein function in lymphocyte development. Annu Rev Immunol 20: 301–322. [DOI] [PubMed] [Google Scholar]
- 12.Yang CY, Best JA, Knell J, Yang E, Sheridan AD, Jesionek AK, Li HS, Rivera RR, Lind KC, D’Cruz LM, Watowich SS, Murre C, and Goldrath AW 2011. The transcriptional regulators Id2 and Id3 control the formation of distinct memory CD8+ T cell subsets. Nat Immunol 12: 1221–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Omilusik KD, Shaw LA, and Goldrath AW 2013. Remembering one’s ID/E-ntity: E/ID protein regulation of T cell memory. Curr Opin Immunol 25: 660–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Miyazaki M, Rivera RR, Miyazaki K, Lin YC, Agata Y, and Murre C 2011. The opposing roles of the transcription factor E2A and its antagonist Id3 that orchestrate and enforce the naive fate of T cells. Nat Immunol 12: 992–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Omilusik KD, Nadjsombati MS, Shaw LA, Yu B, Milner JJ, and Goldrath AW 2018. Sustained Id2 regulation of E proteins is required for terminal differentiation of effector CD8(+) T cells. J Exp Med 215: 773–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Miyazaki M, Miyazaki K, Chen S, Itoi M, Miller M, Lu LF, Varki N, Chang AN, Broide DH, and Murre C 2014. Id2 and Id3 maintain the regulatory T cell pool to suppress inflammatory disease. Nat Immunol 15: 767–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rauch KS, Hils M, Lupar E, Minguet S, Sigvardsson M, Rottenberg ME, Izcue A, Schachtrup C, and Schachtrup K 2016. Id3 Maintains Foxp3 Expression in Regulatory T Cells by Controlling a Transcriptional Network of E47, Spi-B, and SOCS3. Cell Rep 17: 2827–2836. [DOI] [PubMed] [Google Scholar]
- 18.Rauch KS, Hils M, Menner AJ, Sigvardsson M, Minguet S, Aichele P, Schachtrup C, and Schachtrup K 2017. Regulatory T cells characterized by low Id3 expression are highly suppressive and accumulate during chronic infection. Oncotarget 8: 102835–102851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Couter CJ, and Surana NK 2016. Isolation and Flow Cytometric Characterization of Murine Small Intestinal Lymphocytes. JoVE: e54114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Srivastava S, Koch MA, Pepper M, and Campbell DJ 2014. Type I interferons directly inhibit regulatory T cells to allow optimal antiviral T cell responses during acute LCMV infection. J Exp Med 211: 961–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Koch MA, Tucker-Heard G, Perdue NR, Killebrew JR, Urdahl KB, and Campbell DJ 2009. The transcription factor T-bet controls regulatory T cell homeostasis and function during type 1 inflammation. Nat Immunol 10: 595–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sun IH, Oh MH, Zhao L, Patel CH, Arwood ML, Xu W, Tam AJ, Blosser RL, Wen J, and Powell JD 2018. mTOR Complex 1 Signaling Regulates the Generation and Function of Central and Effector Foxp3(+) Regulatory T Cells. J Immunol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gavin MA, Clarke SR, Negrou E, Gallegos A, and Rudensky A 2002. Homeostasis and anergy of CD4(+)CD25(+) suppressor T cells in vivo. Nat Immunol 3: 33–41. [DOI] [PubMed] [Google Scholar]
- 24.Lee JH, Kang SG, and Kim CH 2006. FoxP3+ T Cells Undergo Conventional First Switch to Lymphoid Tissue Homing Receptors in Thymus but Accelerated Second Switch to Nonlymphoid Tissue Homing Receptors in Secondary Lymphoid Tissues. The Journal of Immunology 178: 301–311. [DOI] [PubMed] [Google Scholar]
- 25.Mackay LK, Rahimpour A, Ma JZ, Collins N, Stock AT, Hafon ML, Vega-Ramos J, Lauzurica P, Mueller SN, Stefanovic T, Tscharke DC, Heath WR, Inouye M, Carbone FR, and Gebhardt T 2013. The developmental pathway for CD103(+)CD8+ tissue-resident memory T cells of skin. Nat Immunol 14: 1294–1301. [DOI] [PubMed] [Google Scholar]
- 26.Schenkel JM, and Masopust D 2014. Tissue-resident memory T cells. Immunity 41: 886–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Milner JJ, Toma C, Yu B, Zhang K, Omilusik K, Phan AT, Wang D, Getzler AJ, Nguyen T, Crotty S, Wang W, Pipkin ME, and Goldrath AW 2017. Runx3 programs CD8(+) T cell residency in non-lymphoid tissues and tumours. Nature 552: 253–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li C, DiSpirito JR, Zemmour D, Spallanzani RG, Kuswanto W, Benoist C, and Mathis D 2018. TCR Transgenic Mice Reveal Stepwise, Multi-site Acquisition of the Distinctive Fat-Treg Phenotype. Cell. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Arvey A, van der Veeken J, Samstein RM, Feng Y, Stamatoyannopoulos JA, and Rudensky AY 2014. Inflammation-induced repression of chromatin bound by the transcription factor Foxp3 in regulatory T cells. Nat Immunol 15: 580–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.