

Abstract
Immune-mediated thrombotic thrombocytopenic purpura is characterized by severe thrombocytopenia and microangiopathic hemolytic anemia. It is primarily caused by immunoglobin G type autoantibodies against ADAMTS13, a plasma metalloprotease that cleaves von Willebrand factor. However, reliable markers predictive of patient outcomes are yet to be identified. Seventy-three unique patients with a confirmed diagnosis of immune-mediated thrombotic thrombocytopenic purpura between April 2006 and December 2017 were enrolled from the Univeristy of Alabama at Birmingham Medical Center. Clinical information, laboratory values, and a panel of special biomarkers were collected and/or determined. The results demonstrated that the biomarkers associated with endothelial injury (e.g., von Willebrand factor antigen and collagen-binding activity), acute inflammation (e.g., human neutrophil peptides 1-3 and histone/deoxyribonucleic acid complexes), and activation of the complement alternative pathway (e.g., factors Bb and iC3b) were all significantly increased in patients with acute immune-mediated thrombotic thrombocytopenic purpura compared to those in the healthy controls. Moreover, failure to normalize platelet counts within 7 days or failure to markedly reduce serum lactate dehydrogenase by day 5, low total serum protein or albumin, and high serum troponin levels were also predictive of mortality, as were the prolonged activated partial thromboplastin time, high fibrinogen, and elevated serum lactate dehydrogenase, Bb, and sC5b-9 on admission. These results may help to stratify patients for more intensive management. The findings may also provide a framework for future multicenter studies to identify valuable prognostic markers for immune-mediated thrombotic thrombocytopenic purpura.
Introduction
Immune-mediated thrombotic thrombocytopenic purpura (iTTP) is a rare, but life-threatening, hematologic disorder.1,2 It is characterized by severe thrombocytopenia and microangiopathic hemolytic anemia (MAHA), with or without end organ damage. The underlying pathophysiology of iTTP is a functional deficiency of plasma ADAMTS13 activity, resulting from autoantibodies targeting plasma ADAMTS13, a metalloprotease that cleaves von Willebrand factor (VWF).3–5 Therapeutic plasma exchange (TPE) remains the standard of care, in conjunction with immunosuppressive therapies that include corticosteroids and rituximab to inhibit acute inflammation and autoantibody production.1,6 However, an in-hospital mortality rate remains as high as ~20%7,8 or less than 10% following the introduction of a novel therapy caplacizumab, an anti-VWF nanobody;9 nearly 30% of surviving patients may experience disease exacerbation10 and/or relapse.7 Currently, clinical factors and biomarkers predictive of clinical course/outcome are scanty, and their predictive values have yet to be established in diverse patient populations.
Demographic features such as race, gender and age are shown to associate with disease prevalence and severity. For instance, iTTP occurs more commonly in African-American females11,12 and, perhaps not surprisingly, older age (>60 years) is associated with an increased mortality.12,13 Additionally, serum levels of creatine kinase-muscle/brain (CK-MB), troponin I,14 lactate dehydrogenase (LDH), ADAMTS13 antigen or activity levels, anti-ADAMTS13 antibody levels13 and, more recently, the platelet recovery rate15 are shown to be associated with increased mortality.
In this study, we describe the Alabama cohort of 73 unique patients with confirmed diagnosis of iTTP selected from a total of 142 admissions. This cohort of patients was primarily from the Southeastern United States. Clinical information, laboratory values, and various biomarkers were collected and analysed with respect to their associations with admission type, disease severity, and mortality.
Methods
Patients
The Institutional Review Board (IRB) of the University of Alabama at Birmingham (UAB) has approved the study protocol. UAB medical center serves as a referral center for the diagnosis and management of patients with thrombotic microangiopathy (TMA) for the state of Alabama and several neighboring states in the Southeast United State of America. Some patients were initially seen by a primary care physician, local internist, or hematologist. If TMA was suspected, patients were referred to the UAB Medical Center for further evaluation and treatment, which may have involved a delay in diagnosis and treatment of one to several days. There were also patients who came directly to the UAB Emergency Department (ED). Within hours of arrival at UAB, a central intravenous catheter was inserted, blood samples were collected for laboratory tests including ADAMTS13 activity and inhibitors, and therapeutic plasma exchange (TPE) was urgently initiated. Seventy-three patients at the UAB Medical Center, between April 2006 and December 2017, were included in this study. Control samples were collected from healthy individuals (age 27-69 years), both male (1/3) and female (2/3), representing the local ethnic population, who did not have a history of hematological diseases, malignancy, and acute inflammatory disorders.
Whole blood was anticoagulated with 3.2% sodium citrate; plasma was separated within four hours of collection, and stored at −80°C prior to analysis. Clinical data pertinent to each patient, including demographic information, past and current medical history, signs and symptoms on admission, laboratory test results, presumptive and final diagnosis, hospital-course, outcome and long-term follow up, were collected by a physician and maintained in the Alabama Registry Database.
Exclusion and inclusion criteria
Patients were excluded from analysis if their final diagnosis was determined to be an alternative TMA, for example: atypical hemolytic uremic syndrome (aHUS), congenital TTP, HIV-related thrombocytopenia, HELLP syndrome, a life-threatening condition during pregnancy with clinical features of hemolysis, elevated liver enzymes, and low platelet count,16 TMA following solid organ or hematopoietic stem cell transplantation, drug-induced TTP (i.e., clopidogrel, ticlopidine or gemcitabine) and/or sepsis. Additionally, we excluded patients who were treated prior to sample drawn with plasma infusion (>3L) and/or TPE and those in remission. Thus, this cohort includes patients experiencing their first episode or an exacerbation, or a relapse (only if the sample from the initial episode was not available). Confirmatory tests for ADAMTS13 activity and inhibitors were performed at the Blood Center of Wisconsin (Milwaukee, WI, USA).
Assays for ADAMTS13 activity, inhibitors, and anti-ADAMTS13 IgG
Plasma ADAMTS13 activity and inhibitor titers were determined using a commerical FRETS-VWF73 assay17 and an in-house FRETS-based assay as previously described.18 Plasma anti-ADAMTS13 IgG was determined by an enzyme-linked immunosorbent assay (ELISA) (Diapharma, West Chester, Ohio) in accordance with the manufacturer’s recommendations.
Assays for plasma VWF antigen and collagen-binding activity
Plasma VWF antigen (VWF-Ag) and collagen-binding activity (VWF-CBA) levels were determined using in-house ELISA-based assays as previously described.19
Assays for complement activation and inflammatory markers
Plasma levels of complement activation markers including iC3b, sC5b-9, Bb, and C4d were determined using a commercial ELISA assays (MicroVue, San Diego, CA, USA) following manufacturer’s instructions.8 Plasma HNP1-3 levels were also determined by an ELISA assay, which recognizes all HNP1-3 (Hycult Biotech, Plymouth Meeting, PA).8 Finally, plasma histone-DNA complexes were quantified by the ELISA assay previously described (Roche, Indianapolis, IN, USA).20
Statistical Analysis
The data were expressed as the medians and 95% confidence intervals (95% CI) for most parameters unless specified otherwise. Mann-Whitney (for two groups) and Kruskal-Willis tests (for multiple groups) for continuous variables and Fisher exact test for categorical variables were performed, with descriptive statistics to summarize both quantitative and categorical variables. Association between various demographic, clinical, and laboratory parameters with clinical outcomes such as mortality, remission, exacerbation, relapse, etc. were also determined using univariate analysis and spearman correlation tests. Furthermore, Cox proportional hazard regression was used to determine the hazard ratios for the predictive variables. P-values of <0.05 and <0.01 were deemed statistically significant and highly significantly, respectively.
Results
Clinical and admission laboratory characteristics of the Alabama iTTP cohort
The majority of our 73 iTTP patients came from the surrounding areas of the cities of Montgomery (n=16, 22%), Birmingham (n=13, 17.8%), and Tuscaloosa (n=9, 12.3%). The remaining patients were from other locations in the state of Alabama with a small number coming from other states including Mississippi, Georgia, New York, Oregon, and Ohio (Figure 1A). Thus, this cohort represents a patient population primarily from the southeastern part of the United States of America. Since inclusion criteria were designed to collect only unique admission events for a patient experiencing an acute iTTP, only 73 unique patients of 142 admissions for the diagnosis and treatment of iTTP were included in this study (Figure 1B).
Figure 1.

The Alabama TTP Cohort. A. A map of the state of Alabama denotes the geographic distribution of the 73 unique patients with iTTP that were included in the study. Five patients residing in Oregon, Ohio, New York, Georgia, and Mississippi are not shown on the map. The location of the UAB Medical Center is marked with a blue star. B. Algorithm demonstrates the number of admissions, patients being excluded, and the final cohort of patients in the study.
The median age of the patients was 41 years, with 56% being female and 44% male. Seventy-seven percent of patients were of African-American decent, while Caucasian and Hispanic patients comprised only 22% and <1%, respectively. A substantial number of patients had one or several comorbidities, including hypertension (52%), diabetes mellitus (18%), and systemic lupus erythematosus (SLE) (11%). Only 3 patients (4%) were pregnant at the time of the diagnosis. In addition, 49% of patients reported a history of smoking, and 21% reported use of recreational drugs (Table 1).
Table 1.
Demographics, clinical presentations, and comorbidities in 73 unique patients with iTTP.
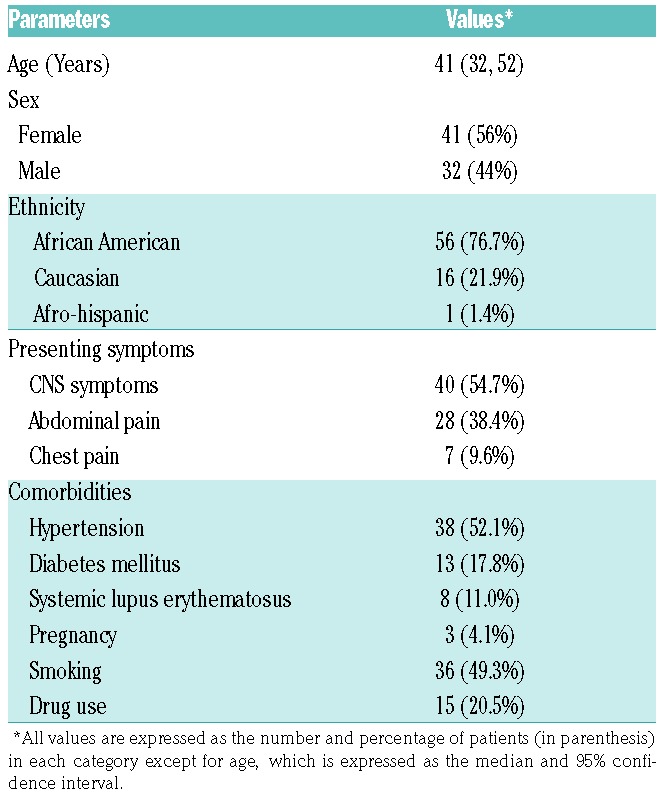
Our blood samples were primarily obtained from patients at their initial presentation (75%) and during a relapse (23%); only one sample was obtained from an exacerbation. This bias is the result of our inclusion criteria designed to prioritize the collection and study of unique patients. The clinical presentation on admission primarily included signs and symptoms of the central nervous system (CNS) (54.7%), abdominal pain (38.4%), and chest pain (9.6%) (Table 1).
The most common blood type in our patient population was type O (63%), followed by A (22%), B (14%), and AB (1%), with 93% being Rh positive. The frequency of type O was statistically higher than that expected (47.9%) (Online Supplementary Table S1), suggesting that patients with type O blood group are not protected from developing iTTP.
All iTTP patients received TPE as primary therapy. The median number of TPEs for all patients was 15 (95% confidence interval [CI] of 12-17). Patients also received corticosteroids (94%), rituximab (38.4%), and vincristine (6.8%) at various times during hospitalization. Only 5.4% of patients underwent splenectomy as part of their treatment.
The outcomes of these patients after treatment were classified as the following: single episode to remission (30.1%), exacerbation or relapse to remission (32.9%), exacerbation and relapse to remission (24.7%), and death (12.3%). Eight out of 9 patients who died within 35 days following admission were unresponsive and/or refractory to TPE; 1 patient died of unrelated disease one year after the initial therapy. Therefore, the overall remission rate was 87.5% despite exacerbation and/or relapse after a median follow up of 828 days (or 2.3 years) (95% CI of 326-1,720 days or 1-4.7 years) (Table 2).
Table 2.
Therapeutic interventions and observed long-term outcomes in 73 patients with iTTP.

ADAMTS13 activity and inhibitors in patients with iTTP
All patients except for 2 having ADAMTS13 activity of 13 and 26 U/dL, respectively) had plasma ADAMTS13 activity <5 U/dL (or <5% normal) (Figure 2A). However, these two samples were drawn by ordering physicians following TPE or a large volume of plasma transfusion. However, the ADAMTS13 activity in pre-treatment samples stored separately for research purposes was <5 U/dL. Therefore, all patients included in the study had severe deficiency of ADAMTS13 activity. ADAMTS13 inhibitors (>0.4 U/mL), measured by the 50/50 mixing study, were found to be present in 65 of 73 (89%) patients. There was no difference in the inhibitor titers in patients presenting during the initial and those with a relapse episode (Figure 2B). All 8 patients who tested negative (<0.4 U/mL) for ADAMTS13 inhibitors had significantly increased levels of anti-ADAMTS13 IgG (Figure 2C). Therefore, all patients were confirmed as having severe ADAMTS13 deficiency and positive antibody against ADAMTS13, hence the diagnosis of iTTP.
Figure 2.

Plasma ADAMTS13 activity and autoantibodies in 73 unique patients with iTTP. Plasma levels of ADAMTS13 activity (A) and functional inhibitor titers (B) in patients with iTTP compared with those in the healthy controls. Additionally, plasma anti-ADAMTS13 IgG levels in iTTP patients with negative (<0.4 U/mL) and positive (>=0.4 U/mL) inhibitors are shown (C). Each individual dot represents a single patient with the median ± 95% confidence intervals (solid red lines). Mann-Whitney was used to determine the statistical significance between two groups. Here *, **, ***, and **** indicate the P values of <0.05, <0.001, 0.0005, and <0.0001, respectively; n.s. stands for no statistical difference between two groups.
Plasma VWF antigen and collagen-binding activity in patients with iTTP
Plasma VWF-Ag, VWF-CBA, and the ratio of VWF-CBA/VWF-Ag were determined in plasma samples obtained from iTTP patients and healthy controls. Plasma levels of VWF-Ag (P<0.0001) (Figure 3A) and VWF-CBA (P<0.005) (Figure 3B) on admission were significantly higher in patients with either an initial or a relapse episode of iTTP than those in the healthy controls. However, the ratio of VWF-CBA to VWF-Ag, a quantitative measurement of VWF multimer size,21 was only significantly different between the initial episodes and controls (P<0.01), but not between the relapse episodes and controls (P>0.05) (Figure 3C). Plasma levels of VWF-Ag and VWF-CBA were moderately associated with each other with a Spearman correlation coefficient (rho) of 0.592 (P<0.0001). These results indicated that despite increased expression and/or release of VWF, the ultra-large VWF multimers may be selectively removed from the circulation in some cases as a result of ongoing microvascular thrombosis during the acute episode. A positive association between plasma levels of VWF-Ag and serum LDH (rho=0.36, P=0.004) supports our hypothesis.
Figure 3.

Plasma levels of VWF antigen and collagen-binding activitity in patients with iTTP. Plasma VWF antigen (VWF-Ag) (A), collagen-binding activity (VWF-CBA) (B), and the ratio of VWF-CBA to VWF-Ag (C) were determined in patients with iTTP (initial vs. exacerbated or relapsed) and the healthy controls. Each dot represents a single patient and solid lines are the median ± 95% confidence intervals. Kruskal-Willis analysis was used to determine the statistical significance among three different groups. Here *, **, ***, and **** indicate the P values of <0.05, <0.001, 0.0005, and <0.0001, respectively; n.s. stands for no statistical difference.
Plasma levels of HNP1-3 and histone/DNA complexes in acute iTTP
Previous studies demonstrated that plasma levels of HNP1-38 and DNA/histone complexes20 were significantly elevated in patients with acute iTTP. However, their prognostic value in iTTP was not determined. Consistent with the results previously reported, plasma levels of HNP1-3 (P<0.0001) (Figure 4A) and histone/DNA complexes (P<0.0001) (Figure 4B) in patients with an initial or exacerbated or relapsed episode of acute iTTP were dramatically increased when compared to those in the healthy controls. Additionally, plasma HNP1-3 levels in iTTP patients were significantly correlated with the levels of histone/DNA complex (rho=0.50, P<0.001). While none of these two biomarkers was predictive of an adverse outcome in iTTP patients (e.g., mortality, exacerbation, and relapse), plasma levels of HNP1-3 appeared to significantly correlate with serum troponin (rho=0.37, P<0.05) and LDH (rho=0.48, P<0.001). The plasma levels of histone/DNA complex were also correlated with serum LDH (rho=0.66, P<0.0001), but not troponin. Noticeably, serum troponin was elevated in 35 of 37 (~95%) patients tested in the ED, and remained elevated in 59 of 68 (~87%) patients tested after being admitted to hospital (prior to TPE). These results suggest that plasma levels of HNP1-3 and histone/DNA complexes may also be markers of organ injury in addition to serum LDH and troponin in patients with acute iTTP.
Figure 4.

Plasma levels of HNP1-3 and histone/DNA complexes in patients with iTTP. Plasma levels of HNP1-3 (A) and histone/DNA complexes (B) in patients with acute iTTP (initial vs. exacerbated or relapsed) and healthy controls are shown as the dot plots for individual patients with the median ± 95% confidence intervals (solid red lines). Kruskal-Willis analysis was used to determine the statistical significance. Here **** indicates P value <0.0001 when compared the values in the healthy controls.
Plasma levels of complement activation markers in acute iTTP
While complement activation through an alternative pathway is the primary cause of aHUS,22 recent studies have suggested that complement activation may also occur in patients with acute iTTP.23–25 To assess the prognostic value of complement activation markers in these patients, we determined plasma levels of C4d (classical pathway), Bb (alternative pathway), and iC3b and sC5b-9 (common pathway) in iTTP patients and healthy controls. As shown, plasma C4d levels were not significantly increased in patients with acute iTTP (P>0.05) (Figure 5A). Plasma levels of Bb (P<0.0001) (Figure 5B), iC3b (P<0.01) (Figure 5C), and sC5b-9 (P<0.0001) (Figure 5D) were significantly elevated when compared to healthy controls. There was a modestly positive correlation between Bb and creatinine (rho=0.3, P<0.05), LDH (rho=0.65, P<0.0001), and troponin (rho=0.329, P<0.05) in iTTP patients. Additionally, there was a positive correlation between sC5b-9 and LDH (rho=0.4, P<001). These results suggest that complement over-activation through the alternative pathway may participate in the pathophysiology of acute iTTP, although a causative role of complement activation in iTTP is yet to be determined in animal models.
Figure 5.

Plasma levels of complement activation markers in patients with iTTP. Plasma levels of complement activation markers including C4d (A), Bb (B), iC3b (C), and sC5b-9 (D) in patients with acute iTTP (initial vs. exacterbated or relapsed) and the healthy controls. Each dot represents the value of each individual subject. The red solid lines are the medians ± 95% confidence interval. Kruskal-Willis analysis was used to determine the statistical significance. Here *, **, and **** indicate the P values of <0.01, and <0.0001, respectively; n.s. stands for no statistical difference.
Predictive values of certain clinical factors and routine laboratory parameters
The associations between demographics, clinical presentation, admission laboratory values, the aforemen tioned biomarkers, and patient outcomes were determined. On univariate analysis, the following variables were significantly associated with in-patient mortality: an inability to normalize platelet count within seven days of TPE (i.e., platelet count of ≥150×109/L) (Online SupplementaryTable S2), failure to significantly reduce serum LDH after five days of treatment (P=0.03), low serum albumin (P=0.03), and high troponin levels at ED (P=0.008) and after being admitted to hospital prior to TPE (P=0.01) (Table 3). Additionally, Cox regression analysis demonstrated that high levels of total serum protein or albumin on admission were associated with a reduced risk of in-hospital mortality (HR, 0.37 or 0.21, P=0.032 or 0.003), while prolonged aPTT (HR 2.03, P=0.02), increased fibrinogen (HR 1.9, P=0.03), elevated LDH at day five (HR 2.93, P=0.04), high plasma Bb (HR 1.3, P=0.04), and sC5b-9 (HR 1.54, P=0.05) were all found to be the significant markers of mortality (Table 4). When the achievement of clinical remission was used as an outcome, the normalization of platelet count within seven days (P<0.001) and absence of SLE diagnosis (P=0.038) were found to be the significant prognostic factors for remission (Online Supplementary Table S2). Together, our results demonstrate that lack of admission coagulopathy, low serum tropin level, absence of lupus, platelet recovery in seven days, and marked reduction of LDH in five days, patient overall well-being, and low levels of complement activation markers appear to associate with a good outcome in patients with acute iTTP.
Table 3.
Univariate analysis identifies the important laboratory parameters that predict mortality in 73 unique patients with iTTP.

Table 4.
COX hadard regression analysis identifies the statistically significant parameters that predict mortality in iTTP patients.

Discussion
The Alabama cohort represents a distinct, yet undescribed, iTTP patient population. Our patients were predominantly of African American descent (77%); currently, African Americans only make up 25% of the current population in Alabama (https://cber.cba.ua.edu/rbriefs/ black.html). This overrepresentation of African-Americans may be in part attributed to the strict inclusion criteria that required the presence of plasma ADAMTS13 activity <5 U/dL and either positive inhibitors or elevated anti-ADAMTS13 IgG in the appropriate clinical context. Patients of African American descent have been shown to have reduced frequency of a protective HLA DRB1*04 allele, which renders them more susceptible to the development of autoantibodies against ADAMTS13.26
Since 2015, we have seen approximately 17 new cases of iTTP per year at the UAB Medical Center, which serves as the major referral center for diagnosis and management of TMA in the Southeastern United States. With a population of ~4.9 millions in the State of Alabama, the incidence rate is estimated to be ~3.5 cases per million per year, fairly similar to that reported in the literature.27,28 This incidence rate is most likely underestimated due to patients who may expire prior to reaching our institution or being enrolled in the study.
Data from the Ohio Registry suggest that there are differences in race, neurological symptoms, platelet counts, LDH, ADAMTS13 activity, and total number of TPE required between patients with an initial episode and those with relapses, despite no major difference in outcome.29 Our data do not show a difference in patients with an initial episode or relapse regarding admission neurologic symptoms, hemoglobin, platelet count, LDH, inhibitor, anti-ADAMTS13 IgG, and/or any of the novel biomarkers evaluated.
To date, the diagnosis of iTTP relies on laboratory findings of severe deficiency of plasma ADAMTS13 activity (i.e., <5 U/dL, or <10 U/dL depending on the lab cut-off) with a positive inhibitor, and/or elevated anti-ADAMTS13 IgG levels in an appropriate clinical context (i.e., thrombocytopenia and microangiopathic hemolytic anemia without other explanations).1,30 All patients in this study had an ADAMTS13 activity <5 U/dL when only the pre-treatment plasma samples were interrogated. Of note, all patients, including 5 with negative inhibitors (<0.4 U/mL), had significantly elevated levels of anti-ADAMTS13 IgG by ELISA. These results suggest that the ELISA-based binding assay may be more sensitive than the functional assays for the diagnosis of iTTP. Therefore, we recommend that any patient with severe deficiency of plasma ADAMTS13 activity (<10 U/dL), but a negative functional inhibitor, should undergo anti-ADAMTS13 IgG ELISA testing. While anti-ADAMTS13 IgG may be detected in plasma of healthy individuals, their levels are not sufficient to inhibit ADAMTS13 activity.31 Findings of high concentrations of anti-ADAMTS13 IgG in addition to low ADAMTS13 antigen levels appear to predict adverse outcomes in patients with iTTP according to the UK registry data.13
The hallmark of iTTP is the autoantibody-mediated inhibition of plasma ADAMTS13 activity. This leads to the inability to cleave newly released ultra-large VWF multimers released from and anchored on the endothelium,32 in the circulating blood,33 or at the site of thrombus formation.34 Subsequently, the accumulated ultra-large VWF multimers may spontaneously agglutinate platelets in small arterioles and capillaries, leading to end organ damage.2 Data supporting this hypothesis include significant elevation of plasma levels of VWF antigen and collagen-binding activity in acute iTTP patients despite the slightly reduced ratios of VWF activity to VWF antigen in these patients during the acute setting – consistent with the partial consumption of ultra large VWF multimers during active thrombus formation. In 1982, Moake et al. showed that ultralarge VWF multimers are only detectable in chronic and relapsing TTP patients during remission, not during the acute disease.33 Plasma from Adamts13−/− mice appear to have ultra-large VWF multimers when the animals are well or not stressed.35 Nevertheless, the increased levels of plasma VWF antigen, but not VWF activity, correlate with increased levels of serum LDH, suggesting that either plasma VWF antigen or serum LDH may be used as a biomarker of end-organ damage.
Infection, inflammation, and pregnancy are known to be complement-amplifying conditions, presumably through activation of the alternative pathway.36 Consistent with our previous findings,8,20 plasma levels of HNP1-3, histone/DNA complexes, Bb, and iC3b were also significantly increased in this cohort of iTTP patients; plasma HNP1-3, histone/DNA complexes, Bb, and iC3b correlate with the markers of organ damage, including serum LDH, creatinine and/or troponin. There was a strong correlation between plasma sC5b and serum LDH. Rapid reduction in LDH was correlated with survival, consistent with a previous report.37 The elevated plasma levels of Bb and sC5b-9 were found to be predictive of mortality in the Cox regression analysis. These results suggest that both neutrophil and complement activation may participate in the pathogenesis of iTTP, although the causative role of HNP1-3, histone, and complement activation in iTTP are yet to be determined. Other clinical and laboratory factors such as the elevated troponin levels and reduced Glasgow Coma Score (GCS) confer a six-fold and nine-fold increase, respectively, in the mortality of patients with iTTP, reported from the UK TTP registry.13 Our univariate analysis also demonstrated the association between an increased troponin level and mortality.
In summary, our study further demonstrates the utility of several clinical and laboratory markers including aPTT, fibrinogen, troponin, the rate of platelet and LDH normalization, and total protein/albumin etc. for predicting outcome in patients with iTTP. Moreover, we identified several novel biomarkers related to inflammation (e.g., VWF, HNP1-3, and histone/DNA complexes) and innate immunity (e.g., Bb and sC5b-9) that may be used to assess dis ease severity and predict long-term outcome in patients with iTTP. Our findings may help stratify patients who may benefit from more intensive care and management including twice daily TPE, early rituximab, and eclulizumab etc. to reduce in-hospital mortality and long-term complications.
Supplementary Material
Acknowledgments
The collection and storage of patient samples could not have occurred without the help of the UAB apheresis nurses, the UAB Coagulation Lab, as well as the UAB Pathology residents and Transfusion Medicine fellows.
Footnotes
Check the online version for the most updated information on this article, online supplements, and information on authorship & disclosures: www.haematologica.org/content/104/1/166
Funding
This work was partially supported by grants from the National Heart, Lung, and Blood Institute (HL-26724 and HL-115187) and the Answering T.T.P. Foundation, Canada.
References
- 1.Saha M, McDaniel JK, Zheng XL. Thrombotic thrombocytopenic purpura: pathogenesis, diagnosis and potential novel therapeutics. J Thromb Haemost. 2017;15(10):1889–1900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Moake JL. Thrombotic thrombocytopenic purpura: the systemic clumping “plague”. Annu Rev Med. 2002;53:75–88. [DOI] [PubMed] [Google Scholar]
- 3.Tsai HM, Lian EC. Antibodies to von Willebrand factor-cleaving protease in acute thrombotic thrombocytopenic purpura. N Eng J Med. 1998;339(22):1585–1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zheng XL, Wu HM, Shang D, Falls E, Skipwith CG, Cataland SR, et al. Multiple domains of ADAMTS13 are targeted by autoantibodies against ADAMTS13 in patients with acquired idiopathic thrombotic thrombocytopenic purpura. Haematologica. 2010;95(9):1555–1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Casina V, Hu WB, Mao JH, et al. High resolution epitope mapping by HX MS reveals the pathogenic mechanism and a possible therapy for autoimmune TTP syndrome. Proc Natl Acad Sci USA. 2015;112(31):9620–9625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.George JN. How I treat patients with thrombotic thrombocytopenic purpura: 2010. Blood. 2010;116(20):4060–4069. [DOI] [PubMed] [Google Scholar]
- 7.Kremer Hovinga JA, Vesely SK, Terrell DR, Lammle B, George JN. Survival and relapse in patients with thrombotic thrombocytopenic purpura. Blood. 2010;115(8):1500–1511. [DOI] [PubMed] [Google Scholar]
- 8.Cao W, Pham HP, Williams LA, et al. Human neutrophil peptides and complement factor Bb in pathogenesis of acquired thrombotic thrombocytopenic purpura. Haematologica. 2016;101(11):1319–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Peyvandi F, Scully M, Kremer Hovinga JA, et al. Caplacizumab reduces the frequency of major thromboembolic events, exacerbations and death in patients with acquired thrombotic thrombocytopenic purpura. J Thromb Haemost. 2017;15(7):1448–1452. [DOI] [PubMed] [Google Scholar]
- 10.Cataland SR, Yang SB, Witkoff L, et al. Demographic and ADAMTS13 biomarker data as predictors of early recurrences of idiopathic thrombotic thrombocytopenic purpura. Eur J Haematol. 2009;83(6):559–564. [DOI] [PubMed] [Google Scholar]
- 11.Zheng XL, Kaufman RM, Goodnough LT, Sadler JE. Effect of plasma exchange on plasma ADAMTS13 metalloprotease activity, inhibitor level, and clinical outcome in patients with idiopathic and nonidiopathic thrombotic thrombocytopenic purpura. Blood. 2004;103(11):4043–4049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Terrell DR, Vesely SK, Kremer Hovinga JA, Lammle B, George JN. Different disparities of gender and race among the thrombotic thrombocytopenic purpura and hemolyticuremic syndromes. Am J Hematol. 2010; 85(11):844–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Alwan F, Vendramin C, Vanhoorelbeke K, et al. Presenting ADAMTS13 antibody and antigen levels predict prognosis in immune-mediated thrombotic thrombocytopenic purpura. Blood. 2017;130(4):466–471. [DOI] [PubMed] [Google Scholar]
- 14.Brazelton J, Oster RA, McCleskey B, Fuller J, Adamski J, Marques MB. Increased troponin I is associated with fatal outcome in acquired thrombotic thrombocytopenic purpura. J Clin Apher. 2017;32(5):311–318. [DOI] [PubMed] [Google Scholar]
- 15.Liu C, Kallogjeri D, Dynis M, Grossman BJ. Platelet recovery rate during plasma exchange predicts early and late responses in patients with thrombotic thrombocytopenic purpura. Transfusion. 2013;53(5):1096–1107. [DOI] [PubMed] [Google Scholar]
- 16.Dusse LM, Alpoim PN, Silva JT, Rios DR, Brandao AH, Cabral AC. Revisiting HELLP syndrome. Clin Chim Acta. 2015;451(Pt B):117–120. [DOI] [PubMed] [Google Scholar]
- 17.Kokame K, Nobe Y, Kokubo Y, Okayama A, Miyata T. FRETS-VWF73, a first fluorogenic substrate for ADAMTS13 assay. Br J Haematol. 2005;129(1):93–100. [DOI] [PubMed] [Google Scholar]
- 18.Zhang L, Lawson HL, Harish VC, Huff JD, Knovich MA, Owen J. Creation of a recombinant peptide substrate for fluorescence resonance energy transfer-based protease assays. Anal Biochem. 2006;358(2):298–300. [DOI] [PubMed] [Google Scholar]
- 19.Kumar M, Cao W, McDaniel JK, et al. Plasma ADAMTS13 activity and von Willebrand factor antigen and activity in patients with subarachnoid haemorrhage. Thromb Haemost. 2017;117(4):691–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fuchs TA, Kremer Hovinga JA, Schatzberg D, Wagner DD, Lammle B. Circulating DNA and myeloperoxidase indicate disease activity in patients with thrombotic microangiopathies. Blood. 2012;120(6): 1157–1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gerritsen HE, Turecek PL, Schwarz HP, Lammle B, Furlan M. Assay of von Willebrand factor (vWF)-cleaving protease based on decreased collagen binding affinity of degraded vWF: a tool for the diagnosis of thrombotic thrombocytopenic purpura (TTP). Thromb Haemost. 1999;82(5):1386–1389. [PubMed] [Google Scholar]
- 22.Afshar-Kharghan V. Atypical hemolytic uremic syndrome. Hematology Am Soc Hematology Am Soc Hematol Educ Program. 2016;2016(1):217–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Westwood JP, Langley K, Heelas E, Machin SJ, Scully M. Complement and cytokine response in acute Thrombotic Thrombocytopenic Purpura. Br J Haematol. 2014;164(6):858–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tati R, Kristoffersson AC, Stahl AL, et al. Complement activation associated with ADAMTS13 deficiency in human and murine thrombotic microangiopathy. J Immunol. 2013;191(5):2184–2193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Westwood JP, Langley K, Heelas E, Machin SJ, Scully M. Complement and cytokine response in acute Thrombotic Thrombocytopenic Purpura. Br J Haematol. 2013;64(6):858–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Martino S, Jamme M, Deligny C, et al. Thrombotic thrombocytopenic purpura in black people: impact of ethnicity on survival and genetic risk factors. PLoS One. 2016;11(7):e0156679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Terrell DR, Williams LA, Vesely SK, Lammle B, Hovinga JA, George JN. The incidence of thrombotic thrombocytopenic purpura-hemolytic uremic syndrome: all patients, idiopathic patients, and patients with severe ADAMTS-13 deficiency. J Thromb Haemost. 2005;3(7):1432–1436.15978100 [Google Scholar]
- 28.Mariotte E, Azoulay E, Galicier L, et al. Epidemiology and pathophysiology of adulthood-onset thrombotic microangiopathy with severe ADAMTS13 deficiency (thrombotic thrombocytopenic purpura): a cross-sectional analysis of the French national registry for thrombotic microangiopathy. Lancet Haematol. 2016;3(5):e237–245. [DOI] [PubMed] [Google Scholar]
- 29.Masias C, Wu H, McGookey M, Jay L, Cataland S, Yang S. No major differences in outcomes between the initial and relapse episodes in patients with thrombotic thrombocytopenic purpura: The experience from the Ohio State University Registry. Am J Hematol. 2018;93(3):E73–E75. [DOI] [PubMed] [Google Scholar]
- 30.Scully M, Cataland S, Coppo P, et al. Consensus on the standardization of terminology in thrombotic thrombocytopenic purpura and related thrombotic microangiopathies. J Thromb Haemost. 2016;15(2):312–322. [DOI] [PubMed] [Google Scholar]
- 31.Grillberger R, Casina VC, Turecek PL, Zheng XL, Rottensteiner H, Scheiflinger F. Anti-ADAMTS13 IgG autoantibodies present in healthy individuals share linear epitopes with those in patients with throm botic thrombocytopenic purpura. Haematologica. 2014;99(4):e58–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dong JF, Moake JL, Nolasco L, et al. ADAMTS-13 rapidly cleaves newly secreted ultralarge von Willebrand factor multimers on the endothelial surface under flowing conditions. Blood. 2002;100(12):4033–4039. [DOI] [PubMed] [Google Scholar]
- 33.Moake JL, Rudy CK, Troll JH, et al. Unusually large plasma factor VIII:von Willebrand factor multimers in chronic relapsing thrombotic thrombocytopenic purpura. N Engl J Med. 1982;307(23):1432–1435. [DOI] [PubMed] [Google Scholar]
- 34.Sadler JE. A new name in thrombosis, ADAMTS13. Proc Natl Acad Sci USA. 2002;99(18):11552–11554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jin SY, Xiao J, Bao J, Zhou S, Wright JF, Zheng XL. AAV-mediated expression of an ADAMTS13 variant prevents shigatoxin-induced thrombotic thrombocytopenic purpura. Blood. 2013;121(19):3825–3829, S1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Noris M, Mescia F, Remuzzi G. STEC-HUS, atypical HUS and TTP are all diseases of complement activation. Nat Rev Nephrol. 2012;8(11):622–633. [DOI] [PubMed] [Google Scholar]
- 37.Patton JF, Manning KR, Case D, Owen J. Serum lactate dehydrogenase and platelet count predict survival in thrombotic thrombocytopenic purpura. Am J Hematol. 1994; 47(2):94–99. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.