

Abstract
C-1027 is a potent antitumor antibiotic composed of an apoprotein (CagA) and a reactive enediyne chromophore. The chromophore has four distinct chemical moieties, including an (S)-3-chloro-5-hydroxy- β-tyrosine moiety, the biosynthesis of which from L-α-tyrosine requires five proteins: SgcC, SgcC1, SgcC2, SgcC3, and SgcC4; a sixth protein, SgcC5, catalyzes the incorporation of this β-amino acid moiety into C-1027. Biochemical characterization of SgcC has now revealed that (i) SgcC is a two-component, flavin adenine dinucleotide (FAD)-dependent monooxygenase, (ii) SgcC is only active with SgcC2 (peptidyl carrier protein)-tethered substrates, (iii) SgcC-catalyzed hydroxylation requires O2 and FADH2, the latter supplied by the C-1027 pathway-specific flavin reductase SgcE6 or Escherichia coli flavin reductase Fre, and (iv) SgcC efficiently catalyzes regioselective hydroxylation of 3-substituted β-tyrosyl-S-SgcC2 analogues, including the chloro-, bromo-, iodo-, fluoro-, and methyl-substituted analogues, but does not accept 3-hydroxy-β-tyrosyl-S-SgcC2 as a substrate. Together with the in vitro data for SgcC4, SgcCI, and SgcC3, the results establish that SgcC catalyzes the hydroxylation of (S)-3-chloro-β-tyrosyl-S-SgcC2 as the final step in the biosynthesis of the (S)-3-chloro-5-hydroxy-β-tyrosine moiety prior to incorporation into C-1027. SgcC now represents the first biochemically characterized two-component, FAD-dependent monooxygenase that acts on a carrier-protein-tethered aromatic substrate.
Introduction
C-1027 is a chromoprotein antitumor antibiotic produced by Streptomyces globisporus and is isolated as a noncovalent complex consisting of an apoprotein (CagA) and the C-1027 chromophore (1). The structure of 1 consists of four distinct moieties: an enediyne core, a deoxy aminosugar, a benzoxazo- linate, and an (S)-3-chloro-5-hydroxy-β-tyrosine moiety (Figure 1).1–3 Upon release from CagA, the enediyne core of 1 readily undergoes a Bergman cycloaromatization to generate a highly reactive diradical intermediate that is capable of abstracting hydrogen atoms from DNA, leading to both double-stranded breaks (DSBs) and interstrand cross-links (ICLs), and hence ultimately cell death.4–9 In view of its unique structure, mechanism of action, and potent cytotoxicity, C-1027 has attracted intense interest from chemists and biologists alike in search of novel C-1027 analogues as potential cancer chemo-therapeutic agents. We recently reported that engineered C-1027 analogues with a single substitution to the chromophore can shift the mechanism of DNA damage to primary DSBs or ICLs, suggesting that the modified enediynes might prove to be therapeutically advantageous.8,9
Figure 1.
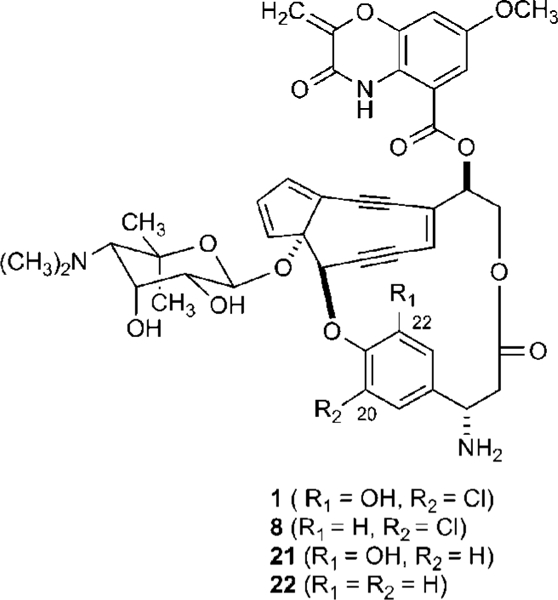
Structures of the enediyne chromophore of C-1027 (1) and engineered analogues 22-deshydroxy-C-1027 (8), 20-deschloro-C-1027 (21), and 20-deschloro-22-deshydroxy-C-1027 (22).
The biosynthetic gene cluster for C-1027 was previously cloned and sequenced.7 Bioinformatics analysis of the open reading frames led to the proposal that the (S)-3-chloro-5- hydroxy-β-tyrosine moiety originates from L-α-tyrosine (2) by virtue of five proteins: SgcC, SgcCl, SgcC2, SgcC3, and SgcC4. Once produced, the (S)-3-chloro-5-hydroxy-β-tyrosine moiety is incorporated into C-1027 by the condensation enzyme SgcC5 via a β-aminoacyl-S-peptidyl carrier protein (SgcC2) intermediate (3) (Figure 2). Biochemical characterization using recombinant enzymes has already confirmed that (i) SgcC4 catalyzes the conversion of L-a-tyrosine to (S)-β-tyrosine (4) as the first step,10–13 (ii) SgcCl activates (S)-β-tyrosine as the (S)-β-tyrosyl adenylate (5) and subsequently loads (S)-β-tyrosine onto holo- SgcC2, a type II peptidyl carrier protein (PCP), to yield (S)-β- tyrosyl-S-SgcC2 (6) as the second step,13,14 and (iii) SgcC3 is a FAD-dependent halogenase catalyzing regioselective chlorination of (S)-β-tyrosyl-S-SgcC2 to form (S)-3-chloro-β-tyrosyl- S-SgcC2 (7).15 Furthermore, in vivo data have suggested that the last transformation, C-5 hydroxylation of (S)-3-chloro-β- tyrosyl-S-SgcC2, is catalyzed by SgcC since 22-deshydroxy- C-1027 (8) was isolated from the fermentation of a ΔsgcC inactivation mutant.7,13
Figure 2.
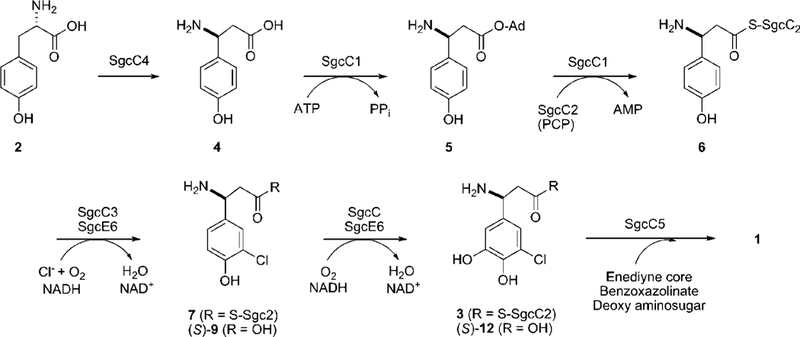
Biosynthetic pathway for the (S)-3-chloro-5-hydroxy-β-tyrosine moiety of the C-1027 enediyne chromophore from L-tyrosine (2).
A variety of enzyme families catalyze hydroxylation of aromatic compounds, including cytochrome P-450-dependent monooxygenases, non-heme Fe3+-dependent monooxygenases, and flavin-dependent monooxygenases, among others.16 The last family, the flavin-dependent monooxygenases, consists of two different classes of enzymes that have no structural or sequence similarities.17–19 The first class, or one-component monooxy-genases, are single polypeptides that utilize FAD or FMN as a cofactor and require NADH or NADPH to initiate oxidation of substrates; thus, these monooxygenases have both flavin reductase and monooxygnease activity. The second class, or two- component monooxygenases, utilize reduced FAD or FMN directly as a cosubstrate and therefore require a separate NAD(P)H:flavin reductase to supply the reduced flavin. Thus, the two-component systems consist of two different proteins, one serving as a reductase and the other as a monooxygenase. Although the flavin-dependent monooxygenases typically catalyze hydroxylation using small-molecule substrates, such as p-hydroxybenzoate (by a one-component monooxygenase)20–22 and p-hydroxyphenylacetate (by a two-component monoxyge- nase),23–26 a single example has been reported recently wherein BtrO, a two-component FMN-dependent monooxygenase involved in the biosynthesis of butirosin, requires a carrier protein- tethered aliphalic substrate.27
The gene product of sgcC has high sequence similarity to two-component p-hydroxyphenylacetate monooxygenases such as HpaA from Pseudomonas aeruginosa (accession no. NP_252780, 50% identity/62% similarity)23 and HpaB from Escherichia coli (accession no. CAA82321, 49% identity/61% similarity),24 and, as previously noted, gene inactivation of sgcC led to the isolation of 22-deshydroxy-C-1027, supporting the functional assignment based on sequence analysis.7 To continue our investigations on the biosynthesis of C-1027, particularly to delineate the events leading to the (S)-3-chloro-5-hydroxy-β-tyrosine moiety of 1, we sought to characterize the enzymatic activity of SgcC in vitro to provide insight into the substrate specificity, timing of the hydroxylation step in biosynthesis of the (S)-3-chloro-5-hydroxy-β-tyrosine moiety of 1, and preliminary mechanistic details of this enzyme. In this report, we now establish that SgcC is a two-component, FAD-dependent monooxygenase responsible for the regioselective hydroxylation of (S)-3-chloro-β-tyrosyl-S-SgcC2, requiring O2 and reduced FAD (FADH2) provided by the pathway-specific flavin reductase SgcE6 or E. coli flavin reductase Fre, for the fourth and final enzymatic step of the biosynthesis of 3 from L-α-tyrosine (Figure 2). Similar to BtrO, SgcC is a two-component monoxygenase that is dependent upon a carrier-protein-tethered substrate. But in contrast, SgcC utilizes reduced FAD to hydroxylate an aromatic substrate instead of reduced FMN as for BtrO.27 We also investigated the substrate specificity of SgcC and demonstrated that SgcC is capable of hydroxylating other 3-substituted 7 analogues, including the fluoro-, bromo-, iodo-, and methyl- substituted analogues. The functional assignment and preliminary characterization of SgcC lay the foundation for future biochemical studies on this mechanistically intriguing group of carrier protein-dependent monooxygenases, and the results provided here have clear ramifications with respect to engineering novel 1 analogues by combinatorial biosynthesis.
Experimental Procedures
Materials and Methods.
Adenosine triphosphate disodium salt (ATP), coenzyme A (CoA), flavin adenine dinucleotide disodium salt (FAD), flavin mononucleotide sodium salt (FMN), β-nicoti- namide adenine dinuceotide reduced disodium salt (NADH), and tris(2-carboxyethyl)phosphine hydrochloride (TCEP) were purchased from Sigma-Aldrich (St. Louis, MO). Dithiothreitol (DTT) and isopropyl thiogalactoside (IPTG) were purchased from Research Products International Corp. (Mt. Prospect, IL). Complete protease inhibitor tablet, EDTA-free, was from Roche Applied Science (Indianapolis, IN). The starting materials for the synthesis of β-amino acid analogues, including 3-fluoro-4-hydroxybenzaldehyde, 3-methyl-4-hydroxybenzaldehyde, and 3-iodo-4-hydroxybenzalde- hyde, were purchased from Sigma-Aldrich and used without further purification. 3-Chloro-β-tyrosine (9), 3-bromo-β-tyrosine (10), 3-hydroxy-β-tyrosine (11), and 3-chloro-5-hydroxy-β-tyrosine (12) were synthesized as described.13–15 (S)-3-Amino-3-(4-hydroxyphe- nyl)propionic acid [(S)-β-tyrosine] (4) and (R)-3-amino-3-(4- hydroxyphenyl)propionic acid [(R)-β-tyrosine] were from PepTech Corp. (Burlington, MA). Medium components and chemicals were from Fisher Scientific (Fairlawn, NJ). Chemically competent E. coli DH5α and E. coli BL21(DE3) cells were prepared using standard procedures.28 Synthetic DNA oligonucleotides were purchased from the University of Wisconsin—Madison Biotechnology Center (Madison, WI). PCR was performed with a PerkinElmer GeneAmp 2400 (PerkinElmer Life And Analytical Sciences, Inc., Waltham, MA). Electrospray ionization mass spectroscopy (ESI-MS), high-resolution electrospray ionization mass spectroscopy (HR-ESI-MS), or LC-ESI-MS was performed with an Agilent 1100 HPLC-MSD SL ion trap mass spectrometer (Agilent Technologies, Inc., Santa Clara, CA). Atmospheric pressure chemical ionization mass spectroscopy (APCI-MS) was measured with an Agilent 1100 VL APCI mass spectrometer. NMR data were obtained using a Varian Unity Inova 400 MHz NMR Spectrometer (Varian, Inc., Palo Alto, CA).
Synthesis of 3-Fluoro-β-tyrosine (13), 3-Iodo-β-tyrosine (14), and 3-Methyl-β-tyrosine (15).
Compounds 13, 14, and 15 were prepared following the method reported by Weaver29 (see Supporting Information for details).
Overproduction and Purification of SgcC.
The sgcC gene was amplified with cosmid pBS100530 as a template and platinum Pfx DNA polymerase from Invitrogen (Carlsbad, CA) using the following primers: forward 5’-GGT ATT GAG GGT CGC ATG CCC CAC G GT GCA GAG C-3’ and reverse 5’-AGA GGA GAG TTA GAG CTA CAG CCC TCC GAG AAG G-3’ [the start (ATG) and stop (CTA) codons are underlined]. Purified PCR product was cloned into pET-30Xa/LIC vector following the ligation-independent cloning procedure as described by Novagen (Madison, WI) to give pBS1092, and the identity of sgcC in pBS1092 was confirmed by DNA sequencing.
pBS1092 was transformed into E. coli BL21 (DE3) and grown in LB media supplemented with 50 μg mL—1 kanamycin. Cells were grown at 18 °C and induced with IPTG (final concentration of 0.1 mM) when OD600 reached ~0.5. They were subsequently cultured at 18 °C for an additional 15 h. Cells were harvested by centrifugation (8000 rpm for 15 min at 4 °C) and resuspended in buffer A (100 mM sodium phosphate, pH 7.5, containing 300 mM NaCl) supplemented with a complete protease inhibitor tablet, EDTA-free. The cells were lysed by sonication (4 × 30 s pulsed cycle), and the debris was removed by centrifugation (15 000 rpm for 50 min at 4 °C). The clarified supernatant was loaded onto a pre-equilibrated Ni-NTA agarose (Qiagen, Valencia, CA) column with buffer B (buffer A plus 10% glycerol). The column was washed with five column volumes of buffer B, followed by five column volumes of buffer B containing 20 mM imidazole. The His6-tagged SgcC protein was eluted with six column volumes of buffer B containing 250 mM imidazole. After desalting using a PD-10 column (GE Healthcare, Piscataway, NJ), the purified SgcC protein was concentrated using an Amicon Ultra-4 (10K, GE Healthcare) and stored at —25 °C as 40% glycerol stocks. The purity of isolated SgcC was examined upon 12% SDS—PAGE analysis. Protein concentration was determined using the Bradford protein assay (Bio-Rad, Hercules, CA).
Determination of Cofactor Present in Purified SgcC.
SgcC was denatured by boiling for 3 min or by adding 50% methanol (final concentration).15 After centrifugation, the supernatant was loaded onto an Apollo C18 reverse-phase column (5 μm, 250 × 4.6 mm, Alltech Associates Inc., Deerfield, IL) and analyzed using a linear gradient from 0 to 60% CH3CN in H2O at a flow rate of 1 mL min—1 with UV-vis detection at 266 nm.
Preparation of SgcC2-Tethered Substrates for SgcC.
A general procedure was used to generate all potential substrates for SgcC, including 6, 7, (S)-3-F-β-tyrosyl-S-SgcC2 (16), (S)-3-Br-β- tyrosyl-S-SgcC2 (17), (S)-3-I-β-tyrosyl-S-SgcC2 (18), (S)-3-Me-β-tyrosyl-S-SgcC2 (19), and (S)-3-OH-β-tyrosyl-S-SgcC2 (20). Recombinant Svp,31 apo-SgcC2,15 and SgcC113,14 were prepared as described. Post-translational modification of apo-SgcC2 with the 4’-phosphopantetheine moiety of CoA was achieved in a 1.8 mL reaction mixture containing 100 mM Tris-HCl (pH 7.5), 200 μM apo-SgcC2, 1.0 mM CoA, 12.5 mM MgCl2, 2.0 mM TCEP, and 10 μM Svp.31 After incubation at room temperature for 45 min, a loading solution consisting of 3.5 mM β-tyrosine analogue (4, 9, 10, 11, 13, 14, or 15), 4 mM ATP, 2.0 mM TCEP, and 12.5 mM MgCl2 (all final concentrations) was added to an equal volume of the above solution. SgcC1 was added to a final concentration of 5 μM, and the resulting solution was incubated at room temperature for an additional 60 min.
Purification of SgcC2-Tethered Substrates for SgcC.
Compounds 6, 7, 16, 17, 18, 19, and 20 were purified using anion- exchange chromatography. A 5-mL HiTrap Q column (GE Healthcare) was pre-equilibrated with 20 mM sodium phosphate buffer (pH 7.0), and the SgcC2-tethered substrate preparations were loaded. The SgcC2-tethered products were eluted using a linear gradient from 0 to 100% 1.0 M NaCl in 20 mM sodium phosphate buffer (pH 7.0) for 25 column volumes at a flow rate of 3 mL min-1. The purified substrates, which were eluted between 0.35 and 0.4 M NaCl, were desalted by two cycles of concentration/dilution using an Amicon Ultra-4 concentrator device (5K, GE Healthcare) prior to use in SgcC assays.
Determination of the Stereochemistry of (S)-3-Cl-β-tyrosyl- S-SgcC2 Substrate for SgcC.
After the HiTrap Q anion-exchange column, the purified (S)-3-Cl-β-tyrosyl-S-SgcC2 (7) was subjected to alkaline hydrolysis in 0.1 N KOH solution incubated at 70 °C for 15 min. The resulting solution containing the free 3-Cl-β- tyrosine was injected into an Apollo C18 column (5 μm, 250 × 4.6 mm, Alltech Associates Inc.) using a 20 min linear gradient from 0 to 25% CH3CN in 0.1% TFA-H2O at a flow rate of 1 mL min−1 and UV-vis detection at 280 nm for purification. The peak of 3-Cl-β-tyrosine was collected and concentrated by speed-vac. The resultant solution was analyzed through a CrownPak CR (+) column (5 μm, 150 × 4.0 mm, Chiral Technologies Inc., West Chester, PA) to determine the stereochemistry. The column was run isocratically in aqueous perchloric acid buffer (pH 2.4) at a flow rate of 1 mL min−1 as recommended by the manufacturer, and an authentic standard of (S)-3-Cl-β-tyrosine, made by SgcC3- catalyzed chlorination of (S)-β-tyrosyl-S-SgcC2 as described previously,15 was used for comparison (Figure S2, Supporting Information).
Characterization of the SgcC Hydroxylation Activity in Vitro.
The typical SgcC assay solution contained 250 μM 7 or other indicated substrates, 5 mM NADH, 10 μM FAD, 1 mM TCEP, 5 μM SgcC, and 1.5 μM SgcE6 in 50 mM sodium phosphate buffer (pH 6.0) containing 50 mM NaCl. Reactions were initiated by addition of SgcC and SgcE6, incubated at 25 °C for 1 h, and terminated by the addition of 35 μL of 70% trichloroacetic acid (TCA) to a final concentration of 10%. After incubation on ice for 15 min, the precipitate was separated by centrifugation (14 000 rpm for 15 min at 4 °C). The resulting pellet was washed twice with 200 μL of 5% TCA and once with 200 μL of ice-cold ethanol. After drying by speed-vac for 10 min, the protein pellet was redissolved in 150 μL of 0.1 M KOH containing 50 mM DTT and incubated at 50 °C for 15 min to hydrolyze all thioester bonds. After neutralization of the alkaline hydrolysis solution, the precipitate was removed by centrifugation, and the clarified supernatant was concentrated by speed-vac and analyzed by HPLC with a Varian ProStar 210 HPLC system equipped with an Apollo C18 reverse-phase column (5 μm, 250 × 4.6 mm, Alltech Associates Inc.), using a 24 min linear gradient from 0 to 25% CH3CN in 0.1% TFA-H2O at a flow rate of 1 mL min−1 and UV-vis detection at 280 nm. The identity of the peaks was determined using synthetic standards, and the product peaks were collected and subjected to LC-ESI-MS analysis using an Agilent 1100 HPLC-MSD SL ion trap mass spectrometer. Control reactions were carried out under identical conditions, except with boiled SgcC.
To determine kinetic parameters for SgcC, 7 (60–700 μM) was incubated in a final volume of 200 μL with 3 μM SgcC, 2 SgcE6, 5 mM NADH, 10 μM FAD, 1 mM TCEP, and 50 mM phosphate buffer (pH 6.0) containing 50 mM NaCl at 25 °C. Reactions were initiated by the addition of SgcC and SgcE6, quenched by addition of 35 μL of 70% cold TCA after a 10 min incubation, and carried out in duplicate. The mixture was treated with the same workup procedure and HPLC analysis as described above to determine the product formation using a standard curve generated with synthetic 12.12,13 Plots of product formation were fitted to the Michaelis-Menten equation to extract the Km and kcat parameters.
To determine the specific activity of SgcC with respect to the other putative substrates, the SgcC-catalyzed hydroxylation reaction was carried out in a final volume of 200 μL with 5 mM NADH, 10 μM FAD, 1 mM TCEP, and 50 mM phosphate buffer (pH 6.0), containing 50 mM NaCl at 25 °C. When 7, 17, or 18 (250 μM) was used as a substrate, 1.5 pM SgcC and 2 μM SgcE6 were used. When 6, 16, 19, or 20 (250 μM) was used as a substrate, 6 μM SgcC and 2 μM SgcE6 were employed. The reactions were initiated by the addition of SgcC and SgcE6 and run in duplicate. At different time points (Table S1, Supporting Information), the reactions were quenched by addition of 35 μL of ice-cold 70% TCA. The samples were treated with the same workup procedure and HPLC analysis as described above. The use of differential wavelengths allows for facile detection and determination of the product formation (Table S1). Standard curves were generated with synthetic β-tyrosine analogues in order to correlate peak area with the amount of product formed in each reaction. The product formation was fitted to a linear equation to obtain the initial velocity, and the specific activity was calculated from the initial velocity divided by the concentration of SgcC as determined using the Bradford dye-binding procedure.
Determination of pH Dependence of the SgcC Activity.
Three buffers, 50 mM sodium acetate (pH 5.0 and 5.5), 50 mM sodium phosphate buffer (pH 6.0–8.0), and 50 mM Tris-HCl (pH 9.0), were chosen to determine the optimal pH for the SgcC hydroxylation activity in vitro. The assay solutions, consisting of 200 μM 7, 3 μM SgcC, 2 μM SgcE6, 5 mM NADH, 10 pM FAD, and 1 mM TCEP in 50 mM buffer, varying pH from 5.0 to 9.0, containing 50 mM NaCl, were incubated at 25 °C for 30 min. The product formation was monitored by HPLC after the reactions were terminated and worked up as described above.
Results
Production, Purification, and Properties of SgcC.
The sgcC gene was amplified from the cosmid pBS10057,30 and directly cloned into the pET-30Xa/LIC vector to generate expression plasmid pBS1092, in which SgcC would be overproduced as an N-terminal His6-tagged fusion protein. Standard conditions were used for expression,28 and SgcC was purified with a yield of about 9 mg/L following affinity chromatography. SDS-PAGE analysis revealed that SgcC was purified to near homogeneity with the expected molecular weight of 63.2 kD (Figure S1, Supporting Information). The solution containing the purified protein was colorless, and HPLC analysis showed that no flavin was copurified with SgcC, consistent with the lack of flavin- binding domain based on sequence analysis.
Substrate Preparation, Determination of Stereochemistry of Substrate, and Activity of SgcC.
Prior to testing the activity of SgcC, Fre15 and SgcE615 were overproduced in E. coli, the recombinant proteins were purified, and the reductase activity was confirmed spectroscopically by monitoring the consumption of NADH. The recombinant proteins SgcC1,13,14 a promiscuous 3-tyrosine-activating adenylation enzyme, and SgcC2,15 the PCP, and Svp,31 a promiscuous 4’-phosphopantetheinyl transferase involved in bleomycin biosynthesis, were prepared as previously described. The 3-tyrosine substrate 9 was synthesized starting from the appropriate benzaldehyde precursor as previously described. 15 To generate the PCP-tethered substrate, holo- SgcC2 was first enzymatically prepared using Svp, followed by loading of holo-SgcC2 with 9 catalyzed by SgcC1. The resulting product 7 was purified by anion exchange to eliminate excessive substrates or products for Svp and SgcC1, as well as other proteins, and was desalted prior to use in enzyme assays. Remarkably, only the (S)-3-Cl-β-tyrosine was specifically activated and loaded onto the holo-SgcC2 by SgcC1, although racemic 3-Cl-β-tyrosine was employed as a substrate in the SgcC1-catalyzed loading reaction, as determined by chiral HPLC analysis in comparison with authentic (S)-3-Cl-β-tyrosine made from the SgcC3-catalyzed chlorination of (S)-β-tyrosyl-S-SgcC2 (Figure S2, Supporting Information). While unexpected, this observation is, in fact, consistent with the previous finding that SgcCl shows a 25-fold preference for 4 over its (R)-enantiomer, serving as a “gate-keeper” that permits only the (S)-β-amino acid to be incorporated into 1 in vivo. It is therefore concluded that, under the same conditions, only the (S)-enantiomers were similarly activated and tethered to SgcC2 for all other 3-substituted β-tyrosine analogues used in this study.
Initial activity assays were carried out with 250 μM 7, 5 mM NADH, 10 μM FAD, 1 mM TCEP, 5 μM SgcC, and 1.5 μM SgcE6 in 50 mM sodium phosphate buffer (pH 6.0), containing 50 mM NaCl under aerobic conditions at 25 °C. The reaction was monitored by subjecting the aminoacyl-S-SgcC2 substrate and product to alkaline hydrolysis followed by HPLC analysis. A new peak appeared eluting before 9, and this new peak had a retention time identical to that of authentic 12 (Figure 3A). The identity of the new peak was confirmed by LC-ESI-MS analysis, yielding a pair of [M + H]+ ions at m/z = 232.1 and 234.1 with a ~3:1 ratio, consistent with a monochlorinated species when compared to the authentic standard 12.
Figure 3.
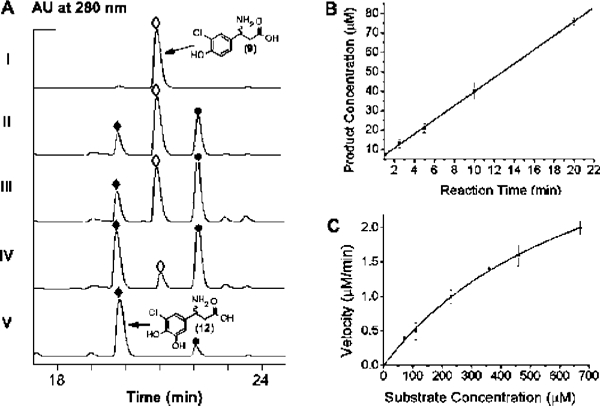
In vitro characterization of SgcC as a two-component, FAD- dependent monooxygenase. (A) HPLC profiles of 3-chloro-β-tyrosine (9, ◊) standard (I), hydrolyzed compounds following incubations of (S)-3- chloro-β-tyrosinyl-S-SgcC2 (7) with SgcC for 10 min (II), 20 min (III), and 60 min (IV), and synthetic 3-chloro-5-hydroxyl-β-tyrosine (12, ♦) standard (V). The other peak in the chromatograms is 4,5-dihydroxy-1,2- dithiane (●) presented in the assay. (B) Time course of SgcC-catalyzed hydroxylation of (S)-3-chloro-β-tyrosinyl-S-SgcC2 (7) as followed by HPLC analysis for the first 20 min. (C) Single-substrate kinetic analysis for SgcC with varying concentration of (S)-3-chloro-β-tyrosinyl-S-SgcC2 (7).
Optimization of SgcC Activity and Kinetic Analysis.
Prior to kinetic analysis, the pH dependence of SgcC-catalyzed hydroxylation was examined. The pH profile exhibited a bell-shaped curve between pH 5.5 and 9.0 with an optimal activity at pH 6.0 (Figure S3A, Supporting Information). As a result, all subsequent assays were performed in 50 mM phosphate buffer at pH 6.0. Since the activity of two-component, flavin- dependent monooxygenases has been shown to be dependent on flavin concentration,17,26,32 the effect of FAD concentration on SgcC activity was next examined (Figure S3B, Supporting Information). SgcC showed a nearly 2-fold increase in activity when the FAD concentration was decreased from 50 μM to 10 μM. Maximum product formation occurred when the FAD concentration was between 1 and 10 μM. Subsequently, all assays were performed in the presence of 10 μM FAD.
Using the optimized conditions and the HPLC-based assay, a time course analysis of the SgcC-catalyzed hydroxylation showed an increase in 12 with the concomitant loss of 9, and under these conditions product formation was linear with respect to time until approximately 20 min (Figure 3B). Finally, preliminary kinetic analysis revealed that SgcC displayed Michaelis-Menten kinetics with respect to 7, yielding a Km of 742 ± 69 μM and kcat of 1.4 ± 0.2 min−1 (Figure 3C).
Cofactor and Cosubstrate Requirements for SgcC Activity.
The requirement of cofactors and cosubstrates for SgcC activity was systematically examined using the HPLC-based assay (summarized in Table 1). No product was observed when SgcE6 was omitted from the assay mixture, consistent with the requirement of FADH2 for hydroxylation (entry 2). The formation of 3 was completely abolished when cofactor FAD or cosubstrates NADH and O2 were excluded from the reaction (entries 3–5). Comparable amounts of 3 were produced by replacing SgcE6 with E. coli flavin reductase Fre (entry 6). Finally, 3 was not detected when FMN was substituted for FAD (entry 7). Thus, these results are consistent with the functional assignment of SgcC as a two-component, FAD-dependent monooxygenase following the known catalytic cycle for these enzymes as shown in Figure 4.
Table 1.
Cofactor and Cosubstrate Requirements for SgcC-Catalyzed Hydroxylation of (S)-3-Chloro-β-tyrosinyl-S-SgcC2 in Vitro
| entry | cofactors or cosubstrates | flavin reductase |
conversion (%)a |
relative activity |
|---|---|---|---|---|
| 1 | NADH, FAD, and O2 | SgcE6 | 16 ± 4.5 | 100 |
| 2 | NADH, FAD, and O2 | no | 0 | 0 |
| 3 | NADH and FAD | SgcE6 | 0 | 0 |
| 4 | FAD and O2 | SgcE6 | 0 | 0 |
| 5 | NADH and O2 | SgcE6 | 0 | 0 |
| 6 | NADH, FAD, and O2 | Fre | 19 ± 2.0 | 119 |
| 7 | NADH, FMN, and O2 | Fre | 0 | 0 |
Reactions were performed under initial velocity conditions, and product formation was quantitated using HPLC peak area with comparisons to an authentic standard.
Figure 4.
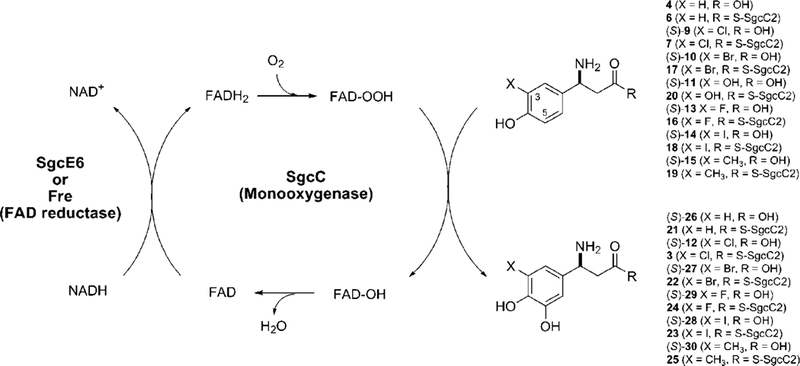
SgcC-catalyzed C-5 hydroxylation of (S)-3-chloro-β-tyrosyl-S-SgcC2 (7) and its (S)-3-substituted β-tyrosyl-S-SgcC2 analogues (6, 7, 16-20) depicting a catalytic cycle involving the flavin reductase SgcE6 or Fre.
Substrate Specificity of SgcC.
The timing of the hydroxylation step in 1 biosynthesis was examined by exploring the substrate specificity of SgcC. Reactions with free amino acids as substrates, including 4, (R)-β-tyrosine, 9, and 11, revealed no enzyme activity regardless of the reaction conditions, consistent with hydroxylation occurring after generation of the β-aminoa- cyl-S-SgcC2 species (Figure 2). Thus, 6 was enzymatically prepared using SgcC1 and tested under the optimized hydroxylation conditions. SgcC showed activity with 6, affording the corresponding hydroxylated product 21, but the specific activity was decreased 120-fold with respect to that of 7 (Figure 4 and Table 2). In combination with the data obtained with SgcC4,10–12 SgcC1,13,14 and SgcC3,15 the results here are consistent with SgcC catalyzing the hydroxylation of 7 to afford 3, the last intermediate prior to attachment to the enediyne core (Figure 2).
Table 2.
Substrate Specificity of SgcC-Catalyzed Hydroxylation of (S)-3-Chloro-β-tyrosinyl-S-SgcC2 in Comparison with (S)-3-Substituted β-Tyrosyl-S-SgcC2 Analogues
| substrate | product formed, specific activity (min−1)a |
relative activity |
|---|---|---|
| (s)-3-Cl-β-tyrosyl-S-SgcC2 (7)c | 3, 2.4 ± 0.2 | 100 |
| (s)-3-F-β-tyrosyl-S-SgcC2 (16) | 24, 0.3 ± 0.1 | 12 |
| (s)-3-Br-β-tyrosyl-S-SgcC2 (17) | 22, 3.1 ± 0.2 | 129 |
| (s)-3-I-β-tyrosyl-S-SgcC2 (18) | 23, 5.5 ± 1.0 | 229 |
| (s)-3-Me-β-tyrosyl-S-SgcC2 (19) | 25, 0.5 ± 0.1 | 21 |
| (s)-β-tyrosyl-S-SgcC2 (6) | 21, 0.02b | 0.8 |
| (s)-3-OH-β-tyrosyl-S-SgcC2 (20) | 0 | 0 |
Reactions were performed under initial velocity conditions, and product formations were quantitated using HPLC peak area with comparisons to authentic standards. See Figure 4 for product structures.
The conversion is so low that specific activity toward (S-β-) tyrosyl-S-SgcC2 (6) is estimated from product formation over incubation at 25 °C for 20, 40, and 60 min.
Finally, the activity of SgcC was tested using C-3-substituted analogues including 16–20, and the results are summarized in Figure 4 and Table 2. SgcC was found to readily hydroxylate 17 and 18, yielding the corresponding products 22 and 23, with specific activities slightly greater than that for the native substrate 7. While both 16 and 19 were converted to the corresponding hydroxylated products 24 and 25, the specific activities of SgcC for these substrates were significantly decreased, by approximately 5-fold, in comparison with that for 7. No product formation was detected when 20 was used as a substrate in the SgcC assay. The identities of these products were all confirmed by LC-ESI-MS analysis of their free acids, (S)-12, (S)-26, (S)-27, (S)-28, (S)-29, and (S)-30, released from SgcC2 upon hydrolytic cleavage (Figure 4 and Table S2, Supporting Information).
Discussion
The enediyne antitumor antibiotics are structurally complex metabolites that are among the most cytotoxic natural products ever described. They all share a similar enediyne core that is directly implicated in bioactivity, and each core is decorated with a variety of chemical moieties that alter the properties, including cytotoxicity, of a given enediyne.33 C-1027, a model nine-membered enediyne, is composed of a deoxy aminosugar, a benzoxazolinate moiety, and the moiety of interest here, a (S)-3-chloro-5-hydroxy-β-tyrosine (Figure 1). This moiety contributes critical interactions with the apoprotein CagA34,35 and modulates the reactivity of the enediyne core via π—π stacking interactions;34 as a consequence, structural permutations of the moiety have been shown to have a clear impact on the bioactivity.8,9
The biosynthetic gene cluster for 1 was previously cloned and sequenced,7 providing the first insights into how such complex metabolites as the enediynes are assembled. We have since shown that the (S)-3-chloro-4,5-dihydroxy-β-phenylalanine moiety is derived from L-α-tyrosine, and we have characterized the first three enzymatic conversions that, together, established (S)-3-chloro-β-tyrosyl-S-SgcC2 as the pathway intermediate most likely to serve as a substrate for hydroxylation (Figure 2). Based on bioinformatics analysis, the best candidate for this transformation was SgcC, and this assignment was validated upon inactivation of the sgcC gene that led to the accumulation of the expected compound 22-deshydroxy-C-1027 from the fermentation broth of the AsgcC mutant strain.7 However, the substrate specificity and enzymatic properties of SgcC remained unclear. To unravel the molecular details of this hydroxylation event, we cloned sgcC and overproduced and purified the recombinant protein for in vitro characterization.
An approach similar to that utilized during the functional characterization of SgcC3 was applied here to generate the carrier protein-tethered substrate for SgcC.15 In short, 3-chloro- β-tyrosine was synthetically prepared, but only (S)-3-chloro-β- tyrosine was directly loaded onto holo-SgcC2 to afford the desired substrate (S)-3-chloro-β-tyrosyl-S-SgcC2, by taking advantage of the high stereospecificity but relaxed substrate specificity of SgcCl,13,14 thus bypassing the halogenase- catalyzed reaction (Figure S2, Supporting Information). 3-Chloro- 5-hydroxy-β-tyrosine, the expected product of hydroxylation of (S)-3-chloro-β-tyrosyl-S-SgcC2 by SgcC following hydrolysis from SgcC2, was also synthesized to serve as an authentic standard. After the optimal conditions were determined for the hydrolytic cleavage of the SgcC2-tethered substrate 7 and product 3 to release the free acids (S)-3-chloro-β-tyrosine and (S)-3-chloro-5-hydroxy-β-tyrosine, chemo-enzymatically prepared substrate (S)-3-chloro-β-tyrosyl-S-SgcC2 was incubated with SgcC using typical reaction conditions for two-component monooxygenases. HPLC analysis of the hydrolyzed product as (S)-3-chloro-5-hydroxy-β-tyrosine unambiguously established that SgcC is a monooxygenase catalyzing the formation of (S)- 3-chloro-4,5-dihydroxy-β-phenylalanyl-S-SgcC2 (3, Figure 3).
Detailed studies with E. coli and P. aeroginosa two- component p-hydroxyphenylacetate monooxygenases have established the catalytic requirements for this relatively new enzyme family.23–27 In short, hydroxylation is dependent on two proteins, a reductase and a monooxygenase, with the former responsible for production of reduced FAD—at the expense of NAD(P)H—that diffuses to the latter protein responsible for O2 activation and aromatic hydroxylation.17,16 The data presented here for SgcC are entirely consistent with the catalytic cycle shown in Figure 4, since excluding SgcE6, NADH, FAD, or O2 resulted in no product formation. Furthermore, SgcE6 is readily substituted by E. coli Fre, thus supporting the prior findings that the reductase and monooxygenase components do not form a complex and that oxygenase activity is independent of the source of FADH2.26 Finally, the SgcE6-SgcC hydroxylation activity decreased in the presence of excess FAD, which has been shown previously for other two-component monooxygenases to be a result of substantial autoxidation of FADH2 to generate H2O2.26,32 Consequently, the SgcE6 (or Fre):SgcC:FAD ratio was first optimized prior to subsequent studies to afford the most efficient coupling possible for the SgcE6-SgcC two- component system.
After assigning the function of SgcC, the timing of the hydroxylation step in 1 biosynthesis and the substrate specificity of SgcC were examined. The former was interrogated by comparing the specific activity of SgcC with that of hypothetical pathway intermediates. While the amount of required substrate and unavoidable limitations of the assay precluded a more rigorous quantification of substrate specificity, the results clearly demonstrate (S)-3-chloro-β-tyrosyl-S-SgcC2 is preferred over (S)-β-tyrosyl-S-SgcC2, supporting the indirect evidence obtained from prior reports that SgcC is the fourth enzyme in the pathway.7 Perhaps most significant, however, is the finding that SgcC cannot convert any free acid to the corresponding hydroxylated product. Thus, like the halogenase SgcC3,15 SgcC hydroxylation requires a carrier protein-tethered substrate, reflecting the essential interactions provided by SgcC2 during substrate recognition and catalysis. These results are intriguing, considering that SgcC and SgcC3 have low sequence homology (12% identity and 22% similarity) yet display remarkable parallels with respect to substrate requirements and mechanism, wherein the only significant difference between the catalytic cycles is the nature of the electrophile species (FAD-OCl for SgcC3 versus FAD-OOH for SgcC) undergoing electrophilic aromatic substitution. Having both enzymes available now sets the stage to explore the structural determinants that lead to similarities and contrasts in substrate recognition and utilization for SgcC3 and SgcC.
The phenomenon of enzyme-catalyzed hydroxylation utilizing carrier protein-tethered substrates has been predicted to be a common occurrence in the assembly of antibiotics containing amino acid derivatives.37 Indeed, rather recently, two heme- dependent monooxygenases involved in novobiocin and nikko- mycin biosynthesis have been shown to hydroxylate the amino acids tyrosine and histidine, respectively, only after tethering to a carrier protein.38,39 To our knowledge, there has been only one carrier protein-dependent, two-component monooxygenase identified: BtrO.27 BtrO is involved in the biosynthesis of the unusual (2S)-4-amino-2-hydroxybutyryl side chain of the aminoglycoside butirosin, and it has been demonstrated that BtrO hydroxylates an aliphatic substrate bound to a carrier protein utilizing FMN as a cosubstrate. The absolute requirement for a carrier protein for all these hydroxylases suggests a common strategy for substrate recognition, and the molecular details and protein dynamics underlying the hydroxylation strategy for SgcC with respect to BtrO and other characterized two-component monooxgyenase systems are subjects of ongoing research.
Finally, in keeping with our goal of using combinatorial biosynthesis to generate new 1 analogues,8,9 a panel of (S)-3- chloro-β-tyrosyl-S-SgcC2 analogues were prepared to interrogate potential substrate flexibility for SgcC. Surprisingly, the 3-iodo- and 3-bromo-β-tyrosine analogues (18 and 17) displayed slightly higher activity with SgcC, and furthermore SgcC turned over both the 3-fluoro- and 3-methyl-β-tyrosine substrate analogues (16 and 19). Therefore, both the adenylation enzyme SgcC1 and now the hydroxylase SgcC seem to have substrate specificity that is amenable to genetic engineering and combinatorial biosynthetic approaches to generate new enediyne compounds. Furthermore, the realization that 20-deschloro- C1027 (21),14 20-deschloro-22-deshydroxy-C-1027 (22),14 and 22-deshydroxy-C-10277 are isolated upon inactivation of sgcC3 and sgcC, respectively, suggests the condensation enzyme SgcC5 also has relaxed substrate specificity, thus clearly setting the stage to manipulate C-1027 biosynthesis to rationally produce novel enediynes using precursor-directed biosynthesis. Further mechanistic studies on SgcC3 in combination with in vitro characterization of SgcC5 will clearly provide additional, valuable insights into such a strategy.
In conclusion, SgcC has been functionally characterized as a two-component, FAD-dependent monooxygenase that regiospe- cifically hydroxylates (S)-3-chloro-β-tyrosyl-S-SgcC2. Similarly to the halogenase SgcC3, SgcC absolutely requires O2, a carrier- protein-tethered substrate, and a separate flavin reductase to provide diffusible FADH2 for catalysis. Along with the results obtained for SgcC4, SgcC1, and SgcC3, the data support SgcC as the fourth enzyme in the pathway leading to biosynthesis of the (S)-3-chloro-4,5-dihydroxy-β-phenylalanine moiety of C-1027 (Figure 2). Finally, SgcC is capable of hydroxylating a variety of β-tyrosine analogues, including all of the 3-halogenated compounds tested (Figure 4). In lieu of the relaxed substrate specificity of other enzymes in the pathway, application of precursor-directed biosynthesis and combinatorial biosynthesis methods to the C-1027 biosynthetic machinery now affords a unique opportunity to generate unnatural C-1027 analogues, some of which may have improved biological activity.8,9
Supplementary Material
Acknowledgment.
We thank Dr. Y. Li, Institute of Medicinal Biotechnology, Chinese Academy of Medical Sciences, Beijing, China, for the wild-type S. globisporus strain and the Analytical Instrumentation Center of the School of Pharmacy, University of Wisconsin—Madison for support in obtaining MS and NMR data. This work is supported in part by NIH grants CA78747 and CA113297. S.V.L. is the recipient of an NIH postdoctoral fellowship (CA1059845).
Footnotes
Supporting Information Available: Full experimental details for the synthesis of 3-fluoro-β-tyrosine, 3-iodo-β-tyrosine, and 3-methyl-β-tyrosine, SDS-PAGE of purified recombinant SgcC (Figure S1), preparation and HPLC chromatography for determining the stereochemistry of the SgcC substrate (S)-3-Cl-β- tyrosyl-S-SgcC2 (Figure S2), pH (Figure S3A) and FAD concentration (Figure S3B) dependence of SgcC, time points and detection wavelengths used to determine the SgcC substrate specificity Table S1), and the ESI-MS data for the products of SgcC-catalyzed hydroxylation of (S)-3-Cl-β-tyrosyl-S-SgcC2 and its analogues (Table S2). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- (1).(1) Hu J; Xue YC; Xie MY; Zhang R; Otani T; Minami Y; Yamada Y; Marunaka T J. Antibiot. 1988, 41, 1575–1579. [DOI] [PubMed] [Google Scholar]
- (2).Otani T; Minami Y; Marunaka T; Zhang R; Xie MY J. Antibiot. 1988, 41, 1580–1585. [DOI] [PubMed] [Google Scholar]
- (3).Otani T; Yasuhara T; Minami Y; Shimazu T; Zhang R; Xie MY Agric. Biol. Chem. 1991, 55, 407–417. [PubMed] [Google Scholar]
- (4).Sugimoto Y; Otani T; Oie S; Wierzba K; Yamada YJ Antibiot. 1990, 43, 417–421. [DOI] [PubMed] [Google Scholar]
- (5).Sugiura Y; Matsumoto T Biochemistry 1993, 32, 5548–5553. [DOI] [PubMed] [Google Scholar]
- (6).Xu Y; Zhen X; Zhen Y; Goldberg IH Biochemistry 1995, 34, 12451–12460. [DOI] [PubMed] [Google Scholar]
- (7).Liu W; Christenson SD; Standage S; Shen B Science 2002, 297, 1170–1173. [DOI] [PubMed] [Google Scholar]
- (8).Kennedy DR; Gawron LS; Ju J; Liu W; Shen B; Beerman TA Cancer Res. 2007, 67, 773–781. [DOI] [PubMed] [Google Scholar]
- (9).Kennedy DR; Ju J; Shen B; Beerman TA Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 17632–17637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Christenson SD; Liu W; Toney MD; Shen BJ Am. Chem. Soc. 2003, 125, 6062–6063. [DOI] [PubMed] [Google Scholar]
- (11).Christenson SD; Wu W; Spies MA; Shen B; Toney MD Biochemistry 2003, 42, 12708–12718. [DOI] [PubMed] [Google Scholar]
- (12).Christianson CV; Montaavon TJ; Van Lanen SG Shen B.; Bruner SD Biochemistry 2007, 42, 7205–7214. [DOI] [PubMed] [Google Scholar]
- (13).Van Lanen SG; Dorrestein PC; Christenson SD; Liu W; Ju J; Kelleher NL; Shen BJ Am. Chem. Soc. 2005, 127, 11594–11595. [DOI] [PubMed] [Google Scholar]
- (14).Van Lanen SG; Lin S; Dorrestein PC; Kelleher NL; Shen BJ Biol. Chem. 2006, 281, 29633–29640. [DOI] [PubMed] [Google Scholar]
- (15).Lin S; Van Lanen SG; Shen BJ Am. Chem. Soc. 2007, 129, 12432–12438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Ullrich R; Hofrichter M Cell. Mol. Life Sci. 2007, 64, 271–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Ballou DP; Entsch B; Cole LJ Biochem. Biophys. Res. Comm. 2005, 338, 590–598. [DOI] [PubMed] [Google Scholar]
- (18).van Berkel WJH; Kamerbeek NM; Fraaije MW J. Biotechnol. 2006, 124, 670–689. [DOI] [PubMed] [Google Scholar]
- (19).Ziegler DM Drug Metab. Rev. 2002, 34, 503–511. [DOI] [PubMed] [Google Scholar]
- (20).Gatti DL; Palfey BA; Lah MS; Entsch B; Massey V; Ballou DP; Ludwig ML Science 1994, 266, 110–114. [DOI] [PubMed] [Google Scholar]
- (21).Schreuder HA; Mattevi A; Obmolova G; Kalk KH; Hol WG; van der Bolt FJ; van Berkel WJ Biochemistry 1994, 33, 10161–10170. [DOI] [PubMed] [Google Scholar]
- (22).Wang J; Ortiz-Maldonado M; Entsch B; Massey V; Ballou D; Gatti DL Proc. Natl. Acad. Sci. U.S.A. 2002, 99, 608–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Cuskey SM; Olsen RH J. Bacteriol. 1988, 176, 393–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Prieto MA; Garcia JL J. Biol. Chem. 1994, 269, 22823–22829. [PubMed] [Google Scholar]
- (25).Galan B; Diaz E; Prieto MA; Garcia JL J. Bacteriol. 2000, 182, 627–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Xun LY; Snadvik ER Appl. Environ. Microbiol. 2000, 66, 481–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Li Y; Llewellyn NM; Giri R; Huang F; Spencer JB Chem. Biol 2005, 12, 665–675. [DOI] [PubMed] [Google Scholar]
- (28).Sambrook J; Fritsch EF; Maniatis T Molecular Cloning: A Laboratory Manual, 3rd ed; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, 2000. [Google Scholar]
- (29).Tan CYK; Weaver DF Tetrahedron 2002, 58, 7449–7461. [Google Scholar]
- (30).Liu W; Shen B Antimicrob. Agents Chemother. 2000, 44, 382–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Sanchez C; Du L-C; Edwards DJ; Toney MD; Shen B Chem.Biol. 2001, 8, 725–738. [DOI] [PubMed] [Google Scholar]
- (32).Entsch B; Chakraborty S; Ortiz M; Ballou D In Flavins and Flavoproteins’, Nishino T, Miura R, Tanokura M., Eds.; Small World Internet Publishing: Japan, 2005. [Google Scholar]
- (33).Shen B; Liu W; Nonaka K Curr. Med. Chem. 2003, 10, 2317–2325. [DOI] [PubMed] [Google Scholar]
- (34).Tanaka T; Fukuda-Ishisaka S; Hirama M; Otani TJ Mol. Biol. 2001, 309, 267–283. [DOI] [PubMed] [Google Scholar]
- (35).Okuno Y; Iwashita T; Sugiura YJ Am. Chem. Soc. 2000, 122, 6848–6854. [Google Scholar]
- (36).Louie TM; Xie XS; Xun L Biochemistry 2003, 42, 7509–7517. [DOI] [PubMed] [Google Scholar]
- (37).Chen H; Thomas MG; O’Connor SE; Hubbard BK; Burkart MD; Walsh CT Biochemistry 2001, 40, 11651–11659. [DOI] [PubMed] [Google Scholar]
- (38).Chen H; Walsh CT Chem. Biol. 2001, 8, 301–312. [DOI] [PubMed] [Google Scholar]
- (39).Chen H; Hubbard BK; O’Connor SE; Walsh CT Chem. Biol. 2002, 9, 103–112. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.