

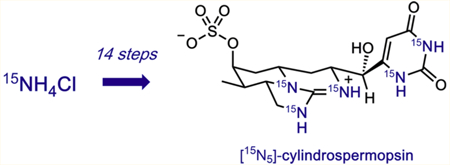
Abstract
Fresh water cyanobacterial algal blooms represent a major health risk because these organisms produce cylindrospermopsin, a toxic, structurally complex, zwitterionic uracil-guanidine alkaloid recognized by the EPA as a dangerous drinking water contaminant. At present, the ability to detect and quantify the presence of cylindrospermospin in water samples is severely hampered by the lack of an isotopically labeled standard for analytical mass spectrometry. Herein, we present a concise, scaled total synthesis of 15N cylindrospermosin from 15N ammonium chloride, which leverages a unique stereoselective intramolecular double conjugate addition step to assemble the tricyclic guanidine core. In addition to providing the first pure isotopically labeled probe for precise quantification of this potent biotoxin in fresh water sources, our results demonstrate how unique constraints associated with isotope incorporation compel novel solutions to synthesis design.
Graphical Abstract
INTRODUCTION
In recent years, cyanobacterial algal blooms have occurred with increasing frequency near population centers worldwide, causing significant socioeconomic impact and risk to human health.1 Cylindrospermopsin (CYN)2 and its naturally occurring 7-epi-3 and 7-deoxy-4 congeners are potent biotoxins released into fresh water sources during such blooms5 (Figure 1a,b). In June 2015, the US Environmental Protection Agency released a drinking water health advisory on cylindrospermop-sins detailing this emerging environmental threat.6 In addition to the acute toxicity risks, exposure to cylindrospermopsin and its derivatives is linked to hepatotoxicity, gastrointestinal inflammation, hemorrhages, anorexia, and liver failure.7
Figure 1.
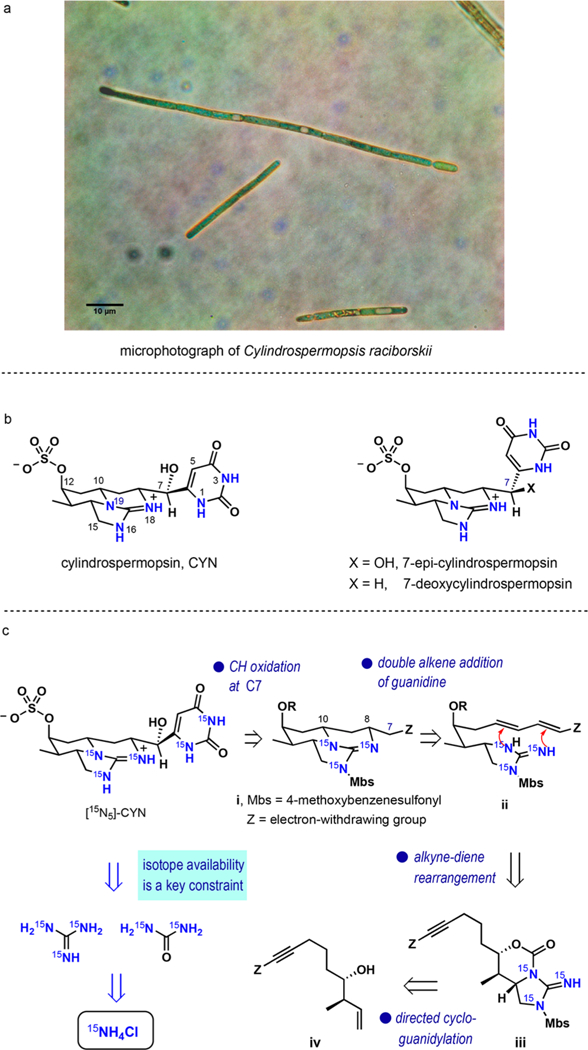
(a) A confocal optical microscopy image of Cylindrospermopsis raciborskii, (b) the structures of cylindrospermopsins, and (c) the outline of the synthetic design for [15N5]-CYN.
High-performance liquid chromatography coupled with mass spectrometry is the preferred method for cylindrospermopsin detection in environmental samples.8–13 In particular, isotopedilution mass spectrometry (IDMS)14 provides an attractive quantification method, as it uses an isotope of the analyte as an internal reference standard to ensure high sensitivity and precision in the measurement. The application of IDMS to cylindrospermopsin analysis, however, is hampered by the lack of isotopically labeled cylindrospermopsins. To solve this important problem, we coupled recent advances in synthetic chemistry and enzymatic bio-oxidation to prepare 7-epi- and 7-deoxy-cylindrospermopsin from [15N]-ammonium chloride, achieving >99.5% isotope incorporation into all five nitrogen atoms of these complex alkaloid natural products.
To facilitate mass spectrometry data interpretation, we sought an isotope of CYN with a mass difference of at least 5 atomic units relative to the most abundant natural isotope. We considered isotopes of hydrogen, carbon, nitrogen, and oxygen for this purpose. To ensure structural integrity of the analytical standard, isotope insertion at nonexchangeable positions throughout the synthesis is essential. Based on these two key requirements, we chose to pursue the total synthesis of [15N1,15N3,15N16,15N18,15N19]-cylindrospermopsin ([15N5]-CYN, [15N5]-1, Figure 1c). The decision to incorporate 15N in the cylindrospermopsin scaffold required us to rethink past synthetic strategies applied to the total synthesis of CYN and structurally related alkaloids, as these approaches are incompatible with efficient label installation at nonexchangeable atomic positions. In addition, these prior strategies, while elegant in design and execution, are laborious, requiring 20 or more steps, and present uncertainties regarding scalability.15–22 Similarly, preparation of labeled cylindrospermospins via fermentation using enriched nutrients such as Na15NO3 has proven untenable.23 These key concerns prompted the development of a de novo approach, whereby we explored whether isotope incorporation at the design stage could lead to an improved synthetic strategy that is more effective than biosynthetic or synthetic strategies reported previously.
Several principal considerations factored into the synthesis planning. Clearly, the effective introduction of the 15N isotope becomes a major factor. Compared to a typical total synthesis effort, the payoff of a convergent versus linear, atom-by-atom assembly is now substantially enhanced because all but the most elemental sources of the 15N isotope (15NH4Cl or aqueous 15NH3) become impractical.24 Thus, a convergent design with preassembled intermediates containing several 15N isotopes is essential. The common metric of synthetic efficiency, namely, minimizing the number of individual steps based on robust, reproducible chemical transformations, becomes especially critical given the costs of the starting isotopes.25–27 Finally, all intended transformations must ensure retention of the isotopic label.
RESULTS AND DISCUSSION
Synthesis Design.
Following the logic outlined above, we formulated a synthesis plan depicted in Figure 1c. [15N3]-Guanidine and [15N2]-urea, each derived from 15NH4Cl, serve as precursors of the tricyclic guanidine and uracil subunits of cylindrospermopsin, respectively. The synthesis plan requires a late stage conversion of the electron-withdrawing group Z in intermediate i to uracil and C—H oxidation at C7. The electron-withdrawing group Z enables a key simplification to diene ii by disconnection of two C—N bonds in tricyclic guanidine i at C8 and C10. Stereoselective intramolecular addition of guanidine to diene, although lacking precedent, is expected to ensure rapid assembly of the tricyclic guanidine subunit of cylindrospermopsins. Alkyne iii was intended as a precursor to diene ii via a transition-metal-mediated rearrangement. As an essential element of the plan, homoallylic alcohol iv serves as the substrate for a hydroxyl-directed stereoselective delivery of free [15N3]-guanidine that we previously developed for this application.28
The success of this plan rests on identifying a suitable substituent Z, which must first activate the diene for addition of guanidine, then facilitate C7 hydroxylation, and eventually be converted to uracil. Although several options can be considered, heterocyclic substituents such as 2,4-dimethoxypyr-imid-6-yl appeared to be especially enticing as precursors of uracil. Various electron-deficient heterocylic groups are known to stabilize carbanions, and activation of double bonds by substitution with heterocycles for nucleophilic addition has been reported.29–32 What remained uncertain was whether diene activation for double addition of guanidine, a centerpiece of our strategy, was feasible. After a careful analysis supported by experimentation, we identified 2,4-dimethoxypyrimid-6-yl as group Z because of its potential to meet these criteria most effectively.
Preparation of [15N]-Labeled Precursors.
While the initial validation of the entire total synthesis was carried out with the natural isotope, we performed the scaled synthesis starting with 15NH4Cl as depicted in Scheme 1. [15N]-Ammonium chloride proved to be the most economical source of dry liquid [15N]-ammonia on the laboratory scale, affording >95% yields of the reagent reproducibly upon treatment with NaOH in solid phase as long as the scale exceeds 0.5 g. [15N2]-2,4-Dimethoxy-6-bromopyrimidine [15N2]-5 was obtained by the initial conversion of 15NH3 to urea,33 its condensation with diethyl malonate to [15N2]-barbituric acid [15N2]-4, treatment with POBr3, and methanolysis with 2 equiv of NaOMe. In contrast to the synthesis of urea, the reaction of 15NH3 with diphenyl thiocarbonate gave ammonium thiocyanate, pyrolysis of which was required to access [15N2]-thiourea.34 The preparation of [15N3]-guanidine hydrochloride was accomplished in four steps by S-methylation, treatment with Boc2O, displacement of the MeS group with 15NH3 in chloroform, and complete hydrolysis with 12 M HCl at reflux. The resulting [15N3]-guanidine hydrochloride and [15N2]-5 constitute the source of all 15N isotopes in the target alkaloid.
Scheme 1.
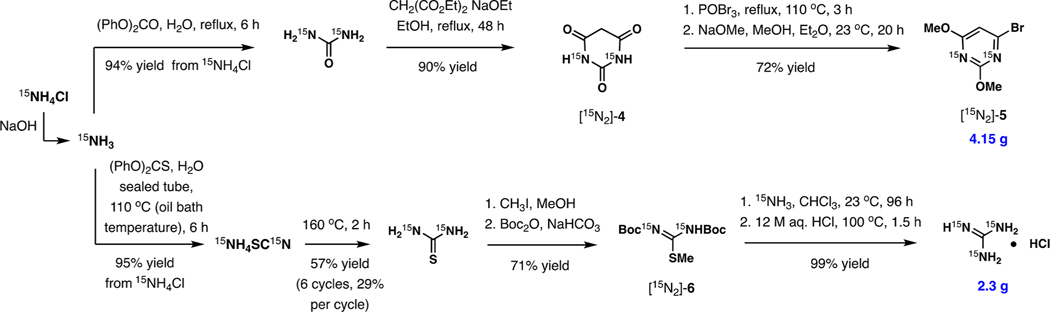
Preparation of [15N2]-2,4-Dimethoxy-6-bromopyrimidine and [15N3]-Guanidine Hydrochloride from 15NH4Cl, the Key Precursors Constituting the Source of All 15N Isotopes in Cylindrospermopsins
Total Synthesis of [15N5]-Cylindrospermopsins.
Advancing the synthesis of [15N5]-cylindrospermopsin, bromopyr-imidine [15N2]-5 was subjected to Sonogashira coupling with 5-hexyn-1-ol (Scheme 2). After Swern oxidation of [15N2]-7 to [15N2]-8, efficient enantioselective crotylation was achieved using Leighton’s reagent 935 (99% yield, dr 10:1, er 97:3). To shorten the synthesis, we explored direct conversion of [15N2]-7 to [15N2]-10 using a powerful method developed by Krische and co-workers.36 Although the enantiocontrol was excellent (er 98:2), lower diastereoselectivity (4:1) and yield (21%) thwarted its application for the scaled synthesis of [15N2]-10 (see the Supporting Information for additional details). The next objective was the appendage of guanidine unit to the unactivated terminal alkene in [15N2]-10. This was achieved by a two-stage approach described previously.28 First, free [15N3]-guanidine was attached to the hydroxy group by a carbonyl linker in good yield using 1.2 equiv of 15N-labeled reagent. Second, electrophilic cycloguanidylation was performed in the presence of N-iodosuccinimide, delivering [15N5]-12 after treatment with 4-methoxybenzenesulfonyl chloride (MbsCl) in 71% yield and 2.5:1 dr favoring the requisite diastereomer.
Scheme 2.
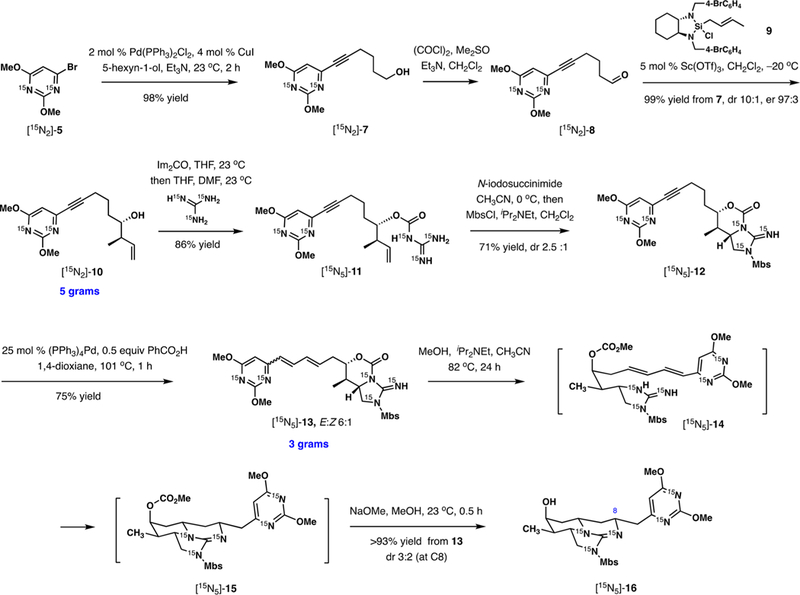
Incorporation of All Five 15N Isotopes into Advanced Intermediate [15N5]-16 from [15N2]-2,4-Dimethoxy-6-bromopyrimidine [15N2]-5 and [15N3]-Guanidine Hydrochloride
Alkyne [15N5]-12 served as an effective precursor of the 1,3-diene required for the key double addition of guanidine.37–40
We identified (Ph3P)4Pd and benzoic acid at reflux in dioxane as the optimal reaction system for the challenging substrate containing two heterocyclic substituents. Diene [15N5]-13, obtained in 75% yield as a 6:1 mixture of E and Z isomers, was now poised to implement the key cyclization to the tricyclic guanidine ring system of CYNs. Simple heating of [15N5]-13 in 14% methanol in acetonitrile (v/v) in the presence of iPr2NEt at reflux resulted in rapid methanolysis to [15N5]-14. Remarkably, the key addition of guanidine to the diene took place concurrently with methanolysis under these mild reaction conditions, and subsequent removal of methyl carbonate in [15N5]-14 was accomplished with NaOMe in situ, affording [15N5]-16. The success of this process was essential to ensure the brevity of the total synthesis and demonstrated the activating properties of the 2,6-dimethoxy-6-pyrimidinyl substituent for addition of nitrogen nucleophiles to alkenes.
To address one of the most challenging phases of the synthesis, the stereospecific hydroxylation of the C7 methylene group of [15N5]-7-deoxy-CYN ([15N5]-3), we explored enzymatic biooxidation. This conversion represents the final step of CYN biosynthesis and is catalyzed by CyrI, an α-ketoglutarate-dependent nonheme iron oxygenase. We produced pure, recombinant CyrI (Oscillatoria strain PCC7926) in E. coli and applied the enzyme in the direct oxidation of ([15N5]-3), which was readily generated from [15N5]-16 by acidic hydrolysis (12 M HCl, reflux) and O-sulfonation (SO3- Py, DMF, 95% overall yield) (Scheme 3a).41 The enzyme showed high selectivity and did not accept a substrate lacking the O12 sulfate, suggesting that O-sulfonation must precede C7 hydroxylation in the biosynthesis of CYN. Importantly, CyrI enabled a catalytic, protecting-group-free oxidation to afford the final natural product in 93% yield with >90:1 diastereoslectivity achievable with high enzyme loading. The biocatalytic synthesis produced uniformly 15N labeled CYN in 10 total steps with 15% overall yield, demonstrating the advantages of combining traditional organic chemistry with enzymatic transformations.
Scheme 3.
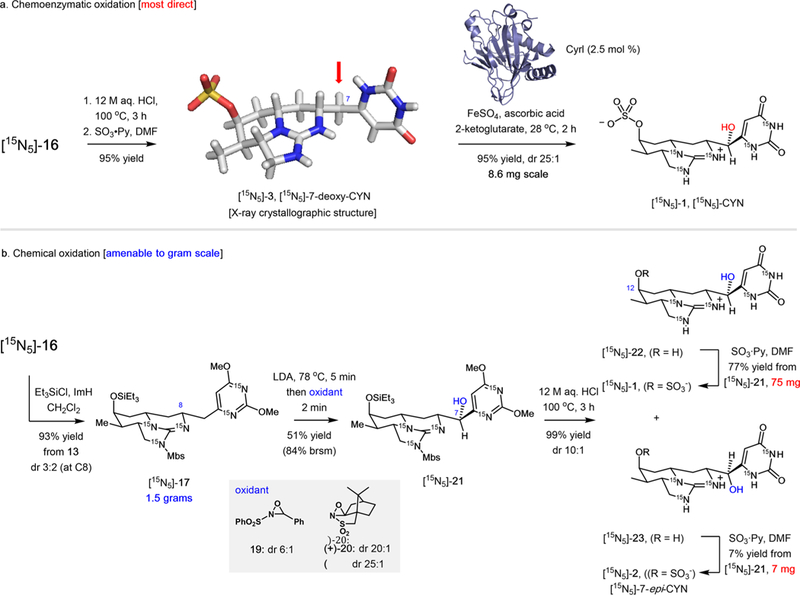
Total Synthesis of [15N5]-CYN, [15N5]-7-epi-CYN, and 7-Deoxy-CYN by (a) C7 Bio-hydroxylation with Cyrl and (b)Stereoselective Chemical Oxidation via Lithiation at C7
As a comparison to the enzymatic approach, and to gain access to the 7-epi congener of cylindrospermospin, we also investigated the chemical oxidation of [15N5]-16 (Scheme 3b). Silylation of [15N5]-16 with Et3SiCl (TESCl) afforded tricyclic guanidine [15N5]-17 in 93% overall yield from diene [15N5]-13 as a separable 3:2 mixture of diastereomers at C8, favoring the required R configuration. Replacement of methyl carbonate with the TES group in [15N5]-17 facilitated two practical objectives: (l) enabling a simple chromatographic separation of C8 diastereomers and (2) compatibility with basic conditions required for the ensuing C7 metalation and hydroxylation. Although the substrate was sensitive to the nature and stoichiometry of the base, we found that rapid lithiation with LDA (5 min, −78 °C) followed by a quench with an oxaziridine afforded the diastereomeric C7 hydroxylation products in good yields along with recovered starting material. The diastereomer ratio is dependent on the oxaziridine: rac-19 afforded a 6:1 dr, while camphor based reagent 20 or ent-20 gave 25:1 and 20:1 dr in favor of [15N5]-1, respectively. The optimized hydroxylation was scaled to 1.50 g of [15N5]-17, providing [15N5]-21 as a pure isomer in 46% yield along with 41% of starting material. Complete hydrolysis to uracil along with the removal of the Mbs group was achieved on 0.350 g scale of [15N5]-21 by reflux in 12 M HCl for 3 h, resulting in a minor epimerization at C7 (10:1). The mixture of epimers [15N5]-22 and [15N5]-23 was submitted to O-sulfonylation with the SO3-Py complex in DMF.18 After optimizing the reagent loading, we observed highly selective monosulfation of the diols at the C12 OH group. The resulting mixture was readily separated by reversed-phase HPLC, affording 75 mg of [15N5]-CYN (77% yield) and 6.8 mg of [15N5]-7-epi-CYN (7% yield) in a single attempt. By comparison with biooxidation using CyrI, the chemical oxidation shows improved scalability, but introduces one additional step in the total synthesis with slightly diminished overall yield and lower stereoselectivity.
With the supply of [15N5]-CYN secured by total synthesis, its development as an analytical standard for the precise quantification of CYN by LC/ESI-MS/MS was undertaken. Based on reported concentrations of the cyanotoxin in environmental samples, we developed an assay for the detection of cylindrospermopsin in a broad range from 10 ng/L to 100 μ/L. While the EPA limit for cylindrospermopsin in drinking water has been proposed to be 1 μg/L,7,42 concentrations below this level still represent an environmental hazard due to bioaccumulation and uncertain health risks of chronic sublethal exposure.43 The limit of detection (LOD, Signal/Noise = 3) and the limit of quantification (LOQ, Signal/Noise = 10) for the method were determined to be 2.5 and 8.25 ng/L respectively, the highest sensitivity reported to date.8–13 The evaluation of matrix effect for analyte quantification was performed using environmental samples free of native CYN that were spiked with 100 μg/L. In all cases the observed recoveries were nearly 100%. This method was then used to quantify cylindrospermopsin in environmental water samples in 14 locations in Southern California where potential contamination or water resources was suspected.44 Notably, availability of [15N5]-CYN afforded the reliable and precise detection of cylindrospermopsin in Lake Casitas, a previously undescribed location of cyanotoxin contamination, where we detected the concentration of 157 ng/L of the toxin.
■ CONCLUDING REMARKS
In summary, we developed a strategy for the concise total synthesis of cylindrospermopsins that is directly applicable for 15N isotope incorporation. We demonstrated the scalability of the synthesis by preparing 0.125 g of [15N5]-CYN in 14 steps (longest linear sequence) and 9% overall yield from [15N]-ammonium chloride, along with 7.5 mg of [15N5]-7-epi-CYN and 20 mg of [15N5]-7-deoxy-CYN. In the natural isotope series, the total synthesis of CYN was completed from commercially available 2,4-dimethoxy-6-bromopyrimidine (5) in only 10 steps via the enzymatic oxidation route (15% yield from 5), and 11 steps by chemical C7 hydroxylation (11% yield), representing a substantial improvement in brevity and scalability compared to the previous efforts. Furthermore, these efforts provided a high-quality isotopically labeled probe for the reliable detection and quantification of cyanotoxins in fresh water sources with the highest limits of detection reported to date. Our results demonstrate how atypical constraints associated with isotope incorporation empower the development of novel approaches to organic synthesis design.
■ ACKNOWLEDGMENTS
This work was supported by the NIH (NIGMS, R01–077379, R01–115388 and NIEHS, R03 ES025345–01). Dr. Hongjun Zhou is acknowledged for assistance with NMR spectroscopy. Dr. Dmitriy Uchenik and the UCSB mass spectroscopy facility are thanked for assistance with mass spectral analysis. Dr. Guang Wu and the UCSB X-ray analytical facility are acknowledged for assistance with the X-ray crystallography. We are especially grateful to Anastasiia S. Minakova for assistance with the enzymatic oxidation experiment. Dr. Ben Lopez and the NRI-MCDB Microscopy Facility at UCSB are acknowledged for obtaining microphotographs of Cylindrosper-mopsis raciborskii.
Footnotes
Notes
The authors declare no competing financial interest.
■ ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.8b03071.
Full experimental procedures and copies of 1H, 13C, 15N NMR spectra (PDF)
Crystallographic data for 3 (CIF)
■ REFERENCES
- (1).Cheung MY; Liang S; Lee J J.Microbiol. 2013, 51, 1–10. [DOI] [PubMed] [Google Scholar]
- (2).Ohtani I; Moore RE; Runnegar MTC J. Am. Chem. Soc. 1992, 114, 7941–7942. [Google Scholar]
- (3).Banker R; Teltsch B; Sukenik A; Carmeli S J. Nat. Prod. 2000, 63, 387–389. [DOI] [PubMed] [Google Scholar]
- (4).Norris RL; Eaglesham GK; Pierens G; Shaw GR; Smith MJ; Chiswell RK; Seawright AA; Moore MR Environ. Toxicol. 1999, 14, 163–165. [Google Scholar]
- (5).De la Cruz AA; Hiskia A; Kaloudis T; Chernoff N; Hill D; Antoniou MG; He X; Loftin K; O’Shea K; Zhao C; Pelaez M; Han C; Lynch TJ; Dionysiou DD Environ. Sci: Processes Impacts 2013, 15, 1979–2003. [DOI] [PubMed] [Google Scholar]
- (6).Drinking water health advisory for the cyanobacterial toxin cylindrospermopsin. EPA-820R15101. https://www.epa.gov/sites/production/files/2017–06/documents/cylindrospermopsin-report-2015.pdf.
- (7).Falconer IR; Humpage AR Environ. Toxicol. 2006, 21, 299–304. [DOI] [PubMed] [Google Scholar]
- (8).Guzmán-Guillén R; Prieto AI; González AG; Soria-Díaz ME; Cameán AM. Environ. Toxicol. Chem. 2012, 31, 2233–2238. [DOI] [PubMed] [Google Scholar]
- (9).Graham JL; Loftin KA; Meyer MT; Ziegler AC Environ. Sci. Technol. 2010, 44, 7361–7368. [DOI] [PubMed] [Google Scholar]
- (10).Kikuchi S; Kubo T; Kaya K Anal. Chim. Acta 2007, 583, 124–127. [DOI] [PubMed] [Google Scholar]
- (11).Bláhová L.; Oravec M; Marsálek B; Sejnohová L.; Simek Z.; Bláha L. Toxicon 2009, 53, 519–524. [DOI] [PubMed] [Google Scholar]
- (12).Gallo P; Fabbrocino S; Grazia Cerulo M; Ferranti P; Bruno M; Serpe L Rapid Commun. Mass Spectrom. 2009, 23, 3279–3284. [DOI] [PubMed] [Google Scholar]
- (13).Kubo T; Sano T; Hosoya K; Tanaka N; Kaya K Toxicon 2005, 46, 104–107. [DOI] [PubMed] [Google Scholar]
- (14).De Bièvre P. Tech. Instrum. Anal. Chem. 1994, 15, 169–183. [Google Scholar]
- (15).Xie C; Runnegar MTC; Snider BB J. Am. Chem. Soc. 2000, 122, 5017–5024. [Google Scholar]
- (16).Heintzelman GR; Fang W-K; Keen SP; Wallace GA; Weinreb SM J. Am. Chem. Soc. 2001, 123, 8851–8853. [DOI] [PubMed] [Google Scholar]
- (17).Heintzelman GR; Fang W-K; Keen SP; Wallace GA; Weinreb SM J. Am. Chem. Soc. 2002, 124, 3939–3945. [DOI] [PubMed] [Google Scholar]
- (18).White JD; Hansen JD J. Am. Chem. Soc. 2002, 124, 4950–4951. [DOI] [PubMed] [Google Scholar]
- (19).White JD; Hansen JD J. Org. Chem. 2005, 70, 1963–1977. [DOI] [PubMed] [Google Scholar]
- (20).Looper RE; Williams RM Angew. Chem., Int. Ed. 2004, 43, 2930–2933. [DOI] [PubMed] [Google Scholar]
- (21).Looper RE; Runnegar MTC; Williams RM Angew. Chem., Int. Ed. 2005, 44, 3879–3881. [DOI] [PubMed] [Google Scholar]
- (22).Looper RE; Runnegar MTC; Williams RM Tetrahedron 2006, 62, 4549–4562. [Google Scholar]
- (23).Kittler K; Hoffmann H; Lindemann F; Koch M; Rohn S; Maul R Anal. Bioanal. Chem. 2014, 406, 5765–5774. [DOI] [PubMed] [Google Scholar]
- (24).Cambridge Isotope Laboratories, Inc., listed prices: NH4Cl, $490/10g; aqueous 15NH3, $168/5g (3.3N solution); 15NH3, $454/1 L of gas; (15NH2)iCO, $305/1g; [15N3]-guanidine, no longer available.
- (25).Trost BM Science 1991, 254, 1471–1477. [DOI] [PubMed] [Google Scholar]
- (26).Kuttruff CA; Eastgate MD; Baran PS Nat. Prod. Rep. 2014, 31, 419–432. [DOI] [PubMed] [Google Scholar]
- (27).Tian M; Yan M; Baran PS J. Am. Chem. Soc. 2016, 138, 14234–14237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Mailyan AK; Young K; Chen JL; Reid BT; Zakarian A Org. Lett. 2016, 18, 5532–5535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Gray AP; Platz RD; Chang TCP; Leverone TR; Ferrick DA; Kramer DN J. Med. Chem. 1985, 28, 111–116. [DOI] [PubMed] [Google Scholar]
- (30).Macor JE; Ordway T; Smith RL; Verhoest PR; Mack RA J. Org. Chem. 1996, 61, 3228–3229. [Google Scholar]
- (31).Djung JF; Hart DJ; Young ERR J. Org. Chem. 2000, 65, 5668–5675. [DOI] [PubMed] [Google Scholar]
- (32).Schroeder AC; Bloch A; Perman JL; Bobek M J.Med. Chem. 1982, 25, 1255–1258. [DOI] [PubMed] [Google Scholar]
- (33).Leitch LC; Davidson WM Can. J. Agr. Sci. 1949, 29, 189–190. [Google Scholar]
- (34).Laxer A; Major DT; Gottlieb HE; Fischer B J.Org. Chem. 2001,66, 5463–5481. [DOI] [PubMed] [Google Scholar]
- (35).Kim H; Ho S; Leighton JL J. Am. Chem. Soc. 2011, 133, 6517–6520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Kim IS; Han SB; Krische M J. Am. Chem. Soc. 2009, 131, 2514–2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Kwong CK-W; Fu MY; Lam CS-L; Toy PH Synthesis 2008, 2008, 2307–2317. [Google Scholar]
- (38).Shintani R; Duan W-L; Park S; Hayashi T Chem. Commun. 2006, 3646–3647. [DOI] [PubMed] [Google Scholar]
- (39).Kadota I; Shibuya A; Mpaka Lutete L; Yamamoto Y J.Org. Chem. 1999, 64, 4570–4571. [DOI] [PubMed] [Google Scholar]
- (40).Patil NT; Huo Z; Bajracharya GB; Yamamoto Y J. Org. Chem. 2006, 71, 3612–3614. [DOI] [PubMed] [Google Scholar]
- (41).Mazmouz R; Chapuis-Hugon F; Pichon V; Méjean A; Ploux O. ChemBioChem 2011, 12, 858–862. [DOI] [PubMed] [Google Scholar]
- (42).Humpage AR; Falconer IR Environ. Toxicol. 2003, 18, 94–103. [DOI] [PubMed] [Google Scholar]
- (43).Kinnear S Mar. Drugs 2010, 8, 542–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Tatters AO; Howard MDA; Nagoda C; Busse L; Gellene AG; Caron DA Toxins 2017, 9, 95–111. [DOI] [PMC free article] [PubMed] [Google Scholar]