

Abstract
Key points
A single bout of exercise is capable of increasing insulin sensitivity in human skeletal muscle. Whether this ability is affected by training status is not clear.
Studies in mice suggest that the AMPK‐TBC1D4 signalling axis is important for the increased insulin‐stimulated glucose uptake after a single bout of exercise.
The present study is the first longitudinal intervention study to show that, although exercise training increases insulin‐stimulated glucose uptake in skeletal muscle at rest, it diminishes the ability of a single bout of exercise to enhance muscle insulin‐stimulated glucose uptake.
The present study provides novel data indicating that AMPK in human skeletal muscle is important for the insulin‐sensitizing effect of a single bout of exercise.
Abstract
Not only chronic exercise training, but also a single bout of exercise, increases insulin‐stimulated glucose uptake in skeletal muscle. However, it is not well described how adaptations to exercise training affect the ability of a single bout of exercise to increase insulin sensitivity. Rodent studies suggest that the insulin‐sensitizing effect of a single bout of exercise is AMPK‐dependent (presumably via the α2β2γ3 AMPK complex). Whether this is also the case in humans is unknown. Previous studies have shown that exercise training decreases the expression of the α2β2γ3 AMPK complex and diminishes the activation of this complex during exercise. Thus, we hypothesized that exercise training diminishes the ability of a single bout of exercise to enhance muscle insulin sensitivity. We investigated nine healthy male subjects who performed one‐legged knee‐extensor exercise at the same relative intensity before and after 12 weeks of exercise training. Training increased and expression of mitochondrial proteins in muscle, whereas the expression of AMPKγ3 was decreased. Training also increased whole body and muscle insulin sensitivity. Interestingly, insulin‐stimulated glucose uptake in the acutely exercised leg was not enhanced further by training. Thus, the increase in insulin‐stimulated glucose uptake following a single bout of one‐legged exercise was lower in the trained vs. untrained state. This was associated with reduced signalling via confirmed α2β2γ3 AMPK downstream targets (ACC and TBC1D4). These results suggest that the insulin‐sensitizing effect of a single bout of exercise is also AMPK‐dependent in human skeletal muscle.
Keywords: AMP‐activated protein kinase, TBC1D4, glucose uptake
Key points
A single bout of exercise is capable of increasing insulin sensitivity in human skeletal muscle. Whether this ability is affected by training status is not clear.
Studies in mice suggest that the AMPK‐TBC1D4 signalling axis is important for the increased insulin‐stimulated glucose uptake after a single bout of exercise.
The present study is the first longitudinal intervention study to show that, although exercise training increases insulin‐stimulated glucose uptake in skeletal muscle at rest, it diminishes the ability of a single bout of exercise to enhance muscle insulin‐stimulated glucose uptake.
The present study provides novel data indicating that AMPK in human skeletal muscle is important for the insulin‐sensitizing effect of a single bout of exercise.
Introduction
Muscle insulin sensitivity increases as an adaptive response to chronic exercise training (Dela et al. 1992; Holten et al. 2004; Frøsig et al. 2007a) involving changes in muscle size, morphology, capillarization and protein composition. In addition, in response to a single exercise bout, the prior exercised muscle responds with an acute increase in insulin sensitivity to stimulate glucose uptake lasting for up to 48 h (Mikines et al. 1988). Together, these adaptations secure enhanced insulin sensitivity during a period of exercise training and may partly explain the health‐promoting effects of physical activity. Improved insulin sensitivity following a single bout of exercise is described in skeletal muscle of various species (Richter et al. 1982, 1989; Bonen et al. 1984; Garetto et al. 1984; McConell et al. 2015). Observations from isolated rodent muscle preparations (Richter et al. 1982; Garetto et al. 1984) and one‐legged exercise models in humans (Richter et al. 1989; Wojtaszewski et al. 1997; Frøsig et al. 2007b) indicate a central role for local contraction‐induced mechanisms within the skeletal muscle in the improved insulin sensitivity after a single bout of exercise (Richter et al. 1984; Cartee, 2015). In rodents, the improved insulin sensitivity associates with an increased abundance of glucose transporter 4 (GLUT4) at the muscle cell surface membrane (Hansen et al. 1998). Accordingly, the point of convergence in cellular signalling events leading to GLUT4 translocation elicited by exercise and insulin has been a matter of active research for years. Because prior exercise does not alter the proximal insulin signalling (Bonen et al. 1984; Wojtaszewski et al. 1997, 2000; Hamada et al. 2006; Frøsig et al. 2007b), current hypotheses suggest that more distal signalling molecules might be involved. In this context, TBC1D4, which is involved in insulin‐stimulated glucose transport (Sano et al. 2003; Kramer et al. 2006), has been proposed as a signalling point of convergence between exercise and insulin (Cartee & Wojtaszewski, 2007; Cartee, 2015). In support, both human and rodent studies find increased phosphor‐regulation of TBC1D4 by insulin in the recovery period from acute exercise concomitantly with increased insulin sensitivity (Funai et al. 2009; Treebak et al. 2009; Castorena et al. 2014).
We recently demonstrated, in the skeletal muscle of mice, that both AICAR, a potent AMPK activator, and contraction/exercise increased insulin‐stimulated glucose uptake in an AMPK‐dependent manner (Kjøbsted et al. 2015, 2017). This was associated with site‐specific phosphorylation of TBC1D4, which specifically depended on the AMPK heterotrimeric complex, α2β2γ3 (Kjøbsted et al. 2017). Whether this also applies to humans is unclear. In human skeletal muscle, three heterotrimeric complexes are detectable (α1β2γ1, α2β2γ1 and α2β2γ3). Of these, the α2β2γ3 complex is activated potently and rapidly in response to acute exercise (Birk & Wojtaszewski, 2006). Intriguingly, the expression of the γ3 subunit is highly responsive to muscle use (expression decreases) (Frøsig et al. 2004; Wojtaszewski et al. 2005; Mortensen et al. 2013) and disuse (expression increases) (Kostovski et al. 2013). Accordingly, the AMPK α2β2γ3 complex is much less activated during exercise in the trained compared to the untrained muscle, even when exercise is performed at the same relative intensity (Mortensen et al. 2013). If the α2β2γ3 complex is central for the insulin‐sensitizing effect of acute exercise also in human skeletal muscle, a consequence of the above observations would be that the ability of the trained muscle to improve insulin sensitivity in response to acute exercise would be diminished.
Only two human studies bring some but inconclusive insights to this scenario. Mikines et al. (1989) did not find an increased insulin sensitivity 1 h after acute cycling exercise (75% ) in a group of well‐trained subjects. However, the insulin sensitivity measured after acute exercise was compared with a ‘control/resting’ condition, which was only 15 h after an exercise training session. It is thus conceivable that insulin sensitivity under both conditions were improved by the prior exercise bout (Mikines et al. 1988). Furthermore, in the ‘control’ condition, muscle glycogen stores were not fully replenished (Mikines et al. 1989). As glycogen levels have been reported to influence the ability of insulin to increase glucose uptake (Jensen et al. 1997; Derave et al. 2000; Richter et al. 2001; Wojtaszewski et al. 2003a), this could also contribute to the absence of improved insulin sensitivity after acute exercise reported in their study (Mikines et al. 1989). In another study, whole‐body insulin sensitivity index was increased after acute exercise in a group of sedentary obese subjects but not in a group of regularly physically active obese subjects (Nelson & Horowitz, 2014). The physical fitness was, however, low in both groups and did not differ significantly between the groups. Together, these two studies suggest that training status might affect the ability of acute exercise to enhance insulin sensitivity. Yet the results are inconclusive and these observations at the whole‐body level cannot be related specifically to skeletal muscle.
Thus, in the present study, we tested the hypothesis that exercise training diminishes the ability of acute exercise to enhance insulin‐stimulated glucose uptake. We predicted that this was associated with decreased AMPKα2β2γ3 expression/activation and thus lesser exercise‐ and insulin‐induced TBC1D4 signalling after training. Such observations will strengthen the case in favour for AMPKα2β2γ3 being a key regulator of insulin sensitivity in human skeletal muscle. We explored this hypothesis by comparing muscle insulin‐stimulated glucose uptake 4 h after one‐legged knee‐extensor exercise before and after 12 weeks of whole‐body cycling exercise training.
Methods
Ethical approval
Nine young (aged 25 ± 1 years), lean (body mass index 23.5 ± 0.5 kg m–2) and healthy men gave their written, informed consent to participate in the study approved by the Regional Ethics Committee for Copenhagen (H‐6‐2014‐038) and complied with the ethical guidelines of the Declaration of Helsinki II, except for registration in a database.
Experimental protocol
The experimental protocol consisted of two experimental days separated by 12 weeks of endurance cycling training (Fig. 1). Minimum one week prior to the PRE training experimental day, peak oxygen uptake () was determined by an incremental test to exhaustion on a Monark ergometer cycle (Ergomedic 839E; Monark Exercise AB, Vansbro, Sweden) using breath by breath measurements of (Masterscreen CPX; IntraMedic, Gentofte, Denmark). Body composition was measured by dual x‐ray absorptiometry (DPX‐IQ Lunar; Lunar Corporation, Madison, WI, USA). After familiarization to the one‐legged knee‐extensor ergometer (Andersen et al. 1985), peak workload (PWL) of the knee‐extensors was determined in both legs by an incremental test. Subjects were instructed to record food intake for 3 days and to abstain from alcohol, caffeine and strenuous physical activity for 48 h prior to the PRE training experimental day.
Figure 1. Experimental study design.
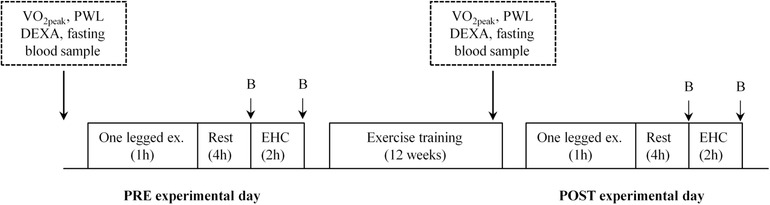
Subjects underwent two experimental days separated by 12 weeks of endurance cycling training (PRE and POST training experimental day). On each experimental day, subjects performed an acute bout of one‐legged exercise (80% peak workload (PWL) interspersed with 3 × 5 min intervals at 100% PWL). After 4 h of rest, a 2 h euglycemic hyperinsulinaemic clamp (EHC) was initiated. Biopsies (B) in the prior exercised and rested legs were taken immediately before and after EHC. Prior to the PRE experimental day and in the 12th training week, body composition, and PWL were determined and fasting plasma glucose and plasma insulin levels were measured.
On the morning of the experimental day, subjects arrived at the laboratory 1 h after having ingested a small breakfast (oatmeal, skimmed milk, sugar; 5% of daily energy intake) (Henry CJ, 2005). Upon arrival, they performed 1 h of dynamic knee‐extensor exercise, with a randomized leg, at 80% of PWL interspersed with 3 × 5 min at 100% of PWL. After exercise, subjects rested in the supine position and catheters (Pediatric Jugular Catherization set; Arrow International, Reading, PA, USA) were inserted into the femoral vein of both legs and in a dorsal hand vein (Venflon Pro Safety; Mediq, Brøndby, Denmark) for sampling of arterialized venous blood (heated hand vein). After 4 h of rest, a euglycaemic hyperinsulinaemic clamp (EHC) was initiated with a bolus of insulin (9 mU kg−1; Atrapid; Novo Nordisk, Bagsværd, Denmark) followed by 120 min of constant insulin infusion (1.4 mU min−1 kg−1). Blood samples were drawn simultaneously from all three catheters before (−60, −30 and 0 min) and during the EHC (15, 30, 45, 60, 80, 100 and 120 min). Prior to each blood sampling, femoral arterial blood flow was measured using the ultrasound Doppler technique (Philips iU22; ViCare Medical A/S, Birkerød, Denmark). This allowed for calculation of the glucose uptake using Fick's principle across the previously exercised and rested leg, respectively. Muscle biopsies of musculus vastus lateralis were obtained in both legs immediately before and after the clamp under local anaesthesia (∼3 mL of xylocaine 2%; AstraZeneca, Copenhagen, Denmark) using the Bergström needle technique with suction (Bergström, 1962).
The experimental day was repeated in the same way after completing 12 weeks of training (POST training experimental day). For the one‐legged knee‐extensor exercise to be performed at the same relative intensity, the absolute workload was increased compared to the PRE training experimental day. Subjects repeated their 3 day diet regime and abstained from alcohol, caffeine and strenuous physical activity for 48 h prior to the POST training experimental day. Both PRE and POST training, a venous blood sample was taken on a separate day after an overnight fast to measure fasting plasma glucose and insulin concentrations (antecubital vein). By using the one‐legged knee extensor model, the resting leg serves as a within subject control leg. This design also gives the advantages that the subjects only had to go through the invasive procedure twice (one before and one after training).
Training regime
To ensure feasibility and high compliance to the training regime, we chose to use indoor cycling for the 12 weeks of training consisting of 4 × 1 h of indoor cycling exercise per week (both legs) (Body bike supreme classic; Pedan, Køge, Denmark). The intensity of the training sessions ranged from 75% to 90% of maximal heart rate measured by Polar heart rate monitors (Polar CS400; Polar, Kempele, Finland). Three of the four weekly training sessions were performed at the subjects' home residence, whereas the fourth training session was performed at our laboratory. Throughout the 12 weeks of training, subjects were instructed to continue their habitual diet, remain weight stable and measure resting heart rate in the morning (3 days a week). In the 12th training week, two of the training sessions were substituted with a and PWL test, respectively. The last training session was performed 48–72 h prior to the POST training experimental day.
Analysis of plasma samples
Plasma glucose concentration was measured by a blood‐gas analyser (ABL800 FLEX; Radiometer, Copenhagen, Denmark). Plasma insulin concentration was measured using an insulin enzyme‐linked immunosorbent assay kit (ALPCO, Salem, NH, USA). The concentration of plasma fatty acids (NEFA C kit; Wako Chemicals GmbH, Neuss, Germany) and triacylglycerol (GPO‐PAP kit; Roche Diagnostics, Mannheim, Germany) were measured using enzymatic colorimetric methods (Hitachi 912 automatic analyser; Hitachi, Mannheim, Germany).
Muscle homogenate and lysate preparation
Muscle biopsies were freeze dried for 48 h and dissected free of visible blood, fat and connective tissue. Muscle homogenates were generated as described previously (Kristensen et al. 2015). Lysates were recovered by centrifuging the homogenates (18,320 g for 20 min at 4°C). Homogenate and lysate protein content were determined by the bicinchoninic acid method (Pierce Biotechnology, Rockford, IL, USA).
SDS‐PAGE and Western blotting
To measure protein expression and phosphorylation, samples were separated on self‐cast gels using SDS‐PAGE followed by semi‐dry transfer of proteins on polyvinylidene fluoride membranes. Membranes were blocked for 15 min in 2% skimmed milk in TBS containing 0.05% Tween‐20 followed by overnight incubation at 4°C in primary antibodies against: anti‐ACC (streptavidin) Dako, Glostrup, Denmark); anti‐phospho‐ACCSer221, anti‐phospho‐AktSer473, anti‐phospho‐AktThr308, anti‐Akt2, anti‐phospho‐AMPKThr172, anti‐HKII, anti‐phospho‐TBC1D4Thr642 and anti‐phospho‐TBC1D4Ser588 (Cell Signaling Technology, Beverly, MA, USA). Anti‐GLUT4 (Thermo Fisher Scientific, Waltham, MA, USA); anti‐α1 AMPK, anti‐γ1 AMPK, anti‐CS and anti‐OXPHOS total cocktail (human) (Abcam, Cambridge, UK); anti‐α2 AMPK, anti‐β1 AMPK (Santa Cruz Biotechnology, Dallas, TX, USA); anti‐β2 AMPK (kindly provided by Dr D. G. Hardie, University of Dundee, Dundee, UK); anti‐GS (custom made, Oluf Pedersen, University of Copenhagen, Copenhagen, Denmark); anti‐γ3 AMPK (Zymed Laboratories Inc., San Francisco, CA, USA); anti phospho‐TBC1D4Ser704 (custom made, Professor Laurie Goodyear, Joslin Diabetes Centre and Harvard Medical School, Boston, MA, USA); anti‐TBC1D4 (Upstate; Millipore, Billerica, MA, USA). The next day, membranes were incubated with horseradish peroxidase‐conjugated secondary antibodies (Jackson ImmunoResearch, West Grove, PA, USA) for 1 h at room temperature before visualizing protein bands with chemiluminescence (Millipore) and a ChemiDoc MP imaging system (Bio‐Rad, Hercules, CA, USA). Some membranes were stripped and re‐probed with a new primary antibody against another phosphorylation site or corresponding total protein after removal of the first antibody by incubation in stripping buffer (62.3 mm Tris‐HCl, 69.4 mm SDS, ddH2O and 0.08% β‐mercaptoethanol, pH 6.7). The membranes were checked for successful removal of the initial primary antibody before re‐probing.
Muscle glycogen
Muscle glycogen content was measured in homogenates (150 μg of protein) as glycosyl units after acid hydrolysis determined by a fluorometric method (Lowry & Passoneau, 1972).
AMPK and glycogen synthase (GS) activity
Isoform‐specific AMPK activity was measured in muscle lysate (250 μg of protein) by sequential immunoprecipitation of the γ3, α2 and α1 subunit. AMPKγ3 antibody used for immunoprecipitation was custom made at Yenzym Antibodies (San Francisco, CA, USA); AMPK α2 was obtained from Santa Cruz Biotechnology and α1 was custom made at Genscript USA Inc. (Piscataway, NJ, USA). AMPK activity was measured in the presence of 200 μm AMP and 100 μm AMARA‐peptide (Schafer‐N, Copenhagen, Denmark) as substrate, as described previously (Birk & Wojtaszewski, 2018). GS activity was measured in homogenates (duplicates) in the presence of 0.02, 0.17 and 8 mm glucose‐6‐phosphate (G6P), as described previously (Højlund et al. 2009).
Statistical analyses
Data are presented as the mean ± SEM. Subject characteristics, fold changes in muscle protein expression, Δ glycogen content between legs and Δ glucose uptake between legs were evaluated by paired t tests. To evaluate changes during the last 40 min of the EHC on leg glucose uptake, arterial blood flow and plasma glucose concentration arterial–venous (A–V) difference, two‐way repeated‐measures (RM) ANOVAs were performed. A two‐way RM ANOVA was also applied to evaluate changes in clamp parameters. For all protein phosphorylation and activity measurements, four two‐way RM ANOVAs were applied; one two‐way RM ANOVA was used to test the factors ‘acute exercise’ (rested leg vs. acutely exercised leg) and ‘insulin’ (basal vs. insulin) for the PRE training experimental day alone. Similarly, a second two‐way RM ANOVA was used to test the factors ‘acute exercise’ and ‘insulin’ for the POST training experimental day alone. A third and fourth two‐way RM ANOVA tested the factors ‘acute exercise’ and ‘training’ (PRE training vs. POST training) for the basal samples and the insulin‐stimulated samples, separately. Significant interactions were evaluated by Tukey's post hoc test. P < 0.05 was considered statistically significant. N = 9, except otherwise stated. All statistical analyses were performed using Sigma Plot, version 13 (Systat Software Inc., Chicago, IL, USA).
Results
Adaptations to 12 weeks of endurance training
All subjects completed 94 ± 1% of all training sessions and remained weight stable throughout the 12 weeks of training (Table 1). Adaptations in several parameters signify an effective training regime i.e. increased (17%) and peak knee‐extensor workload (PWL, 18%), as well as decreased fat mass (−6%) and resting heart rate (−5 beats min–1) (Table 1). In skeletal muscle, protein expression of glucose handling enzymes (hexokinase II, GS) and mitochondrial markers (citrate synthase, complex II, III and V) was increased, whereas GLUT4 remained unchanged (Fig. 2). Protein expression of some regulatory signalling components (AMPKα1, AMPKβ1, AMPKβ2, AMPKγ1 and TBC1D4) increased in response to training, whereas others (AMPKα2, acetyl‐CoA carboxylase) remained unchanged (Fig. 2). As expected, AMPKγ3 protein expression decreased with training (Fig. 2).
Table 1.
Subject characteristics PRE and POST training
| PRE training | POST training | |
|---|---|---|
| Age (years) | 25 ± 1 | |
| Weight (kg) | 78.3 ± 1.7 | 78.0 ± 1.8 |
| Body mass index | 23.5 ± 0.5 | 23.4 ± 0.6 |
| Lean mass (kg) | 56.6 ± 1.9 | 57.5 ± 1.9 |
| Fat mass (kg) | 19.0 ± 1.2 | 17.8 ± 0.9** |
| Fat mass (%) | 24.2 ± 1.5 | 22.8 ± 1.2** |
| Visceral adipose tissue (g) | 464 ± 75 | 351 ± 74* |
| (mL min−1 kg−1) | 43.6 ± 1.6 | 50.9 ± 1.2*** |
| HRmax | 197 ± 2 | 194 ± 1 |
| HRrest | 60 ± 2 | 55 ± 2* |
| PWL (W) | 39 ± 4 | 46 ± 3** |
| Fasting glucose (mmol L−1) | 5.2 ± 0.1 | 5.4 ± 0.1* |
| Fasting insulin (μIU mL−1) | 5.1 ± 0.5 | 5.6 ± 0.5 |
Values are the mean ± SEM. * P < 0.05, ** P < 0.01 and *** P < 0.001 vs. PRE training. HR, heart rate; PWL, peak workload for the knee‐extensors of one leg. Lean mass, fat mass and visceral adipose tissue were determined by dual X‐ray absorptiometry.
Figure 2. Change in protein expression after 12 weeks of exercise training.
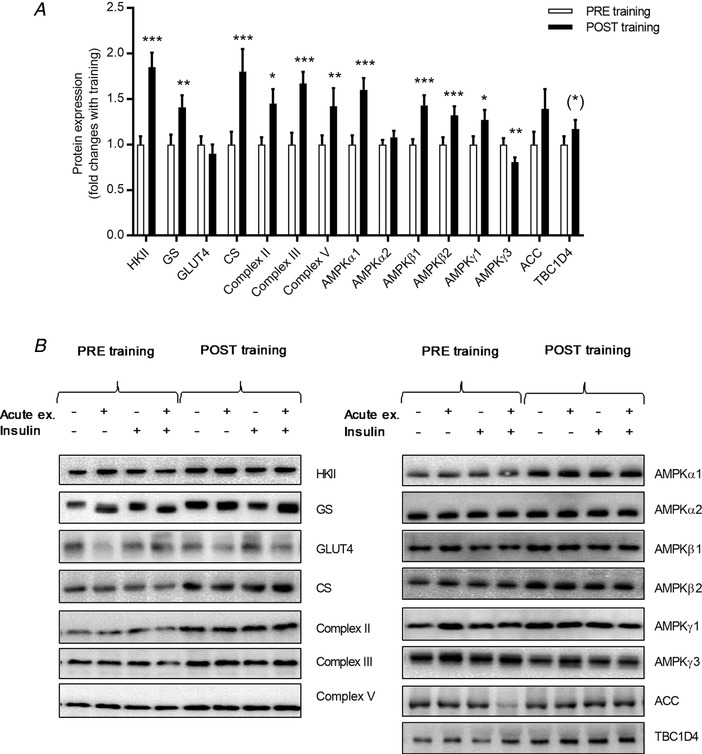
Protein expression of ACC, AMPK α1, α2, β1, β2, γ1 and γ3, CS, GLUT4, GS, HKII, TBC1D4 and complex II, III and V of the electron transport chain was evaluated by Western blotting PRE training (white bars) and POST training (black bars) (A). Data are calculated as the mean value of protein expression measured in the prior exercised and rested leg before and after insulin stimulation. Representative blots (B). Data are expressed as the mean ± SEM. (*) = 0.058, * P < 0.05, ** P < 0.01 and *** P < 0.001 PRE vs. POST training.
Glucose infusion rate during the euglycaemic hyperinsulinaemic conditions was increased by training from 4.2 ± 0.3 mg min−1 kg−1 to 5.0 ± 0.3 mg min−1 kg−1 (Table 2) reflecting increased whole body insulin‐stimulated glucose uptake. Plasma glucose (∼5 mmol L−1) and insulin (∼120 μIU mL−1) concentrations were similar during the insulin clamp performed before and after the training period (Table 2). Plasma concentrations of fatty acids and triacylglycerol decreased in response to insulin but were unaffected by training (Table 2). Improved insulin‐stimulated glucose uptake was also evident in the skeletal muscle by an increased insulin‐stimulated glucose uptake in the rested leg after training compared to before training (Fig. 3 A). This was largely a result of enhanced glucose extraction rather than changes in muscle blood perfusion (Fig. 3 C and D). The increased glucose extraction was likely associated with the increased capacity for glucose handling within the muscle cell by increased HKII and GS protein expression (Fig. 2)
Table 2.
Clamp parameters PRE and POST training
| PRE training | POST training | |||
|---|---|---|---|---|
| Glucose infusion rate (mg min−1 kg−1) | 4.2 ± 0.3 | 5.0 ± 0.4* | ||
| Pre clamp | End of clamp | Pre clamp | End of clamp | |
| Arterial plasma glucose (mmol L−1) | 5.1 ± 0.1 | 5.1 ± 0.1 | 5.1 ± 0.1 | 5.0 ± 0.1 |
| Arterial plasma Insulin (μIU mL−1) | 5.7 ± 0.7 | 117.2 ± 7.7‡‡‡ | 5.7 ± 0.6 | 121.8 ± 5.6‡‡‡ |
| Arterial plasma fatty acids (μmol L−1) | 641 ± 42 | 37 ± 5‡‡‡ | 557 ± 66 | 37 ± 1‡‡‡ |
| Arterial plasma triacylglycerol (mmol L−1) | 1.1 ± 0.3 | 0.9 ± 0.2‡‡‡ | 1.1 ± 0.1 | 0.9 ± 0.2‡‡‡ |
| RER | 0.75 ± 0.02 | 0.83 ± 0.02‡‡‡ | 0.76 ± 0.01 | 0.84 ± 0.01‡‡‡ |
Values are the mean ± SEM. * P < 0.05 vs. PRE training. ‡‡‡ Main effect of insulin (P < 0.001). RER, respiratory exchange ratio. Glucose infusion rate is expressed as weighted mean of 120 min of euglycaemic hyperinsulinaemic clamp (EHC). Pre clamp: mean of blood samples taken 30 min and 0 min before EHC. End of clamp: mean last 40 min of EHC. RER pre clamp: 60 min before EHC, RER end of clamp: 110 min into the EHC.
Figure 3. Acute one‐legged exercise on leg glucose uptake, glycogen utilization, A–V difference and arterial blood flow.
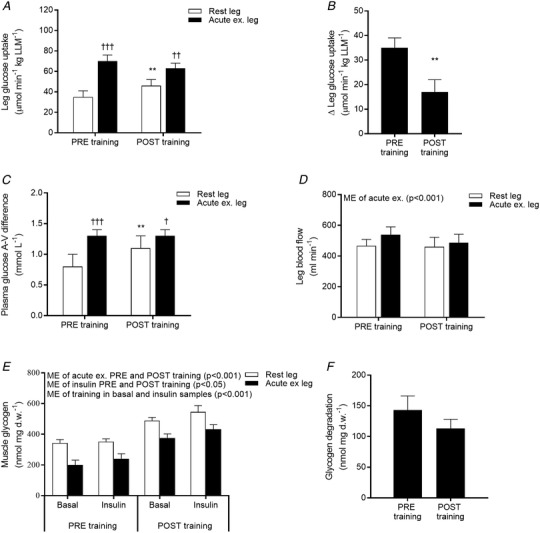
Leg glucose uptake (A) was calculated using Fick's principle by multiplying glucose A–V difference (C) with leg arterial blood flow (D) and divided by lean leg mass (LLM). The difference in leg glucose uptake between the rested and acutely exercised leg is expressed as Δ leg glucose uptake (B). Leg glucose uptake, glucose A–V difference and arterial blood flow are expressed as mean values of the last 40 min of the 2 h EHC in the rested (white bars) and prior exercised leg (black bars), PRE and POST training. Muscle glycogen measured in the rested (white bars) and prior exercised leg (black bars) 4 h after acute one‐legged exercise (basal) and after a 2 h EHC (insulin) PRE and POST training (E). The difference in glycogen content between the rested and acutely exercised leg 4 h after acute exercise is depicted as glycogen degradation (F). Data are expressed as the mean ± SEM. ME, main effect. ** P < 0.01 PRE vs. POST training; †P < 0.05, ††P < 0.01 and †††P < 0.001 rested leg vs. acutely exercised leg.
Acute one‐legged exercise was performed at the same relative intensity
Both PRE and POST training muscle glycogen was reduced in response to acute exercise in the prior exercised leg compared to the rested leg (Fig. 3 E). Although training increased the glycogen content within the muscles (Fig. 3 E), the absolute glycogen utilization during acute one‐legged exercise was similar PRE and POST training (Fig. 3 F). In a subset of subjects (n = 6), the percentage maximum heart rate (53 ± 3% vs. 50 ± 2%) and rate of perceived exertion (16 ± 0 vs. 16 ± 0 on the Borg (6–20) scale) were measured during acute one‐legged exercise with no difference between the PRE and POST training experimental day. Together, these results support that the subjects (as per the design) performed the acute bout of one‐legged exercise at the same relative intensity PRE and POST training (absolute workload was 33 ± 3 W PRE training and 39 ± 2 W POST training).
Diminished ability of acute exercise to enhance insulin‐stimulated leg glucose uptake after training
Both PRE and POST training, insulin increased leg glucose uptake to a greater extent in the prior exercised leg compared to the rested leg (Fig. 3 A). However, the difference in insulin‐stimulated glucose uptake between the prior exercised and rested leg was markedly smaller (−50 ± 12%) POST training compared to PRE training (Fig. 3 B). This reflects a diminished ability of acute exercise to enhance muscle insulin‐stimulated glucose uptake in the trained state.
During the insulin clamp, the plasma glucose concentration A–V difference was higher in the prior exercised leg compared to the rested leg both PRE and POST training. In response to training, the plasma glucose concentration A–V difference was increased in the rested leg, whereas the A–V difference in the prior exercised leg was unaffected by training (Fig. 3 C). Femoral arterial blood flow was slightly higher in the prior exercised leg compared to the rested leg and was unaffected by training (Fig. 3 D). Thus, in the prior exercised leg, lower glucose extraction rather than glucose delivery was responsible for the diminished ability to increase insulin‐stimulated glucose uptake after training, reflecting important intramuscular changes.
Muscle signalling
To investigate the effects of prior exercise on insulin signalling to GLUT4 translocation, phosphorylation of Akt and TBC1D4 was measured. Insulin increased, as expected, the phosphorylation of AktSer473 and AktThr308, whereas prior exercise did not, nor did exercise improve the effect of insulin on Akt phosphorylation (Fig. 4 A and B). Akt2 protein expression increased in response to training (Fig. 4 C).
Figure 4. Phosphorylation and expression of Akt2.
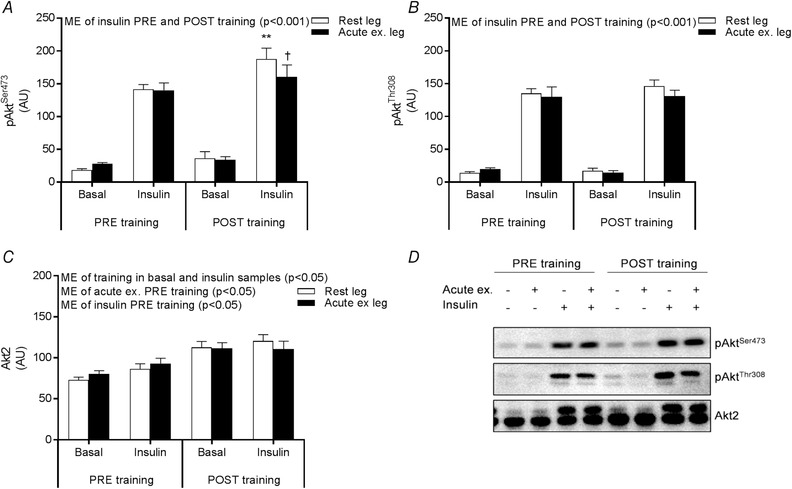
Site‐specific phosphorylation of AktSer473 (A) and AktThr308 (B) and Akt2 protein expression (C) measured by Western blotting in the rested (white bars) and prior exercised leg (black bars) 4 h after acute one‐legged exercise (basal) and after a 2 h EHC (insulin) PRE and POST training. Representative blots (D). Data are expressed as the mean ± SEM. AU, arbitrary units; ME, main effect. ** P < 0.01 PRE vs. POST training; † P < 0.05, rested leg vs. acutely exercised leg.
Phosphorylation of TBC1D4Ser704, a confirmed AMPKα2β2γ3 target site (Treebak et al. 2010), was significantly higher in the prior exercised vs. rested muscle both PRE and POST training before the clamp was initiated (Fig. 5 A). However, the TBC1D4Ser704phosphorylation in the prior exercised muscle was significantly lower POST training compared to PRE training. In response to insulin, the phosphorylation increased in the rested muscle but did not increase further in prior exercised muscle PRE training. Phosphorylation of TBC1D4Ser704 increased in response to insulin in the muscles of both legs POST training (Fig. 5 A).
Figure 5. Phosphorylation of TBC1D4.

Site‐specific phosphorylation of TBC1D4Ser704 (A), TBC1D4Thr642 (B) and TBC1D4Ser588 (C) measured by Western blotting in the rested (white bars) and prior exercised leg (black bars) 4 h after acute one‐legged exercise (basal) and after a 2 h EHC (insulin) PRE and POST training. Representative blots (D). Data are expressed as the mean ± SEM. AU, arbitrary units; ME, main effect. * P < 0.05, ** P < 0.01 PRE vs. POST training; † P < 0.05 and ††† P < 0.001 rested leg vs. acutely exercised leg; ‡ P < 0.05 and ‡‡‡ P < 0.001 basal vs. insulin.
TBC1D4Thr642 is a confirmed Akt target site and, although this site is not a direct AMPK target site, some data suggest that phosphorylation of TBC1D4Thr642 is dependent on TBC1D4ser704 phosphorylation and thus becomes indirectly dependent of AMPK (Kjøbsted et al. 2015). Accordingly, phosphorylation of TBC1D4Thr642 was higher in the prior exercised muscle compared to rested muscle 4 h after acute exercise PRE training but not POST training. In response to insulin, phosphorylation of TBC1D4Thr642 was increased in muscles of both legs both PRE and POST training (Fig. 5 B).
TBC1D4Ser588 is also a confirmed Akt target site reported to be independent of TBC1D4Ser704 phosphorylation. TBC1D4Ser588 phosphorylation was modestly higher in the prior exercised vs. rested muscle 4 h after acute one‐legged exercise both PRE and POST training prior to insulin stimulation (Fig. 5 C). In response to insulin, phosphorylation of TBC1D4Ser588 increased in muscles of both legs both at the PRE and POST training experimental day (Fig. 5 C).
Thus, a major difference between the PRE and POST experimental day is the markedly elevated TBC1D4Ser704 phosphorylation in the prior exercised muscle observed PRE training but not POST training. To gain further support for a potential role of AMPK, we measured phosphorylation of ACCSer221 a confirmed AMPK target site (Abu‐Elheiga et al. 1997; Munday, 2002). Accordingly, phosphorylation of ACCSer221 was only increased by exercise PRE training and not POST training (Fig. 6 A). Together, the phosphorylation patterns of both TBC1D4Ser704 and ACCSer221 suggest a lower activation of AMPK in response to acute exercise in the trained state compared to the untrained state. As a result of the study design, biopsies were not obtained during exercise in the present study. Thus, we cannot directly confirm the reduced AMPK activation during exercise POST training compared to PRE training. Four hours after exercise, phosphorylation of AMPKThr172 and activity of the AMPKα2β2γ3 complex showed a rather fast reversal toward basal levels (Fig. 6 B and C), confirming previous observations (Mortensen et al. 2013).
Figure 6. ACC and AMPK activation.
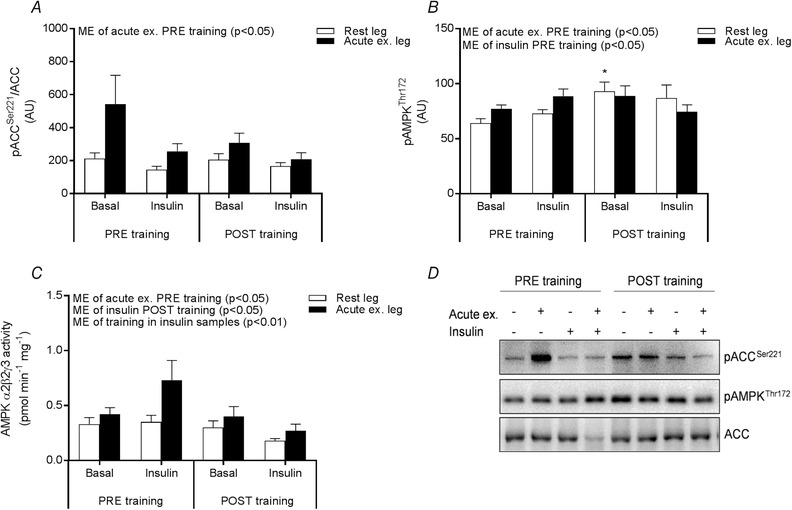
Site‐specific phosphorylation of ACCSer221 (A) and AMPKThr172 (B) measured by Western blotting in the rested (white bars) and prior exercised leg (black bars) 4 h after acute one‐legged exercise (basal) and after a 2 h EHC (insulin) PRE and POST training. Representative blots (C). Activity of AMPKα2β2γ3 in the rested (white bars) and prior exercised leg (black bars) 4 h after acute one‐legged exercise (basal) and after a 2 h EHC (insulin) PRE and POST training (D). Data are expressed as the mean ± SEM. AU, arbitrary units; ME, main effect. * P < 0.05 PRE vs. POST training.
Activation of GS in response to acute exercise and insulin PRE and POST training
As the major part of muscle glucose metabolism is non‐oxidative during insulin stimulation, the activity of GS (rate‐limiting enzyme in glycogenesis) was measured. In accordance with the higher GS protein expression (Fig. 2), total GS activity was also increased by training (Fig. 7 A). The activity of GS measured in the presence of 0.02 and 0.17 mm G6P (Fig. 7 B and C) was increased by prior exercise and insulin both PRE and POST training. Yet, the ability of insulin to activate GS was not dependent of prior exercise either at the PRE or POST training experimental day. Thus, the activity pattern of GS does not explain the reduced effect of acute exercise to enhance insulin‐stimulated glucose uptake in the trained state.
Figure 7. GS activity.
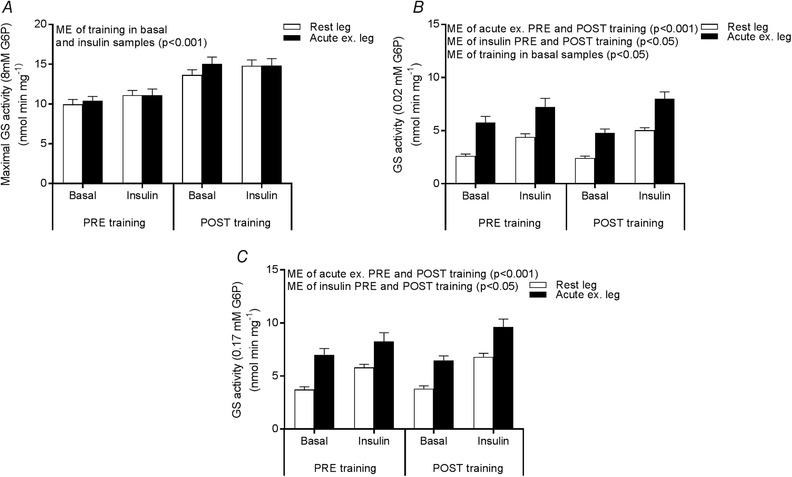
GS activity measured in the presence of 8 mm G6P (A) 0.02 mm G6P (B) and 0.17 mm G6P (C) in the rested (white bars) and prior exercised leg (black bars) 4 h after acute one‐legged exercise (basal) and after a 2 h EHC (insulin) PRE and POST training. Data are expressed as the mean ± SEM. ME, main effect.
Discussion
In the present study, we found that 12 weeks of exercise training increased insulin‐stimulated glucose uptake on a whole body level, as well as in skeletal muscle. Interestingly, insulin‐stimulated glucose uptake in the acutely exercised leg was not further enhanced by training. This led to our primary finding that the ability of acute exercise to enhance insulin‐stimulated glucose uptake is reduced in the trained state. Importantly, this was seen despite that the acute bout of exercise was performed at the same relative intensity eliciting similar absolute glycogen degradation during exercise before and after training.
In contrast to the findings reported by Mikines et al (1989) and Nelson and Horowitz (2014), we found increased insulin‐stimulated glucose uptake following acute exercise even in the trained muscle. This could be a result of differences in study design (cross‐sectional vs. intervention study, study population, etc.), although it indeed reflects a lack of knowledge about the dynamics and magnitude of the response to acute exercise and to the mechanisms regulating these factors, including regulation of microvascular perfusion (Sjøberg et al. 2017). It may be speculated that an upper limit of muscle insulin sensitivity is obtainable in the highly trained muscle leaving only very little or no additional effects of a single exercise bout in these subjects. An important premise for such an interpretation is that leg glucose uptake has not reached the maximal obtainable level. We did not evaluate this in the present study. However previous studies, on comparable groups of subjects, have reported higher (∼60–80%) levels of leg glucose uptake in response to a maximal dose of insulin (Richter et al. 1989; Dela et al. 1992). Furthermore, in studies in which leg glucose uptake has been evaluated under hyperinsulinaemic–hyperglycaemic conditions (Hansen et al. 1999) or when insulin and exercise have been superimposed (Dela et al. 1994), rates of leg glucose uptake exceeded by far (∼4‐fold) the values obtained in the present study. Thus, we assume that, under the conditions applied, leg glucose uptake has not reached saturation.
From an applied view, the observation that the same exercise bout elicits markedly different responses in insulin‐stimulated glucose uptake depending on the person's fitness level may add additional explanatory insight to the complexity of controlling glucose homeostasis in patients dependent on insulin treatment. Indeed, it may add insights that are important for prediction of the optimal insulin dose algorithm (Riddle, 2008).
The present study provided the opportunity to identify possible molecular mechanisms in skeletal muscle explaining the reduced ability of acute exercise to enhance insulin‐stimulated glucose uptake in the trained state. A series of studies in rodents and transgenic animal models have signified the importance of the signalling node at TBC1D4 integrating inputs from AMPK (‘exercise’) and Akt (‘insulin’), indicating both the necessity and sufficiency of AMPK in muscle insulin sensitization (Fisher et al. 2002; Kjøbsted et al. 2015, 2017; Wang et al. 2018). More specifically, these studies point to interactions between AMPKα2β2γ3 and Akt2 and the phosphor‐regulation of TBC1D4Ser711/Thr642. Indications of similar interactions in human muscle have also been reported. Thus, the level of phosphorylation of the equivalent TBC1D4Ser704 (in humans) is associated with AMPKα2β2γ3 activity during exercise in samples of whole muscle as well as in samples representing either type I or type II muscle fibres (Kristensen et al. 2015). Also, we recently reported enhanced phosphor‐regulation of TBC1D4 by insulin concomitantly with enhanced insulin‐stimulated glucose uptake following exercise (Treebak et al. 2009; Pehmøller et al. 2012; Sjøberg et al. 2017; Hingst et al. 2018). In the present study, we found reduced phosphorylation of TBC1D4Ser704/Thr642 4 h after exercise in the acutely exercised leg after training compared to before training, whereas phosphorylation of TBC1D4Ser704/Thr642 during insulin stimulation reached similar levels before and after training. Exactly how this may relay to the regulation of insulin action following exercise is still unclear. Rodent studies suggest that the localization of TBC1D4 and protection against dephosphorylation mediated by prior exercise might be of importance (Zheng & Cartee, 2016; Arias et al. 2018). In the present study, we showed decreased AMPKγ3 expression in skeletal muscle in response to 12 weeks of cycling exercise, confirming previous observations (Frøsig et al. 2004; Wojtaszewski et al. 2005; Mortensen et al. 2013). We have previously shown that the AMPKα2β2γ3 is the primary complex activated during acute exercise (Birk & Wojtaszewski, 2006) and that the protein expression of AMPKγ3 correlates with the AMPKα2β2γ3 activity (Mortensen et al. 2009), suggesting that alterations in AMPKγ3 expression probably affect AMPKα2β2γ3 activation during acute exercise. As a result of the study design, we did not obtain muscle biopsies immediately after exercise. Therefore, we were unable to directly confirm such differences in AMPKα2β2γ3 activation during the acute one‐legged exercise before and after training. However, two arguments suggest that, also in the present study, AMPKα2β2γ3 activation has been higher during exercise before training vs. after training. First, in the present study, the phosphor‐regulation of the confirmed AMPK targets ACCSer221 and TBC1D4Ser704 was increased in the previously exercised muscle before but not after training. Second, previous cross‐sectional and training intervention studies have reported lesser activation of the AMPK‐ACC signalling axis in response to acute exercise in the trained vs. the untrained state. (Nielsen et al. 2003; McConell et al. 2005; Lee‐Young et al. 2009; Mortensen et al. 2013). The mechanism responsible for the lesser AMPK activation in the trained muscle, even by exercise loads eliciting the same relative intensity, is unclear. Glycogen might be a regulator of AMPK activity (Wojtaszewski et al. 2003b; McConell et al. 2005) via the ability of AMPK to bind to the glycogen particles (McBride et al. 2009). Rodent studies also suggest that the absolute level of glycogen influences the ability of insulin to increase glucose uptake (Jensen et al. 1997; Derave et al. 2000) and the insulin‐sensitizing effect of exercise has previously been proposed to be dependent on the glycogen utilization during exercise (Richter et al. 2001). Thus, the higher absolute levels of glycogen in muscle after training might reduce the ability of acute exercise to enhance insulin‐stimulated glucose uptake by reducing activation of AMPK.
In the present study, phosphorylation of TBC1D4Thr642 responded similarly to acute exercise as TBC1D4Ser704. This is in line with previous studies suggesting a mutual dependency of these two phosphor‐sites (Kjøbsted et al. 2015, 2017), such that, even though TBC1D4Thr642 is not an AMPK target site, its regulation is dependent on the degree of TBC1D4Ser704 phosphorylation (Kjøbsted et al. 2015). By contrast, such an interaction is not seen in the phosphor‐regulation of TBC1D4Ser588 (Kjøbsted et al. 2015), probably explaining why the phosphorylation of TBC1D4Ser588 increased to the same extent in response to acute exercise before and after training in the present study. As TBC1D4Ser588 is not an AMPK target site (Kjøbsted et al. 2015), it is plausible that the increased phosphorylation of TBC1D4Ser704 and Thr642 in response to acute exercise before but not after training was mediated via AMPK and not another upstream kinase. In line with this, the activation (by phosphorylation) of the insulin‐regulated TBC1D4 kinase Akt2 was not enhanced in response to acute exercise either before or after training.
In conclusion, although exercise training increases insulin‐stimulated glucose uptake in skeletal muscle at rest, we show that it is not enhanced further by training in the acutely exercised leg. This leads to a diminished insulin‐sensitizing effect of a single bout of exercise by exercise training. Prior exercised muscle thus displays a similar absolute level of glucose uptake during insulin stimulation before and after training. Whether this reflects an upper limit for muscle insulin sensitivity or a strong dependency on the absolute muscle glycogen levels remains to be seen. Our data suggest that a reduced AMPKγ3 expression with exercise training may contribute to a lesser AMPKα2β2γ3 mediated signalling following a single bout of exercise. We propose this to be responsible for the reduced phosphorylation of TBC1D4 and thus the reduced ability of acute exercise to enhance insulin‐stimulated muscle glucose uptake in the trained state. Together with previous evidence in humans and rodents, these findings contribute to the idea that AMPK is an important regulator of insulin action in skeletal muscle.
Additional information
Competing interests
The authors declare that they have no competing interests.
Author contributions
The experiments were performed at the section of Molecular Physiology, Department of Nutrition, Exercise and Sports, University of Copenhagen, Denmark. DES and JFPW were responsible for the conception and design of the research. DES, NBJ, KAS, BK, EAR and JFPW performed the experiments. DES, NBJ and JBB performed the analyses. DES, NBJ, JBB, KAS, BK, EAR and JFPW interpreted the results. DES and JFPW drafted the manuscript. DES, NBJ, JBB, KAS, BK, EAR and JFPW edited and revised manuscript. All authors read and approved the final version submitted for publication. JFPW is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis
Funding
This study was supported by grants from the Danish Council for Independent Research Medical Sciences (FSS 6110‐00498B); The Novo Nordisk Foundation (NNF16OC0023046, NNF17OC0027224), The Lundbeck Foundation (R266‐2017‐4358); and the research program (2016) ‘Physical Activity and Nutrition for Improvement of Health’ funded by the University of Copenhagen Excellence Program for Interdisciplinary Research. DES was supported by a research grant from the Danish Diabetes Academy funded by the Novo Nordisk Foundation. KAS was supported by a postdoctoral research grant from the Council for Independent Research/ Medicine (4092‐00309).
Acknowledgements
We acknowledge the skilled technical assistance of Betina Bolmgren, Irene B. Nielsen and Nicoline R. Andersen (University of Copenhagen, Denmark), as well as the kind donation of material essential for this work by L. J. Goodyear (Joslin Diabetes Centre, Boston, MA, USA) and O. B. Pedersen (University of Copenhagen, Denmark)
Biography
Dorte E. Steenberg received her master's degree in human physiology from the Department of Nutrition, Exercise and Sports, UCPH. Her master thesis focused on the effects of acute exercise on AMPK signalling in skeletal muscle fibres. Subsequently, she continued into the fascinating world of science as a PhD student investigating the effects of acute exercise on insulin sensitivity under different conditions in humans. One aspect of this is whether training status affects the insulin‐sensitizing effect of acute exercise, as investigated in the present study. She considers that this will provide new knowledge to the important ongoing question of ‘how physical activity improves insulin sensitivity and health’.

Edited by: Michael Hogan & Paul Greenhaff
Linked articles: This article is highlighted in a Perspectives article by Cartee. To read the Perspectives article, visit https://doi.org/10.1113/JP277302.
References
- Abu‐Elheiga L, Almarza‐Ortega DB, Baldini A & Wakil SJ (1997). Human acetyl‐CoA carboxylase 2. Molecular cloning, characterization, chromosomal mapping, and evidence for two isoforms. J Biol Chem 272, 10669–10677. [DOI] [PubMed] [Google Scholar]
- Andersen P, Adams RP, Sjøgaard G, Thorboe A & Saltin B (1985). Dynamic knee extension as model for study of isolated exercising muscle in humans. J Appl Physiol 59, 1647–1653. [DOI] [PubMed] [Google Scholar]
- Arias EB, Wang H & Cartee GD (2018). Akt substrate of 160 kDa dephosphorylation rate is reduced in insulin‐stimulated rat skeletal muscle after acuter exercise. Physiol Res 67, 143–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergström J (1962). Muscle electrolytes in man determined by neutron activation analysis on needle biopsy specimens. Scand J Clin Lab Invest 14, 7–110.13862378 [Google Scholar]
- Birk JB & Wojtaszewski JFP (2006). Predominant alpha2/beta2/gamma3 AMPK activation during exercise in human skeletal muscle. J Physiol 577, 1021–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birk JB & Wojtaszewski JFP (2018). Kinase activity determination of specific AMPK complexes/heterotrimers in skeletal muscle In AMPK: Methods and Protocols. Methods in Molecular Biology, Vol. 1732, ed. Neumann D. & Viollet B, pp. 215–228. Humana Press, New York, NY. [DOI] [PubMed] [Google Scholar]
- Bonen A, Tan MH & Watson‐Wright WM (1984). Effect of exercise on insulin binding and glucose metabolism in muscle. Can J Physiol Pharmacol 62, 1500–1504. [DOI] [PubMed] [Google Scholar]
- Cartee GD (2015). Mechanisms for greater insulin‐stimulated glucose uptake in normal and insulin resistant skeletal muscle after acute exercise. Am J Physiol Endocrinol Metab 309, E949–E959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartee GD & Wojtaszewski JFP (2007). Role of Akt substrate of 160 kDa in insulin‐stimulated and contraction‐stimulated glucose transport. Appl Physiol Nutr Metab 32, 557–566. [DOI] [PubMed] [Google Scholar]
- Castorena CM, Arias EB, Sharma N & Cartee GD (2014). Postexercise improvement in insulin‐stimulated glucose uptake occurs concomitant with greater AS160 phosphorylation in muscle from normal and insulin‐resistant rats. Diabetes 63, 2297–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dela F, Mikines KJ, Von Linstow M, Secher NH & Galbo H (1992). Effect of training on insulin‐mediated glucose uptake in human muscle. Am J Physiol Endocrinol Metab 263, E1134–E1143. [DOI] [PubMed] [Google Scholar]
- Dela F, Mikines KJ, Sonne B & Galbo H (1994). Effect of training on interaction between insulin and exercise in human muscle. J Appl Physiol 76, 2386–2393. [DOI] [PubMed] [Google Scholar]
- Derave W, Hansen BF, Lund S, Kristiansen S & Richter EA (2000). Muscle glycogen content affects insulin‐stimulated glucose transport and protein kinase B activity. Am J Physiol Endocrinol Metab 279, E947–E955. [DOI] [PubMed] [Google Scholar]
- Fisher JS, Gao J, Han DH, Holloszy JO & Nolte LA (2002). Activation of AMP kinase enhances sensitivity of muscle glucose transport to insulin. Am J Physiol Endocrinol Metab 282, E18–E23. [DOI] [PubMed] [Google Scholar]
- Frøsig C, Jørgensen S, Hardie D, Richter E & Wojtaszewski J (2004). 5′‐AMP‐activated protein kinase activity and protein expression are regulated by endurance training in human skeletal muscle. Am J Physiol Endocrinol Metab 286, E411–E417. [DOI] [PubMed] [Google Scholar]
- Frøsig C, Rose AJ, Treebak JT, Kiens B, Richter EA & Wojtaszewski JFP (2007a). Effects of endurance exercise training on insulin signaling in human skeletal muscle: Interactions at the level of phosphatidylinositol 3‐kinase, Akt, and AS160. Diabetes 56, 2093–2102. [DOI] [PubMed] [Google Scholar]
- Frøsig C, Sajan MP, Maarbjerg SJ, Brandt N, Roepstorff C, Wojtaszewski JFP, Kiens B, Farese RV & Richter Ea (2007b). Exercise improves phosphatidylinositol‐3,4,5‐trisphosphate responsiveness of atypical protein kinase C and interacts with insulin signalling to peptide elongation in human skeletal muscle. J Physiol 582, 1289–1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funai K, Schweitzer GG, Sharma N, Kanzaki M & Cartee GD (2009). Increased AS160 phosphorylation, but not TBC1D1 phosphorylation, with increased postexercise insulin sensitivity in rat skeletal muscle. Am J Physiol Endocrinol Metab 297, E242–E251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garetto LP, a Richter E, Goodman MN & Ruderman NB (1984). Enhanced muscle glucose metabolism after exercise in the rat: the two phases. Am J Physiol Endocrinol Metab 246, E471–E475. [DOI] [PubMed] [Google Scholar]
- Hamada T, Arias EB & Cartee GD (2006). Increased submaximal insulin‐stimulated glucose uptake in mouse skeletal muscle after treadmill exercise. J Appl Physiol 101, 1368–1376. [DOI] [PubMed] [Google Scholar]
- Hansen BF, Asp S, Kiens B & Richter EA (1999). Glycogen concentration in human skeletal muscle: effect of prolonged insulin and glucose infusion. Scand J Med Sci Sports 9, 209–213. [DOI] [PubMed] [Google Scholar]
- Hansen PA, Nolte LA, Chen MM & Holloszy JO (1998). Increased GLUT‐4 translocation mediates enhanced insulin sensitivity of muscle glucose transport after exercise. J Appl Physiol 85, 1218–1222. [DOI] [PubMed] [Google Scholar]
- Henry CJ (2005). Basal metabolic rate studies in humans: measurement and development of new equations. Public Heal Nutr 8, 1133–1152. [DOI] [PubMed] [Google Scholar]
- Hingst JR, Bruhn L, Hansen MB, Rosschou MF, Birk JB, Fentz J, Foretz M, Viollet B, Sakamoto K, Færgeman NJ, Havelund JF, Parker BL, James DE, Kiens B, Richter EA, Jensen J & Wojtaszewski JFP (2018). Exercise‐induced molecular mechanisms promoting glycogen supercompensation in human skeletal muscle. Mol Metab; 16, 24–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Højlund K, Birk JB, Klein DK, Levin K, Rose AJ, Hansen BF, Nielsen JN, Beck‐Nielsen H & Wojtaszewski JF (2009). Dysregulation of glycogen synthase COOH‐ and NH2‐terminal phosphorylation by insulin in obesity and type 2 diabetes mellitus. J Clin Endocrinol Metab 94, 4547–4556. [DOI] [PubMed] [Google Scholar]
- Holten MK, Zacho M, Gaster M, Juel C, Wojtaszewski JFP & Dela F (2004). Uptake, GLUT4 content, and insulin signaling in skeletal muscle in patients with type 2 diabetes. Diabetes 53, 294–305. [DOI] [PubMed] [Google Scholar]
- Jensen J, Aslesen R, Ivy JL & Brørs O (1997). Role of glycogen concentration and epinephrine on glucose uptake in rat epitrochlearis muscle. Am J Physiol Endocrinol Metab 272, E649–E655. [DOI] [PubMed] [Google Scholar]
- Kjøbsted R, Munk‐Hansen N, Birk JB, Foretz M, Viollet B, Bjornholm M, Zierath JR, Treebak JT & Wojtaszewski JFP (2017). Enhanced muscle insulin sensitivity after contraction/exercise is mediated by AMPK. Diabetes 66, 598–612. [DOI] [PubMed] [Google Scholar]
- Kjøbsted R, Treebak JT, Fentz J, Lantier L, Viollet B, Birk JB, Schjerling P, Björnholm M, Zierath JR & Wojtaszewski JFP (2015). Prior AICAR stimulation increases insulin sensitivity in mouse skeletal muscle in an AMPK‐dependent manner. Diabetes 64, 2042–2055. [DOI] [PubMed] [Google Scholar]
- Kostovski E, Boon H, Hjeltnes N, Lundell LS, Ahlsén M, Chibalin AV, Krook A, Iversen PO & Widegren U (2013). Altered content of AMP‐activated protein kinase isoforms in skeletal muscle from spinal cord injured subjects. Am J Physiol Endocrinol Metab 305, E1071–E1080. [DOI] [PubMed] [Google Scholar]
- Kramer HF, Witczak CA, Taylor EB, Fujii N, Hirshman MF & Goodyear LJ (2006). AS160 regulates insulin‐ and contraction‐stimulated glucose uptake in mouse skeletal muscle. J Biol Chem 281, 31478–31485. [DOI] [PubMed] [Google Scholar]
- Kristensen DE, Albers PH, Prats C, Baba O, Birk JB & Wojtaszewski JFP (2015). Human muscle fibre type‐specific regulation of AMPK and downstream targets by exercise. J Physiol 593, 2053–2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee‐Young RS, Canny BJ, Myers DE & McConell GK (2009). AMPK activation is fiber type specific in human skeletal muscle: effects of exercise and short‐term exercise training. J Appl Physiol 107, 283–289. [DOI] [PubMed] [Google Scholar]
- Lowry OH & Passoneau JV (1972). A flexible system of enzymatic analysis. A Flex Syst Enzym Anal 1, 263–282. [Google Scholar]
- McBride A, Ghilagaber S, Nikolaev A & Hardie DG (2009). The glycogen‐binding domain on the AMPK β subunit allows the kinase to act as a glycogen sensor. Cell Metab 9, 23–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McConell GK, Kaur G, Falcão‐Tebas F, Hong YH & Gatford KL (2015). Acute exercise increases insulin sensitivity in adult sheep: a new preclinical model. Am J Physiol Regul Integr Comp Physiol 308, R500–R506. [DOI] [PubMed] [Google Scholar]
- McConell GK, Lee‐Young RS, Chen Z‐P, Stepto NK, Huynh NN, Stephens TJ, Canny BJ & Kemp BE (2005). Short‐term exercise training in humans reduces AMPK signalling during prolonged exercise independent of muscle glycogen. J Physiol 568, 665–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikines KJ, Sonne B, a Farrell P, Tronier B & Galbo H (1988). Effect of physical exercise on sensitivity and responsiveness to insulin in humans. Am J Physiol Endocrinol Metab 254, E248–E259. [DOI] [PubMed] [Google Scholar]
- Mikines KJ, Sonne B, Tronier B & Galbo H (1989). Effects of acute exercise and detraining on insulin action in trained men. J Appl Physiol 66, 704–711. [DOI] [PubMed] [Google Scholar]
- Mortensen B, Hingst JR, Frederiksen N, Hansen RWW, Christiansen CS, Iversen N, Friedrichsen M, Birk JB, Pilegaard H, Hellsten Y, Vaag A & Wojtaszewski JFP (2013). Effect of birth weight and 12 weeks of exercise training on exercise‐induced AMPK signaling in human skeletal muscle. Am J Physiol Endocrinol Metab 304, E1379–E1390. [DOI] [PubMed] [Google Scholar]
- Mortensen B, Poulsen P, Wegner L, Stender‐Petersen KL, Ribel‐Madsen R, Friedrichsen M, Birk JB, Vaag A & Wojtaszewski JFP (2009). Genetic and metabolic effects on skeletal muscle AMPK in young and older twins. Am J Physiol Endocrinol Metab 297, E956–E964. [DOI] [PubMed] [Google Scholar]
- Munday MR (2002). Regulation of mammalian acetyl‐CoA carboxylase. Biochem Soc Trans 30, 1059–1064. [DOI] [PubMed] [Google Scholar]
- Nelson RK & Horowitz JF (2014). Acute exercise ameliorates differences in insulin resistance between physically active and sedentary overweight adults. Appl Physiol Nutr Metab 39, 811–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen JN, Mustard KJW, Graham DA, Yu H, MacDonald CS, Pilegaard H, Goodyear LJ, Hardie DG, Richter Ea & Wojtaszewski JFP (2003). 5′‐AMP‐activated protein kinase activity and subunit expression in exercise‐trained human skeletal muscle. J Appl Physiol 94, 631–641. [DOI] [PubMed] [Google Scholar]
- Pehmøller C, Brandt N, Birk JB, Høeg LD, Sjøberg KA, Goodyear LJ, Kiens B, Richter EA & Wojtaszewski JFP (2012). Exercise alleviates lipid‐induced insulin resistance in human skeletal muscle‐signaling interaction at the level of TBC1 domain family member 4. Diabetes 61, 2743–2752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter EA, Garetto LP, Goodman MN & Ruderman NB (1982). Muscle glucose metabolism following exercise in the rat: increased sensitivity to insulin. J Clin Invest 69, 785–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter EA, Garetto LP, Goodman MN & Ruderman NB (1984). Enhanced muscle glucose metabolism after exercise: modulation by local factors. Am J Physiol Endocrinol Metab 246, E476–E482. [DOI] [PubMed] [Google Scholar]
- Richter EA, Mikines KJ, Galbo H & Kiens B (1989). Effect of exercise on insulin action in human skeletal muscle. J Appl Physiol 66, 876–885. [DOI] [PubMed] [Google Scholar]
- Richter EA, Derave W & Wojtaszewski JFP (2001). Glucose, exercise and insulin: emerging concepts. J Physiol 535, 313–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riddle MC (2008). Starting and advancing insulin for type 2 diabetes: algorithms and individualized methods are both necessary. J Clin Endocrinol Metab 93, 372–374. [DOI] [PubMed] [Google Scholar]
- Sano H, Kane S, Sano E, Miinea CP, Asara JM, Lane WS, Garner CW & Lienhard GE (2003). Insulin‐stimualted phosphorylation of a Rab GTPase‐activiating protein regulates GLUT4 translocation. J Biol Chem 278, 14599–14602. [DOI] [PubMed] [Google Scholar]
- Sjøberg KA, Frøsig C, Kjøbsted R, Sylow L, Kleinert M, Betik AC, Shaw CS, Kiens B, Wojtaszewski JFP, Rattigan S, Richter EA & McConell GK (2017). Exercise increases human skeletal muscle insulin sensitivity via coordinated increases in microvascular perfusion and molecular signaling. Diabetes 66, 1501–1510. [DOI] [PubMed] [Google Scholar]
- Treebak JT, Frøsig C, Pehmøller C, Chen S, Maarbjerg SJ, Brandt N, MacKintosh C, Zierath JR, Hardie DG, Kiens B, Richter EA, Pilegaard H & Wojtaszewski JF. (2009). Potential role of TBC1D4 in enhanced post‐exercise insulin action in human skeletal muscle. Diabetologia 52, 891–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treebak JT, Taylor EB, Witczak CA, An D, Toyoda T, Koh H‐J, Xie J, Feener EP, Wojtaszewski JFP, Hirshman MF & Goodyear LJ (2010). Identification of a novel phosphorylation site on TBC1D4 regulated by AMP‐activated protein kinase in skeletal muscle. AJP Cell Physiol 298, C377–C385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Arias EB, Pataky MW, Goodyear LJ & Cartee GD (2018). Postexercise improvement in glucose uptake occurs concomitant with greater γ3‐AMPK activation and AS160 phosphorylation in rat skeletal muscle. Am J Physiol Metab. DOI: 10.1152/ajpendo.00020.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Arias EB, Pataky MW, Goodyear LJ & Cartee GD (2018). Postexercise improvement in glucose uptake occurs concomitant with greater γ3‐AMPK activation and AS160 phosphorylation in rat skeletal muscle. Am J Physiol Metab. DOI: 10.1152/ajpendo.00020.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojtaszewski JF, Hansen BF, Gade, Kiens B , Markuns JF, Goodyear LJ & Richter Ea (2000). Insulin signaling and insulin sensitivity after exercise in human skeletal muscle. Diabetes 49, 325–331. [DOI] [PubMed] [Google Scholar]
- Wojtaszewski JFP, Birk JB, Frøsig C, Holten M, Pilegaard H & Dela F (2005). 5′ AMP activated protein kinase expression in human skeletal muscle: Effects of strength training and type 2 diabetes. J Physiol 564, 563–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojtaszewski JFP, Hansen BF, Kiens B & Richter EA (1997). Insulin signaling in human skeletal muscle: time course and effect of exercise. Diabetes 46, 1775–1781. [DOI] [PubMed] [Google Scholar]
- Wojtaszewski JFP, Jørgensen SB, Frøsig C, MacDonald C, Birk JB & Richter EA (2003a). Insulin signalling: effects of prior exercise. Acta Physiol Scand 178, 321–328. [DOI] [PubMed] [Google Scholar]
- Wojtaszewski JFP, MacDonald C, Nielsen JN, Hellsten Y, Hardie DG, Kemp BE, Kiens B & Richter EA (2003b). Regulation of 5′AMP‐activated protein kinase activity and substrate utilization in exercising human skeletal muscle. Am J Physiol Endocrinol Metab 284, E813–E822. [DOI] [PubMed] [Google Scholar]
- Zheng X & Cartee GD (2016). Insulin‐induced effects on the subcellular localization of AKT1, AKT2 and AS160 in Rat Skeletal Muscle. Sci Rep DOI: 10.1038/srep39230. [DOI] [PMC free article] [PubMed] [Google Scholar]