

Abstract
Key points
Leptin is a potent respiratory stimulant.
A long functional isoform of leptin receptor, LepRb, was detected in the carotid body (CB), a key peripheral hypoxia sensor. However, the effect of leptin on minute ventilation (V E) and the hypoxic ventilatory response (HVR) has not been sufficiently studied.
We report that LepRb is present in approximately 74% of the CB glomus cells.
Leptin increased carotid sinus nerve activity at baseline and in response to hypoxia in vivo.
Subcutaneous infusion of leptin increased V E and HVR in C57BL/6J mice and this effect was abolished by CB denervation.
Expression of LepRb in the carotid bodies of LepRb deficient obese db/db mice increased VE during wakefulness and sleep and augmented the HVR.
We conclude that leptin acts on LepRb in the CBs to stimulate breathing and HVR, which may protect against sleep disordered breathing in obesity.
Abstract
Leptin is a potent respiratory stimulant. The carotid bodies (CB) express the long functional isoform of leptin receptor, LepRb, but the role of leptin in CB has not been fully elucidated. The objectives of the current study were (1) to examine the effect of subcutaneous leptin infusion on minute ventilation (V E) and the hypoxic ventilatory response to 10% O2 (HVR) in C57BL/6J mice before and after CB denervation; (2) to express LepRb in CB of LepRb‐deficient obese db/db mice and examine its effects on breathing during sleep and wakefulness and on HVR. We found that leptin enhanced carotid sinus nerve activity at baseline and in response to 10% O2 in vivo. In C57BL/6J mice, leptin increased V E from 1.1 to 1.5 mL/min/g during normoxia (P < 0.01) and from 3.6 to 4.7 mL/min/g during hypoxia (P < 0.001), augmenting HVR from 0.23 to 0.31 mL/min/g/Δ (P < 0.001). The effects of leptin on V E and HVR were abolished by CB denervation. In db/db mice, LepR b expression in CB increased V E from 1.1 to 1.3 mL/min/g during normoxia (P < 0.05) and from 2.8 to 3.2 mL/min/g during hypoxia (P < 0.02), increasing HVR. Compared to control db/db mice, LepRb transfected mice showed significantly higher V E throughout non‐rapid eye movement (20.1 vs. −27.7 mL/min respectively, P < 0.05) and rapid eye movement sleep (16.5 vs 23.4 mL/min, P < 0.05). We conclude that leptin acts in CB to augment V E and HVR, which may protect against sleep disordered breathing in obesity.
Keywords: Leptin, Carotid Body, Sleep Apnoea
Key points
Leptin is a potent respiratory stimulant.
A long functional isoform of leptin receptor, LepRb, was detected in the carotid body (CB), a key peripheral hypoxia sensor. However, the effect of leptin on minute ventilation (V E) and the hypoxic ventilatory response (HVR) has not been sufficiently studied.
We report that LepRb is present in approximately 74% of the CB glomus cells.
Leptin increased carotid sinus nerve activity at baseline and in response to hypoxia in vivo.
Subcutaneous infusion of leptin increased V E and HVR in C57BL/6J mice and this effect was abolished by CB denervation.
Expression of LepRb in the carotid bodies of LepRb deficient obese db/db mice increased VE during wakefulness and sleep and augmented the HVR.
We conclude that leptin acts on LepRb in the CBs to stimulate breathing and HVR, which may protect against sleep disordered breathing in obesity.
Introduction
The body has several compensatory mechanisms in response to hypoxia. One of these is the ability to increase ventilation, which is called the hypoxic ventilatory response (HVR) (Teppema & Dahan, 2010). The HVR is a complex phenomenon. Peripheral sensing of hypoxia occurs predominantly in the carotid bodies (CB), which are located in the carotid artery bifurcation with the aortic bodies playing a secondary role. The CB glomus (type I) cells sense hypoxia and transmit chemosensory input via the carotid sinus nerve (CSN), a branch of the glossopharyngeal nerve, to the nucleus of the solitary tract (NTS) and to the brainstem respiratory network (Gonzalez‐Martin et al. 2011; Silva & Schreihofer, 2011; Prabhakar et al. 2012; Nurse & Piskuric, 2013; Prabhakar, 2013). The HVR is time dependent and the isocapnic HVR can be divided in two phases, a first phase (0–5 min) of hyperventilation followed by a second phase of a slow decline (5–20 min). The first phase is predominantly peripheral governed by the CB activity and the initial 1–2 min is exclusively peripheral, whereas both peripheral and central chemoreception contribute to the second phase (Powell et al. 1998; Duffin, 2007; Teppema & Dahan, 2010).
Abnormal HVR plays an important role in the pathogenesis of sleep disordered breathing (SDB), which is one of the most common complications of obesity. Obstructive sleep apnoea, i.e. recurrent closure of the upper airway during sleep, is observed in >50% of obese individuals (Tufik et al. 2010; Peppard et al. 2013), and is associated with an abnormally augmented HVR (Younes et al. 2007; Pialoux et al. 2009; Trombetta et al. 2013; Younes, 2014; Mateika, 2015). Obesity may also lead to alveolar hypoventilation during sleep due to respiratory depression, which can be present independent of obstructive sleep apnoea (Mokhlesi, 2010). Obesity hypoventilation is characterized by suppressed HVR (Zwillich et al. 1975). The mechanisms by which obesity affects the HVR are not fully understood.
Recent reports suggest that the CB express receptors for leptin. Leptin is an adipocyte‐produced hormone, levels of which increase exponentially with increased obesity (Maffei et al. 1995; Considine et al. 1996). LepRb, the longest isoform of leptin receptor (LepR) primarily responsible for leptin signalling (Chen et al. 1996), is abundantly expressed in the CB (Porzionato et al. 2011; Messenger & Ciriello, 2013; Messenger et al. 2013; Ciriello & Caverson, 2014). Leptin acts in medullary centres to regulate responses to hypercapnia (O'Donnell et al. 1999; Inyushkina et al. 2010; Bassi et al. 2012, 2014; Yao et al. 2016). Leptin deficiency causes central alveolar hypoventilation and obstructive sleep apnoea in rodents (O'Donnell et al. 1999; Polotsky et al. 2001, 2012; Pho et al. 2016; Yao et al. 2016) and may predispose obese humans to sleep apnoea (Shapiro et al. 2014). Leptin increases minute ventilation (V E) in rats and mice during normoxia (O'Donnell et al. 1999; Yao et al. 2016; Ribeiro et al. 2018) and during hypoxic exposure (Ribeiro et al. 2018; Yuan et al. 2018) and augments CSN activity in response to anoxia ex vivo (Shirahata et al. 2015; Ribeiro et al. 2018). In awake lean Zucker rats, leptin did not change baseline V E, but increased HVR and the effect of leptin on HVR was abolished by CB denervation (Yuan et al. 2018). However, many aspects of leptin's action remain unknown. It is unclear if leptin acts in the CB to augment baseline V E or only the HVR and whether leptin regulates CSN activity in vivo during exposure to physiologically relevant levels of hypoxia. The role of leptin's action in the CB in the pathogenesis of obesity‐induced SDB is unknown.
We hypothesized that leptin acts in the CB to increase the HVR and minute ventilation during sleep and wakefulness. We elucidated the effects of leptin in the CB by several complementary approaches. First, we examined the effects of intravenous leptin on CSN activity during 100% hyperoxia, when CB are inactivated, and in response to 10% O2 hypoxia in vivo in C57BL/6J mice. Second, we performed continuous infusion of leptin in lean C57BL/6J mice, raising its levels to the values observed in obese mice and measured minute ventilation and the HVR in unanaesthetized unrestrained animals before and after CNS dissection (CSND). Third, we expressed LepRb in the CB of LepRb‐deficient obese db/db mice and evaluated minute ventilation and HVR during wakefulness and breathing during sleep.
Methods
Ethical approval
All animal surgical procedures were done using aseptic techniques under 1–2% isoflurane anaesthesia. After survival surgeries, mice were housed in a recovery chamber, and their behaviour was monitored. Buprenorphine 0.05–0.1 mg/kg (SQ) was administered as required based on signs of distress or pain (e.g. audible noises, no movement, eating or drinking behaviours). Upon completion of the experimental protocols or in case of ill appearance and/or persistent distress mice were killed under isoflurane anaesthesia by an overdose of pentobarbital (60 mg i.p.). The study was approved by the Johns Hopkins University Animal Use and Care Committee (Protocols MO15M257 and MO16M161) and complied with the American Physiological Society Guidelines for Animal Studies.
Animals and overall design
In total, 42 adult male C57BL/6J mice (Stock #000664), 30 adult male db/db mice on the C57BL/6J background (Stock #000697) 8–10 weeks of age, from the Jackson Laboratory (Bar Harbor, MA, USA), and five male LepRb‐EGFP mice were used in our experiments. LepRb‐EGFP mice were generated by breeding LepRb‐Cre [B6.129(Cg)‐Leprtm2(cre)Rck/J, Stock #008320] and enhanced green fluorescent protein (EGFP) reporter mice [B6;129‐Gt(ROSA)26Sortm2Sho/J, Stock #004077] from the Jackson Laboratory. All mice had free access to food and water and were housed under standard laboratory conditions at 22°C with the 12:12‐h light–dark cycle (0900–2100 h lights on/2100–0900 h lights off). C57BL/6J mice were used for the HVR measurements (n = 17), for arterial blood gas (ABG) measurements (n = 7) and for CSN activity measurements (n = 18, including ten mice treated with leptin and eight mice treated with vehicle, i.e. saline). For the HVR experiments, mice were acclimatized for 4–7 days to the plethysmography chamber. A subcutaneous osmotic pump (DURECT, Cupertino, CA, USA) filled with saline was implanted and after 2 days of recovery, the HVR was measured (see the HVR protocol below). Subsequently, a subcutaneous osmotic pump for leptin infusion (120 μg/day for 2 days) was implanted and 48 h later the HVR was measured. After this, CSND (n = 12) or CSND sham surgery (n = 5) was performed and, after 5–7 days of recovery, the HVR was determined following the same protocol (Fig. 1 A). Weight, temperature and food intake were measured during leptin and saline infusions and blood samples were taken by retro‐orbital puncture under 1–2% isoflurane anaesthesia to measure leptin levels in plasma. LepRb receptor deficient db/db mice were used for the HVR experiment before and after virus infection (n = 15), for sleep studies (n = 10) and for staining and CB morphometry (n = 5). For the HVR, mice were acclimatized for 3–5 days to the plethysmography chamber, and baseline measurements were then taken. Subsequently, the CB area was infected with Ad‐LacZ (n = 6) or Ad‐ LepRb (n = 9) adenoviral vectors (see protocol below) and the HVR measurements were repeated over 15 days and blood samples were taken to measure plasma leptin levels. Sleep studies were performed 15 days after Ad‐LacZ (n = 5) and after Ad LepRb infections (n = 5).
Figure 1. The effect of leptin on the hypoxic ventilatory response in C57BL/6J mice.
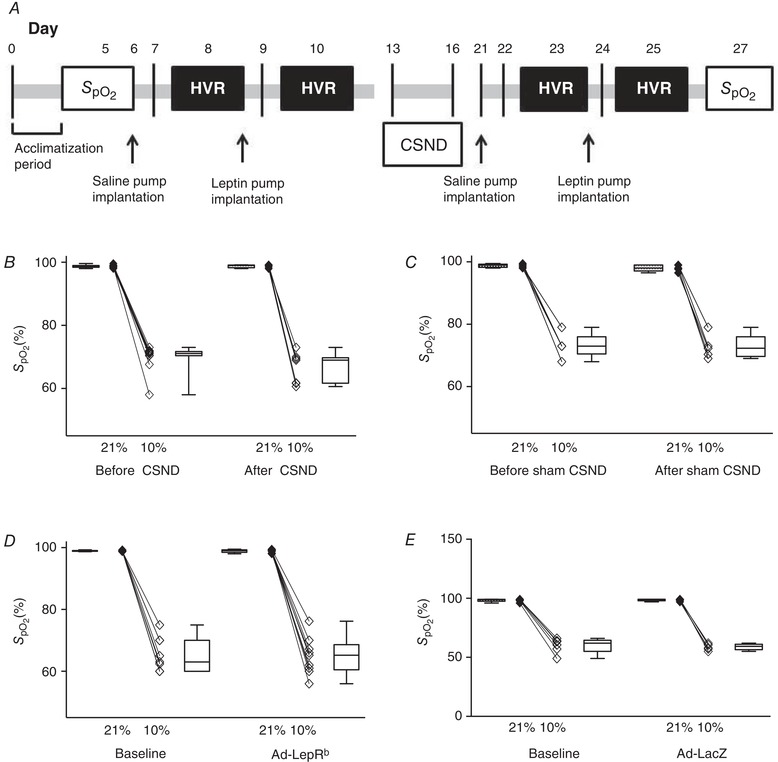
A, experimental design of the hypoxic ventilatory response (HVR) experiment in C57BL/6J mice before and after subcutaneous leptin infusion via an osmotic pump (120 μg/day) before and after carotid sinus nerve denervation (CSND); B and C, oxyhaemoglobin saturation () in C57BL/6J mice exposed to normoxia and hypoxia before and after CSND (n = 9, B) or sham surgery (n = 5, D); D and E, in db/db mice exposed to normoxia and hypoxia at baseline and after carotid body area infection with Ad‐LacZ (n = 6, D) or Ad‐LepRb (n = 9, E).
Carotid sinus nerve dissection protocol
CSND was performed as previously described by our group (Shin et al. 2014) with modifications. Briefly, the surgery was performed under 1–2% isoflurane anaesthesia and body temperature was maintained at 37°C. The CSNs were bilaterally dissected and painted with a solution of 2% phenol diluted in ethanol at the points of branching from the glossopharyngeal nerve to the cranial pole of the CB. To prevent discomfort, buprenorphine at 0.05 mg/kg/day s.c. was administered. Mice recovered for at least 5–7 days. Sham surgery was performed in a similar manner except that the CSNs were not severed.
Ad‐LacZ and Ad‐LepR infection of the CB area in db/db mice
For the leptin receptor long LepRb isoform expression in the CB, db/db mice were anaesthetized with 2% isoflurane, and the carotid artery bifurcation areas were carefully exposed bilaterally. The adenovirus harbouring the LepRb gene (Ad‐LepRb) or control virus Ad‐LacZ, provided by Dr Christopher Rhodes (University of Chicago) were suspended in Matrigel matrix (BD Biosciences, Bedford, MA, USA) at 1:5 and 5 μL of the viral suspension was applied to the CB area bilaterally at a final concentration of 0.86 × 1012 plaque‐forming units (pfu)/mL. Following this procedure, skin was re‐apposed and the incision was carefully closed. The HVR measurements were performed 15 days later, allowing for recovery and adenovirus expression in the CB area. In addition, five db/db mice were transfected with Ad‐LepRb‐GFP (Vector Biolabs, Malvern, PA., USA; 2–5 × 1010 pfu/mL) for morphometry.
Arterial blood gases experiment
An arterial catheter was implanted in the left femoral artery under 1–2% isoflurane anaesthesia. The femoral artery was carefully exposed via a 0.5 cm incision and an arterial catheter was inserted 5–8 mm deep into the femoral artery, glued in place and routed under the skin. The skin was sutured with 6.0 silicon‐coated silk sutures. The catheter was attached to a single‐channel fluid swivel (model 375/25, Instech Laboratories, Plymouth Meeting, PA, USA) and perfused slowly by an infusion pump (0.5 mL/day) with a sterile saline solution containing heparin (1000 U heparin/L saline). Mice were allowed to recover for a minimum of 48 h prior to blood gas testing, which was performed in awake anaesthetized unrestrained mice removing 150 μL of arterial blood, which was processed in a blood analyser (Radiometer ABL 800 Flex, Diamond Diagnostics, Holliston, MA, USA). The alveolar–arterial gradient for partial pressure of O2 (P AO2 − ) was calculated at room air conditions before and after CSND (n = 7) assuming that P AO2 = (P atm − P H2O) − /0.8.
Body temperature measurements
Body temperature was measured at 10.00 h after overnight acclimation to thermoneutral conditions by a rectal probe in awake C57BL/6J mice at baseline and during saline and leptin infusions and in db/db mice before and 15 days after Ad‐LepRb or Ad‐LacZ control CB infection. Body temperature measurements were repeated before and after the HVR measurements and sleep studies.
Pulse oximetry
Oxyhaemoglobin saturation () was measured by pulse oximetry in 10% and 20.9% O2 by applying a Mouse CollarClip (Starr Life Sciences, Oakmont, PA, USA).
Minute ventilation and the hypoxic ventilatory response
HVR measurements were performed at thermoneutral conditions (30°C) in a neonatal incubator (Draeger 8000 IC), which has been adapted for respiratory and metabolic measurements in our laboratory (Jun et al. 2013). Mice were acclimatized overnight prior to the measurements. Tidal volume (V T), respiratory rate and minute ventilation (V E) were measured in C57BL/6J mice before and after CSND during saline and leptin infusions, and in db/db mice before and after virus infection as previously described (Polotsky et al. 2001, 2004) with modifications according to Hernandez et al. (2012). The methodology of our ventilatory measurements has been previously described in detail (Hernandez et al. 2012). Briefly, the animals were placed in a whole body barometric plethysmography (WBP) chamber (Buxco, Wilmington, NC, USA) with internal diameter of 80 mm, height of 50.5 mm and volume approximately of 0.4 L. The chamber consisted of a sealed animal chamber, a reference chamber and a platform inside the chamber to support the mouse (Pho et al. 2016). The chamber was equipped with two high‐resistance ports on the upper surface and one large side port and three small side ports at the base. The inflow and outflow high‐resistance ports were connected to positive and negative pressure sources to generate a steady bias flow through the mouse chamber preventing CO2 accumulation. The bias flow inlet and outlet ports were adjusted to maintain chamber at atmospheric pressure and to keep the time constant of the chamber much longer than the inspiratory time of the animal, thereby preventing pressure leakage during respiration (Hernandez et al. 2012). This approach, based on previous work (Pappenheimer, 1977; Jacky, 1978), allowed us to use the chamber as if it was completely sealed. The Drorbaugh and Fenn equation was used to calculate WBP tidal volume signal from the WBP chamber pressure signal (Drorbaugh & Fenn, 1955). Application of this formula required measurement of the following variables during each WBP recording session: mouse rectal temperature, chamber temperature, room temperature and relative humidity, and chamber gas constant, which was calculated by utilizing a known‐volume injection and the resultant WBP pressure deflection. Calibration injections of fixed 1 mL volume of room air were made with the animal inside the constant‐volume chamber. All signals were digitized at 1000 Hz (sampling frequency per channel) and recorded in LabChart 7 Pro (version 7.2). WBP pressure was recorded with a differential pressure transducer (no. 3613, Emka Technologies, Falls Church, VA, USA). A first‐order derivative (dV/dt) was applied to the WBP tidal volume signal to yield a WBP tidal airflow signal (Hernandez et al. 2012). All respiratory measurements in our systems were previously validated against gold standard pneumotachographic measures of tracheal pressure in anaesthetized mice (Hernandez et al. 2012).
During both normoxia (20.9% O2) and hypoxia (10% O2 + 3% CO2), air flow was constant. and was regulated by the mass flow controller. Once the chamber was at constant volume, V T (tidal volume) and respiratory rate (RR) were derived from changes in pressure (Statham Gould PM15E differential pressure transducer, Hato Ray, Puerto Rico). Minute ventilation () was reported as (V T × RR)/body weight and was measured during quiet wakefulness at baseline, 20.99% O2 for 25 min three times at least 30 min apart, and in response to 10% O2 + 3% CO2 for 5 min three times at least 30 min apart. To eliminate inhibitory effects of hypocapnia on the HVR, we used a 10% O2 and 3% CO2 mixture for our measurements. During normoxia was 20.9% and CO2 was 0.4%. After switching to hypoxia, decreased to 10% and CO2 increased to 3% within a 30 s interval.
In C57BL/6J mice, the HVR was measured before and after CSND or sham surgery and in db/db mice, it was measured before and after Ad‐LacZ or Ad‐ LepRb infection. Relationships between and in C57BL/6J mice before and after CSND or sham surgery are presented in Fig. 1 B and C, and the same relationships in db/db mice before and after Ad‐LacZ or Ad‐ LepRb infection are presented in Fig. 1 D and E. In C57BL/6J mice, the bulk of the osmotic pump prevented accurate measurements during leptin or saline infusion. Therefore, the HVR was calculated as the ratio of [V E (10% O2) − V E (20.9% O2) to the change in (∆ = 10.9% between normoxia and hypoxia) and reported as ΔV E/Δ. In db/db mice, the HVR was reported both as ∆ V E/Δ and as a ratio of [V E (10% O2) − V E (20.9% O2) the change in oxyhaemoglobin saturation ( (20.9% O2) − (10% O2)] and reported as ΔV E/Δ. Given that the peripheral chemoreflex is more brisk and predominates during the first 2 min of hypoxic exposure (Powell et al. 1998; Teppema & Dahan, 2010; Duffin, 2007), we reported values during the first 2 min of hypoxic exposure. We also performed ABG measurements at normoxic conditions and during the first 2 min of hypoxic exposure to ensure isocapnoea during hypoxic exposure. In mice with intact and denervated CB, ABG showed similar levels of partial pressure of CO2 () at normoxic and hypoxic conditions (Table 1).
Table 1.
Arterial blood gas values in awake unrestrained C57BL/6J mice exposed to 21% O2 and 10% O2 + 3% CO2 before and after carotid sinus nerve dissection
| Intact carotid bodies | Carotid sinus nerve dissection | |||
|---|---|---|---|---|
| 21% O2 | 10% O2 + 3% CO2 | 21% O2 | 10% O2 + 3% CO2 | |
| pH | 7.4 ± 0.01 | 7.4 ± 0.01 | 7.4 ± 0.02 | 7.4 ± 0.001 |
| (mmHg) | 96.0 ± 4.6 | 52.3 ± 2.9 | 106.5 ± 7.7 | 53.0 ± 10.4 |
| (mmHg) | 28.1 ± 1.5 | 29.7 ± 1.5 | 31.3 ± 0.9 | 29.07 ± 0.7 |
Results are presented as mean ± SEM.
Sleep studies
EEG and EMG electrodes were implanted with an EEG/EMG Headmount (Pinnacle Technology, Lawrence, KS, USA) as previously described (Pho et al. 2016). Mice were allowed to recover for 3–4 days prior to polysomnography in a designated room where sleep recordings were later performed. This climate control sound‐proof room had a constant temperature of 27–28°C and the 12:12 h light‐dark cycle (lights on 09.00–21.00 h/lights off 21.00–09.00 h). Animals were placed in a modified whole body plethysmography open‐system chamber designed to record tidal airflow, respiratory effort and sleep–wake state continuously (Hernandez et al. 2012) as described above. During full polysomnographic recording sessions, the chamber was humidified to 90% relative humidity. Mice were habituated to the WBP chamber from 10.00 to 16.00 h 1 day before the recording session and 45 min to acclimatize to the chamber before recordings were initiated on the day of recording. Sleep recordings were performed for 6 h during the light phase (10.00 to 16.00 h). Tidal volume and respiratory movements were measured as described above, and EEG/EMG signals were recorded as previously described (Pho et al. 2016; Fleury Curado et al. 2018).
Sleep–wake state was scored visually in 5‐s epochs from 10.00 to 16.00 h. Standard criteria were employed to score sleep–wake state based on EEG and EMG frequency content and amplitude, as previously described (Pho et al. 2016). Studies were scored by two independent scorers (CCE and HP), who were blinded to study conditions. Wakefulness was characterized by low‐amplitude, high‐frequency (∼10 to 20 Hz) EEG waves and high levels of EMG activity compared with the sleep states. Non‐rapid eye movement (NREM) sleep was characterized by high‐amplitude, low‐frequency (∼2–5 Hz) EEG waves with EMG activity considerably less than during wakefulness. Rapid eye movement (REM) sleep was characterized by low‐amplitude, mixed‐frequency (∼5–10 Hz) EEG waves with EMG amplitude either below or equal to that during NREM sleep.
Ventilation was assessed by V E and its components, tidal volume and respiratory rate, during non‐flow‐limited and flow‐limited breathing. Obstruction was characterized by the development of inspiratory flow limitation as previously described (Pho et al. 2016). Briefly, the severity of airflow obstruction was defined by maximal inspiratory flow at the onset of flow limitation (V Imax), measured at the point of peak inspiratory airflow. Breaths were considered to be flow limited if (1) breaths had an early peak of inspiratory flow followed by a plateau or a decrease in flow, (2) a mid‐inspiratory plateau occupied ≥20% of inspiratory time between early and late inspiratory peaks in flow, or (3) when the late inspiratory airflow peak exceeded the early peak (Pho et al. 2016). Mean characterized the severity of gas exchange abnormalities across sleep/wake states. Because sleep stage can markedly affect SDB severity, we stratified analyses by NREM vs. REM sleep. The oxygen desaturation index (ODI) was defined as ≥ 4% oxyhaemoglobin desaturation from the baseline (ODI) for at least two breaths. We also measured the frequency of sighs defined as breaths with amplitude exceeding two tidal volumes as an indicator of respiratory control stability during sleep (Yamauchi et al. 2008).
Measurements of CSN activity in ventilated mice
Mice were anaesthetized with urethane 1.25 g kg−1 (i.p.) and ventilated via a tracheal tube by a modified volume‐controlled Harvard animal ventilator (Model 687, Harvard Apparatus, Holliston, MA, USA) under hyperoxia (100% O2) as previously described (Tankersley et al. 1994; Pichard et al. 2015). The right common carotid artery was cannulated for monitoring blood pressure and the right jugular vein was cannulated for leptin infusion. Subsequently, for neuromuscular blockade mice were given pancuronium bromide (0.03–0.04 mg kg−1, i.p.). The neck was dissected to expose the left carotid bifurcation and the glossopharyngeal nerve. CSN was identified and the central end was sectioned. The area including the CB and CSN was filled with Krebs solution. The sectioned end was then placed into a glass suction pipette containing a silver–silver chloride electrode and Krebs solution. Additionally, a silver–silver chloride electrode was placed in the surrounding solution to serve as the ground electrode. The signal was amplified (AC‐Preamplifier P15, Grass Instruments Co., West Warwick, RI, USA) and collected using digital recording software (AcqKnowledge, BioPac Systems, Inc., Goleta, CA, USA) together with airway pressure and arterial pressure. Baroreceptors were mechanically denervated in a manner similar to those previously described (Shirahata & Fitzgerald, 1991; Van Vliet et al. 1999). Successful removal of baroreceptors was confirmed when increased blood pressure did not accompany an increase in CSN activity. Functional viability of the CB and CSN was assessed prior to the experiment and at the end by a brief exposure to 100% inspired N2, which leads to a vigorous increase in nerve activity. Following confirmation of a viable preparation, mice were left undisturbed for 5 min under hyperoxic conditions (100% O2). Following this 5‐min baseline, two hypoxic challenges (10% O2) of 90 s were performed with a 5‐min inter‐challenge period for recovery under hyperoxic conditions. Following the second hypoxic challenge and 5‐min recovery period, intravenous leptin (3 μg in 300 μL of saline over 45–60 s) was given and allowed to stabilize under hyperoxic conditions for 10 min. Following this stabilization period the above‐mentioned protocol was repeated to test hypoxic responses under leptin. Control experiments were conducted in a similar fashion expect that the same volume of saline was infused over the same time instead of leptin. For the analysis, each continuous 120‐s challenge (15‐s baseline, 90‐s challenge and 15‐s post‐challenge recovery period) was filtered using both low‐pass and high‐pass window‐based filters of 1000 and 250 Hz, respectively. Filters were selected based on data presented by Pichard, et al. (2015), which demonstrated that this approach would adequately capture the activity of the in vivo murine CSN activity in response to 10% hypoxia. Subsequently, nerve activity in each continuous challenge was broken down into adjacent 5‐s segments. Signal in each segment was then detrended, rectified and finally integrated. The maximum of the integrated signal represents the total neural activity for each 5‐s segment. For each hypoxic period, the sum of all 5‐s segments was taken as total neural output for the hypoxic challenge.
Plasma leptin levels
Plasma leptin levels in C57BL/6J and db/db mice were measured by Mouse Leptin ELISA kits (catalogue number EZML_82K) from Millipore (Billerica, MA, USA).
Immunohistochemistry in tissue sections
Mice were anaesthetized with 1–2% isoflurane and perfused transcardially using 500 mL ice‐cold phosphate buffered saline (PBS; 0.01 m, pH 7.4) followed by 4% paraformaldehyde (PF) in 0.1 m PBS. The carotid artery bifurcations with the CB were removed bilaterally, kept for 2 h in PF 4%, washed in PBS 0.1 m, cryoprotected in PBS with 30% sucrose and embedded in Tissue‐Tek O.C.T. compound, and frozen on dry ice. Coronal cryosections of 10 μm were obtained on Superfrost plus slides (Thermo Scientific, Waltham, MA, USA) using a cryostat (HM 560, Thermo Scientific) and tissue was stored at −20°C. For staining, cryosections were brought to room temperature for 10 min and then rehydrated with PBS. Antigen retrieval was subsequently performed using sodium citrate buffer (10 mm sodium citrate, 0.05% Tween 20, pH 6.0) at 95°C for 20 min followed by three washes for 10 min with PBS with 0.3% Triton X‐100. The sections were blocked at room temperature with 10% normal goat serum, 1% bovine serum albumin, 20 μL Triton X‐100 and 10 μL Tween 20 for 1 h followed by incubation with primary antibodies diluted in blocking solution overnight at 4°C. On the following day, sections were washed with PBS with 0.1% Triton X‐100 and incubated with the secondary antibodies for 1 h at room temperature and washed with PBS. Then, slides were coverslipped with mounting medium for fluorescence with DAPI (4,6‐diamidino‐2‐phenylindole; Vectashield, Vector Labs, Burlingame, CA, USA).
LepRb protein expression in the CB of LepRb‐EGFP mice was detected based on the presence of a reporter, EGFP (Scott et al. 2009). LepRb protein expression in LepRb‐deficient db/db mice after infection with Ad‐LepRb was also detected based on the presence of the reporter. We used tyrosine hydroxylase (TH) as a type I cell marker and glial fibrillary acidic protein (GFAP) as a type II cell marker (Lopez‐Barneo et al. 2009) For the anti‐TH staining in LepRb‐EGFP and in db/db transfected mice we used a chicken anti‐TH primary antibody (Abcam 1:500, catalogue number ab76442; Cambridge, MA, USA) and goat anti‐Chicken IgY secondary antibody, Alexa Fluor 568 (1:500, catalogue number A‐11041). For GFAP we used a rabbit anti‐GFAP antibody (Dako, 1:500, catalogue number Z0334) and goat anti‐rabbit secondary antibody, Alex Fluor 647 (1:500, catalogue number A‐21245). Fluorescence images were examined with an Inverted Axio Observer 3 microscope (Carl Zeiss, Jena, Germany) equipped with an Axiocam 512 camera. The images in LepRb‐EGFP mice were acquired at a resolution of 2363 × 2544 pixels with an LD A‐Plan 20×/0.35 Ph 1 objective and a resolution of 4248 × 2832 pixels with an LD A‐Plan 40×/0.55 Ph 1 objective. The images in db/db mice photos were acquired at a resolution of 2179 × 2194 pixels with an LD A‐Plan 20×/0.35 Ph 1 objective and a resolution of 4248 × 2832 pixels with an LD A‐Plan 40×/0.55 Ph 1 objective. The following filters were used: for DAPI, the led module‐385 nm filter; for Alexa 647 the led module‐630 nm filter, for Alexa 568 the led module‐ 567 nm filter, and for EGFP the led module‐475 nm filter. All images were processed to merge with the software Carl Zeiss Image.
We quantified the number of glomus (type I) cells and type II cells and the proportion of cells expressing leptin receptor (LepRb+ cells) from the immunohistochemistry images of CB of LepRb‐EGFP mice and Ad‐LepRb‐infected db/db mice. Cells were manually counted from images taken under a 20× or 40× microscope objective. All measurements were performed by the same investigator using the count tool of the Adobe Photoshop CS5.1 (Adobe Systems) software. First, we counted the total number of type I and II cells in the CB, merging the pictures of DAPI with GFAP (Alexa 647) and TH (Alexa 568). Quantification of LepRb cells was performed by merging the pictures of EGFP staining with a type I (glomus) cell marker TH (Alexa 568) and a type II cell marker GFAP (Alexa 647).
Statistical analyses
All analyses were performed using STATA version 14.0 (StataCorp LLC, College Station, TX, USA) and prism 7.0 (GraphPad Software, La Jolla, CA, USA). We used the Wilcoxon matched‐paired signed rank test, paired t test and Mann–Whitney U test to compare variables before and after specific treatments, and two‐way ANOVA to examine interactions and the independent effect of two treatments. Mixed effects regression models were performed to investigate the independent effects of treatments with random intercepts to account for differences between mice. P values < 0.05 were considered statistically significant.
Results
Leptin increased baseline minute ventilation and HVR in C57BL/6J mice and leptin's effects were abolished by CSND
The cellular distribution of LepRb in mouse CB was first determined in LepRb‐EGFP mice. Quantitative analysis of immunofluorescence images showed that TH+ type I cells represented 90.4 ± 1.1%, whereas GFAP+ type II cells represented 9.6 ± 1.3% of the CB cellular population (Fig. 2 A–C). The expression of LepRb in type I and type II CB cells was determined by EGFP fluorescence, which was present in 73.8 ± 5.9% of type I cells and 61.1 ± 9.3% of type II cells (Fig. 2). Of note, LepRb was not detected in the superior cervical ganglia (Fig. 2 A).
Figure 2. The long isoform of leptin receptor (LepRb) was expressed in tyrosine hydroxylase (TH)‐positive type I (glomus) cells and glial fibrillary acidic protein (GFAP)‐positive type II cells of the carotid bodies of LepRb‐EGFP mice.
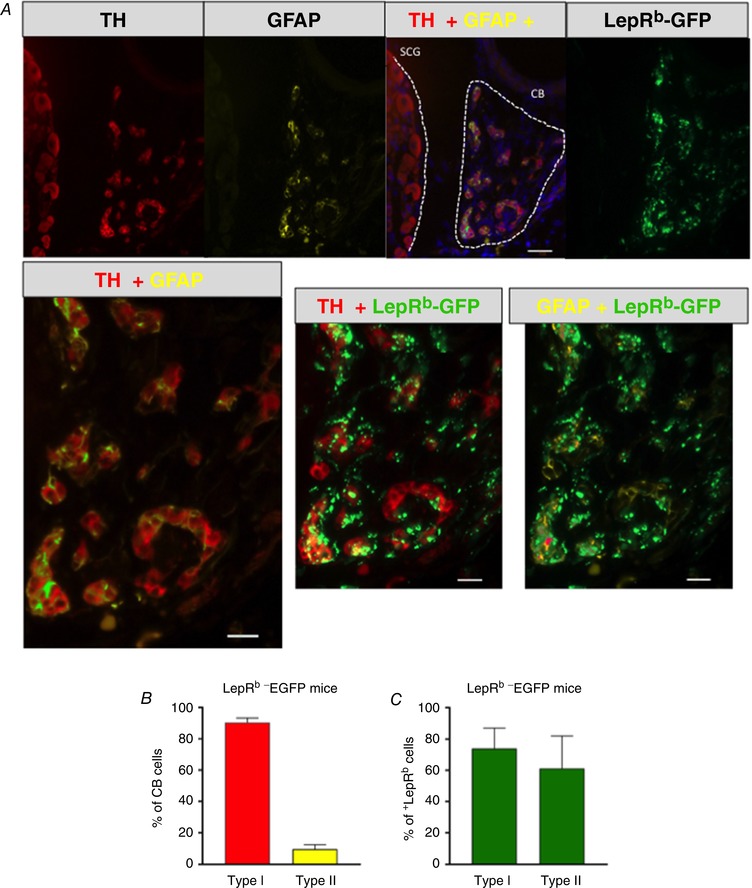
A, co‐localization of TH (red), GFAP (yellow) and LepRb‐EGFP (green) in the carotid bodies (CB) and superior cervical ganglion (SCG). Upper panel: 20, scale bar = 20 μm; lower panel, 40×, scale bar = 50 μm. B, percentage of type I (TH‐positive cells) and type II (GFAP‐positive cells) cells in the carotid bodies. C, percentage of type I and type II cells expressing LepRb (n = 5).
The effect of leptin on CSN activity was assessed in vivo in anaesthetized mechanically ventilated mice during 100% O2 hyperoxia and 10% O2 hypoxia. Leptin significantly increased CSN activity during hyperoxia and augmented CSN response to hypoxia (Fig. 3). In contrast, vehicle (saline) injections did not have an effect.
Figure 3. Leptin augmented the carotid sinus nerve (CSN) response to hypoxia in vivo .
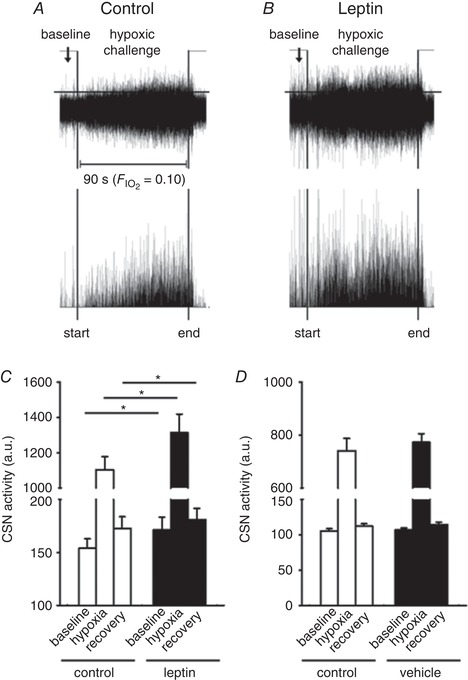
CSN was dissected in a sedated, tracheostomized and mechanically ventilated C57BL/6J mouse subject to neuromuscular blockade, which was exposed to 100% O2 (hyperoxia) followed by 10% O2 hypoxia before and after intravenous infusion of leptin (0.3 μg). Representative tracings of the CSN recordings before and after leptin infusions are shown in the upper panels of A and B, respectively. For clarity, the lower panels are plotted to show activities above the user‐defined threshold (horizontal lines). C, mean total CSN activity recorded before (baseline), during (hypoxia) and after (recovery) the hypoxic challenges in mice before and after leptin infusions (n = 10, * P < 0.05); D, mean total CSN activity recorded before and after vehicle (saline) infusions (n = 8).
HVR measurements were performed in unrestrained unanaesthetized C57BL/6J mice during quiet wakefulness before and after CSND or sham surgery. CSND or sham CSND did not affect mouse body weight or food intake, whereas, as expected, leptin increased body temperature and suppressed food intake resulting in weight loss, regardless of the CSND status (Tables 2 and 3). There was no significant change in body temperature during HVR measurements. For example, in mice with intact CB rectal temperature was 35.8 ± 0.1°C and 35.7 ± 0.1°C immediately before and after the measurements, respectively.
Table 2.
Basic characteristics of C57BL/6J mice during saline and leptin infusion before and after carotid sinus nerve dissection
| Intact carotid bodies | Carotid sinus nerve dissection | |||||||
|---|---|---|---|---|---|---|---|---|
| Leptin infusion (n = 12) | Saline infusion (n = 12) | Leptin (n = 9) | Saline infusion (n = 9) | |||||
| Baseline | 48 h Leptin pump | Baseline | 48 h Saline pump | Baseline | 48 h Leptin pump | Baseline | 48 h Saline pump | |
| Body weight (g) | 25.42 ± 0.7 | 24.13 ± 0.8*** | 25.31 ± 0.7 | 25.6 ± 07 | 25.9 ± 1.4 | 24.9 ± 1.1* | 26.5 ± 0.7 | 27.1 ± 0.6 |
| Body temperature (°C) | 35.2 ± 0.1 | 36.6 ± 0.2*** | 35.5 ± 0.1 | 35.7 ± 0.1 | 35.5 ± 0.2 | 36.6 ± 0.2* | 35.6 ± 0.2 | 35.6 ± 0.2 |
| Food intake (g/day) | 3 ± 0.1 | 1.35 ± 0.1** | 3.2 ± 0.1 | 3.4 ± 0.06 | 3.7 ± 0.01 | 1.5 ± 0.8*** | 3.6 ± 0.04 | 3.2 ± 0.8 |
| Leptin (ng/mL) | ND | 34.9 ± 4.3 | ND | 1.2 ± 0.3††† | ND | 36.1 ± 8.3 | ND | 1.5 ± 0.6†† |
* P < 0.05, ** P < 0.01, *** P < 0.001 for differences between baseline and leptin pump; †† P < 0.01 and ††† P < 0.001 for the difference between saline and leptin infusion. ND, not done. Results are presented as mean ± SEM.
Table 3.
Basic characteristics of C57BL/6J mice during saline and leptin infusion before and after sham surgery
| Intact carotid bodies | Sham carotid sinus nerve dissection | |||||||
|---|---|---|---|---|---|---|---|---|
| Leptin infusion (n = 5) | Saline infusion (n = 5) | Leptin (n = 5) | Saline infusion (n = 5) | |||||
| Baseline | 48 h Leptin pump | Baseline | 48 h Saline pump | Baseline | 48 h Leptin pump | Baseline | 48 h Saline pump | |
| Body weight (g) | 23.2 ± 0.8 | 21.7 ± 0.8*** | 22.5 ± 0.6 | 22.5 ± 0.6 | 24.6 ± 0.7 | 20.8 ± 0.4* | 23.2 ± 0.5 | 23.1 ± 0.5 |
| Body temperature (°C) | 35.9 ± 0.3 | 37.5 ± 0.3** | 35.5 ± 0.1 | 35.9 ± 0.3 | 35.8 ± 0.2 | 37.2 ± 0.07* | 35.4 ± 0.1 | 35.7 ± 0.1 |
| Food intake (g/day) | 3.9 ± 0.0 | 0.78 ± 0.03*** | 3.5 ± 0.18 | 3.2 ± 0.2 | 3.7 ± 0.15 | 0.9 ± 0.1*** | 4.6 ± 0.15 | 4.15 ± 0.15 |
| Leptin (ng/mL) | ND | 73.8 ± 30.2†† | ND | 1.1 ± 0.5 | ND | 143 ± 16.4†† | ND | 1.1 ± 0.6 |
* P < 0.05, ** P < 0.01, *** P < 0.001 for differences between baseline and leptin pump; †† P < 0.01 for the difference between saline and leptin infusion. ND, not done. Results are presented as mean ± SEM.
Hypoxic exposure resulted in severe oxyhaemoglobin desaturation with declining from a median value of 98.6% at normoxic conditions to 71% during hypoxia (Fig. 1 B). Prior to CSND, continuous infusion of leptin increased V E from 1.1 to 1.5 mL/min/g during normoxia (20.9% O2, P < 0.01 vs. saline infusion, Fig. 4 A) and from 3.6 to 4.7 mL/min/g during isocapnic hypoxia (10% O2 + 3% CO2, P < 0.001, Fig. 5 A). Leptin increased both respiratory rate, from 125 to 138 during normoxia (P < 0.05, Fig. 4 B) and from 224 to 234 during hypoxia (P < 0.05, Fig. 5 B), and tidal volume, from 0.009 to 0.011 mL/g during normoxia (P < 0.05, Fig. 4 C) and from 0.015 to 0.020 mL/g during hypoxia (P < 0.01, Fig. 5 C). Leptin increased the HVR, from 0.23 to 0.31 mL/min/g/Δ (P < 0.001, Fig. 5 D).
Figure 4. Leptin augmented minute ventilation (V E) at normoxic conditions (21% O2) and this effect was abolished by carotid sinus nerve dissection (CSND) in C57BL/6J mice during quiet wakefulness.
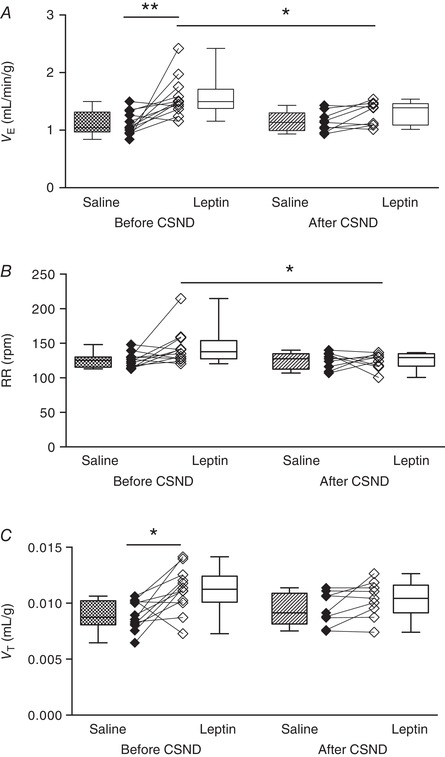
A, V E was calculated as tidal volume per body weight (V T) × respiratory rate (RR) at 21% O2 during saline and leptin continuous infusions (120 μg/day, s.c.) measured before (n = 12) and after (n = 9) CSND. B and C, RR (B) and V T (C) values at the same conditions. * P < 0.05, ** P < 0.01 using the Wilcoxon test for paired comparisons and Mann–Whitney test for unpaired comparisons.
Figure 5. Leptin augmented minute ventilation (V E) at isocapnic hypoxia (10% O2 + 3% CO2) and the hypoxic ventilatory response (HVR) and the effects were abolished by carotid sinus nerve dissection (CSND) in C57BL/6J mice during quiet wakefulness.
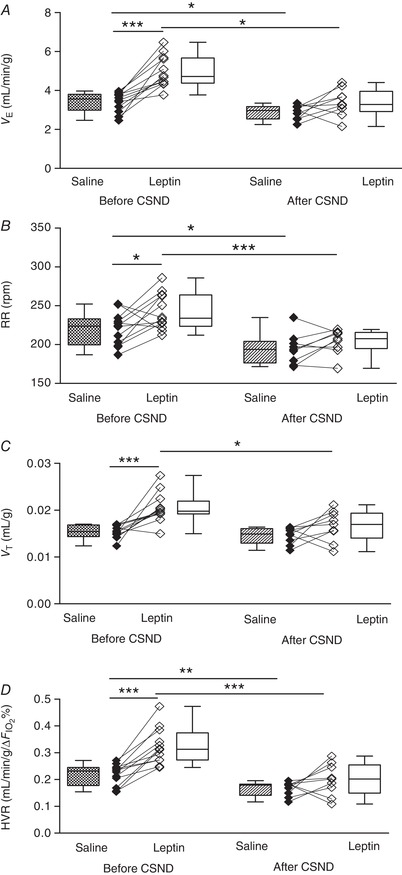
A, V E was calculated as tidal volume per body weight (V T) × respiratory rate (RR) at 10% O2 during saline and leptin continuous infusion (120 μg/day, s.c.) measured before (n = 12) and after (n = 9) CSND. B and C, RR (B) and V T (C) values at the same conditions. D, HVR represents the ratio of a change in V E to a change in the fraction of inspired oxygen (HVR = ΔV E/Δ) at the conditions described above. * P < 0.05, ** P < 0.01, *** P < 0.001 using the Wilcoxon test for paired comparisons and Mann–Whitney test for unpaired comparisons.
CSND had no effect on , or (Fig. 1 C, Table 1), although there was a significant decline in the alveolar–arterial O2 gradient at room air conditions, from 18.6 ± 4.8 mmHg before CSND to 0 ± 5.7 mmHg after CSND (P < 0.05), suggesting improvement in ventilation/perfusion matching. CB denervation did not affect V E at normoxic conditions (Fig. 4). In contrast, CSND significantly decreased V E and respiratory rate during hypoxic exposure and markedly attenuated the HVR (Fig. 5). CSND abolished the effects of leptin on minute ventilation during normoxia (Fig. 4 A) and hypoxia (Fig. 5 A) and abolished the leptin‐induced increase in HVR (Fig. 5 D). In contrast to CSND, sham surgery did not affect minute ventilation or HVR and did not attenuate the effect of leptin (Fig. 6).
Figure 6. Leptin augmented minute ventilation (V E) and the hypoxic ventilatory response (HVR) and this effect was not modified by sham carotid sinus nerve dissection (CSND) in C57BL/6J mice during quiet wakefulness.
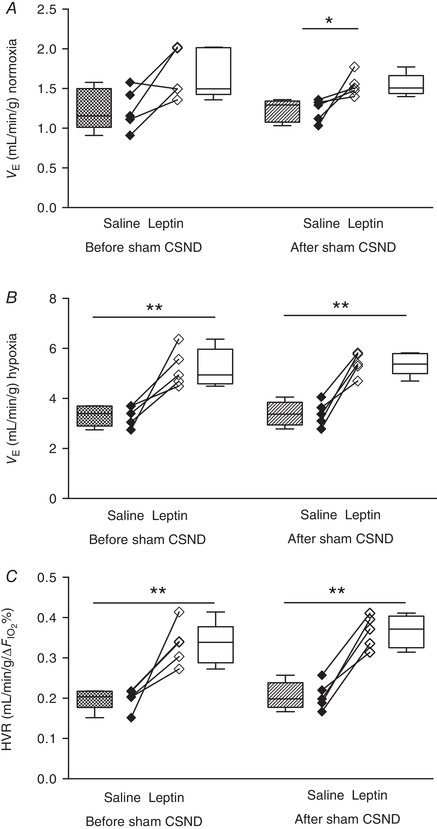
A and B, V E was calculated as tidal volume per body weight (V T) × respiratory rate (RR) at 21% O2 (A) and 10% O2 (B) during saline and leptin continuous infusion (120 μg/day, s.c.) measured before (n = 5) and after (n = 5) sham CSND. C, HVR represents the ratio of a change in V E to a change in the fraction of inspired oxygen (HVR = ΔV E/Δ). * P < 0.05, ** P < 0.01 using the Mann–Whitney test for unpaired comparisons.
LepRb expression in the CB of LepRb‐deficient db/db mice increased baseline minute ventilation in awake and sleeping mice and increased the HVR
The cellular composition of the CB in db/db mice was similar to that in LepRb‐EGFP mice; 86.5 ± 1.5% of all cells were TH+ type I cells and 13.5 ± 1.5% were GFAP+ type II cells (Fig. 7). Ad‐LepR b infection of the CB sites resulted in abundant expression of the long isoform of leptin receptor (LepRb), which was present in 100% of type I and type II cells (Fig. 7). Of note, Ad‐LepRb infection was not specific to the CB, involving other adjacent tissues such as the superior cervical ganglion neurons (Fig. 7).
Figure 7. The long functional isoform of leptin receptor (LepRb) was expressed in tyrosine hydroxylase (TH)‐positive type I (glomus) cells and glial fibrillary acidic protein (GFAP)‐positive type II cells of the carotid body of LepRb‐deficient obese db/db mice after Ad‐LepRb‐ GFP infection.
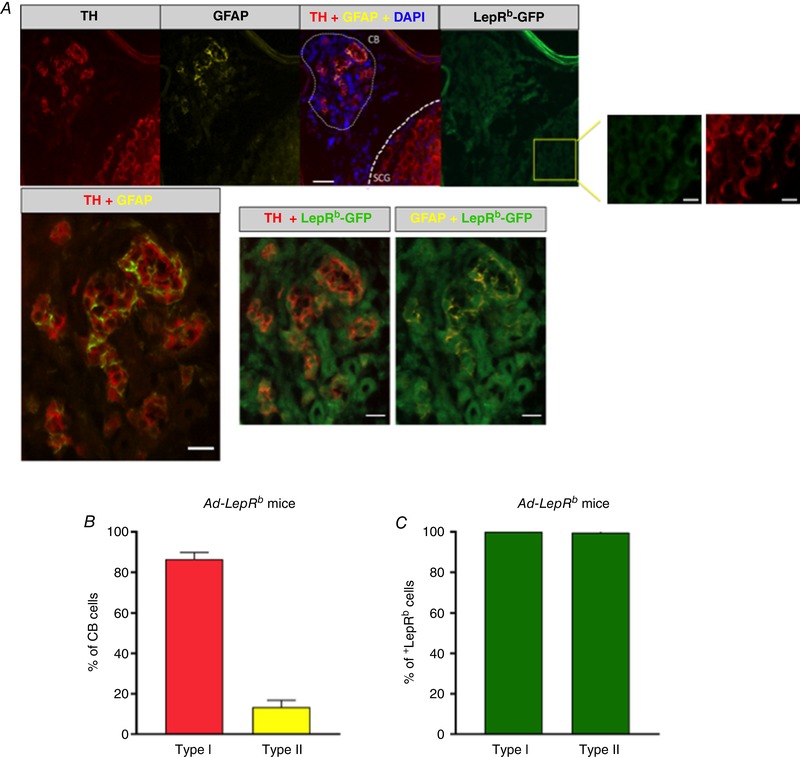
A, co‐localization of TH (red), GFAP (yellow) and LepRb‐GFP (green) in the carotid bodies (CB) and superior cervical ganglion (SCG). The enlarged insert on the right shows LepRb‐GFP‐TH‐positive cells in SCG. Upper panel: 20×, scale bar = 50 μm, scale bar for the inserts is 20 μm; lower panel: 40×, scale bar = 20 μm. B, percentage of type I (TH‐positive) cells and type II (GFAP‐positive) cells in the carotid bodies. C, percentage of type I and type II cells expressing LepRb (n = 5).
LepRb expression in the CB did not affect food intake, rectal temperature or body weight and there was no difference in circulating leptin levels between Ad‐LepRb‐ and Ad‐LacZ‐treated db/db mice (Table 4). As expected (Handa et al. 2014), circulating leptin levels in db/db mice were similar to those in C57BL/6J mice during infusion of leptin (compare Tables 2 and 4).
Table 4.
Basic characteristics of Ad‐LepRb and Ad‐LacZ infected db/db mice
| Ad‐LacZ | Ad‐LepRb | |||
|---|---|---|---|---|
| Baseline | After infection | Baseline | After infection | |
| Sample number (N) | 11 | 11 | 14 | 14 |
| Body weight (g) | 51.0 ± 1.9 | 54.7 ± 2.9 | 50.3 ± 1.7 | 52.4 ± 1.7 |
| Body temperature (°C) | 34.6 ± 0.2 | 34.7 ± 0.3 | 34.4 ± 0.2 | 34.5 ± 0.3 |
| Food intake (g/day) | 5.5 ± 0.5 | 5.5 ± 1.1 | 5.2 ± 0.2 | 5.2 ± 0.2 |
| Leptin (ng/mL) | ND | 62.1 ± 3.24 | ND | 57.2 ± 5.9 |
ND, not done. Results are presented as mean ± SEM.
V E was measured in db/db mice during quiet wakefulness before and 15 days after Ad‐LepRb or Ad‐LacZ infections. Hypoxic exposure resulted in severe oxyhaemoglobin desaturations with median decreasing from 98.8% to 63% and there was no significant effect of Ad‐LepRb (Fig. 1 D and E). LepRb expression in the CB increased minute ventilation from 1.1 to 1.3 mL/min/g during normoxia (P < 0.05, Fig. 8 A) and from 2.8 to 3.2 mL/min/g during hypoxia (P < 0.05, Fig. 9 A). The expression of the long isoform of leptin receptor increased tidal volumes (Figs. 8 E and 9 E), while respiratory rate was not significantly changed (Figs. 8 C and 9 C). LepRb expression consistently increased the HVR when quantified with either Δ or Δ (P < 0.05, Fig. 10 A and C). Control Ad‐LacZ infection did not affect any of the respiratory parameters (Figs. 8, 9, 10). Short hypoxic exposures during HVR measurements did not significantly decrease body temperature.
Figure 8. LepRb expression in the carotid bodies (CB) of LepRb‐deficient db/db mice increased minute ventilation (V E) at normoxic conditions (21% O2).
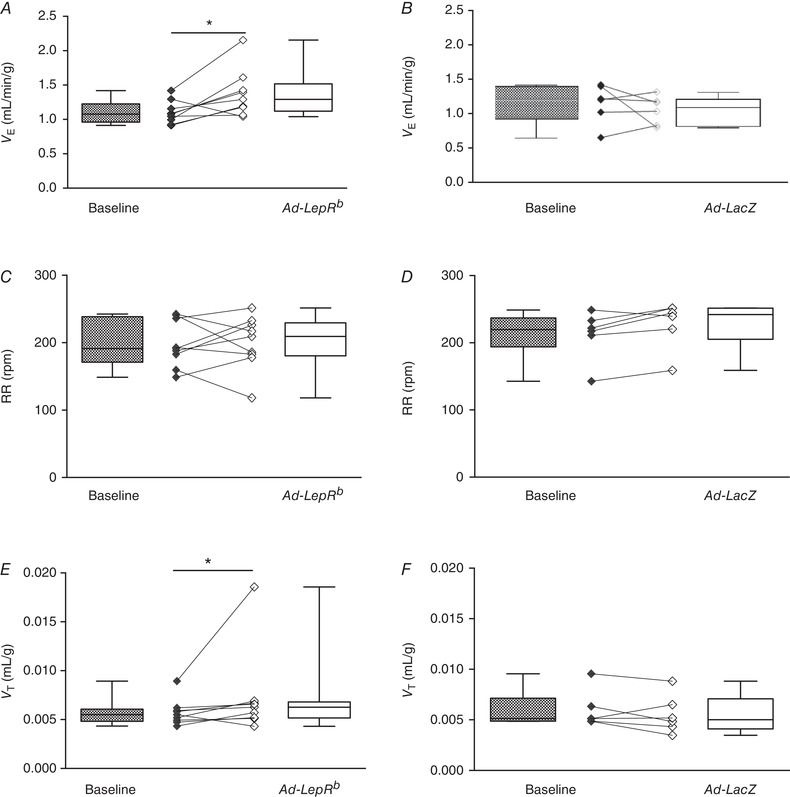
A and B, V E calculated as tidal volume per body weight (V T) × respiratory rate (RR), RR (C and D) and V T (E and F) before (baseline) and 15 days after Ad‐LepRb (n = 9) or Ad‐LacZ (n = 6) infection, at the same conditions. * P < 0.05 using the Wilcoxon test.
Figure 9. LepRb expression in the carotid bodies (CB) of LepRb‐deficient db/db mice increased minute ventilation at isocapnic hypoxia (10% O2 + 3% CO2).
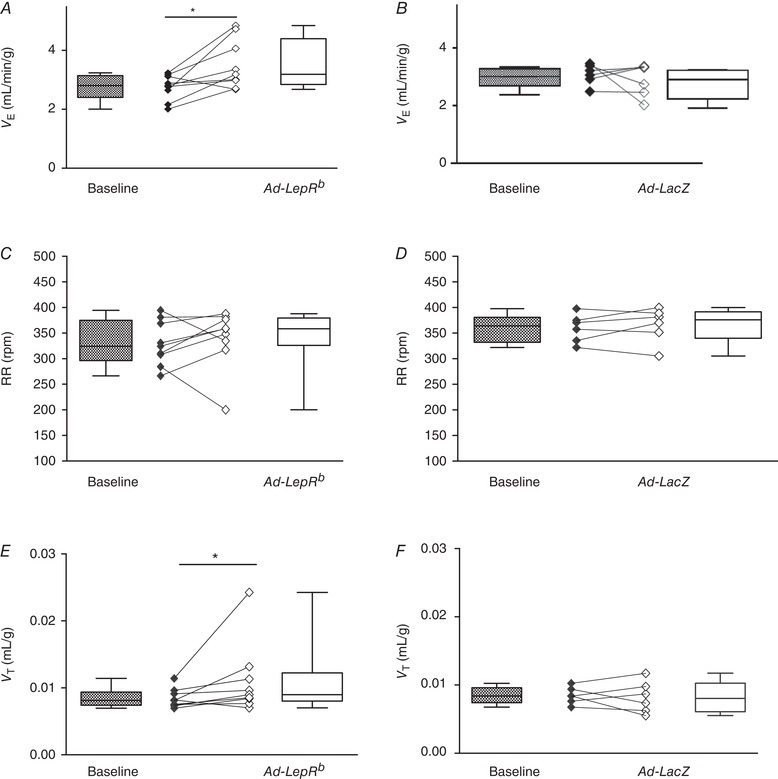
A and B, V E calculated as tidal volume per body weight (V T) × respiratory rate (RR). RR (C and D) and V T (E and F) before (baseline) and 15 days after Ad‐LepRb (n = 9) or Ad‐LacZ (n = 6) infection, at the same conditions. * P < 0.05 and ** P < 0.01 using the Wilcoxon test.
Figure 10. LepRb expression in the carotid bodies (CB) of LepRb‐deficient db/db mice increased the hypoxic ventilatory response (HVR).
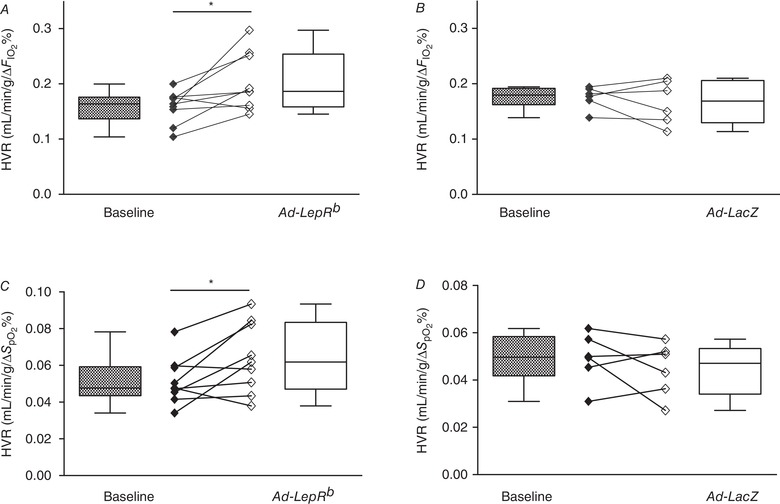
HVR calculated as ΔV E/Δ (A and B) and as ΔV E/Δ (C and D) before (baseline) and 15 days after Ad‐LepRb (A and C, n = 9) or Ad‐LacZ (B and D, n = 6) infection. * P < 0.05 and ** P < 0.01 using the Wilcoxon test.
Sleep recordings showed that LepRb expression in the CB decreased the amount of NREM sleep in db/db mice compared to LacZ control, whereas other sleep metrics (sleep efficiency, amount of REM sleep, number and length of sleep bouts) were unaffected (Table 5). Expression of LepRb in the CB of db/db mice did not influence the prevalence of upper airway obstruction. In Ad‐LepRb‐infected mice, the percentage of breaths with inspiratory flow limitation was 0.1 ± 0.1% in NREM sleep and 23 ± 6% in REM sleep, whereas in Ad‐LacZ mice, inspiratory flow limitation was observed in 2 ± 2% and 25 ± 11% of breaths, respectively. However, LepRb expression significantly increased minute ventilation, maximal and mean inspiratory flow and tidal volume compared to control (Figs. 11 and 12). Specifically, in LepRb mice minute ventilation in NREM sleep was 27.7 mL/min vs. 20.1 mL/min in control (P < 0.05) and in REM sleep it was 23.4 mL/min vs. 16.5 mL/min in control (P < 0.05) (Fig. 12 A). In LepRb mice, mean inspiratory flow in NREM sleep was 1.3 mL/s vs. 0.9 mL/s in control (P < 0.05), and in REM sleep it was 1.0 mL/s vs. 0.7 mL/s in control (P < 0.05) (Fig. 12 B). Maximal inspiratory flow in NREM sleep was 2.35 mL/s vs. 1.6 mL/s in control (P < 0.05), and in REM sleep it was 1.6 mL/s vs. 1.2 mL/s in control (P < 0.05) (Fig. 12 C). LepRb expression increased tidal volumes (Fig. 12 D) but not respiratory rate (Fig. 12 E). There was no significant effect of leptin on the ODI (Fig. 12 F). Sigh frequency varied greatly between mice. In the Ad‐LepRb group it fluctuated from 0 to 15.1/h, with a median of 7.4/h, whereas in the Ad‐LacZ group it varied from 0 to 8.5/h, with a median of 4.3/h; there was no difference in sigh frequency between the groups (P > 0.05).
Table 5.
Sleep architecture in db/db mice after Ad‐LacZ and Ad‐LepR infection of the carotid bodies
| Sleep bouts | ||||||||
|---|---|---|---|---|---|---|---|---|
| Sleep duration (min) | Number | Length (min) | ||||||
| Sleep efficiency (% of total time) | Total | NREM | REM | NREM | REM | NREM | REM | |
| Ad‐LacZ | 54.40 ± 0.05 | 215.4 ± 26 | 208 ± 27 | 7.51 ± 4.0 | 107.8 ± 10.7 | 6 ± 3.2 | 1.8 ± 0.3 | 1.02 ± 0.3 |
| Ad‐LepR | 42.40 ± 0.05 | 146.6 ± 24 | 139.2 ± 23* | 7.46 ± 1.07 | 80.8 ± 8.2 | 6.2± 1.6 | 1.6 ± 0.3 | 1.4 ± 0.2 |
* P < 0.05 vs. Ad‐LacZ. Results are presented as mean ± SEM. NREM, non‐rapid eye movement sleep; REM, rapid eye movement sleep.
Figure 11. A representative recording of non‐rapid eye movement sleep 15 days after infection of the carotid bodies with control virus (left, Ad‐LacZ, n = 6) or after LepRb (right, Ad‐LepRb) expression in the carotid bodies of db/db mice.
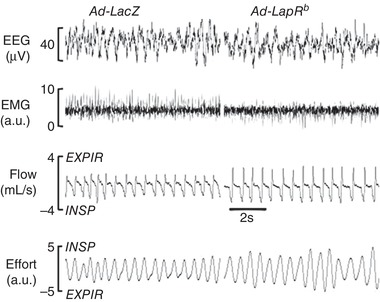
Ad‐LepRb infection shows increased minute ventilation compared to the Ad‐LacZ control. EEG, electroencephalography; EMG, electromyography of nuchal muscles; EXPIR, expiration; INSPIR, inspiration.
Figure 12. LepRb expression in the carotid bodies of LepRb‐deficient db/db mice increased minute ventilation.
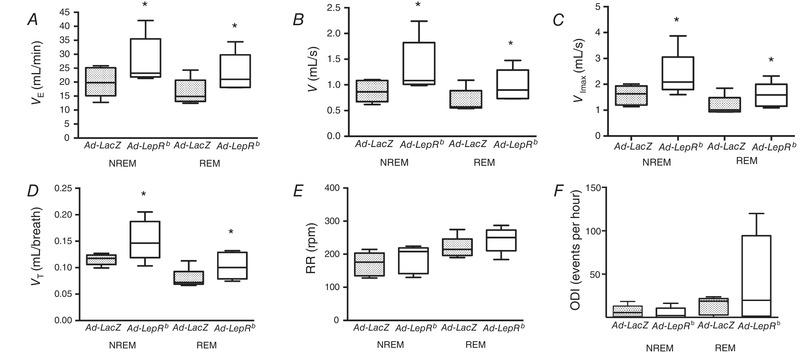
V E (A), mean inspiratory flow rate (V, B), maximal inspiratory flow rate (V Imax, C), and tidal volumes (V T, D) during non‐rapid eye movement (NREM) and rapid eye movement (REM) sleep, but not respiratory rate (RR, E), or oxygen desaturation index (ODI, F). Ad‐LepR infected mice. * P < 0.05 shows the independent effect of LepRb compared to the Ad‐LacZ control by ANOVA.
Discussion
The main novel finding of our study was that leptin acted in the CB to increase minute ventilation at normoxic conditions in awake and sleeping mice. This finding was confirmed by several independent lines of evidence. First, leptin increased minute ventilation at normoxic conditions in awake C57BL/6J mice and this increase was abolished by CB denervation, but not by sham surgery. Second, expression of the long isoform of leptin receptor LepRb in the CB increased minute ventilation at normoxic conditions in awake and sleeping LepRb‐deficient db/db mice. Third, intravenous infusion of leptin increased CSN activity in vivo at hyperoxic conditions, when CB activity is suppressed (Ling et al. 1997). In addition, we report several other important findings. We have shown that leptin acts in the CB to augment the HVR in C56BL/6J mice and this effect was abolished by CB denervation, similar to a recent report in rats (Yuan et al. 2018). Parallel experiments in db/db mice confirmed that this effect is mediated by the LepRb receptor. Finally, we are first to report on the quantitative morphometry of LepRb in the CB, showing its ubiquitous distribution in both type I and type II cells.
Leptin's effect on CB and respiratory function during normoxia
We have shown that leptin infusion increases baseline ventilation in C57BL/6J mice. The stimulating impact of leptin on minute ventilation was described two decades ago in ob/ob mice (O'Donnell et al. 1999). Leptin's action can be attributed in part to an increase in metabolic rate and CO2 production with ensuing increases in minute ventilation (Gautron & Elmquist, 2011). However, leptin also acts as a respiratory stimulant, independent of its metabolic effects (O'Donnell et al. 1999). Leptin‐deficient ob/ob mice show a suppressed minute ventilation and hypercapnic ventilatory response, which are normalized by leptin infusion (O'Donnell et al. 1999). Leptin infusion did not increase minute ventilation in lean Zucher rats, but the investigators induced a very modest increase in circulating leptin level (1.6‐fold from baseline, Yuan et al. 2018), whereas we induced an ∼ 30‐fold increase in leptin, similar to that observed in obese mice (Fleury Curado et al. 2018) and humans (Phipps et al. 2002; Shapiro et al. 2014). Intracerebroventricular administration of leptin instantaneously increased respiration in ob/ob mice (Bassi et al. 2012; Yao et al. 2016), whereas microinjections of leptin into the retrotrapezoid nucleus/parafacial respiratory group in ob/ob mice (Bassi et al. 2014) and into the NTS in rats (Inyushkina et al. 2010) localized respiratory effects of leptin to CO2 sensing areas. Thus, leptin is a potent respiratory stimulant acting on respiratory control centres in the medulla.
What evidence is there that leptin regulates breathing at normoxic conditions via peripheral chemoreception? LepRb, the only LepR isoform capable of intracellular signalling, has been previously identified in the CB of rats (Porzionato et al. 2011; Messenger et al. 2013; Messenger & Ciriello, 2013). We report that LepRb was present in > 70% of the CB glomus cells and > 60% of type II cells. Intracarotid administration of leptin increased minute ventilation in anaesthetized rats (Ribeiro et al. 2018). Leptin increased baseline CSN activity in vitro (Ribeiro et al. 2018) and in vivo according to our current data. We have shown that leptin increased minute ventilation in awake unrestrained C57BL/6J mice at normoxic conditions and its effect was abolished by CB denervation. LepRb expression in the CB of hyperleptinaemic db/db mice also increased minute ventilation, during both sleep and wakefulness. We provide two independent lines of evidence that leptin regulates respiratory function in the CB, independent of its metabolic effects. First, in C57BL/6J mice, leptin infusion suppressed food intake and increased metabolic rate and these effects were not attenuated by CSND (Table 2), whereas leptin‐induced augmentation of minute ventilation was abolished by CB denervation. Second, LepRb expression in the CB of LepRb‐deficient db/db mice did not affect food intake or body temperature (Table 4), whereas minute ventilation was increased, during both wakefulness and sleep (Figs. 8 and 12). Our data suggest that leptin's action in the CB plays an important role in the CB afferent input to the brainstem respiratory centres. Thus, our study provides first evidence that leptin acts in the CB to augment minute ventilation at normoxic conditions in unanaesthetized awake and sleeping mice.
Leptin's effect on CB and the HVR
The effect of leptin on the hypercapnic ventilatory response is well recognized (Tankersley et al. 1996; O'Donnell et al. 1999; Polotsky et al. 2001, 2004; Bassi et al. 2014). In contrast, the role of leptin in regulation of the HVR has not been convincingly defined. Tankersley et al. (1996) studied young leptin‐deficient ob/ob mice, mice heterozygous for the leptin gene and weight‐matched wild‐type mice and found similar HVR in all groups. However, leptin was not administered and leptin‐deficient mice were not compared to mice with severe diet‐induced obesity with very high circulating leptin levels. Our data provide first evidence in mice that leptin augments CSN activity in response to physiologically relevant levels of hypoxia in vivo and confirm previous findings in rats (Yuan et al. 2018) that leptin increases the HVR in awake unrestrained spontaneously breathing rodents. The HVR varied significantly between mice in each group, which is not surprising given that HVR measurements are typically variable within a subject (Teppema & Dahan, 2010). Nevertheless, our study overcame this intrinsic variability using within‐animal comparisons (before and after CSND, before and after Ad‐LepRb transfection) and appropriate controls (sham surgery and Ad‐LacZ transfection). Our findings are consistent with previously published data that leptin induced CSN activity in response to anoxia in the ex vivo preparations (Shirahata et al. 2015; Ribeiro et al. 2018) and augmented ventilation in anaesthetized tracheostomized rats during bilateral carotid artery occlusion (Ribeiro et al. 2018).
The current study has demonstrated the importance of leptin receptor in the CB for control of breathing in three complementary experiments. First, in awake wild‐type mice with ubiquitously expressed LepRb, leptin dramatically augmented minute ventilation in the 10% O2 environment and the HVR and both effects were abolished by CB denervation. Second, selective expression of LepRb in the CB of LepRb‐deficient db/db mice significantly enhanced minute ventilation at hypoxic conditions and augmented the HVR. Third, db/db mice transfected with LepRb in the CB showed significant increases in both maximal and mean inspiratory flow during NREM and REM sleep, which suggests that leptin signalling in the CB stimulates the respiratory drive (O'Donnell et al. 1999; Pho et al. 2016). Thus, the leptin‐dependent mechanism in the CB stimulates the hypoxic ventilatory response and regulates respiratory drive during sleep.
Clinical implications
Although breathing during sleep in leptin‐resistant db/db mice has not been sufficiently studied, phenotypically similar leptin‐deficient ob/ob mice exhibit both hypoventilation during sleep (O'Donnell et al. 1999) and obstructive sleep apnoea (Pho et al. 2016; Yao et al. 2016), which can be treated with leptin. Our current data showed that a leptin‐dependent mechanism in the CB stimulates minute ventilation throughout NREM and REM sleep without any effect on upper airway obstruction, i.e. obstructive sleep apnoea. It is conceivable that leptin signalling in the CB protects the respiratory function in severe obesity, preventing the development of obesity hypoventilation syndrome. Leptin may also play a role in maintaining oxygenation at the range of hypoxic conditions, including exposure to high altitude and hypoxaemia due to cardiorespiratory disorders.
Pathologically augmented HVR may destabilize breathing leading to central apnoea and aggravating obstructive apnoea (Younes et al. 2007; Pialoux et al. 2009; Trombetta et al. 2013; Younes, 2014; Mateika, 2015). According to our sleep data, leptin's action in the CB did not lead to apnoea, probably because mice were not hypoxaemic in the absence of exposure to hypoxic. Nevertheless, leptin‐induced hypoxic hyperventilation during sleep may destabilize breathing by decreasing CO2 below the apnoea threshold. Thus, leptin's action in the CB during sleep in the absence of significant hypoxaemia may be protective, whereas in the presence of hypoxaemia it may be destabilizing. In addition, leptin's action on the CSN may activate the sympathetic nervous system leading to a variety of poor outcomes including insomnia (note a decrease in NREM sleep time in Table 5) and cardiovascular complications (Prabhakar et al. 2012).
Limitations
Our study had potential limitations. (1) Hypoxia decreases and body temperature, which could affect respiratory measurements and their interpretations. Additionally, some animals had low rectal temperature at baseline, which could also affect measurements. However, short exposures to hypoxia during HVR measurements did not decrease rectal temperature in our experiments. Hence, metabolic changes induced by hypoxia were unlikely to have an impact on ventilatory measurements during first 2 min of the hypoxic exposure. (2) We did not measure and arterial blood gas during leptin infusion due to technical limitations. (3) CSND did not affect , or , despite a lack of an appropriate ventilatory response to hypoxia. This phenomenon could be attributed, at least in part, to improved ventilation/perfusion match in the lungs as indicated by a decrease in the alveolar–arterial O2 gradient, probably as a result of a decrease in sympathetic vasoconstriction. (4) We did not examine downstream mechanisms by which leptin acts in the CB. Given that LepRb is abundantly expressed in both oxygen sensing type I and type II cells, leptin's effect may occur in either cell type. In fact, Ribeiro et al. (2018) showed that leptin does not affect intracellular Ca2+ in type I cells in vitro, which questions the role of oxygen sensing glomus cells. Nevertheless, leptin's effects can differ between in vitro and in vivo conditions. Leptin may have chronic transcriptional effects mediated via Janus kinase 2/signal transducers and the activator of transcription 3 pathway or the phosphoinositide 3‐kinase pathway (Villanueva & Myers, 2008). Leptin can stabilize hypoxia inducible factor 1α (Calgani et al. 2016) in the glomus cells, which would enhance the HVR (Peng et al. 2006; Prabhakar & Semenza, 2012). Ribeiro et al. (2018) showed that leptin mediates adenosine release in the whole CB ex vivo preparation. Leptin may also act acutely to affect specific ion channels (Shirahata et al. 2015). Yuan et al. (2018) recently demonstrated that the effect of leptin on chemoreception may be mediated by oxygen sensing TASK channels. (5) Ad‐LepRb infection was not cell‐specific, affecting both type I and type II cells of the CB as well as adjacent tissues such as the superior cervical ganglion. While the non‐selective transfection technique is a limitation of the study, together with leptin studies in wild‐type mice with denervated and intact (sham surgery) CB, and with CSN recordings in vivo in the absence and presence of leptin, our study provides evidence that leptin acts in the CB to regulate HVR. (6) We did not investigate whether the effects of leptin in the CB are modified by the leptin‐resistant state such as diet‐induced obesity (Ribeiro et al. 2018) or sexual differences (Polotsky et al. 2001). (7) We did not measure the HVR during sleep and did not determine whether leptin signalling in the CB in the presence of hypoxia has a destabilizing effect on breathing during sleep.
Conclusions
Leptin acts in the CB to augment minute ventilation and the hypoxic ventilatory response, which may protect against SDB in obesity.
Additional information
Competing interests
The authors have no competing financial and non‐financial interests or other interests that might be perceived to influence the results and/or discussion reported in this paper.
Author contributions
M.S*, J.S.K.S., A.R.S. and V.Y.P conceived and designed research; C.C.E., M.‐K. S., H.P., L.J.K., S.B., L.E.P., ZJ.W and H.‐Y. Y. performed experiments; C.C.E., M.‐K. S., H.P., L.J.K., L.E.P., L.P., C.G., H.‐Y. Y., W.‐Y. T., and V.Y.P. analysed data; C.C.E., M.‐K. S., H.P., L.E.P., H.‐Y. Y., W.‐Y. T., J.S.K.S. and V.Y.P. interpreted results of experiments; C.C.E., H.P., L.J.K., L.E.P., H.‐Y. Y., W.‐Y. T. and V.Y.P. prepared figures; C.C.E and V.Y.P. drafted the manuscript, J.S.K.S., A.R.S. and V.Y.P edited and revised manuscript; C.C.E., M.‐K. S., H.P., L.J.K., L.E.P., ZJ.W, L.P., C.G., S.B., H.‐Y. Y., W.‐Y. T., J.S.K.S. and V.Y.P. approved the final version of the manuscript. *M.S. is deceased.
Funding
This work was funded by the NIH R01s HL133100 to V.Y.P. and J.S.K.S., R01 HL128970 to V.Y.P. and A.R.S., R01 HL138932 to V.Y.P. V.Y.P. is also supported by NIEHS grant P50 ES018176 and EPA Agreements 83615201 & 83451001. C.C.E. was supported in part by ‘Beca de estancias formativas de la Consejería de Salud de Andalucía 2017. C. Caballero Eraso. Número de expediente: EF‐0128‐2016’. This publication has not been formally reviewed by EPA. The views expressed in this document are solely those of the authors and do not necessarily reflect those of the Agency. EPA does not endorse any products or commercial services mentioned in this publication.
Biography
Candela Caballero Eraso's research focuses on understanding the role of the carotid body in the setting of intermittent hypoxia/sleep apnoea with special interest in its regulatory mechanisms modifying specific cardiovascular or respiratory responses. Candela received her degree in Medicine from the Faculty of Medicine, University of Extremadura, in 2007 and her MD, PhD in 2015 from the University of Seville. Since 2013 she has combined her clinical activity as a specialist in respiratory medicine at the University Hospital Virgen del Rocío with her biomedical research at the Institute of Biomedicine of Seville and the Johns Hopkins University in Baltimore, USA.

Edited by: Harold Schultz & Benedito Machado
References
- Bassi M, Furuya WI, Menani JV, Colombari DS, do Carmo JM, da Silva AA, Hall JE, Moreira TS, Wenker IC, Mulkey DK & Colombari E (2014). Leptin into the ventrolateral medulla facilitates chemorespiratory response in leptin‐deficient (ob/ob) mice. Acta Physiol (Oxf) 211, 240–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassi M, Giusti H, Leite CM, Anselmo‐Franci JA, do Carmo JM, da Silva AA, Hall JE, Colombari E & Glass ML (2012). Central leptin replacement enhances chemorespiratory responses in leptin‐deficient mice independent of changes in body weight. Pflugers Arch 464, 145–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calgani A, Monache SD, Cesare P, Vicenti C, Bologna M & Angelucci A (2016). Leptin contributes to long‐term stabilization of HIF‐1α in cancer cells subjected to oxygen limiting conditions. Cancer Lett 376, 1–9. [DOI] [PubMed] [Google Scholar]
- Chen H, Charlat O, Tartaglia LA, Woolf EA, Weng X, Ellis SJ, Lakey ND, Culpepper J, Moore KJ, Breitbart RE, Duyk GM, Tepper RI & Morgenstern JP (1996). Evidence that the diabetes gene encodes the leptin receptor: identification of a mutation in the leptin receptor gene in db/db mice. Cell 84, 491–495. [DOI] [PubMed] [Google Scholar]
- Ciriello J & Caverson MM (2014). Carotid chemoreceptor afferent projections to leptin receptor containing neurons in nucleus of the solitary tract. Peptides 58, 30–35. [DOI] [PubMed] [Google Scholar]
- Considine RV, Sinha MK, Heiman ML, Kriauciunas A, Stephens TW, Nyce MR, Ohannesian JP, Marco CC, McKee LJ & Bauer TL (1996). Serum immunoreactive‐leptin concentrations in normal‐weight and obese humans. N Engl J Med 334, 292–295. [DOI] [PubMed] [Google Scholar]
- Drorbaugh JE & Fenn WO (1955). A barometric method for measuring ventilation in newborn infants. Pediatrics 16, 81–87. [PubMed] [Google Scholar]
- Duffin J (2007). Measuring the ventilatory response to hypoxia. J Physiol 584, 285–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleury Curado T, Pho H, Berger S, Caballero‐Eraso C, Shin M‐K, Sennes LU, Pham L, Schwartz AR & Polotsky VY (2018). Sleep‐disordered breathing in C57BL/6J mice with diet‐induced obesity. Sleep 41 https://doi.org/1093/sleep/zsy089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautron L & Elmquist JK (2011). Sixteen years and counting: an update on leptin in energy balance. J Clin Invest 121, 2087–2093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez‐Martin MC, Vega‐Agapito MV, Conde SV, Castaneda J, Bustamante R, Olea E, Perez‐Vizcaino F, Gonzalez C & Obeso A (2011). Carotid body function and ventilatory responses in intermittent hypoxia. Evidence for anomalous brainstem integration of arterial chemoreceptor input. J Cell Physiol 226, 1961–1969. [DOI] [PubMed] [Google Scholar]
- Handa P, Maliken BD, Nelson JE, Morgan‐Stevenson V, Messner DJ, Dhillon BK, Klintworth HM, Beauchamp M, Yeh MM, Elfers CT, Roth CL & Kowdley KV (2014). Reduced adiponectin signaling due to weight gain results in nonalcoholic steatohepatitis through impaired mitochondrial biogenesis. Hepatology 60, 133–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez AB, Kirkness JP, Smith PL, Schneider H, Polotsky M, Richardson RA, Hernandez WC & Schwartz AR (2012). Novel whole body plethysmography system for the continuous characterization of sleep and breathing in a mouse. J Appl Physiol (1985) 112, 671–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inyushkina EM, Merkulova NA & Inyushkin AN (2010). Mechanisms of the respiratory activity of leptin at the level of the solitary tract nucleus. Neurosci Behav Physiol 40, 707–713. [DOI] [PubMed] [Google Scholar]
- Jacky JP (1978). A plethysmograph for long‐term measurements of ventilation in unrestrained animals. J Appl Physiol 45, 644–647. [DOI] [PubMed] [Google Scholar]
- Jun JC, Shin MK, Yao Q, Devera R, Bevans‐Fonti S & Polotsky VY (2013). Thermoneutrality modifies the impact of hypoxia on lipid metabolism. Am J Physiol Endocrinol Metab 304, E424–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling L, Olson EB, Vidruk EH & Mitchell GS (1997). Integrated phrenic responses to carotid afferent stimulation in adult rats following perinatal hyperoxia. J Physiol 500, 787–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez‐Barneo J, Ortega‐Saenz P, Pardal R, Pascual A, Piruat JI, Duran R & Gomez‐Diaz R (2009). Oxygen sensing in the carotid body. Ann N Y Acad Sci 1177, 119–131. [DOI] [PubMed] [Google Scholar]
- Maffei M, Halaas J, Ravussin E, Pratley RE, Lee GH, Zhang Y, Fei H, Kim S, Lallone R & Ranganathan S (1995). Leptin levels in human and rodent: measurement of plasma leptin and ob RNA in obese and weight‐reduced subjects. Nat Med 1, 1155–1161. [DOI] [PubMed] [Google Scholar]
- Mateika JH (2015). The role of high loop gain induced by intermittent hypoxia in the pathophysiology of obstructive sleep apnea. Sleep Med Rev 22, 1–2. [DOI] [PubMed] [Google Scholar]
- Messenger SA & Ciriello J (2013). Effects of intermittent hypoxia on leptin signalling in the carotid body. Neuroscience 232, 216–225. [DOI] [PubMed] [Google Scholar]
- Messenger SA, Moreau JM & Ciriello J (2013). Effect of chronic intermittent hypoxia on leptin and leptin receptor protein expression in the carotid body. Brain Res 1513, 51–60. [DOI] [PubMed] [Google Scholar]
- Mokhlesi B (2010). Obesity hypoventilation syndrome: a state‐of‐the‐art review. Respir Care 55, 1347–1362. [PubMed] [Google Scholar]
- Nurse CA & Piskuric NA (2013). Signal processing at mammalian carotid body chemoreceptors. Semin Cell Dev Biol 24, 22–30. [DOI] [PubMed] [Google Scholar]
- O'Donnell CP, Schaub CD, Haines AS, Berkowitz DE, Tankersley CG, Schwartz AR & Smith PL (1999). Leptin prevents respiratory depression in obesity. Am J Respir Crit Care Med 159, 1477–1484. [DOI] [PubMed] [Google Scholar]
- Pappenheimer JR (1977). Sleep and respiration of rats during hypoxia. J Physiol 266, 191–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peppard PE, Young T, Barnet JH, Palta M, Hagen EW & Hla KM (2013). Increased prevalence of sleep‐disordered breathing in adults. Am J Epidemiol 177, 1006–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng YJ, Yuan G, Ramakrishnan D, Sharma SD, Bosc‐Marce M, Kumar GK, Semenza GL & Prabhakar NR (2006). Heterozygous HIF‐1 alpha deficiency impairs carotid body‐mediated systemic responses and reactive oxygen species generation in mice exposed to intermittent hypoxia. J Physiol 577, 707–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phipps PR, Starritt E, Caterson I & Grunstein RR (2002). Association of serum leptin with hypoventilation in human obesity. Thorax 57, 75–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pho H, Hernandez AB, Arias RS, Leitner EB, Van Kooten S, Kirkness JP, Schneider H, Smith PL, Polotsky VY & Schwartz AR (2016). The effect of leptin replacement on sleep‐disordered breathing in the leptin‐deficient ob/ob mouse. J Appl Physiol 120, 78–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pialoux V, Hanly PJ, Foster GE, Brugniaux JV, Beaudin AE, Hartmann SE, Pun M, Duggan CT & Poulin MJ (2009). Effects of exposure to intermittent hypoxia on oxidative stress and acute hypoxic ventilatory response in humans. Am J Respir Crit Care Med 180, 1002–1009. [DOI] [PubMed] [Google Scholar]
- Pichard LE, Crainiceanu CM, Pashai P, Kostuk EW, Fujioka A & Shirahata M (2015). Role of BK channels in murine carotid body neural responses in vivo. Adv Exp Med Biol 860, 325–333. [DOI] [PubMed] [Google Scholar]
- Polotsky M, Elsayed‐Ahmed AS, Pichard L, Harris CC, Smith PL, Schneider H, Kirkness JP, Polotsky V & Schwartz AR (2012). Effects of leptin and obesity on the upper airway function. J Appl Physiol 112, 1637–1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polotsky VY, Smaldone MC, Scharf MT, Li J, Tankersley CG, Smith PL, Schwartz AR & O'Donnell CP (2004). Impact of interrupted leptin pathways on ventilatory control. J Appl Physiol 96, 991–998. [DOI] [PubMed] [Google Scholar]
- Polotsky VY, Wilson JA, Smaldone MC, Haines AS, Hurn PD, Tankersley CG, Smith PL, Schwartz AR & O'Donnell CP (2001). Female gender exacerbates respiratory depression in leptin‐deficient obesity. Am J Respir Crit Care Med 164, 1470–1475. [DOI] [PubMed] [Google Scholar]
- Porzionato A, Rucinski M, Macchi V, Stecco C, Castagliuolo I, Malendowicz LK & De CR (2011). Expression of leptin and leptin receptor isoforms in the rat and human carotid body. Brain Res 1385, 56–67. [DOI] [PubMed] [Google Scholar]
- Powell FL, Milsom WK & Mitchell GS (1998). Time domains of the hypoxic ventilatory response. Respir Physiol 112, 123–134. [DOI] [PubMed] [Google Scholar]
- Prabhakar NR (2013). Sensing hypoxia: physiology, genetics and epigenetics. J Physiol 591, 2245–2257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prabhakar NR, Kumar GK & Peng YJ (2012). Sympatho‐adrenal activation by chronic intermittent hypoxia. J Appl Physiol 113, 1304–1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prabhakar NR & Semenza GL (2012). Adaptive and maladaptive cardiorespiratory responses to continuous and intermittent hypoxia mediated by hypoxia‐inducible factors 1 and 2. Physiol Rev 92, 967–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribeiro MJ, Sacramento JF, Gallego‐Martin T, Olea E, Melo BF, Guarino MP, Yubero S, Obeso A & Conde SV (2018). High fat diet blunts the effects of leptin on ventilation and on carotid body activity. J Physiol 96, 3187–3199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott MM, Lachey JL, Sternson SM, Lee CE, Elias CF, Friedman JM & Elmquist JK (2009). Leptin targets in the mouse brain. J Comp Neurol 514, 518–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro SD, Chin CH, Kirkness JP, McGinley BM, Patil SP, Polotsky VY, Biselli PJ, Smith PL, Schneider H & Schwartz AR (2014). Leptin and the control of pharyngeal patency during sleep in severe obesity. J Appl Physiol 116, 1334–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin M‐K, Yao Q, Jun JC, Bevans‐Fonti S, Yoo D‐Y, Han W, Mesarwi O, Richardson R, Fu Y‐Y, Pasricha PJ, Schwartz AR, Shirahata M & Polotsky VY (2014). Carotid body denervation prevents fasting hyperglycemia during chronic intermittent hypoxia. J Appl Physiol 117, 765–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirahata M & Fitzgerald RS (1991). Dependency of hypoxic chemotransduction in cat carotid body on voltage‐gated calcium channels. J Appl Physiol (1985) 71, 1062–1069. [DOI] [PubMed] [Google Scholar]
- Shirahata M, Tang WY, Shin MK & Polotsky VY (2015). Is the carotid body a metabolic monitor? Adv Exp Med Biol 860, 153–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva AQ & Schreihofer AM (2011). Altered sympathetic reflexes and vascular reactivity in rats after exposure to chronic intermittent hypoxia. J Physiol 589, 1463–1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tankersley C, Kleeberger S, Russ B, Schwartz A & Smith P (1996). Modified control of breathing in genetically obese (ob/ob) mice. J Appl Physiol 81, 716–723. [DOI] [PubMed] [Google Scholar]
- Tankersley CG, Fitzgerald RS & Kleeberger SR (1994). Differential control of ventilation among inbred strains of mice. Am J Physiol Regul Integr Comp Physiol 36, R1371–R1377. [DOI] [PubMed] [Google Scholar]
- Teppema LJ & Dahan A (2010). The ventilatory response to hypoxia in mammals: mechanisms, measurement, and analysis. Physiol Rev 90, 675–754. [DOI] [PubMed] [Google Scholar]
- Trombetta IC, Maki‐Nunes C, Toschi‐Dias E, Alves MJ, Rondon MU, Cepeda FX, Drager LF, Braga AM, Lorenzi‐Filho G & Negrao CE (2013). Obstructive sleep apnea is associated with increased chemoreflex sensitivity in patients with metabolic syndrome. Sleep 36, 41–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tufik S, Santos‐Silva R, Taddei JA & Bittencourt LR (2010). Obstructive sleep apnea syndrome in the Sao Paulo Epidemiologic Sleep Study. Sleep Med 11, 441–446. [DOI] [PubMed] [Google Scholar]
- Van Vliet BN, Chafe LL & Montani JP (1999). Contribution of baroreceptors and chemoreceptors to ventricular hypertrophy produced by sino‐aortic denervation in rats. J Physiol 516, 885–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villanueva EC & Myers MG Jr (2008). Leptin receptor signaling and the regulation of mammalian physiology. Int J Obes (Lond) 32(Suppl 7): S8–S12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamauchi M, Hasan O, Dostal J, Jacono FJ, Loparo KA & Strohl KP (2008). Post‐sigh breathing behavior and spontaneous pauses in the C57BL/6J (B6) mouse. Respir Physiol Neurobiol 162, 117–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Q, Pho H, Kirkness J, Ladenheim EE, Bi S, Moran TH, Fuller DD, Schwartz AR & Polotsky VY (2016). Localizing effects of leptin on upper airway and respiratory control during sleep. Sleep 39, 1097–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Younes M (2014). CrossTalk proposal: elevated loop gain is a consequence of obstructive sleep apnoea. J Physiol 592, 2899–2901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Younes M, Ostrowski M, Atkar R, Laprairie J, Siemens A & Hanly P (2007). Mechanisms of breathing instability in patients with obstructive sleep apnea. J Appl Physiol 103, 1929–1941. [DOI] [PubMed] [Google Scholar]
- Yuan F, Wang H, Feng J, Wei Z, Yu H, Zhang X, Zhang Y & Wang S (2018). Leptin signaling in the carotid body regulates a hypoxic ventilatory response through altering TASK channel expression. Front Physiol 9, 249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwillich CW, Sutton FD, Pierson DJ, Greagh EM & Weil JV (1975). Decreased hypoxic ventilatory drive in the obesity‐hypoventilation syndrome. Am J Med 59, 343–348. [DOI] [PubMed] [Google Scholar]