

Abstract
Until recently, in vivo and ex vivo experiments were the only means to determine factors and pathways involved in disease pathophysiology. After the generation of characterized human embryonic stem cell lines, human diseases could readily be studied in an extensively controllable setting. The introduction of human‐induced pluripotent stem cells, a decade ago, allowed the investigation of hereditary diseases in vitro. In the field of cardiology, diseases linked to known genes have successfully been studied, revealing novel disease mechanisms. The direct effects of various mutations leading to hypertrophic cardiomyopathy, dilated cardiomyopathy, arrythmogenic cardiomyopathy, or left ventricular noncompaction cardiomyopathy are discovered as a result of in vitro disease modeling. Researchers are currently applying more advanced techniques to unravel more complex phenotypes, resulting in state‐of‐the‐art models that better mimic in vivo physiology. The continued improvement of tissue engineering techniques and new insights into epigenetics resulted in more reliable and feasible platforms for disease modeling and the development of novel therapeutic strategies. The introduction of CRISPR‐Cas9 gene editing granted the ability to model diseases in vitro independent of induced pluripotent stem cells. In addition to highlighting recent developments in the field of human in vitro cardiomyopathy modeling, this review also aims to emphasize limitations that remain to be addressed; including residual somatic epigenetic signatures induced pluripotent stem cells, and modeling diseases with unknown genetic causes. Stem Cells Translational Medicine 2019;8:66–74
Keywords: In vitro disease models, Stem cells, Cardiac disease, Heart failure
Significance Statement.
Before human cardiomyocytes could be generated from stem cells, the only means to disease mechanics was via difficult and labor‐intensive methods. The introduction of human induced pluripotent stem cells provided a new means to obtain virtually unlimited amounts of patient‐derived cardiomyocytes. Major advances in gene editing techniques enabled the targeted mutation of specific genes, which could result in the introduction of aberrant or restored gene function. Collectively, these novel methods formed the basis for a new era of in vitro cardiac disease modeling. This review highlights the impact and applications of these state‐of‐the‐art techniques in the field of heart failure.
Introduction
Heart failure is a clinical syndrome that is caused by a wide variety of factors, and between 2011 and 2014, an estimated 6.5 million adults were diagnosed with heart failure 1. The number of heart failure patients is rising markedly. Dysfunctionality of the cardiac muscle leading to heart failure can be caused by different cardiomyopathies. The most common forms are hypertrophic cardiomyopathy (HCM) and dilated cardiomyopathy (DCM), followed by arrhythmogenic cardiomyopathy (ACM) and left ventricular noncompaction cardiomyopathy (LVNC) 2, 3, 4, 5. They result from a complex and diverse mechanism that is often a mix of functional, structural, and biological adaptions specific for each cardiomyopathy. This makes studying heart failure pathophysiology a daunting task.
Technological advances that were made during the last decades enabled researchers to noninvasively study cardiac function in detail. Nevertheless, studying pathological molecular mechanisms occurring in the failing heart of patients primarily involves invasive methods. Taking any form of biopsy from cardiac tissue comes with the risk of perforation. The amount of material is often insufficient for extensive molecular analyses and biopsies are only taken in very few patients with severe (end‐stage) cardiac pathology. Moreover, cardiomyocytes are nonproliferative, which makes in vitro culturing of primary cardiomyocytes complicated. Alternatively, standardized cell lines were used (e.g., H9C2, HL‐1, or immortalized cardiomyocytes), while these cells proliferate indefinitely and resemble cardiomyocytes to some extent, each line also has major disadvantages (e.g., nonhuman cells or tumor‐like properties). In addition, using animals to isolate (neonatal) cardiomyocytes requires a large number of animals to acquire sufficient amounts of cells.
The emergence of human embryonic stem cells (hESC) and the development of appropriate culturing techniques quickly made them a potent tool to study previously rare tissues and mechanisms 6. In 2007, the pioneering methods for generating human induced pluripotent stem cells (hiPSC) were published and provided the means to conduct patient‐specific in vitro studies 7. The development of these cell‐based tools enabled researchers to attempt recapitulating various aspects of a disease through in vitro disease modeling.
This review aims to highlight the current status of in vitro cardiomyopathy models while focusing on tissue engineering and gene editing to recapitulate human cardiomyopathies.
Cardiac Disease Modeling—Translation to the Clinical Setting
Cellular Sources for in vitro Cardiomyopathy Models
Early in vitro cardiac tissue models were based on either immortalized human cell lines or cells isolated from animals. The immortalized human ventricular AC16 cell line was developed using fusion of primary ventricular cardiomyocytes with an SV‐40 transformed fibroblast cell line 8. These cells resemble human cardiomyocytes to great extent (e.g., these cells contract and express main cardiac genes), but the proliferative capacity of these cells remains the main disadvantage as proliferating cardiomyocytes cannot maintain stable myofibrils.
Primary cardiomyocytes isolated from neonatal mice, rats, and chicken embryos were popular cell sources for in vitro cardiac models 9, 10, 11, but research based on these primary cells demonstrated that animal cell‐based models cannot truly recapitulate human physiology. Consequently, more sophisticated cell models were developed to create human‐like tissue models 12, 13. However, establishing human models proved to be challenging as cardiac tissue or isolated cardiomyocytes from patients are difficult to obtain and cannot survive long‐term culture 14.
Human Pluripotent Stem Cells
Cardiomyocytes were considered a rare cell type for in vitro studies, until hESC‐derived cardiomyocytes (hESC‐CM) were the first source of human heart cells for large‐scale experimental set‐ups 15. Since the introduction of hESC‐CM in 2001, the use of hESC as a source for in vitro cardiac disease modeling has been copious 16. Additionally, hiPSC‐derived cardiomyocytes (hiPSC‐CM) were found to recapitulate phenotypic characteristics caused by genetic variations 17, which render these cells an suitable source for human disease models. Furthermore, hiPSC‐CM was found to be a powerful tool for patient stratification in regard to drug safety and responsiveness 18. To date, artificially matured patient‐derived hiPSC‐CM proved to be similar in to isolated primary human cardiomyocytes molecular, mechanical, electrophysiological, metabolic, and ultrastructural properties 19, 20. However, hiPSC‐CM exhibits various fetal characteristics as opposed to mature (isolated) cardiomyocytes. To resolve these issues, hiPSC‐CM can be cultured for extended periods or subjected to specific bioengineering approaches. Protocols using hormone stimulation 19 or conditioning with mechanical stress and electrical pacing 21, 22 have collectively led to a more mature phenotype, but the exact mechanisms that induce maturation remain only partially understood 23, 24, 25, 26. Diverse epigenetic processes, including long‐noncoding RNA (lncRNA) 27, microRNAs 28, chromatin, and histone proteins 29, and DNA methylation 29 have been suggested as crucial mediators in both developmental processes and in disease.
Inherited Cardiomyopathies—hiPSC to Model Genetic Causality
A plethora of genetic mutations have been associated with the pathogenesis of genetic heart diseases, including the main inherited cardiomyopathies (i.e., HCM, DCM, ACM, and LVNC). Investigating how genetic mutations explain causality in the pathophysiology of cardiomyopathies and how they interact with secondary genetic and environmental factors is imperative to improving diagnosis and decision‐making regarding treatment strategies. The introduction of patient‐specific hiPSC‐CM provides a versatile new tool that may tremendously improve our understanding of the disease mechanisms. Consequently, these cells have been widely applied to study the complexity of cardiac disease. However, cardiomyopathies are divided into four classes, each with a distinct pathophysiology, resulting in various types of heart failure. The most common cardiomyopathy, HCM, is characterized by increased cardiac mass due to left ventricular wall thickening (hypertrophy) that most often is asymmetric, with particular involvement of the interventricular septum, myocytes disarray, and cardiac fibrosis 30. DCM is characterized by left ventricular chamber enlargement and systolic dysfunction, which often leads to heart failure, arrhythmia, and sudden death. ACM predominantly affects right ventricular cardiomyocytes and occurs due to defects in the cardiac desmosome as a consequence of mutations in key desmosomal components, but also because of ion channel defects. Consequently, ACM hallmarks include right ventricular dilation, scarring, exaggerated lipogenesis and lipid infiltration, and arrhythmias. Finally, LVNC is characterized by cardiac noncompaction, primarily resulting in trabeculation and deep recesses in the left ventricle. Many studies performed in patient‐derived hiPSC‐CM have often recapitulated these respective hallmarks of inherited cardiomyopathies and thereby markedly increased our understanding of underlying molecular mechanisms, as summarized in Table 1. In addition to cardiomyopathies, inherited arrhythmias are generally caused by a pathological mutation in a gene encoding an ion channel or an associated protein. However, this review focusses on cardiomyopathies, whereas arrhythmias are beyond the scope of this review. A recent review highlights the recent advances in the use genome editing to study cardiotoxicity and model inherited arrhythmia 31.
Table 1.
Summary of cardiomyopathy‐associated mutations that have been studied in hiPSC‐based in vitro models
| Gene | Mutation | Main phenotype | Ref | |
|---|---|---|---|---|
| HCM | MYH7 | p.R442G | Enlarged cellular size, disorganized myofibrils, disrupted sarcomere structure, dysfunctional ion channel homeostasis. | 32 |
| p.R663H | Enlarged cellular size, contractile arrhythmia, dysfunctional Ca2+‐handling, increased [Ca2+]i | 33 | ||
| MYBPC3 | c.1358‐1359insC | Enlarged cellular size, disrupted gene expression profile | 34 | |
| p.Q1061X | Enlarged cellular size, aberrant electrophysiological properties, dysfunctional Ca2+ ‐handling, and disrupted gene expression profile | 35 | ||
| p.G999‐Q1004del | Enlarged cellular size, disorganized myofibrils | 36 | ||
| c.2373dupG | Aberrant electrophysiological properties, reduced contractile force generation, aberrant bioenergetics | 37 | ||
| TPM1 | p.D175N | Enlarged cellular size, aberrant electrophysiological properties, dysfunctional Ca2+ ‐handling, disrupted gene expression profile | 35 | |
| DCM | TTN | p.A22352fs+/− p.P22582fs+/− p.W976R+/− |
Reduced contractile force generation, disrupted sarcomere structure, impaired response to mechanical and β‐adrenergic stress | 38 |
| LMNA | p.R225X p.Q354X p.T518 fs |
Nuclear blebbing, increased senescence, increased apoptosis | 39 | |
| TNNT2 | p.R173W | Dysfunctional Ca2+‐handling, reduced contractile force generation, disrupted sarcomere structure | 40,41 | |
| DES | p.A285V | Disrupted sarcomere structure, ultrastructural disarray | 42 | |
| RBM20 | p.R636S | Sarcomeric remodeling, dysfunctional Ca2+‐handling, increased [Ca2+]i, disrupted gene expression profile | 43,44 | |
| PLN | p.R14del | Dysfunctional Ca2+‐handling, aberrant electrophysiological properties, increased hypertrophy markers | 45,46 | |
| ACM | PKP2 | c.2484C > T c.2013delC |
Increased lipogenesis, increased apoptosis, dysfunctional Ca2+‐handling, disrupted desmosome structure | 47 |
| c.1841 T > C | Increased lipogenesis, disrupted desmosome structure | 48 | ||
| c.972InsT/N | Increased lipogenesis, disrupted desmosome structure | 49 | ||
| SCN5A | p.R1898H | Dysfunctional Na+‐handling | 50 | |
| LVNC | TBX20 | c. 951C > A | Reduced proliferative capacity, disrupted gene expression profile | 51 |
Disease Modeling Utilizing Known Versus Unknown Genetic Variations
In vitro disease modeling has proven to be a valuable tool to study molecular pathophysiological mechanisms and disease etiology for diseases with a known genetic cause. Indeed, modeling disease without a known genetic defect is challenging. Nevertheless, these in vitro models have also been successfully applied to cardiac diseases that develop without a known causing genetic variant. For example, Burridge et al. have recently demonstrated that it is possible to determine the underlying genetic aberrations found in heart failure patients that experienced doxorubicin‐induced cardiotoxicity 52. Furthermore, hiPSC were used to screen for cardiovascular toxicity of anticancer tyrosine kinase inhibitors using multiple healthy controls and two patients receiving cancer treatment 53. Additionally, studies have identified genetic targets in hypoplastic left heart syndrome in hiPSC with previously unknown mutations 54, 55. These examples show that the use of hiPSC for in vitro cardiac disease modeling without the presence of a known genetic defect are thus challenging, albeit not impracticable. This has also been demonstrated for other fields of disease, where hiPSC have been used to model noncardiac diseases like sporadic Alzheimer's disease 56, chemotherapy‐induced neuroticxicity 57, and was shown as a valuable tool in cancer research and precision oncology 58.
In vitro modeling of multifactorial diseases that are mechanistically complex or diseases that arise because of environmental causes is challenging and unrealistic. HiPSC are patient‐derived and harbor all relevant genetic factors that may contribute to the disease. Hence, even when the exact underlying mechanisms of a disease are unknown, hiPSC provide a reliable platform for disease modeling. A disease of the heart is often assumed to arise from cardiomyocytes themselves. However, due to tightly regulated cell‐autonomous versus noncell‐autonomous responses (e.g., interactions between cardiomyocytes and neighboring fibroblasts and endothelial cells), this may not be the case. A disease may very well originate in a nonmyocyte cell type and functionally disrupt cardiomyocyte function (for example: endothelial dysfunction and subsequent disturbed perfusion). As hiPSC can differentiate toward virtually all cell types, researchers can quickly change protocols and obtain these other relevant cell types based on hiPSC derived from a single patient. This potential of plasticity highlights the significance of hiPSC as a platform for disease modeling.
Gene Editing in Cardiomyopathies
Traditional genome editing methods have been mostly based on zinc‐finger nucleases (ZFNs) and transcription activator‐like effector nucleases (TALENs). Both ZNFs and TALENs use DNA binding motifs that can be designed and combined to target any nucleotide sequence for cleavage. ZNFs target trinucleotide sequences, while TALENs can recognize a single nucleotide. This makes the use of TALENs generally more straightforward. Recent technological breakthroughs for targeted gene editing using site‐specific nucleases primarily related to clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR‐associated protein 9 (Cas9) systems allow for genome engineering, reverse genetics, and targeted transgene integration experiments that can be performed in an accurate and reproducible fashion 59. The CRISPR/Cas9 system is based on site targeting based on guide RNA design and results in improved efficiency compared to earlier methods 60, 61. Furthermore, site targeting is more flexible with the CRISPR/Cas9 system than with ZNFs and TALENs and offers the possibility to introduce multiple mutations at the same time by injecting different guide RNAs. By applying these tools, genes have been functionally removed from specific loci, thereby creating disease‐causing mutations in hiPSC‐CM or other cardiovascular disease models in vitro 62. Vice versa, genetic mutations could be corrected in patient‐derived cells, resulting in the generation of an isogenic control cell line by exclusively eliminating the disease‐causing genetic variation.
Correcting or silencing a pathological genetic variant can be used to develop future therapies. However, when applying this to human cardiomyopathies, many different, site‐specific corrective strategies need to be designed and tested. This feat is challenging from a clinical trial and regulatory perspective. Each antisense oligonucleotide or guide RNA can only target a very specific nucleotide sequence and is therefore useful for a very small number of patients, which makes placebo‐controlled trials, the regulatory standard, nearly impossible. This has prompted the evaluation of the possibility of broader genetic therapeutic avenues that can target normal genes to enhance cardiac function. For example: gene therapy (i.e., induced overexpression) has been applied to upregulate SERCA2a and as a result enhances myocardial contraction in heart failure patients with reduced ejection fraction 63, 64, 65. However, with respect to disease models, various studies have been successful in recapitulating specific diseases in vitro as well as reverting disease phenotypes by correcting a genetic variant as presented in Figure 1. These studies have been summarized in Table 2.
Figure 1.
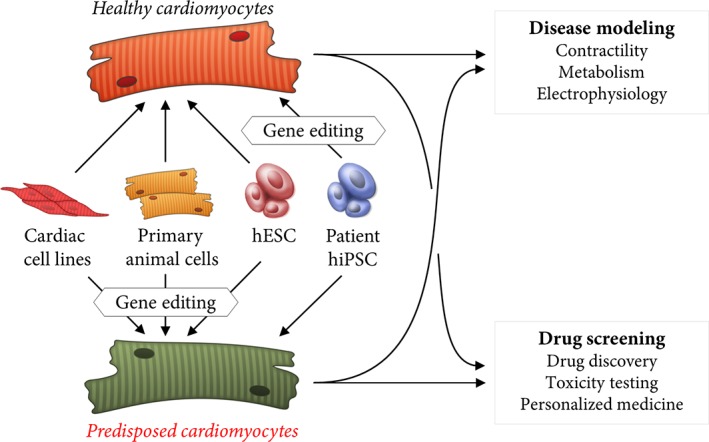
Schematic representation of cell types as a basis for human in vitro models. Primary cells, cell lines and stem cells can be utilized as a basis for in vitro disease models to study cardiomyopathies. State‐of‐the‐art gene editing techniques allow for the introduction of specific disease‐causing mutations. Alternatively, gene editing can also be harnessed to generate isogenic control lines from patient‐derived cells.
Table 2.
Studies that have generated in vitro disease models and studies that have repaired and rescued in vitro disease phenotypes
| Gene | Mutation | Strategy | Ref | |
|---|---|---|---|---|
| Gene repair | SCN5A | p.R1898H | CRISPR/Cas9‐mediated gene repair | 66 |
| PRKAG2 | c.905G > A (p.R302Q) | CRISPR/Cas9‐mediated gene repair | 67 | |
| PRKAG2 | p.R302Q | CRISPR/Cas9‐mediated gene repair | 68 | |
| DMD | Exon 3–6 del | CRISPR/Cas9‐mediated exon deletion | 69 | |
| CALM2 | p.D130G | CRISPR interference | 70 | |
| CALM2 | p.N98S | CRISPR/Cas9‐mediated allele knock out | 71 | |
| Introduction of mutation | ADRB2 GRK5 RYR2 ACTC1 |
Multiple c.122A > T c.6737C > T c.301G > A |
PiggyBac‐mediated gene editing | 72 |
| TNNT2 | p.I79N | CRISPR/Cas9‐mediated gene editing | 73 |
Generation of hESC‐Based Disease Models
While hiPSC are currently a popular choice for many cell‐based studies, recent advances in the CRISPR‐Cas9 technology have rendered hESC a valid and feasible alternative as well. Any wild‐type cell can be altered to harbor a specific mutation using CRISPR‐Cas9 mediated gene editing. Indeed, CRISPR‐Cas9 can be applied to create the perfect experimental controls in hiPSC and hESC: a pathogenic mutation can be corrected in patient‐derived hiPSC, while a putative pathogenic mutation can be inserted in otherwise wild‐type hESC. As result, genetically edited stem cells are the same as their original cell line in all aspects except the edited genes. It is important to note that any method facilitating gene editing can result in off‐target effects in various genomic regions. Following its introduction, studies demonstrated that this was also relevant for CRISPR‐Cas9 74, 75. However, in recent years, new nucleases have been discovered and have been verified to induce no off‐target effects 76, 77, 78. These new techniques allow for the generation of edited cell lines from a single source that only differ in the edited gene. This way, difference found between those cell lines can directly be attributed to a single mutation and can then be further studied in more complex models (e.g., patient‐derived hiPSC‐based models with familial controls).
Epigenetics and Environmental Influence
In contrast to a disease resulting from genetic variants, diseases can also arise from environmental factors, such as malnutrition, drug‐related effects, exogenous toxins, or maternal disease during gestation 79, 80, 81, 82, 83, 84, 85. Some of these environmental factors can lead to epigenetic changes, like DNA methylation. In this case, chances of obtaining a phenotype will be extremely small in a hiPSC‐based experimental setup. During reprogramming of somatic cells to hiPSC, most epigenetic features characteristic for a specific cell type are removed while cell type‐specific marks remain 86. More specifically, every cell type has a unique DNA methylation pattern. Importantly, epigenetic profiles that are linked to disease progression are lost during reprogramming. While losing disease‐causing epigenetic marks due to reprogramming may result in a model without a phenotype, which directly emphasizes the need to focus on (and possibly attenuate) the epigenetics factors in a specific patient 87.
To conclude, the patient‐derived aspect of hiPSC‐based disease models enables studies to be designed that may unravel pathological mechanisms caused by genetic as well as epigenetic anomalies. Due to the precision with which all other (in vitro and in vivo) models are designed, it can be expected that not all disease‐causing factors, for example, DNA methylation, are included and are therefore overlooked.
Cardiac Tissue Engineering
The heart is a complex organ composed of various cell types (e.g., cardiomyocytes, fibroblasts and endothelial cells) in a three‐dimensional (3D) organization. While many studies are performed with two‐dimensional (2D) in vitro cultures, previous studies showed that cells better recapitulate in vivo physiology when cultured in a 3D system 88, 89. Additionally, generation of cardiac tissue containing an appropriate mix of cell types improved feasibility of studies that were previously challenging, such as studies involving electrophysiology, cell–cell or cell‐extracellular matrix (ECM) interactions, cocultures, or drug screening 90. Subsequently, it provides an adaptable platform with the ability to replace various animal‐based studies, ultimately reducing the number of laboratory animals. To achieve such tissues for cardiac disease modeling, various techniques have been employed. Seminal work by Moscona in 1959 demonstrated that embryonic chicken cardiomyocytes spontaneously form beating cardiospheres. This was the basis for the currently most commonly used and adapted model: the engineered heart tissue model 91, 92, where hESC‐CM are seeded in a hydrogel. The hydrogel matrix casted in a mold, which can be cultured under mechanical strain between fixed anchoring points 92. The effects of various growth factors, cyclic uniaxial or multiaxial mechanical stretching, cardiomyocyte maturation, and electrical pacing 93 were studied using this model. The finding that nonmyocytes promote contractile force generation while also better reflecting the composition of the human heart, compared to tissue consisting of purified cardiomyocytes, has led to the standardization of adding various nonmyocytes to the tissue 94.
A second model of engineered cardiac tissue is based on the same principle demonstrated by Moscona in which various cell types can aggregate into spheroids (or microtissues) under the right conditions. Nonadhesive surfaces, hanging droplets and rotation systems are used to generate spheroids 95. While spheroids are generally small and challenging to physically manipulate, they are very suitable to study 3D behavior of cells and cell–cell interactions, drug testing, and can be used as building blocks to create larger tissues 96. A third and alternative approach to make tissues is the formation of cell sheets. By utilization of a coating, that dissolves at room temperature, intact detached cell‐monolayers that can be stacked to create thicker tissues for transplantation or drug screening 97.
However, the main limitation to these methods is the lack of vascularization and consequently low perfusion of oxygen and nutrients. Prefabricated channels and tubes have been incorporated in tissue constructs to address to improve tissue perfusion 98. As opposed to using self‐assembly and artificial matrices as a basis for tissue engineering, decellularized explanted hearts were also demonstrated to be viable scaffolds 99. Although, the main goal was to create fully functional hearts for transplantation, this has been largely unsuccessful to date. However, decellularized tissues retain hierarchical large and smaller vascular structures 100. These studies have set a precedent to use decellularized explanted tissues (i.e., small pieces of tissue) as a scaffold for tissue engineering. Remarkably, this can also be done with plant‐derived scaffolds, as was recently demonstrated by Gerschlak et al. 101. The overarching goal is to develop a high throughput screening platform with highly representative cardiac tissue. Aforementioned, there have been many advances in this field recently. To reach this goal, there have been various seminal studies published recently. The study by Mills et al. has elegantly demonstrated a procedure to generate high throughput screening platform based on human cardiac organoids 102. Additionally, to induce maturation in these organoids, Mills et al. have activated the proliferation pathways mediated by β‐catenin and Yes‐associated protein 1 (YAP1). As a result, matured human cardiac organoids can be applied for high throughput screening. Alternatively, Foo et al. have recently introduced a method for the generation of vascularized cardiac tissues by transplanting human stem cell‐derived cardiac precursors subcapsularly onto kidneys in mice 13. Furthermore, Lind et al. demonstrated that the popular “Heart‐on‐a‐chip” concept can now be obtained by a combination of a 3D printed flexible chip and tissue engineering 103. These state‐of‐the‐art tissue engineering techniques are summarized in Figure 2.
Figure 2.
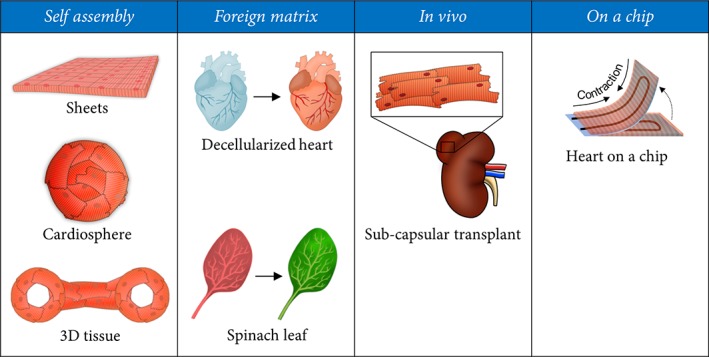
Summary of different technical approaches to cardiac tissue engineering. Cardiac tissues can be generated by allowing cardiac cells to spontaneously form a tissue by self‐assembly. Other approaches include the introduction of a decellularized matrix as a basis for reconstituted cardiac tissue, injecting human cardiac precursor cells into the murine kidney and machine‐guided generation of cardiac tissue on a chip.
Conclusions and Future Perspectives
In summary, to study a disease with incredible detail, target cells from various sources can be collected and cultured in 2D or 3D. These in vitro cultures can be manipulated very precisely, allowing researchers to pinpoint key factors of disease origin and progression. Building on these findings, novel drugs can be discovered and tested, driving the progression toward personalized medicine.
Depending on the field of study, in vitro disease models can be based on any cell type and source. However, to study cardiomyocytes, the cell sources are largely limited to pluripotent stem cells. An argument against the use of hiPSC is the residual epigenetic landscape that remains after reprogramming of any somatic cell type to hiPSC. Indeed, hiPSC can be cultured in pluripotent states similar to hESC and can be differentiated to virtually any cell type, but the effects of these residual epigenetic marks are unknown and depend to great extent on the source. This is a strong argument to use edited hESC instead of patient‐specific hiPS cells, especially since each patient‐derived cell line has a very different genetic background from any other hiPSC line. Therefore, a familial control has to be used for every patient line, as was indicated by Matsa et al. 18. In contrast, a single well defined hESC line (e.g., H9, H1, or HUES9) can be used as a basis for studies based on known mutations in which the unedited line can be a control for all introduced mutations.
Diseases often manifest as the result of one or multiple organs failing with a complex pathophysiology. A single organ contains various cell types with different functions, which often makes studying a disease challenging. By using in vitro disease models, it is possible to study specific cell types, study cocultures of involved cell types, and manipulate tightly regulated mechanisms. Consequently, this approach disregards confounding factors and all systemic effects (i.e., interorgan signaling) as seen with in vivo models. In contrast, this also entails that every aspect of the in vitro culturing method must be optimal for the specific model to be representative. Ultimately, it is no longer a near‐impossible task to recapitulate patient‐specific cardiomyopathies in vitro. As described in this review, recent technological advances have paved the way to more accessible culturing and engineering methods that will drive the field toward crucial insights into disease mechanisms and treatment options.
Presumably safe drugs have been withdrawn from the market more than once due to toxic effects in patients that were unobserved in the respective animal studies. Reasons vary from false negative results to off‐ and on‐target toxicity (including unexpected cardiotoxicity). Typically, drug safety assessment and efficacy testing are performed in animal models followed by expensive clinical trials. To make drug discovery and testing more cost‐effective, it is imperative that reliable alternative strategies are developed; human in vitro disease modeling will improve this process greatly.
Author Contributions
All authors wrote the manuscript and all author critically reviewed the manuscript.
Disclosure of Potential Conflicts of Interest
P.V.D.M. discloses consultant role for Vifor Pharma, Novartis and Astra Zeneca; and unrestricted grant from Vifor Pharma and Astra Zeneca. All other authors have no conflict to disclose.
Acknowledgments
This work was supported by the following grants to PvdM: ZonMW clinical fellow grant (90700436), ERC Stg grant (715732) and Dutch Heart Foundation grant (2012T47).
References
- 1. Benjamin EJ, Virani SS, Callaway CW et al. Heart disease and stroke statistics—2018 update: A report from the American heart association. Circulation 2018;137:e67–e492. [DOI] [PubMed] [Google Scholar]
- 2. Maron BJ, Maron MS. Hypertrophic cardiomyopathy. Lancet 2013;381:242–255. [DOI] [PubMed] [Google Scholar]
- 3. Weintraub RG, Semsarian C, Macdonald P. Dilated cardiomyopathy. Lancet 2017;390:400–414. [DOI] [PubMed] [Google Scholar]
- 4. Rampazzo A, Nava A, Danieli GA et al. The gene for arrhythmogenic right ventricular cardiomyopathy maps to chromosome 14q23–q24. Hum Mol Genet 1994;3:959–962. [DOI] [PubMed] [Google Scholar]
- 5. Jefferies JL, Towbin JA. Dilated cardiomyopathy. Lancet 2010;375:752–762. [DOI] [PubMed] [Google Scholar]
- 6. Gearhart J. New potential for human embryonic stem cells. Science 1998;282:1061–1062. [DOI] [PubMed] [Google Scholar]
- 7. Takahashi K, Tanabe K, Ohnuki M et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007;131:861–872. [DOI] [PubMed] [Google Scholar]
- 8. Davidson MM, Nesti C, Palenzuela L et al. Novel cell lines derived from adult human ventricular cardiomyocytes. J Mol Cell Cardiol 2005;39:133–147. [DOI] [PubMed] [Google Scholar]
- 9. Badie N, Bursac N. Novel micropatterned cardiac cell cultures with realistic ventricular microstructure. Biophys J 2009;96:3873–3885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Parker KK, Tan J, Chen CS et al. Myofibrillar architecture in engineered cardiac myocytes. Circ Res 2008;103:340–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kaneko T, Kojima K, Yasuda K. An on‐chip cardiomyocyte cell network assay for stable drug screening regarding community effect of cell network size. Analyst 2007;132:892–898. [DOI] [PubMed] [Google Scholar]
- 12. Giacomelli E, Bellin M, Sala L et al. Three‐dimensional cardiac microtissues composed of cardiomyocytes and endothelial cells co‐differentiated from human pluripotent stem cells. Development 2017;144:1008–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Foo KS, Lehtinen ML, Leung CY et al. Human ISL1 + ventricular progenitors self‐assemble into an in vivo functional heart patch and preserve cardiac function post infarction. Mol Ther 2018;26:1644–1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Beqqali A, Van Eldik W, Mummery C et al. Human stem cells as a model for cardiac differentiation and disease. Cell Mol Life Sci 2009;66:800–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jiang J, Han P, Zhang Q et al. Cardiac differentiation of human pluripotent stem cells. J Cell Mol Med 2012;16:1663–1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kehat I, Kenyagin‐Karsenti D, Snir M et al. Human embryonic stem cells can differentiate into myocytes with structural and functional properties of cardiomyocytes. J Clin Invest 2001;108:407–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yoshida Y, Yamanaka S. Induced pluripotent stem cells 10 years later. Circ Res 2017;120:1958–1968. [DOI] [PubMed] [Google Scholar]
- 18. Matsa E, Burridge PW, Yu KH et al. Transcriptome profiling of patient‐specific human iPSC‐cardiomyocytes predicts individual drug safety and efficacy responses in vitro. Cell Stem Cell 2016;19:311–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yang X, Rodriguez M, Pabon L et al. Tri‐iodo‐l‐thyronine promotes the maturation of human cardiomyocytes‐derived from induced pluripotent stem cells. J Mol Cell Cardiol 2014;72:296–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ribeiro MC, Tertoolen LG, Guadix JA et al. Functional maturation of human pluripotent stem cell derived cardiomyocytes in vitro–correlation between contraction force and electrophysiology. Biomaterials 2015;51:138–150. [DOI] [PubMed] [Google Scholar]
- 21. Ruan JL, Tulloch NL, Razumova MV et al. Mechanical stress conditioning and electrical stimulation promote contractility and force maturation of induced pluripotent stem cell‐derived human cardiac tissue. Circulation 2016;134:1557–1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ronaldson‐Bouchard K, Ma SP, Yeager K et al. Advanced maturation of human cardiac tissue grown from pluripotent stem cells. Nature 2018;556:239–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Guyette JP, Charest JM, Mills RW et al. Bioengineering human myocardium on native extracellular matrix. Circ Res 2016;118:56–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Caspi O, Lesman A, Basevitch Y et al. Tissue engineering of vascularized cardiac muscle from human embryonic stem cells. Circ Res 2007;100:263–272. [DOI] [PubMed] [Google Scholar]
- 25. Shadrin IY, Allen BW, Qian Y et al. Cardiopatch platform enables maturation and scale‐up of human pluripotent stem cell‐derived engineered heart tissues. Nat Commun 2017;8:1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhang D, Pu WT. Exercising engineered heart muscle to maturity. Nat Rev Cardiol 2018;15:383–384. [DOI] [PubMed] [Google Scholar]
- 27. Han P, Li W, Lin CH et al. A long noncoding RNA protects the heart from pathological hypertrophy. Nature 2014;514:102–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Van Rooij E, Marshall WS, Olson EN. Toward microRNA‐based therapeutics for heart disease: The sense in antisense. Circ Res 2008;103:919–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Anand P, Brown JD, Lin CY et al. BET bromodomains mediate transcriptional pause release in heart failure. Cell 2013;154:569–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Seidman JG, Seidman C. The genetic basis for cardiomyopathy: From mutation identification to mechanistic paradigms. Cell 2001;104:557–567. [DOI] [PubMed] [Google Scholar]
- 31. Christidi, E. , Huang, H. (Margaret) & Brunham, L. R. CRISPR/Cas9‐mediated genome editing in human stem cell‐derived cardiomyocytes: Applications for cardiovascular disease modelling and cardiotoxicity screening. Drug Discov Today Technol (2018). [DOI] [PubMed] [Google Scholar]
- 32. Han L, Li Y, Tchao J et al. Study familial hypertrophic cardiomyopathy using patient‐specific induced pluripotent stem cells. Cardiovasc Res 2014;104:258–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lan F, Lee AS, Liang P et al. Abnormal calcium handling properties underlie familial hypertrophic cardiomyopathy pathology in patient‐specific induced pluripotent stem cells. Cell Stem Cell 2013;12:101–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Prondzynski M, Krämer E, Laufer SD et al. Evaluation of MYBPC3 trans ‐splicing and gene replacement as therapeutic options in human iPSC‐Derived cardiomyocytes. Mol Ther Nucleic Acids 2017;7:475–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ojala M, Prajapati C, Pölönen RP et al. Mutation‐specific phenotypes in hiPSC‐derived cardiomyocytes carrying either myosin‐binding protein C Or α ‐tropomyosin mutation for hypertrophic cardiomyopathy. Stem Cells Int 2016;2016:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tanaka A, Yuasa S, Mearini G et al. Endothelin‐1 induces myofibrillar disarray and contractile vector variability in hypertrophic cardiomyopathy‐induced pluripotent stem cell‐derived cardiomyocytes. J Am Heart Assoc 2014;3:e001263–e001263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Birket MJ, Ribeiro MC, Kosmidis G et al. Contractile defect caused by mutation in MYBPC3 revealed under conditions optimized for human PSC‐cardiomyocyte function. Cell Rep 2015;13:733–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hinson JT, Chopra A, Nafissi N et al. Titin mutations in iPS cells define sarcomere insufficiency as a cause of dilated cardiomyopathy. Science 2015;349:982–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lee Y, Lau YM, Cai ZJ et al. Modeling treatment response for lamin A/C related dilated cardiomyopathy in human induced pluripotent stem cells. J Am Heart Assoc 2017;6:e005677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sun N, Yazawa M, Liu J et al. Patient‐specific induced pluripotent stem cells as a model for familial dilated cardiomyopathy. Sci Transl Med 2012;4:130ra47–130ra47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Broughton KM, Li J, Sarmah E et al. A myosin activator improves actin assembly and sarcomere function of human‐induced pluripotent stem cell‐derived cardiomyocytes with a troponin T point mutation. Am J Physiol Heart Circ Physiol 2016;311:H107–H117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tse HF, Ho JCY, Choi SW et al. Patient‐specific induced‐pluripotent stem cells‐derived cardiomyocytes recapitulate the pathogenic phenotypes of dilated cardiomyopathy due to a novel DES mutation identified by whole exome sequencing. Hum Mol Genet 2013;22:1395–1403. [DOI] [PubMed] [Google Scholar]
- 43. Wyles SP, Li X, Hrstka SC et al. Modeling structural and functional deficiencies of RBM20 familial dilated cardiomyopathy using human induced pluripotent stem cells. Hum Mol Genet 2016;25:254–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Streckfuss‐Bömeke K, Tiburcy M, Fomin A et al. Severe DCM phenotype of patient harboring RBM20 mutation S635A can be modeled by patient‐specific induced pluripotent stem cell‐derived cardiomyocytes. J Mol Cell Cardiol 2017;113:9–21. [DOI] [PubMed] [Google Scholar]
- 45. Stillitano F, Turnbull IC, Karakikes I et al. Genomic correction of familial cardiomyopathy in human engineered cardiac tissues. Eur Heart J 2016;37:3282–3284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Karakikes I, Stillitano F, Nonnenmacher M et al. Correction of human phospholamban R14del mutation associated with cardiomyopathy using targeted nucleases and combination therapy. Nat Commun 2015;6:6955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kim C, Wong J, Wen J et al. Studying arrhythmogenic right ventricular dysplasia with patient‐specific iPSCs. Nature 2013;494:105–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ma D, Wei H, Lu J et al. Generation of patient‐specific induced pluripotent stem cell‐derived cardiomyocytes as a cellular model of arrhythmogenic right ventricular cardiomyopathy. Eur Heart J 2013;34:1122–1133. [DOI] [PubMed] [Google Scholar]
- 49. Caspi O, Huber I, Gepstein A et al. Modeling of arrhythmogenic right ventricular cardiomyopathy with human induced pluripotent stem cells. Circ Cardiovasc Genet 2013;6:557–568. [DOI] [PubMed] [Google Scholar]
- 50. Te Riele AS, Agullo‐Pascual E, James CA et al. Multilevel analyses of SCN5A mutations in arrhythmogenic right ventricular dysplasia/cardiomyopathy suggest non‐canonical mechanisms for disease pathogenesis. Cardiovasc Res 2017;113:102–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kodo K, Ong SG, Jahanbani F et al. iPSC‐derived cardiomyocytes reveal abnormal TGF‐β signalling in left ventricular non‐compaction cardiomyopathy. Nat Cell Biol 2016;18:1031–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Burridge PW, Li YF, Matsa E et al. Human induced pluripotent stem cell–derived cardiomyocytes recapitulate the predilection of breast cancer patients to doxorubicin‐induced cardiotoxicity. Nat Med 2016;22:547–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Sharma A, Burridge PW, WL MK et al. High‐throughput screening of tyrosine kinase inhibitor cardiotoxicity with human induced pluripotent stem cells. Sci Transl Med 2017;9:eaaf2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Jiang Y, Habibollah S, Tilgner K et al. An induced pluripotent stem cell model of hypoplastic left heart syndrome (HLHS) reveals multiple expression and functional differences in HLHS‐derived cardiac myocytes. Stem Cells Transl Med 2014;3:416–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kobayashi J, Yoshida M, Tarui S et al. Directed differentiation of patient‐specific induced pluripotent stem cells identifies the transcriptional repression and epigenetic modification of NKX2‐5, HAND1, and NOTCH1 in hypoplastic left heart syndrome. PLoS One 2014;9:e102796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Israel MA, Yuan SH, Bardy C et al. Probing sporadic and familial Alzheimer's disease using induced pluripotent stem cells. Nature 2012;482:216–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Wing C, Komatsu M, Delaney SM et al. Application of stem cell derived neuronal cells to evaluate neurotoxic chemotherapy. Stem Cell Res 2017;22:79–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Papapetrou EP. Patient‐derived induced pluripotent stem cells in cancer research and precision oncology. Nat Med 2016;22:1392–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Hockemeyer D, Jaenisch R. Induced pluripotent stem cells meet genome editing. Cell Stem Cell 2016;18:573–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ran FA, Hsu PD, Wright J et al. Genome engineering using the CRISPR‐Cas9 system. Nat Protoc 2013;8:2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Cong L, Ran FA, Cox D et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013;339:819–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Karakikes I, Termglinchan V, Cepeda DA et al. A comprehensive TALEN‐based knockout library for generating human‐induced pluripotent stem cell‐based models for cardiovascular diseases. Circ Res 2017;120:1561–1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Hulot J‐S, Salem JE, Redheuil A et al. Effect of intracoronary administration of AAV1/SERCA2a on ventricular remodelling in patients with advanced systolic heart failure: results from the AGENT‐HF randomized phase 2 trial on behalf of the AGENT‐HF Investigators. Eur J Heart Fail 2017;19:1534–1541. [DOI] [PubMed] [Google Scholar]
- 64. Jessup M, Greenberg B, Mancini D et al. Calcium upregulation by percutaneous administration of gene therapy in cardiac disease (CUPID): A phase 2 trial of intracoronary gene therapy of sarcoplasmic reticulum Ca2+‐ATPase in patients with advanced heart failure. Circulation 2011;124:304–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Ma H, Marti‐Gutierrez N, Park SW et al. Correction of a pathogenic gene mutation in human embryos. Nature 2017;548:413–419. [DOI] [PubMed] [Google Scholar]
- 66. te Riele ASJM, Agullo‐Pascual E, James CA et al. Multilevel analyses of SCN5A mutations in arrhythmogenic right ventricular dysplasia/cardiomyopathy suggest non‐canonical mechanisms for disease pathogenesis. Cardiovasc Res 2017;113:102–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Zhan Y, Sun X, Li B et al. Establishment of a PRKAG2 cardiac syndrome disease model and mechanism study using human induced pluripotent stem cells. J Mol Cell Cardiol 2018;117:49–61. [DOI] [PubMed] [Google Scholar]
- 68. Ben Jehuda R, Eisen B, Shemer Y et al. CRISPR correction of the PRKAG2 gene mutation in the patient's induced pluripotent stem cell‐derived cardiomyocytes eliminates electrophysiological and structural abnormalities. Heart Rhythm 2018;15:267–276. [DOI] [PubMed] [Google Scholar]
- 69. Kyrychenko V, Kyrychenko S, Tiburcy M et al. Functional correction of dystrophin actin binding domain mutations by genome editing. JCI Insight 2017;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Limpitikul WB, Dick IE, Tester DJ et al. A precision medicine approach to the rescue of function on malignant calmodulinopathic long‐QT syndromenovelty and significance. Circ Res 2017;120:39–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Yamamoto Y, Makiyama T, Harita T et al. Allele‐specific ablation rescues electrophysiological abnormalities in a human iPS cell model of long‐QT syndrome with a CALM2 mutation. Hum Mol Genet 2017;26:1670–1677. [DOI] [PubMed] [Google Scholar]
- 72. Kondrashov A, Duc Hoang M, Smith JGW et al. Simplified Footprint‐Free Cas9/CRISPR Editing of Cardiac‐Associated Genes in Human Pluripotent Stem Cells. Stem Cells Dev 2018;27:391–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Wang L, Kim K, Parikh S et al. Hypertrophic cardiomyopathy‐linked mutation in troponin T causes myofibrillar disarray and pro‐arrhythmic action potential changes in human iPSC cardiomyocytes. J Mol Cell Cardiol 2018;114:320–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Kim D, Bae S, Park J et al. Digenome‐seq: genome‐wide profiling of CRISPR‐Cas9 off‐target effects in human cells. Nat Methods 2015;12:237–243. [DOI] [PubMed] [Google Scholar]
- 75. Fu Y, Foden JA, Khayter C et al. High‐frequency off‐target mutagenesis induced by CRISPR‐Cas nucleases in human cells. Nat Biotechnol 2013;31:822–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Kleinstiver BP, Pattanayak V, Prew MS et al. High‐fidelity CRISPR–Cas9 nucleases with no detectable genome‐wide off‐target effects. Nature 2016;529:490–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Cox DBT, Gootenberg JS, Abudayyeh OO et al. RNA editing with CRISPR‐Cas13. Science 2017;358:1019–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Abudayyeh OO, Gootenberg JS, Essletzbichler P et al. RNA targeting with CRISPR–Cas13. Nature 2017;550:280–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. de Boer RA, Meems LMG, van Veldhuisen DJ. Vitamin D supplementation in heart failure: case closed? Eur Heart J 2017;38:2287–2289. [DOI] [PubMed] [Google Scholar]
- 80. Hoes MF, Grote Beverborg N, Kijlstra JD et al. Iron deficiency impairs contractility of human cardiomyocytes through decreased mitochondrial function. Eur J Heart Fail 2018;20:910–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Klip IT, Comin‐Colet J, Voors AA et al. Iron deficiency in chronic heart failure: an international pooled analysis. Am Heart J 2013;165:575–582.e3. [DOI] [PubMed] [Google Scholar]
- 82. Ovchinnikova E, Hoes M, Ustyantsev K et al. Modeling human cardiac hypertrophy in stem cell‐derived cardiomyocytes. Stem Cell Rep 2018;10:794–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Cosselman KE, Navas‐Acien A, Kaufman JD. Environmental factors in cardiovascular disease. Nat Rev Cardiol 2015;12:627–642. [DOI] [PubMed] [Google Scholar]
- 84. Gunderson EP, Chiang V, Pletcher MJ et al. History of gestational diabetes mellitus and future risk of atherosclerosis in mid‐life: the coronary artery risk development in young adults study. J Am Heart Assoc 2014;3:e000490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Catalano PM, Tyzbir ED, Roman NM et al. Longitudinal changes in insulin release and insulin resistance in nonobese pregnant women. Am J Obstet Gynecol 1991;165:1667–1672. [DOI] [PubMed] [Google Scholar]
- 86. Kim K, Doi A, Wen B et al. Epigenetic memory in induced pluripotent stem cells. Nature 2010;467:285–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Hewitt KJ, Garlick JA. Cellular reprogramming to reset epigenetic signatures. Mol Asp Med 2013;34:841–848. [DOI] [PubMed] [Google Scholar]
- 88. Elliott NT, Yuan F. A review of three‐dimensional in vitro tissue models for drug discovery and transport studies. J Pharm Sci 2011;100:59–74. [DOI] [PubMed] [Google Scholar]
- 89. Kijlstra JD, Hu D, Mittal N et al. Integrated analysis of contractile kinetics, force generation, and electrical activity in single human stem cell‐derived cardiomyocytes. Stem Cell Rep 2015;5:1226–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Bursac N, Papadaki M, Cohen RJ et al. Cardiac muscle tissue engineering: toward an in vitro model for electrophysiological studies. Am J Phys 1999;277:H433–H444. [DOI] [PubMed] [Google Scholar]
- 91. Moscona, A. Tissues from dissociated cells. Sci Am 1959;200:132–134. [DOI] [PubMed] [Google Scholar]
- 92. Eschenhagen T, Fink C, Remmers U et al. Three‐dimensional reconstitution of embryonic cardiomyocytes in a collagen matrix: a new heart muscle model system. FASEB J 1997;11:683–694. [DOI] [PubMed] [Google Scholar]
- 93. Zuppinger C. 3D culture for cardiac cells. Biochim Biophys Acta, Mol Cell Res 2016;1863:1873–1881. [DOI] [PubMed] [Google Scholar]
- 94. Lesman A, Habib M, Caspi O et al. Transplantation of a tissue‐engineered human vascularized cardiac muscle. Tissue Eng Part A 2010;16:115–125. [DOI] [PubMed] [Google Scholar]
- 95. Fennema E, Rivron N, Rouwkema J et al. Spheroid culture as a tool for creating 3D complex tissues. Trends Biotechnol 2013;31:108–115. [DOI] [PubMed] [Google Scholar]
- 96. Edmondson R, Broglie JJ, Adcock AF et al. Three‐dimensional cell culture systems and their applications in drug discovery and cell‐based biosensors. Assay Drug Dev Technol 2014;12:207–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Fleischer S, Shapira A, Feiner R et al. Modular assembly of thick multifunctional cardiac patches. Proc Natl Acad Sci U S A 2017;114:1898–1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Hasan A, Paul A, Vrana NE et al. Microfluidic techniques for development of 3D vascularized tissue. Biomaterials 2014;35:7308–7325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Ott HC, Matthiesen TS, Goh SK et al. Perfusion‐decellularized matrix: using nature's platform to engineer a bioartificial heart. Nat Med 2008;14:213–221. [DOI] [PubMed] [Google Scholar]
- 100. Guyette JP, Charest JM, Mills RW et al. Bioengineering human myocardium on native extracellular matrixnovelty and significance. Circ Res 2016;118:56–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Gershlak JR, Hernandez S, Fontana G et al. Crossing kingdoms: using decellularized plants as perfusable tissue engineering scaffolds. Biomaterials 2017;125:13–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Mills RJ, Titmarsh DM, Koenig X et al. Functional screening in human cardiac organoids reveals a metabolic mechanism for cardiomyocyte cell cycle arrest. Proc Natl Acad Sci U S A 2017;114:E8372–E8381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Lind JU, Busbee TA, Valentine AD et al. Instrumented cardiac microphysiological devices via multimaterial three‐dimensional printing. Nat Mater 2017;16:303–308. [DOI] [PMC free article] [PubMed] [Google Scholar]