

Summary
Memory was traditionally considered an exclusive hallmark of adaptive immunity. This dogma was challenged by recent reports that myeloid cells can retain ‘memory’ of earlier challenges enabling them to strongly respond to a secondary stimulus. This process, designated ‘trained immunity’, is initiated by modulation of precursors of myeloid cells in the bone marrow. The ancestral innate immune system of lower organisms, e.g. Caenorhabditis elegans, can build long-lasting memory that modifies responses to secondary pathogen encounters. We posit that changes in cellular metabolism may be a common denominator of innate immune memory from lower animals to mammals. We discuss evidence from C. elegans and murine/human systems supporting the concept of an ancestral principle regulating innate immune memory by controlling cellular metabolism.
Innate Immune Training and Metabolism
Adaptive immunity has traditionally been described in contrasting terms with regards to innate immunity, such as the presence, or lack thereof, of immunological memory. However, recent advances from experiments in human and murine immune systems, show that innate immune cells can retain ‘memory’ of past inflammatory events (e.g., owing to infection), which enables them to elicit a heightened immune response upon a secondary challenge, irrespective of adaptive immunity [1]. This enhanced state of immune activation has been designated ‘trained innate immunity’; it lacks specificity to the initial stimulus and thus, can confer cross-protection against subsequent challenges with unrelated fungal, bacterial, or viral pathogens [1].
Given the survival advantage of imprinted memory of past infectious encounters, it could be reasoned that trained immunity represents an evolutionarily conserved way of memory-based enhanced preparedness for future challenges. In support of this notion, forms of innate immune memory have also been documented in non-vertebrate animals and in plants [2]. For instance, the immune system of insects can be primed for long-lasting protection against future infections [3–5]. The nematode Caenorhabditis elegans can build long-lasting and even transgenerational innate immune memory that enables the worms to effectively avoid or resist secondary pathogen encounters [6,7]. The so-called ‘systemic acquired resistance’ of plants is a mechanism of inducible defense that primes even remote tissues for enhanced resistance to subsequent infections [8]. Accordingly, plants inoculated with attenuated microorganisms acquire long-term protection against a wide range of plant pathogens [8,9].
Induction of trained immunity appears to be mediated by long-term adaptations in chromatin (of trained cells), rendered more accessible to the transcriptional machinery [1,2]. This enhanced accessibility persists over time despite the loss/cessation of the inductive stimulus (e.g., microorganisms or certain components thereof, such as the fungal cell wall constituent β-glucan) [1,2]. Accompanying epigenetic chromatin modifications can be regulated by immune signaling and metabolic pathways (e.g., glycolysis, glutaminolysis and changes in the mevalonate metabolism), as metabolites may impact on the epigenetic landscape, serving as signaling molecules or as substrates and/or co-factors for chromatin-modifying enzymes [10–16]. The remarkable persistence of trained immunity despite the rather short lifespan of mature myeloid cells in peripheral blood [17] might be explained by long-term rewiring (metabolic, epigenetic, and transcriptional) of precursors of differentiated innate immune cells, such as hematopoietic stem and progenitor cells (HSPC) in the bone marrow [13,14]. Trained immunity initiated by modulation of progenitors in the bone marrow can enhance the replenishment of innate immune cell populations upon stress associated with infectious, inflammatory or chemotherapeutic challenges [13,14].
Although trained immunity can mediate protective immunity against subsequent systemic or mucosal infections [13,14,18–20], there are settings (for instance, age-related inflammatory diseases, such as atherosclerosis, or neurodegenerative diseases), in which trained immunity (inappropriately induced by microbial or even endogenous stimuli) could promote maladaptive immune/inflammatory responses that aggravate pathology [21–24].
In the present article, we summarize and discuss recent findings on trained innate immunity in mammalian and nematode systems. Epigenetic and transcriptional reprogramming underlying trained immunity in mice and humans may require prior metabolic alterations [11,12,15,25]. Metabolic and neuroendocrine changes may also enhance the resistance to stress and infection in C. elegans as well [26–28]. Consequently, we focus on highlighting the crosstalk that exists between immunity and metabolism, which may provide a unifying principle of innate immune training across species. Specifically, we propose that changes in cellular metabolism in response to an infectious stimulus or immunological stress represent an evolutionarily conserved common denominator in the induction of trained innate immunity from nematodes to mammals.
Metabolic Regulation of Trained Immunity in Mice, Humans, and Nematodes
Immunometabolic Circuits in Mature Myeloid Cell Training
Several stimuli, such as the fungal cell-wall constituent β-glucan and the Bacillus Calmette–Guérin (BCG) vaccine, can induce training of mature innate immune cells (Figure 1), mainly monocytes and macrophages, thereby promoting an enhanced response, as assessed by the release of pro-inflammatory cytokines, to secondary infectious stimuli, such as Candida albicans or Staphylococcus aureus [2]. Trained immunity is closely linked to sustained alterations in gene expression mostly resulting from epigenetic modifications [10,19]. Such epigenetic modifications may involve DNA hypermethylation, which generally results in transcriptional silencing, or histone modifications that facilitate or inhibit transcription factor binding, thereby regulating transcriptional activity [29]. Of note, such immunological imprinting can take two distinct and opposed forms, trained innate immunity, and innate immune tolerance, the latter leading to attenuated immune responses to secondary stimuli [30]. Specific genome-wide histone modifications in human monocytes and macrophages have been associated with trained innate immunity or tolerance upon stimulation with β-glucan or lipopolysaccharide (LPS), respectively [10]. Indeed, the epigenetic signature of LPS-induced tolerance is reversed upon “training” with β-glucan; thereby, innate immune training can reinstate proper cytokine responses of human macrophages after LPS-induced immunological tolerance [31]. This implies that trained immunity might be harnessed therapeutically to normalize innate immune function that is severely impaired during sepsis [32].
Figure 1. Trained immunity in human monocytes.
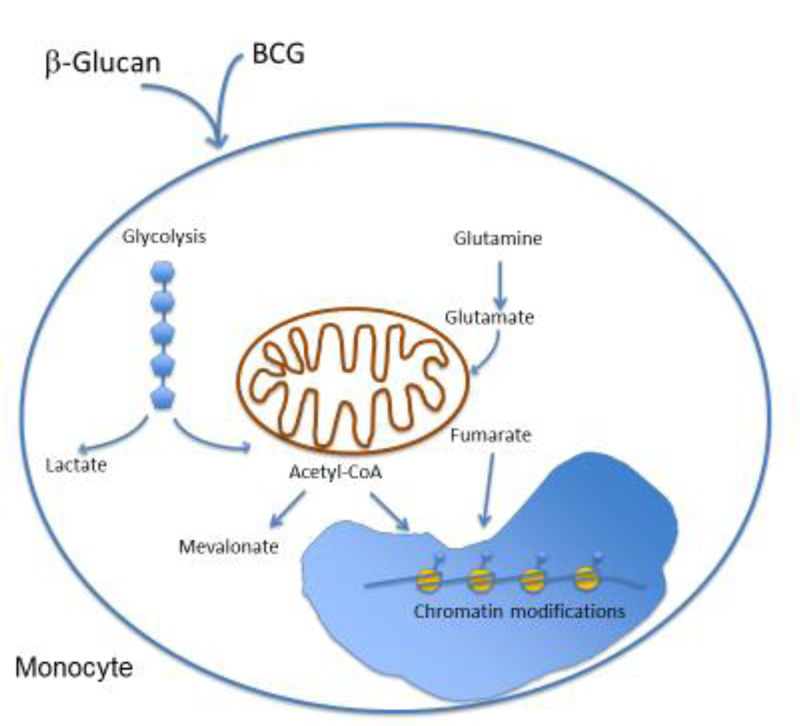
β-Glucan or BCG can lead to immune-metabolic changes in human monocytes, particularly the induction of glycolysis, glutaminolysis and cholesterol metabolism. The generation of metabolic intermediates, including acetyl-CoA, fumarate or mevalonate, can induce epigenetic alterations that drive innate immune memory, thereby improving the response of monocytes to secondary infectious stimuli.
Epigenetic modifications in trained immunity have been associated with metabolic reprogramming of innate immune cells, since metabolic intermediates can act as substrates, co-factors or inhibitors for chromatin-modifying enzymes [33]. This well-documented link between cellular energy metabolism and epigenetic modifications suggests that metabolic reprogramming of immune cells may regulate the phenotypical and functional plasticity of these cells. For example, the production of tricarboxylic acid (TCA) cycle intermediate α-ketoglutarate via glutaminolysis can mediate macrophage polarization towards an alternative (M2) phenotype, through epigenetic changes in M2-related genes in mouse macrophages in a manner dependent on the function of the H3K27 demethylase JMJD3 [34]. A recent study demonstrated that the metabolite itaconate could exert an anti-inflammatory function in human and mouse macrophages via alkylation of cysteine residues on the protein KEAP1, thereby allowing the anti-inflammatory transcription factor Nrf2 to increase expression of anti-inflammatory genes [35]. The itaconate pathway can also play a role in the development of immune tolerance; in this regard, β-glucan-induced trained immunity reduced the expression of immune-responsive gene 1 (IRG1)-- the enzyme that regulates itaconate synthesis -- thereby reversing tolerance induced by endotoxemia in human monocytes ex vivo relative to untreated monocytes [36]. The link between cell metabolism and epigenetic reprogramming also contributes to induction of trained immunity. Indeed, a pioneer study demonstrated that elevated aerobic glycolysis was the metabolic basis of β-glucan-induced trained immunity, by providing energy and the necessary metabolic substrates for the activation of trained innate immune cells [11]. Further integrated transcriptomic, metabolic and epigenetic analyses of human monocytes upon training with β-glucan revealed an interplay between glycolysis, glutamine and cholesterol metabolism in innate immune training [25]. In the latter study, induction of epigenetic modifications in trained cells could be ascribed to fumarate, which inhibits KDM5 histone demethylases [25]. Subsequent studies in human monocytes upon β-glucan-dependent training, suggested that the accumulation of mevalonate, an initial component of cholesterol biosynthesis, could promote monocyte training via activation of the insulin-like growth factor 1 receptor and mTOR signaling [12]. Similar to β-glucan, BCG treatment also induced reprogramming of monocytes towards glycolysis, further supporting the central role of glycolysis as a metabolic basis for trained immunity [37].
The danger signal LPS, interacting with Toll-like receptor 4 (TLR4), has been shown to upregulate glycolysis and fatty acid synthesis, while suppressing oxidative phosphorylation [38]. However, the suppressive effect on oxidative phosphorylation appears to be specific for higher concentrations of LPS, which are linked to the induction of immune tolerance, whereas lower doses of LPS or activation of other TLRs can increase both glycolysis and oxidative phosphorylation in human monocytes [39]. In this context, low-dose LPS can result in the activation of innate immune cells rather than immune tolerance, as supported by several studies [40–42]. Similarly, activation of TLR9 by unmethylated cytosine phosphate guanidine (CpG) oligodeoxynucleotides can confer heterologous protection against intracerebral infection by Escherichia coli in mice [43]. Of note, TLR5 activation by flagellin can also protect mice against Streptococcus pneumoniae lung infection [44] or rotavirus infection [45].
Taken together, the rewiring of cellular metabolism toward aerobic glycolysis and cholesterol biosynthesis and the accumulation of intermediate metabolites (including mevalonate and fumarate) may regulate the activity of chromatin-modifying enzymes. Modulation of cellular metabolism may be integral to innate immune training, but further studies are warranted to better elucidate this point.
Immunometabolic Circuits in Progenitor Cells Linked to Trained Immunity
Two recent studies demonstrated that trained innate immunity was initiated at the level of hematopoietic progenitors. In one of the studies, intravenous BCG administration enhanced myelopoiesis in the bone marrow of mice by inducing transcriptional reprogramming of HSPCs [13]. Noteworthy, macrophages derived from these trained progenitors displayed an enhanced protective response against mycobacterial infection, as shown by improved bacterial clearance [13]. In a parallel study, our group showed that administration of β-glucan to mice, by acting through induction of interleukin-1β (IL-1β), promoted a bias towards myeloid lineage differentiation in HSPCs and increased numbers of myeloid progenitor cells, as compared to vehicle control treatment. Myelopoiesis, induced by β-glucan-dependent training, as compared to vehicle control treatment, was mediated by enhanced glycolysis and cholesterol biosynthesis in progenitors (Figure 2), and could protect against chemotherapy-induced myelosuppression, upon repeated rounds of cyclophosphamide or 5-fluorouracil administration in mice [14]. These data suggest that β-glucan-dependent training might be beneficial for the reversal of neutropenia in patients under chemotherapeutic regimens.
Figure 2. Trained immunity in HSPCs in mice.

Western-type diet and microbe-derived stimuli (e.g. β-glucan) linked to trained immunity can induce the upregulation of pro-inflammatory cytokines, such as IL-1β, which trigger metabolic changes, particularly alterations in glucose and lipid metabolism in HSPCs. Cholesterol accumulation in HSPCs, for instance due to its increased biosynthesis or due to decreased cholesterol efflux through exporters such as ABCA1 or ABCG1, can drive myeloid differentiation bias. These alterations in HSPCs associated with trained immunity can mediate on the one hand, a hematopoietic improved response to chemotherapy, and on the other hand, may promote atherosclerosis and cardiovascular inflammation.
Innate immune training of hematopoietic progenitors shares common features with the expansion of myelopoiesis upon systemic infection, termed emergency myelopoiesis [46]. Systemic infection or LPS administration to mice can activate HSPC proliferation and their myeloid differentiation [47–49]. However, HSPC expansion in emergency myelopoiesis may compromise HSPC functionality, as shown by their decreased repopulation activity tested in competitive transplantation assays. Specifically, high-dose LPS can lead to extensive cell death of hematopoietic cells in the bone marrow [48], whereas repeated administration of lower doses of LPS can promote a functional decline in self-renewal activity of HSPC associated with proliferative stress [49]. Such detrimental effects have not been observed upon administration of stimuli that induce innate immune training [13,14]. In contrast, innate immune training induced by β-glucan injection to mice protected their hematopoiesis against chemotherapy-induced attrition of HSCs and mitigated LPS-induced DNA damage in HSPCs [14].
However, innate immune training is not solely associated with favorable host responses. Immunometabolic crosstalk at the level of HSPCs may contribute to progression of cardiometabolic disease and may also provide a link between clonal hematopoiesis of indeterminate potential (CHIP) (Box 1) and cardiovascular inflammation [21,50–52]. Enhanced myelopoiesis, mainly presented as monocytosis and neutrophilia, is considered an important feature of metabolic syndrome [53–55]. Accordingly, IL-1β produced by adipose tissue macrophages in obese mice has been proposed as the driving force for the activation and proliferation of myeloid lineage progenitors in the bone marrow and their differentiation toward neutrophils and monocytes, as assessed by experiments involving adipose tissue transplantation to lean recipient mice [54]. In another study, western diet-induced systemic inflammation in Low density lipoprotein receptor deficient (Ldlr−/−) mice promoted inflammasome-mediated innate immune training via transcriptional and epigenetic reprogramming of granulocyte-macrophage progenitors, as shown by integrated transcriptomic and epigenetic analysis of this cell population and engagement of Nlrp3−/− mice [21]. These findings showed that aberrant myelopoiesis potentially induced by trained immunity might be a driving force in atherothrombotic disorders.
Box1. A link between trained immunity and CHIP in humans?
Clonal hematopoiesis of indeterminate potential (CHIP) represents a paradigm of the link between dysfunctional myelopoiesis and cardiovascular risk, suggesting an important role of myelopoiesis in the progression of cardiometabolic disease. Individuals with CHIP have mutations associated with myeloid neoplasms, including mutations in DNMT3A, TET2, ASXL1, and JAK2 genes, without any sign of cytopenia. This condition has been recently associated with a significantly increased risk for atherosclerotic disease [50,78]. Accordingly, Tet2 deficiency in hypercholesterolemic mice has been shown to promote a proinflammatory signature in macrophages associated with upregulation of IL-1β and the development of atherosclerosis [50]. In a parallel study, Tet2-mutated HSPCs displayed a bias toward myeloid lineage differentiation after transplantation into hypercholesterolemic mice, which exhibited accelerated atherosclerosis in a NLRP3 inflammasome/IL-1β pathway-dependent manner [79]. Thus, similar to trained immunity, IL-1β appears to orchestrate immunometabolic crosstalk between myelopoiesis and cardiometabolic disease in a scenario of CHIP. Hence, similarities exist between the molecular pathways linked with CHIP and pathways underlying innate immune training of bone marrow progenitors. Further studies are needed to understand, whether a potential link between CHIP and trained immunity exists.
Cell-intrinsic alterations of cholesterol metabolism in HSPCs have also been linked to changes in myelopoiesis in the context of cardiovascular disease. Deficiency in the adenosine triphosphate-binding cassette (ABC) transporters ABCA1 and ABCG1, which mediate the efflux of cholesterol [56], or in apolipoprotein E, which acts in the same pathway [57], resulted in increased myeloid differentiation of HSPC, as demonstrated by the development of monocytosis and neutrophilia in transgenic mice, suggesting that cholesterol accumulation in HSPCs could promote myelopoiesis (Figure 2). These findings are in line with the recently identified role of cholesterol biosynthesis in mediating innate immune training of both mouse and human monocytes [12] and their progenitors [14]. Additionally, disrupted cholesterol efflux has led to increased expression of CD131, the common β subunit of the IL-3/GMCSF receptor, in murine hematopoietic progenitors [56,57]. Accordingly, upregulation of CD131 together with accumulation of cholesterol esters in HSPCs contributed to the enhanced myelopoiesis related to β-glucan-induced trained immunity in mice, as evidenced by the inhibitory effect of GM-CSF receptor blockade and statin administration in the expansion of hematopoietic progenitors in the bone marrow of the animals [14]. Taken together, alterations in metabolic pathways, such as enhanced glycolysis or cholesterol biosynthesis, can contribute to innate immune training of hematopoietic progenitors.
Immune System and Metabolism Crosstalk in C. elegans
C. elegans is a powerful model system for genetic and cell biology research of, among many fields, metabolism and innate immunity [58–60]. One of the critical factors that determines the fitness of the worm population is the abundance of pathogenic microorganisms potentially mixed with the food sources [58]. Thus, a requirement for reliable host defense mechanisms coordinated with the general responses to changes in the nutrient availability has shaped an immune system modulated by conserved metabolic signaling pathways; as outlined below several metabolic pathways are implicated in the responses to multiple bacterial and fungal pathogens [58].
Direct evidence for an immune-metabolic crosstalk in C. elegans emerged from studies showing that changes in the activity of metabolic pathways could alter immune function. Examples of such pathways included one-carbon metabolism, phospholipid production, fatty acid synthesis and desaturation, lipogenesis, and amino acid catabolism [61–64]. Furthermore, in a manner that mirrors the regulation of innate immunity in mammals, these pathways can generate metabolites that either serve as signaling molecules or as substrates/co-factors participating in chromatin modifications. For instance, S-adenosylmethionine (SAM), a major donor of methyl groups in different biochemical reactions, including methylation of nucleic acids and histones, has been reported to play an immunomodulatory role. Specifically, transcriptional analysis complemented with functional assays showed that SAM-depleted worms grown on a non-pathogenic diet displayed constitutive activation of the PMK-1/p38 MAPK-dependent innate immune pathway resulting from decreased production of the methylated phospholipid phosphatidylcholine (PC) [61]. However, survival assays indicated that the aforementioned immune activation failed to protect the SAM-depleted worms from Pseudomonas aeruginosa intestinal infection; in fact, SAM-depleted worms died more rapidly upon exposure to virulent Pseudomonas [61]. This phenomenon was associated with insufficient SAM-dependent H3K4 histone methylation and diminished expression of infection response genes, such as irg-1 or irg-2 upon exposure to Pseudomonas [61]. Other metabolic processes can also influence resistance of worms to P. aeruginosa. For instance, transcriptional and lipidomic analyses demonstrated that the polyunsaturated fatty acids (PUFAs), gamma-linolenic and stearidonic acid were important for survival upon infection with this pathogen, as assessed by enhanced susceptibility to infection in worms deficient in these fatty acids [62]. Furthermore, catabolism of the amino acid proline promoted pathogen resistance via regulation of homeostasis of reactive oxygen species [63]. Future studies should assess whether the distinct metabolic pathways, including SAM, PUFAs or proline catabolism may also act in a cooperative fashion to mediate resistance of C. elegans to pathogens.
Immune-metabolic crosstalk was also demonstrated in the model of epidermal infection using the fungal pathogen Drechmeria coniospora. An RNAi screen identified a critical role of the G-protein coupled receptor (GPCR) DCAR-1, which activates the p38 MAPK pathway, in the response to fungal infection. Targeted metabolomics demonstrated that hydroxyphenyllactic acid (HPLA), a side product of tyrosine metabolism, acted as a ligand of DCAR-1 [64]. Analysis of mutants with defects in the worm cuticle suggested that HPLA was produced upon disruption of the integrity of the cuticle/hypodermis accompanying pathogen entry into the host, thus coupling wounding and infection responses, and acting as an endogenous damage-associated molecular pattern [64]. A C. elegans ortholog of the first enzyme in the tyrosine catabolic pathway, the tyrosine aminotransferase TATN-1, was implicated not only in the synthesis of HPLA [64] but also in the regulation of worm development and longevity in an manner dependent on the AMP-dependent protein kinase [65]. Hence, TATN-1 and HPLA might also provide a link between development and immune responses. Of note, the epidermal production of antimicrobial factors seems to be negatively regulated by the fatty acid synthesis apparatus in a manner that depends on changes in the osmotic environment [66]. Genetic evidence suggested that the pathway involving fatty acid synthesis did not depend on p38/MAPK signaling but rather, on a pathway involved in osmosensation [66]. Results from these findings collectively suggest that distinct metabolic signals may integrate information from different stimuli to render an orchestrated and effective immune response against fungal pathogens.
The existence of an immunometabolic crosstalk in C. elegans is further supported by the fact that core signaling pathways responsible for metabolic control, including the insulin-like pathway, can serve as regulators of the immune system. Specifically, genetic approaches demonstrated that loss-of-function mutations in the insulin/IGF-1 receptor homologue DAF-2 rendered worms resistant to pathogenic bacteria, in a manner that required intact activity of the transcription factor DAF-16, the nematode ortholog of mammalian FoxO proteins [27]. This is relevant because DAF-16 is known to promote longevity and stress resistance downstream of DAF-2 in C. elegans at least partly through control of metabolism [28,67]. In addition, DAF-16 is essential for the formation of a specialized, developmentally arrested stage of survival under adverse conditions in C. elegans and numerous other nematode species, termed dauer (enduring) larva [68], characterized by low oxidative phosphorylation and TCA cycle activity, enhanced build-up of lipids, and Warburg metabolism-like aerobic glycolysis powered by preceding gluconeogenesis [69]. Conceivably, DAF-16-mediated resistance to pathogenic microorganisms might be, at least in part, attributed to changes in the metabolic landscape. In this regard, the metabolic traits induced by insulin signaling in the context of dauer arrest and longevity may be partially conserved in the responses to infection, although this notion remains to be fully investigated (Figure 3). Indeed, suppression of DAF-16 has been reported during infection with P. aeruginosa, which may point to active targeting of the insulin pathway by the pathogen [70]. However, another interpretation of this adaptation could be that the worms avoid excessive DAF-16 activity, which might result in an uncontrolled, deleterious immune response [71].
Figure 3. Putative insulin signaling-mediated immunometabolic crosstalk in C. elegans.

A complex organismal response to infection involves processes such as pathogen recognition, neuro-endocrine signaling, and general stress responses. Such processes trigger signaling cascades that converge to activate DAF-16/FoxO in effector cells (e.g. intestinal cells and neurons), although the exact mechanisms are unknown. The activation of DAF-16 might result from inhibition of the receptor DAF-2 or to a varying extent, could be mediated by factors acting in parallel to DAF-2 (not depicted). DAF-16 activation can trigger the transcription of pro-inflammatory and metabolic genes (e.g., genes boosting aerobic glycolysis and suppressing the TCA cycle) that promote pathogen resistance. The metabolic alterations induced by DAF-16 can also lead, via yet unknown mechanisms, to chromatin remodeling that may underlie trained immunity. Therefore, the insulin cascade may be involved in both the immediate response to pathogens and in the acquisition of immune memory.
Although the immune system of C. elegans is entirely innate, it has the capability to build immune memory for properly adapting host responses to proximal or future challenges [6,7,72,73]. This points to the existence of mechanisms for encoding the information of earlier pathogen encounters into the wiring of immune signaling pathways and/or the epigenetic background, thereby enabling experience-informed responses to secondary challenges. These mechanisms can involve metabolic control; for instance, insulin signaling is implicated in trained immunity in C. elegans. DAF-16, as well as p38 MAP kinase signaling, are required for the conditioning (priming) with enteropathogenic E. coli that allows worms to survive a secondary pathogen exposure that would otherwise be lethal [6]. Furthermore, as an alternative resistance strategy, worms respond with increased dauer formation to subsequent treatments with pathogenic bacteria (e.g., P. aeruginosa and Salmonella enterica), lasting for multiple generations after the original encounter [7]. Dauer formation might provide an advantage to worms, because dauer larvae are sealed off from the environment [68], and hence might be less susceptible to intestinal colonization. Pathogen-induced dauer formation is associated with DAF-16 activation, thereby providing a potential link between immune conditioning and the DAF-16-mediated metabolic control [7]. The insulin pathway also regulates a behavioral form of conditioning towards avoidance of P. aeruginosa [74]. Genetic and behavioral assays have shown that this conditioning, also known as ‘aversive learning’, is negatively regulated by an insulin ligand, INS-11, a neuropeptide produced in the intestine [74]. Altogether, the different forms of trained immunity in worms are associated with modified insulin signaling and metabolic cues. Direct indication that this pathway is involved in the acquisition of innate immune memory via metabolic rearrangements is yet to be demonstrated (Figure 3); however, the connection between DAF-16 and the control of chromatin state in other processes, such as stress resistance and aging, may render this hypothesis highly plausible [75–77]. Thus, the nematode innate immune system appears to be dependent on coordinated metabolic and transcriptional control, as well as on the regulation of chromatin states, in a fashion that may resemble mammalian trained immunity, although further work is necessary.
Concluding Remarks
Based on the evidence discussed above, we propose that innate immune priming or trained innate immunity constitute ancestral and evolutionarily conserved mechanisms by which living organisms, from lower life forms to humans, can be prepared to effectively cope with potential infectious or other stress challenges [1,2,6,7]. The comparison of innate immune memory in mammals (humans and mice) versus C. elegans suggests that the acquisition of a trained immunity phenotype can involve metabolic pathways that mediate transcriptional reprogramming, sustained for long periods of time [1,2,6,7]. A major role in the regulation of trained innate immunity across evolutionarily separated organisms can be ascribed to glucose and lipid metabolism [11,12,14]. However, even individual pathways mediating trained innate immunity may be conserved between C. elegans and humans. For example, the insulin/IGF-1 receptor homolog DAF-2, mediates C. elegans resistance to pathogenic bacteria [27] and the IGF-1 receptor was recently shown to mediate mevalonate-induced innate immune training of human monocytes [12].
Despite the role of cell metabolism as a common denominator of trained innate immunity in different organisms, several questions remain unanswered (see outstanding questions), for which extensive robust studies are still needed. For instance, on the one hand, trained innate immunity in humans and mice involves epigenetic reprogramming induced by metabolites [1,12,15]; such epigenetic changes are likely to occur also in C. elegans, yet they remain to be shown. On the other hand, whether trained immunity is transgenerational in humans and mice is not clear at all. Furthermore, trained innate immunity may also have detrimental effects. Indeed, if the immune system is epigenetically trained (due to an earlier infection, vaccination, or even sterile injury) eliciting a heightened immune response, this increased responsiveness may aggravate pre-existing inflammatory diseases or prove to be deleterious in hosts that are genetically susceptible to these conditions [21]. Additional studies are evidently required to better understand the mechanisms responsible for the detrimental effects of trained innate immunity. Addressing the aforementioned questions is important for understanding the context-dependent beneficial or detrimental effects of trained innate immunity. This should lead to future studies addressing how trained innate immunity and associated changes in cell metabolism of innate immune cells and their precursors could be therapeutically harnessed (stimulated or inhibited) -- perhaps in the context of chemotherapy-induced myeloablation or inflammatory diseases.
On the basis of the above-discussed studies, we hypothesize that an evolutionarily conserved immunometabolic crosstalk may represent an ancestral principle of innate immune training. Our hypothesis may therefore trigger future focused studies aimed a firmly establishing the mechanisms of metabolic control of innate immune memory that have been presumably conserved throughout evolution. Moreover, future studies using the more tractable nematode models may better inform the molecular mechanisms of trained immunity, ideally facilitating the targeted pharmacologic modulation of metabolic pathways implicated in trained immunity, which may be relevant in a variety of pathological conditions.
Highlights:
Metabolic reprogramming and epigenetic modifications are key for innate immune training in humans/mice.
Hematopoietic stem and progenitor cell differentiation into myeloid lineage can contribute to trained innate immunity in mice.
Glycolysis and cholesterol biosynthesis are cellular metabolic pathways involved in innate immune training in both hematopoietic progenitors and mature myeloid cells in mammals.
Changes in the activity of metabolic pathways can regulate immune functions in C. elegans.
Host defense reactions in C. elegans can involve metabolic signaling pathways such as the DAF-2 insulin/IGF-1 pathway.
Long-lasting immune memory in C. elegans depends, in part, on metabolic signaling.
Changes in cellular metabolism may be a common denominator of innate immune memory across species.
Outstanding questions:
What epigenetic changes underlie the ability of C. elegans to confer immune training? Are these epigenetic changes linked to cellular metabolism?
Are any metabolic pathways required for trained immunity fully conserved between C. elegans and mammals? If so, which ones?
Can we use the more tractable nematode models to understand molecular mechanisms of trained immunity and identify potential pharmacologic targets?
Can trained innate immunity be transgenerational in mammals?
Is metabolic reshaping of hematopoietic progenitors a potential target for the prevention of chemotherapy-induced myelosuppression? If so, which metabolic pathways?
Can cellular metabolic pathways involved in the induction of trained innate immunity be therapeutically targeted in the context of inflammatory diseases? If so, which pathways?
Acknowledgements
Supported by grants from the Deutsche Forschungsgemeinschaft (TR-SFB205 to TC) and from the US National Institutes of Health (DE024716 to GH and DE026152 to GH and TC). Supported also by the BMBF/GSRT German-Greek Bilateral Research and Innovation Programme (BRIDGING-T2DGED-0101 to IM and TC). IM is supported by the NCT Dresden, Germany. SP is supported by funds from TU Dresden’s Institutional Strategy, financed by the Excellence Initiative of the Federal and State Governments.
Glossary
- Aversive learning
A conditioning of the behavior of C. elegans towards avoidance of unwanted responses associated with negative preceding experiences.
- Bacillus Calmette–Guérin (BCG) vaccine
A vaccine used against tuberculosis
- Clonal hematopoiesis of indeterminate potential
A condition characterized by the existence of a somatic mutation related to myeloid neoplasms in blood cells without clinical/diagnostic signs of hematologic malignancy.
- Conditioning (priming)
The ability of organisms to enhance their immune responses following a preceding stimulus, e.g. an earlier pathogen encounter.
- Dauer (German for ‘enduring’) larva
Specialized, developmentally arrested larval stage found in many nematode species, including C. elegans. Dauer larvae are morphologically and physiologically adapted to survive under harsh conditions.
- Emergency myelopoiesis
Activation of hematopoiesis for the production of high numbers of myeloid cells, particularly neutrophils and monocytes, in response to systemic infection or inflammation, or following chemotherapy-induced myeloablation.
- Innate immune tolerance
A status of diminished responsiveness of the innate immune system to stimuli that normally elicit an immune response.
- Metabolic syndrome
A medical condition that increases the risk for diabetes and cardiovascular disease and is characterized by the combination of (three or more of) central obesity, high blood sugar / insulin resistance, high levels of triglycerides, low levels of high-density lipoprotein and hypertension.
- NLRP3 inflammasome
A cytosolic multiprotein complex, including NLRP3 (NACHT, LRR and PYD domains-containing protein 3) protein, which responds to infection or injury by activating pro-inflammatory caspases, mainly caspase-1, leading to the cleavage and release of pro-inflammatory cytokines such as IL-1β and IL-18.
- Systemic acquired resistance
A generalized state of heightened and long-lasting immunity to a broad range of microbial pathogens demonstrated by plants as a result of an earlier localized exposure to a pathogen.
- TET2
Tet methylcytosine dioxygenase 2 (TET2) encodes a protein that promotes DNA demethylation. TET2 mutations in humans are observed in myeloid neoplasms.
- Trained innate immunity
An enhanced state of immunity to a future inflammatory or microbial challenge that is based on innate immune memory, i.e., sustained changes in gene expression owing to earlier encounters with the same or even unrelated stimulus or microbial pathogen.
- Warburg metabolism
Metabolic mode originally described in cancer cells characterized by enhanced utilization of (aerobic) glycolysis rather than oxidative phosphorylation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of interest: The authors disclose no conflicts of interest.
References
- 1.Netea MG et al. (2016) Trained immunity: A program of innate immune memory in health and disease. Science 352, aaf1098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Netea MG and van der Meer JWM (2017) Trained Immunity: An Ancient Way of Remembering. Cell Host Microbe 21, 297–300 [DOI] [PubMed] [Google Scholar]
- 3.Rodrigues J et al. (2010) Hemocyte differentiation mediates innate immune memory in Anopheles gambiae mosquitoes. Science 329, 1353–1355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pham LN et al. (2007) A Specific Primed Immune Response in Drosophila Is Dependent on Phagocytes. PLoS Pathog 3, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moret Y and Siva-Jothy MT (2003) Adaptive innate immunity? Responsive-mode prophylaxis in the mealworm beetle, Tenebrio molitor. Proc. R. Soc. B Biol. Sci 270, 2475–2480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Anyanful A et al. (2009) Conditioning protects C. elegans from lethal effects of enteropathogenic E. coli by activating genes that regulate lifespan and innate immunity. Cell Host Microbe 5, 450–462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Palominos MF et al. (2017) Transgenerational Diapause as an Avoidance Strategy against Bacterial Pathogens in Caenorhabditis elegans. mBio 8, no. 5, e01234–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Durrant WE and Dong X (2004) Systemic acquired resistance. Annu. Rev. Phytopathol 42, 185–209 [DOI] [PubMed] [Google Scholar]
- 9.Fu ZQ and Dong X (2013) Systemic acquired resistance: turning local infection into global defense. Annu. Rev. Plant Biol 64, 839–863 [DOI] [PubMed] [Google Scholar]
- 10.Saeed S et al. (2014) Epigenetic programming of monocyte-to-macrophage differentiation and trained innate immunity. Science 345, 1251086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cheng S-C et al. (2014) mTOR- and HIF-1α-mediated aerobic glycolysis as metabolic basis for trained immunity. Science 345, 1250684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bekkering S et al. (2018) Metabolic Induction of Trained Immunity through the Mevalonate Pathway. Cell 172, 135–146.e9 [DOI] [PubMed] [Google Scholar]
- 13.Kaufmann E et al. (2018) BCG Educates Hematopoietic Stem Cells to Generate Protective Innate Immunity against Tuberculosis. Cell 172, 176–190.e19 [DOI] [PubMed] [Google Scholar]
- 14.Mitroulis I et al. (2018) Modulation of Myelopoiesis Progenitors Is an Integral Component of Trained Immunity. Cell 172, 147–161.e12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Arts RJW et al. (2016) Immunometabolic circuits in trained immunity. Semin. Immunol 28, 425–430 [DOI] [PubMed] [Google Scholar]
- 16.Norata GD et al. (2015) The Cellular and Molecular Basis of Translational Immunometabolism. Immunity 43, 421–434 [DOI] [PubMed] [Google Scholar]
- 17.Yona S et al. (2013) Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity 38, 79–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Arts RJW et al. (2018) BCG Vaccination Protects against Experimental Viral Infection in Humans through the Induction of Cytokines Associated with Trained Immunity. Cell Host Microbe 23, 89–100.e5 [DOI] [PubMed] [Google Scholar]
- 19.Kleinnijenhuis J et al. (2012) Bacille Calmette-Guerin induces NOD2-dependent nonspecific protection from reinfection via epigenetic reprogramming of monocytes. Proc. Natl. Acad. Sci. U. S. A 109, 17537–17542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Quintin J et al. (2012) Candida albicans infection affords protection against reinfection via functional reprogramming of monocytes. Cell Host Microbe 12, 223–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Christ A et al. (2018) Western Diet Triggers NLRP3-Dependent Innate Immune Reprogramming. Cell 172, 162–175.e14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bekkering S et al. (2014) Oxidized low-density lipoprotein induces long-term proinflammatory cytokine production and foam cell formation via epigenetic reprogramming of monocytes. Arterioscler. Thromb. Vasc. Biol 34, 1731–1738 [DOI] [PubMed] [Google Scholar]
- 23.Brasacchio D et al. (2009) Hyperglycemia induces a dynamic cooperativity of histone methylase and demethylase enzymes associated with gene-activating epigenetic marks that coexist on the lysine tail. Diabetes 58, 1229–1236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wendeln A-C et al. (2018) Innate immune memory in the brain shapes neurological disease hallmarks. Nature 556, 332–338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Arts RJW et al. (2016) Glutaminolysis and Fumarate Accumulation Integrate Immunometabolic and Epigenetic Programs in Trained Immunity. Cell Metab 24, 807–819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kawli T and Tan M-W (2008) Neuroendocrine signals modulate the innate immunity of Caenorhabditis elegans through insulin signaling. Nat. Immunol 9, 1415–1424 [DOI] [PubMed] [Google Scholar]
- 27.Garsin DA et al. (2003) Long-lived C. elegans daf-2 mutants are resistant to bacterial pathogens. Science 300, 1921. [DOI] [PubMed] [Google Scholar]
- 28.McElwee JJ et al. (2006) Diapause-associated metabolic traits reiterated in long-lived daf-2 mutants in the nematode Caenorhabditis elegans. Mech. Ageing Dev 127, 458–472 [DOI] [PubMed] [Google Scholar]
- 29.Kouzarides T (2007) Chromatin Modifications and Their Function. Cell 128, 693–705 [DOI] [PubMed] [Google Scholar]
- 30.Foster SL et al. (2007) Gene-specific control of inflammation by TLR-induced chromatin modifications. Nature 447, 972–978 [DOI] [PubMed] [Google Scholar]
- 31.Novakovic B et al. (2016) β-Glucan Reverses the Epigenetic State of LPS-Induced Immunological Tolerance. Cell 167, 1354–1368.e14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cheng S-C et al. (2016) Broad defects in the energy metabolism of leukocytes underlie immunoparalysis in sepsis. Nat. Immunol 17, 406–413 [DOI] [PubMed] [Google Scholar]
- 33.Donohoe DR and Bultman SJ Metaboloepigenetics: Interrelationships between energy metabolism and epigenetic control of gene expression. J. Cell. Physiol 227, 3169–3177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu P-S et al. (2017) α-ketoglutarate orchestrates macrophage activation through metabolic and epigenetic reprogramming. Nat. Immunol 18, 985–994 [DOI] [PubMed] [Google Scholar]
- 35.Mills EL et al. (2018) Itaconate is an anti-inflammatory metabolite that activates Nrf2 via alkylation of KEAP1. Nature 556, 113–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Domínguez-Andrés J et al. (2018) The Itaconate Pathway Is a Central Regulatory Node Linking Innate Immune Tolerance and Trained Immunity. Cell Metab DOI: 10.1016/j.cmet.2018.09.003 [DOI] [PubMed] [Google Scholar]
- 37.Arts RJW et al. (2016) Immunometabolic Pathways in BCG-Induced Trained Immunity. Cell Rep 17, 2562–2571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.O’Neill LAJ and Pearce EJ (2016) Immunometabolism governs dendritic cell and macrophage function. J. Exp. Med 213, 15–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lachmandas E et al. (2016) Microbial stimulation of different Toll-like receptor signalling pathways induces diverse metabolic programmes in human monocytes. Nat. Microbiol 2, 16246. [DOI] [PubMed] [Google Scholar]
- 40.Fu Y et al. (2012) Network topologies and dynamics leading to endotoxin tolerance and priming in innate immune cells. PLoS Comput. Biol 8, e1002526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Morris MC et al. (2014) Dynamic modulation of innate immune response by varying dosages of lipopolysaccharide (LPS) in human monocytic cells. J. Biol. Chem 289, 21584–21590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Geng S et al. (2016) The persistence of low-grade inflammatory monocytes contributes to aggravated atherosclerosis. Nat. Commun 7, 13436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ribes S et al. (2014) Intraperitoneal prophylaxis with CpG oligodeoxynucleotides protects neutropenic mice against intracerebral Escherichia coli K1 infection. J. Neuroinflammation 11, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Muñoz N et al. (2010) Mucosal administration of flagellin protects mice from Streptococcus pneumoniae lung infection. Infect. Immun 78, 4226–4233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang B et al. (2014) Viral infection. Prevention and cure of rotavirus infection via TLR5/NLRC4-mediated production of IL-22 and IL-18. Science 346, 861–865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Boettcher S and Manz MG (2017) Regulation of Inflammation- and Infection-Driven Hematopoiesis. Trends Immunol 38, 345–357 [DOI] [PubMed] [Google Scholar]
- 47.Nagai Y et al. (2006) Toll-like receptors on hematopoietic progenitor cells stimulate innate immune system replenishment. Immunity 24, 801–812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen C et al. (2010) Mammalian target of rapamycin activation underlies HSC defects in autoimmune disease and inflammation in mice. J. Clin. Invest 120, 4091–4101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Takizawa H et al. (2017) Pathogen-Induced TLR4-TRIF Innate Immune Signaling in Hematopoietic Stem Cells Promotes Proliferation but Reduces Competitive Fitness. Cell Stem Cell 21, 225–240.e5 [DOI] [PubMed] [Google Scholar]
- 50.Jaiswal S et al. (2017) Clonal Hematopoiesis and Risk of Atherosclerotic Cardiovascular Disease. N. Engl. J. Med 377, 111–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Abegunde SO et al. (2018) An inflammatory environment containing TNFα favors Tet2-mutant clonal hematopoiesis. Exp. Hematol 59, 60–65 [DOI] [PubMed] [Google Scholar]
- 52.Nahrendorf M (2018) Myeloid cell contributions to cardiovascular health and disease. Nat. Med 24, 711–720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Murphy AJ and Tall AR (2016) Disordered haematopoiesis and athero-thrombosis. Eur. Heart J 37, 1113–1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nagareddy PR et al. (2014) Adipose tissue macrophages promote myelopoiesis and monocytosis in obesity. Cell Metab 19, 821–835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Singer K et al. (2014) Diet-induced obesity promotes myelopoiesis in hematopoietic stem cells. Mol. Metab 3, 664–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yvan-Charvet L et al. (2010) ATP-binding cassette transporters and HDL suppress hematopoietic stem cell proliferation. Science 328, 1689–1693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Murphy AJ et al. (2011) ApoE regulates hematopoietic stem cell proliferation, monocytosis, and monocyte accumulation in atherosclerotic lesions in mice. J. Clin. Invest 121, 4138–4149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kim DH and Ewbank JJ (2018) Signaling in the innate immune response. WormBook Online Rev. C Elegans Biol DOI: 10.1895/wormbook.1.83.2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lemieux GA and Ashrafi K (2016) Investigating Connections between Metabolism, Longevity, and Behavior in Caenorhabditis elegans. Trends Endocrinol. Metab. TEM 27, 586–596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Watts JL and Ristow M (2017) Lipid and Carbohydrate Metabolism in Caenorhabditis elegans. Genetics 207, 413–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ding W et al. (2015) s-Adenosylmethionine Levels Govern Innate Immunity through Distinct Methylation-Dependent Pathways. Cell Metab 22, 633–645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nandakumar M and Tan M-W (2008) Gamma-linolenic and stearidonic acids are required for basal immunity in Caenorhabditis elegans through their effects on p38 MAP kinase activity. PLoS Genet 4, e1000273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tang H and Pang S (2016) Proline Catabolism Modulates Innate Immunity in Caenorhabditis elegans. Cell Rep 17, 2837–2844 [DOI] [PubMed] [Google Scholar]
- 64.Zugasti O et al. (2014) Activation of a G protein-coupled receptor by its endogenous ligand triggers the innate immune response of Caenorhabditis elegans. Nat. Immunol 15, 833–838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ferguson AA et al. (2013) TATN-1 mutations reveal a novel role for tyrosine as a metabolic signal that influences developmental decisions and longevity in Caenorhabditis elegans. PLoS Genet 9, e1004020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lee K-Z et al. (2010) The fatty acid synthase fasn-1 acts upstream of WNK and Ste20/GCK-VI kinases to modulate antimicrobial peptide expression in C. elegans epidermis. Virulence 1, 113–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Murphy CT et al. (2003) Genes that act downstream of DAF-16 to influence the lifespan of Caenorhabditis elegans. Nature 424, 277–283 [DOI] [PubMed] [Google Scholar]
- 68.Fielenbach N and Antebi A (2008) C. elegans dauer formation and the molecular basis of plasticity. Genes Dev 22, 2149–2165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Erkut C and Kurzchalia TV (2015) The C. elegans dauer larva as a paradigm to study metabolic suppression and desiccation tolerance. Planta 242, 389–396 [DOI] [PubMed] [Google Scholar]
- 70.Evans EA et al. (2008) Pseudomonas aeruginosa suppresses host immunity by activating the DAF-2 insulin-like signaling pathway in Caenorhabditis elegans. PLoS Pathog 4, e1000175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Singh V and Aballay A (2009) Regulation of DAF-16-mediated Innate Immunity in Caenorhabditis elegans. J. Biol. Chem 284, 35580–35587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Prithika U et al. (2017) Short term memory of Caenorhabditis elegans against bacterial pathogens involves CREB transcription factor. Immunobiology 222, 684–692 [DOI] [PubMed] [Google Scholar]
- 73.Kim Y and Mylonakis E (2012) Caenorhabditis elegans immune conditioning with the probiotic bacterium Lactobacillus acidophilus strain NCFM enhances gram-positive immune responses. Infect. Immun 80, 2500–2508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lee K and Mylonakis E (2017) An Intestine-Derived Neuropeptide Controls Avoidance Behavior in Caenorhabditis elegans. Cell Rep 20, 2501–2512 [DOI] [PubMed] [Google Scholar]
- 75.Riedel CG et al. (2013) DAF-16 employs the chromatin remodeller SWI/SNF to promote stress resistance and longevity. Nat. Cell Biol 15, 491–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lee SS et al. (2003) DAF-16 target genes that control C. elegans life-span and metabolism. Science 300, 644–647 [DOI] [PubMed] [Google Scholar]
- 77.Maures TJ et al. (2011) The H3K27 demethylase UTX-1 regulates C. elegans lifespan in a germline-independent, insulin-dependent manner. Aging Cell 10, 980–990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Jaiswal S et al. (2014) Age-related clonal hematopoiesis associated with adverse outcomes. N. Engl. J. Med 371, 2488–2498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Fuster JJ et al. (2017) Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science 355, 842–847 [DOI] [PMC free article] [PubMed] [Google Scholar]