

Abstract
Epithelial-to-mesenchymal transition (EMT) enables stationary epithelial cells to exhibit migratory behavior and is the key step that initiates heart valve development. Recent studies suggest that EMT is reactivated in the pathogenesis of myxomatous valve disease (MVD), a condition that involves the progressive degeneration and thickening of valve leaflets. These studies have been limited to in vitro experimentation and reliance on histologic co-staining of epithelial and mesenchymal markers as evidence of EMT in mouse and sheep models of valve disease. However, longitudinal analysis of cell lineage origins and potential pathogenic or reparative contributions of newly generated mesenchymal cells have not been reported previously. In this study, a genetic lineage tracing strategy was pursued by irreversibly labeling valve endothelial cells in the Osteogenesis imperfecta and Marfan syndrome mouse models to determine whether they undergo EMT during valve disease. Tie2-CreERT2 and Cdh5(PAC)-CreERT2 mouse lines were used in combination with colorimetric and fluorescent reporters for longitudinal assessment of endothelial cells. These lineage tracing experiments showed no evidence of EMT during adult valve homeostasis or valve pathogenesis. Additionally, CD31 and smooth muscle α-actin (αSMA) double-positive cells, used as an indicator of EMT, were not detected, and levels of EMT transcription factors were not altered. Interestingly, contrary to the endothelial cell-specific Cdh5(PAC)-CreERT2 driver line, Tie2-CreERT2 lineage-derived cells in diseased heart valves also included CD45+ leukocytes. Altogether, our data indicate that EMT is not a feature of valve homeostasis and disease but that increased immune cells may contribute to MVD.
Keywords: EMT, Heart Valves, Valve Disease, Myxomatous degeneration, Marfan syndrome, Osteogenesis imperfect, Epithelial-to-mesenchymal transition, Hematopoietic cells
INTRODUCTION
Epithelial-to-mesenchymal transition (EMT) is a cellular process by which epithelial cells detach from their neighbors, lose apical-basal polarity, and become invasive (Nieto et al., 2016). During embryonic development, this process contributes to gastrulation, neural crest cell formation, and production of complex anatomy in multiple organ systems (Ocana et al., 2017). Pioneering work by Roger Markwald and colleagues over the last three decades has established a mechanistic framework of heart valve development. This includes the discovery that endocardial endothelial cells of the atrioventricular canal and outflow tract of the primitive heart tube undergo EMT, invading the underlying cardiac jelly matrix to populate the endocardial cushions (Markwald et al., 1977; Bernanke and Markwald, 1982; Runyan and Markwald, 1983; Eisenberg and Markwald, 1995). These cells then proliferate and synthesize components of the valve extracellular matrix (ECM), ultimately giving rise to three-layered leaflets that enable unidirectional blood flow through the heart (Combs and Yutzey, 2009; Hinton and Yutzey, 2011).
Pathological manifestation of EMT has been demonstrated most commonly in cancer metastasis (Nieto, 2013) and pulmonary arterial hypertension (Hopper et al., 2016), although its involvement in various fibrotic diseases remains under intense debate (Nieto et al., 2016). Likewise, reactivation of this critical step in heart valve development has been implicated in adult valve diseases, including calcific aortic valve disease (Hjortnaes et al., 2015) and myxomatous valve disease (MVD), which affects approximately 2–3% of the general population (Levine et al., 2015). MVD involves the progressive thickening and degeneration of the valve leaflets, leading to a loss of unidirectional blood flow (i.e., regurgitation) and impaired heart function. Hallmark histopathological features include loss of the three-layer ECM organization of the leaflets, in addition to proteoglycan expansion and collagen/elastin fragmentation in thickened leaflets. In vitro experimentation and studies involving surgically-induced mitral valve regurgitation in sheep have implicated EMT in valve pathogenesis (Dal-Bianco et al., 2009; Geirsson et al., 2012; Wylie-Sears et al., 2014; Shapero et al., 2015). However, these studies have relied on in vitro assays, along with the histological identification of cells positive for both endothelial and mesenchymal markers, as evidence of EMT. Moreover, the conversion of endothelial cells to newly generated interstitial cells in adult valve homeostasis or disease in vivo has not been reported previously.
In this study, we pursued a genetic lineage tracing strategy to determine whether newly produced valve interstitial cells are derived from endothelial cells via the process of EMT in myxomatous valve disease using the Osteogenesis imperfecta (OI) and Marfan syndrome (MFS) mouse models. These mice carry mutations in ECM genes Collagen1 (Col1a2oim/oim) and Fibrillin1 (Fbn1C1039G/+), respectively, and exhibit myxomatous valve characteristics of human OI and MFS connective tissue disease (Ng et al., 2004; Cheek et al., 2012; Hulin et al., 2017). For the labeling of endothelial cells, Tie2-Cre mice have been used extensively to examine EMT during valve development (de Lange et al., 2004; Lincoln et al., 2004) and other contexts. However, Tie2 is not completely endothelial cell-specific as it is also expressed in a subset of circulating hematopoietic cells (De Palma et al., 2003; De Palma et al., 2005; Lewis et al., 2007). Therefore, we compared the endothelial-specific Cadherin5 (Cdh5)(PAC)-CreERT2 (Wang et al., 2010) with Tie2-CreERT2 mice (Forde et al., 2002) for tamoxifen-inducible lineage tracing of valve endothelial cells (VECs) in adult mice. Tamoxifen-induced irreversible labeling of endothelial cells and their derivatives in normal adult mice, as well as in myxomatous disease models, showed no evidence of endothelial-derived mesenchymal cells, demonstrating that EMT is not a feature of normal valve homeostasis or MVD pathogenesis. Instead, an infiltrating population of CD45+ cells was detected in diseased valves, suggesting that these leukocytes may contribute to MVD.
MATERIALS AND METHODS
Mice
The following mice were purchased from the Jackson Laboratory: Col1a2oim/+ (Stock #001815), Fbn1C1039G/+ (Stock #012885), Rosa26mTmG (Stock #007576), and R26R (Stock #003474) (Soriano, 1999). Tie2-CreERT2 were previously described (Forde et al., 2002) and provided by the European Mouse Mutant Archive. Cdh5(PAC)-CreERT2 mice were previously described (Wang et al., 2010) and obtained with permission from Dr. Ralf Adams (Max Planck Institute, Germany). Male and female mice (totaling 4–6 mice per genotype) were used together in all analyses. All mouse experiments conform to NIH guidelines (Guide for the Care and Use of Laboratory Animals) and were performed with protocols approved by the Institutional Animal Care and Use Committee at the Cincinnati Children’s Research Foundation.
Tamoxifen Administration
For adult CreER induction, 3 month-old mice were fed tamoxifen-containing chow (Envigo #TD.130860) at 40mg/kg/day for one week and sacrificed at 4 months of age. Neonatal CreER induction was performed using intragastric injections of tamoxifen once daily, as previously described (Pitulescu et al., 2010). Briefly, tamoxifen (Sigma #T5648) was dissolved in corn oil to make a 10mg/mL stock solution. Neonatal pups were injected with 50μL of tamoxifen working solution (1mg/mL) using insulin syringes (BD #309301) once daily from P0–P2 (totaling 3 injections). Mice were sacrificed at P7 or 2 months of age.
Tissue Collection, Histology, and Immunohistochemistry
Neonatal and adult mice were sacrificed using isoflurane, cervical dislocation, and thoracotomy. The heart was removed, its apex then cut (exposing left and right ventricles), and placed in ice-cold PBS to facilitate drainage of blood. Hearts were then fixed in 4% paraformaldehyde (PFA) overnight at 4°C, dehydrated through a graded ethanol series, cleared in xylene, embedded in paraffin, and sectioned at 5μm (Gomez-Stallons et al., 2016). Movat’s pentachrome staining was performed using manufacturer’s protocols (American MasterTech). β-galactosidase (β-gal) expression in the transgenic mice was analyzed using X-gal detection of β-gal activity as previously described (Searcy et al., 1998). Images were captured using the Olympus BX51 microscope retro-fitted with the Nikon DS-Ri1 camera and DS-U3 controller, and the NIS-Elements BR (version 3.2) software. For studies involving immunofluorescence with the following primary antibodies, antigen retrieval was performed using Proteinase K (Invitrogen #25530049; 1:1000) for 15 minutes at room temperature. These antibodies include: GFP (Abcam #ab290, 1:1000), CD31 (Abcam #ab56299, 1:100), CD45 (R&D Systems #AF114, 1:200), RFP (Rockland #600-401-379, 1:50). We found it possible to visualize Green Fluorescent Protein (GFP) without a primary antibody if histologic sections from Cre/reporter transgenic hearts were pre-treated with Proteinase K. For co-labeling of GFP (Abcam #ab290, 1:1000) and αSMA (Abcam #ab21027, 1:100), heat-mediated antigen retrieval in Citrate buffer was performed using a pressure cooker for 3 minutes. For fluorescent detection of primary antibodies, Alexa Fluor 488 (Abcam, #ab150061, 1:500), Alexa Fluor 568 (Abcam, #ab175475, #ab175692, #ab175704 1:500), and Alexa Fluor 647 (Abcam, #ab150135, 1:500) were used. Nuclei were counterstained using DAPI (4’, 6-Diamidino-2-Phenylindole) (Life Technologies, #D1306, 1:10,000). Images were captured using a Nikon A1-R confocal system with the NIS-Elements Confocal (version 4.5) software.
RNA isolation and real-time quantitative PCR
Mitral valves (excluding chordae tendinae) were micro-dissected, flash-frozen in liquid nitrogen, and individually stored in −80°C until RNA extraction. Mitral valve tissue from a single mouse was considered to be one biological replicate. Total RNA was then purified from each mitral valve sample using the NucleoSpin RNA XS kit (Macherey-Nagel) following manufacturer’s instructions. Reverse transcription was performed using the SuperScript III First-Strand Synthesis Kit (Invitrogen) according to manufacturer’s instructions. Quantitative real-time PCR analyses were performed using the following Taqman probes (Applied Biosystems): B2M (beta-2-microglobulin) (Mm00437762_m1), Snai1 (Mm00441533_g1), Snai2 (Mm00441531_m1), Twist1 (Mm00442036_m1), Cdh5 (Mm00486938_m1), and Ccl2 (chemokine ligand 2; also known as monocyte chemoattractant protein 1, MCP-1) (Mm00441242_m1). Fold changes in gene expression were calculated using the ΔΔCT method normalized to B2M (Wirrig et al., 2015).
Statistics
Students t-tests or Mann-Whitney U tests were used to determine the significance of observed differences using PRISM7 software package (GraphPad). Data are reported as mean +/− Standard Deviation (SD). A p-value <0.05 was considered statistically significant.
RESULTS
Mouse models of connective tissue disorders exhibit myxomatous valve disease by adulthood
MVD involves the progressive thickening and degeneration of the valve leaflets, characterized by diffuse accumulation of proteoglycan and fragmentation of collagen and elastin. It is observed most commonly in left-sided heart valves, namely the mitral and aortic valves (Fig. 1A). There is evidence that humans with OI have a predisposition to aortic valve disease (Folkestad et al., 2016) and Col1a2oim/oim homozygous mutant mouse model of osteogenesis imperfecta murine (OIM) exhibit thickened aortic valves with myxomatous changes (Cheek et al., 2012). By Movat’s pentachrome staining, myxomatous changes can be observed in the aortic valves of 4 month-old Col1a2oim/oim mice (Fig. 1B’) compared to Col1a2+/+ wildtype controls (Fig. 1B). Proteoglycan accumulation (blue) is most striking along with the thickening of the leaflets and abnormal collagen deposition (arrows; Fig. 1B’). These changes were not observed in the mitral valves, as previously noted (Cheek et al., 2012).
FIGURE 1: Mouse models of myxomatous valve disease.
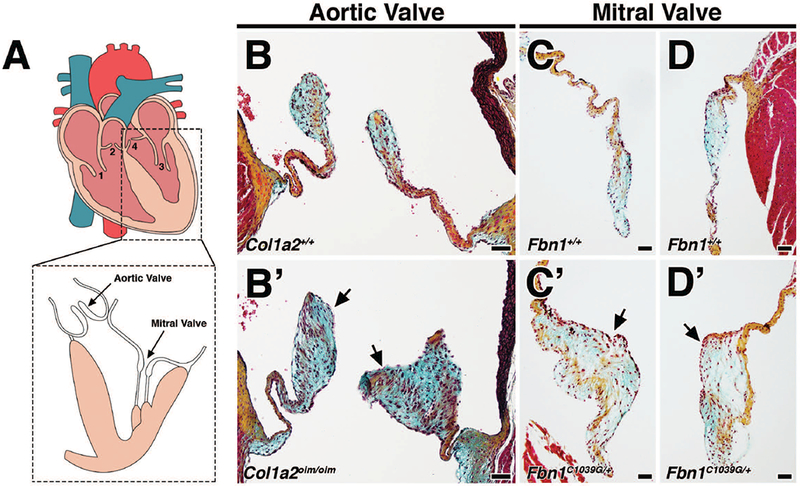
A: The four heart valves consist of the tricuspid valve (1), pulmonic valve (2), mitral valve (3), and aortic valve (4). B-D: Movat’s pentachrome staining of aortic and mitral valves in 4 month-old Col1a2+/+;Fbn1+/+ wildtype mice depict collagen (yellow), proteoglycan (blue), and elastin (black). Myocardium is stained red. B’-D’: Age-matched 4 month-old Col1a2oim/oim and Fbn1C1039G/+ mutant mice exhibit myxomatous aortic and mitral valves, respectively. Myxomatous valves exhibit significant thickening resulting from increased proteoglycan deposition (arrows, blue) and abnormal ECM organization. Scale Bar = 50μm.
Patients with Marfan syndrome (MFS) carry mutations in Fibrillin1 (Fbn1) and frequently present with myxomatous mitral valves. In mice heterozygous for the C1039G mutation in Fbn1 (Fbn1C1039G/+), changes to mitral valve morphology are detected as early as post-natal day 7 (P7), and notably mitral valve leaflets are thickened by 2 months (Ng et al., 2004; Hulin et al., 2017). In contrast to that of Fbn1+/+ wildtype controls (Fig. 1C, D), mitral valves in adult Fbn1C1039G/+ mice (Fig. 1C’, D’) exhibit thickening and increased proteoglycan composition comparable to the aortic valves of Col1a2oim/oim mice (Fig. 1B’) by Movat’s pentachrome. Mitral regurgitation has been reported to occur between 4–9 months of age in Fbn1C1039G/+ mice (Ng et al., 2004; Tae et al., 2016).
Lineage tracing using tamoxifen-inducible Tie2-CreERT2 is suggestive of EMT in myxomatous valves of adult Col1a2oim/oim and Fbn1C1039G/+ mutant mice.
Previous studies of EMT have used Tie2-Cre mice to genetically label endothelial cells and track their fates during valve development (de Lange et al., 2004; Lincoln et al., 2004). In order to address whether reactivation of EMT during adulthood is a critical component to the pathogenesis of myxomatous valve disease, tamoxifen-inducible Tie2-CreERT2 was used to identify derivatives of Tie2-expressing cells in Col1a2oim/oim and Fbn1C1039G/+ adult mice. Because VECs lining the valve leaflets permanently retain β-gal-mediated labeling upon Cre recombination, we reasoned that the presence of β-gal+ cells within the valve interstitium is indicative of an EMT event (Outcome B; Fig. 2A). As expected, β-gal+ cells were detected in the VECs located on the surface of the heart valves in Fbn1+/+ and Col1a2+/+ wildtype mice (Outcome A; Fig. 2A, B, B’). In contrast, β-gal+ cells were detected within the valve interstitium in myxomatous valves of both mutant mouse models (arrows; Fig. 2C, C’, D, D’). Furthermore, quantification in Col1a2oim/oim shows that mutant valves exhibit more β-gal+ cells (Fig. 2E). The observation that Tie2-CreERT2-derived cells are present in the valve interstitium is initial evidence for EMT in MVD.
FIGURE 2: Lineage tracing using Tie2-CreERT2;R26R in Col1a2oim/oim or Fbn1C1039G/+ mutant backgrounds as an indicator of EMT during disease.
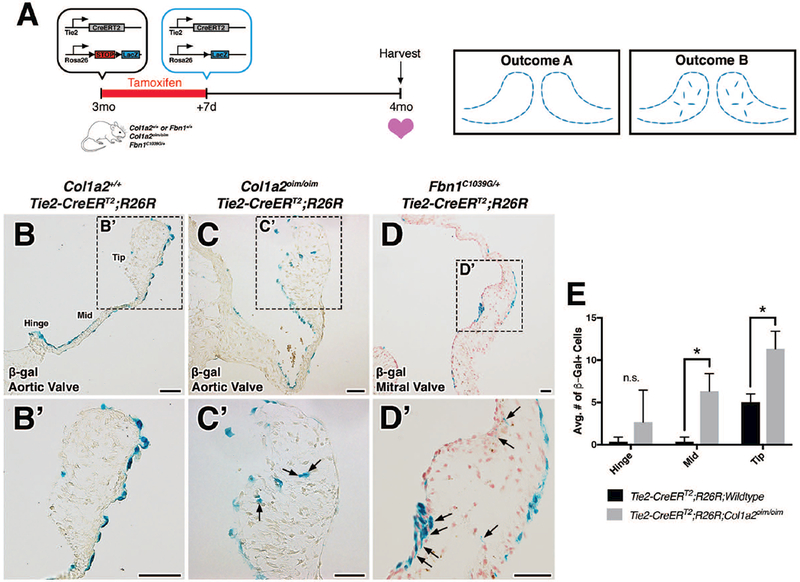
A: Schematic of experiment. Tamoxifen chow was administered to 3 month-old Col1a2+/+;Fbn1+/+ or mutant transgenic mice for 7 days. Cre-mediated recombination results in the deletion of stop codons and expression of β-galactosidase (β-gal) from the lacZ gene. At 4 months-of-age, hearts were harvested, sectioned, and analyzed for β-gal expression. In the absence of EMT, β-gal+ cells will be located solely on the VECs lining the valves (outcome A). In the presence of an EMT event, β-gal+ cells will be detected within the valve interstitium (outcome B). B-D: Histologic sections of aortic (B, B’, C, C’) and mitral (D, D’) valves from β-gal stained wildtype, Col1a2oim/oim, and Fbn1C1039G/+ transgenic mice. In mutant transgenic mice, β-gal+ cells were detected within the valve interstitium (arrows). E: Quantification of β-gal+ cells within the valve interstitium from Col1a2oim/oim and Col1a2+/+ wildtype controls. Student’s t-test was used with statistical significance defined as *p<0.05. Data are reported as mean, and error bars represent standard deviation. Scale Bar = 50μm.
Cells derived from Tie2-expressing cells are not restricted to endothelial lineages
Despite its frequent use as an endothelial marker, Tie2 is also expressed in hematopoietic stem cells and a subpopulation of circulating monocytes (De Palma et al., 2003; De Palma et al., 2005; Lewis et al., 2007). Likewise, atrioventricular valves possess a microvascular network (Weind et al., 2000; I-Ida et al., 2001; Weind et al., 2002; Swanson et al., 2009), a source of endothelial cells located within the valve. In order to differentiate between EMT-derived mesenchymal cells from either endothelial cells of the microvasculature or infiltrating hematopoietic cells known to populate the valves during homeostasis and disease (Hulin et al., 2018), Tie2CreERT2 GFP+ cells were co-labeled with CD31 to detect endothelial cells and CD45 to identify hematopoietic lineages. Cre recombination in a subset of CD31+ endothelial cells was comparable in Tie2-CreERT2;Rosa26mTmG mice (Fig. 3) and Tie2-CreERT2;R26R mice (Fig. 2). A subset of GFP+ cells detected within the Fbn1C1039G/+ mitral valve interstitium was CD31+ (arrow; Fig. 3D-D’’), suggestive of endothelial cells of the valve microvasculature. Interestingly, these cells were arranged in ring-like formation in various sections of the leaflets and were in close proximity to CD45+ cells. The remainder of GFP+ cells in the valve interstitium also express CD45, suggesting that these cells are hematopoietic in origin (Fig. 3E-E’’). Notably, neither GFP+CD31+CD45+ cells, indicative of endothelial-derived CD45+ cells, nor GFP+CD31-CD45-cells, indicative of EMT-derived VICs, were detected in control Fbn1+/+ or mutant Fbn1C1039G/+ mitral valves. Together, these data suggest that Tie2-lineage GFP+ cells in the diseased valve interstitium are either vascular endothelial or hematopoietic lineages and are not derived from EMT of the VECs.
FIGURE 3: Interstitial Tie2-lineage GFP+ cells are not EMT-derived but are endothelial cells of the valve microvasculature or infiltrating hematopoietic cells in Fbn1C1039G/+ mitral valves.
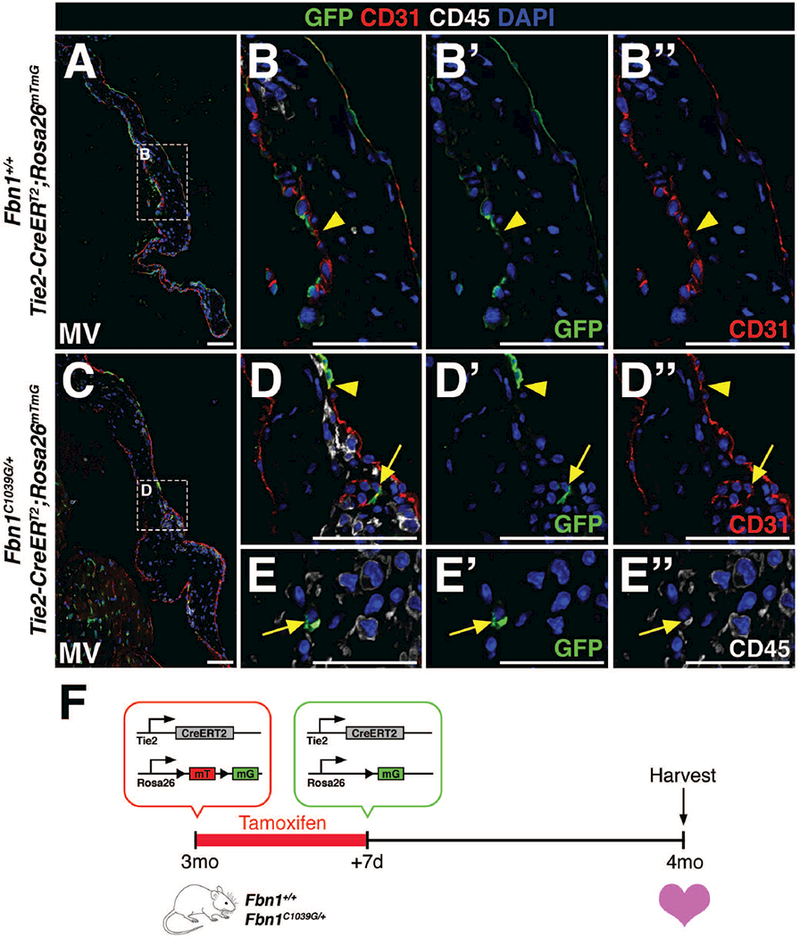
A-D’’: Only a subset of CD31+ (red) endothelial cells in 4 month-old Fbn1+/+ (A-B’’) and Fbn1C1039G/+ (C-D’’) mitral valves are GFP+ (green), demonstrating low recombination efficiency (arrowheads) after tamoxifen administration. C-E’’: GFP+ cells within the mitral valve of Fbn1C1039G/+ mutant mice were either endothelial cells (CD31+, D-D’’) of the valve microvasculature or hematopoietic cells (CD45+, E-E’’). Note that the lumen of valve microvasculature is clearly depicted in panels D-D’’ (arrow). F: Schematic of experiment. Tamoxifen chow was administered to 3 month-old wildtype or mutant transgenic mice for 7 days. Cre-mediated recombination leads to deletion of the mTomato (mT) gene and expression of GFP from the mG locus. At 4 months-of-age, hearts were harvested, sectioned, and analyzed for GFP, CD31, and CD45 protein expression. Scale Bar = 50μm.
Cdh5(PAC)-CreERT2 is restricted to endothelial cells, including valve endothelial cells, within the heart
Due to the observed recombination in a subset of valve endothelial cells and lack of endothelial specificity in Tie2-CreERT2 mice, we sought out alternative inducible Cre-expressing lines for more complete recombination specifically in VECs. The Cdh5(PAC)-CreERT2 mouse was utilized due to its high specificity to vascular endothelial cells (Wang et al., 2010) and was crossed with Rosa26mTmG reporter mice. Subsequent pups were administered intragastric injections of tamoxifen (Pitulescu et al., 2010) once daily from P0–P2 and hearts were collected at either P7 or 2 months of age (Fig. 4O). Remarkably, all four heart valves exhibited robust GFP+ labeling of VECs in neonatal (Fig. 4A-D) and adult (Fig. 4H-K) hearts. As expected, endothelial cells lining the coronary vessels throughout the ventricles, atria (Fig. 4E, F, L, M) and pulmonary vasculature of the lungs (Fig. 4G, N) exhibited strong GFP expression. The lack of detectable GFP in interstitial cells of all four valves suggests that EMT is not a feature of valve homeostasis after birth. Cre induction was similarly robust when using tamoxifen intraperitoneal injections (neonatal or adult) or chow (adult) (data not shown). Thus, Cdh5(PAC)-CreERT2 mice exhibit efficient and comprehensive recombination in heart valve endothelial lineages after birth.
FIGURE 4: Cdh5(PAC)-CreERT2;Rosa26mTmG mice exhibit robust recombination efficiency and specificity to vascular endothelial cells.
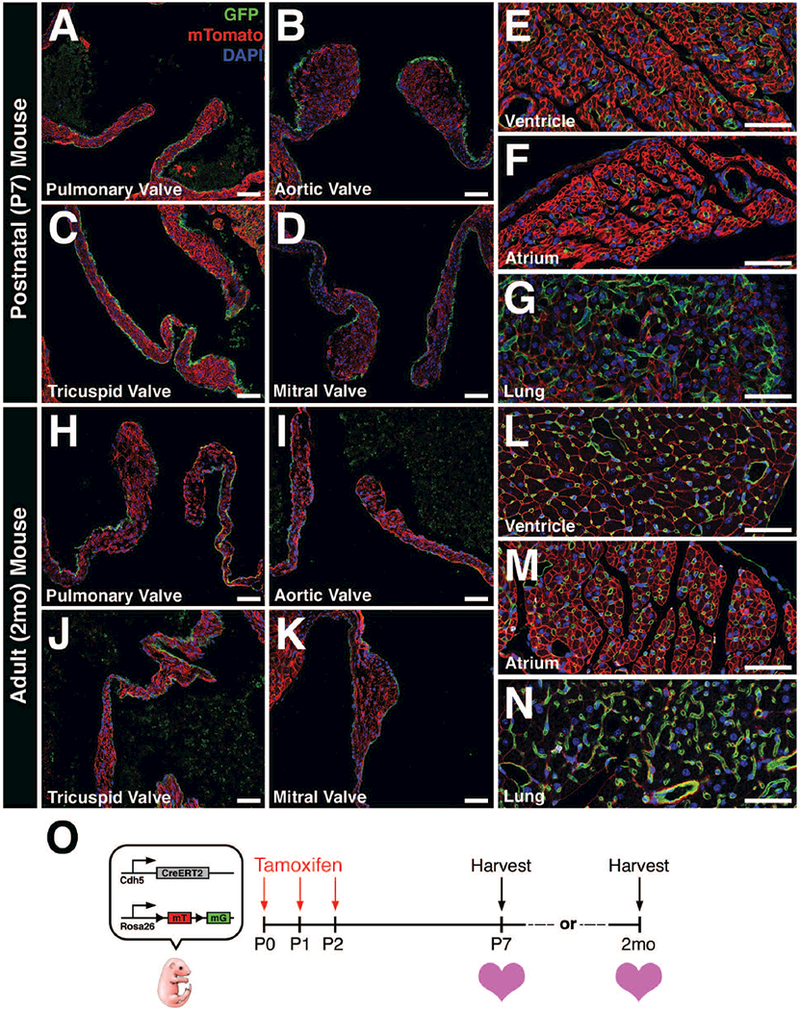
A-D: P7 heart valves exhibit robust GFP expression in valve endothelial cells (VECs) after postnatal tamoxifen induction. E-G: GFP expression is also observed in vascular endothelial cells from other regions of the heart and lungs. H-K: 2 month-old heart valves continue to express GFP in VECs after postnatal tamoxifen induction. L-N: Similarly, GFP expression is also observed in endothelial cells from other regions of the heart and lungs. O: Schematic of experiment. Tamoxifen was administered via intragastric injections once per day from post-natal day 0 (P0) to P2. Hearts were harvested at either P7 or 2 months-of-age, sectioned, and analyzed for GFP expression. Scale Bar = 50μm.
Absence of Cdh5 lineage-derived valve interstitial cells and EMT transcription factors suggest EMT is not a pathogenic feature of myxomatous valve disease in adult Marfan syndrome mice
Cdh5(PAC)-CreERT2;Rosa26mTmG transgenic mice were crossed with Fbn1C1039G/+ mice to re-evaluate EMT during valve disease. Because changes to mitral valve morphology were previously detected as early as P6.5 (Ng et al., 2004), pups were administered intragastric injections of tamoxifen from P0–P2 and harvested at 2 months when myxomatous changes become visibly apparent. Upon confocal microscopy, only valve endothelial cells exhibited GFP in both wildtype controls (Fig 5 A, B) and mutants (Fig. 5F, G) at 2 months of age. As expected, Cdh5-derived GFP+ cells also co-labeled with CD31 (Fig. 5A, B, F, G), confirming their endothelial phenotype.
FIGURE 5: EMT was not detected by endothelial lineage tracing during myxomatous valve disease in adult Marfan syndrome mice.
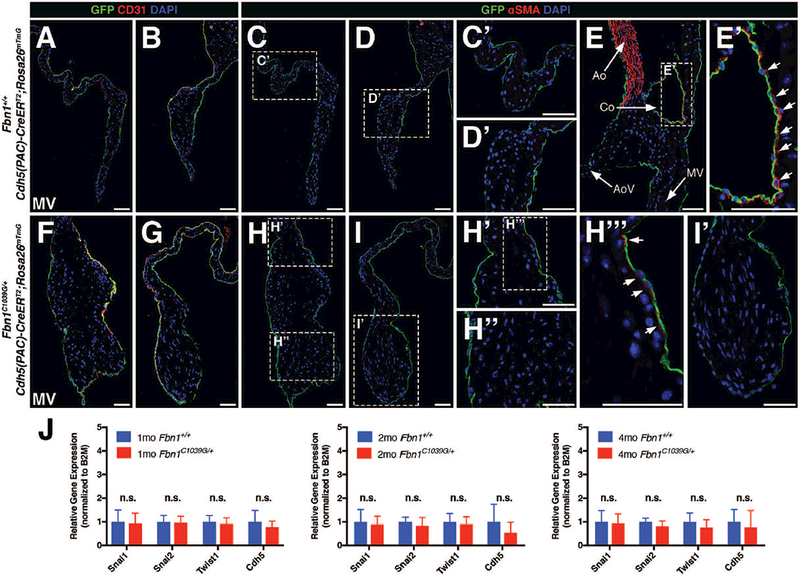
A, B, F, G: Cdh5-derived endothelial cells (GFP+) remain in the periphery of the mitral valve leaflets in both 2 month-old wildtype Fbn1+/+ (A,B) and mutant Fbn1C1039G/+ (F,G) mice. Note that endothelial surface marker, CD31, co-labels with GFP+ cells as indicated by yellow in overlayed confocal images. C, D, H, I: GFP+ Cdh5-derived endothelial cells do not express αSMA in 2 month-old wildtype Fbn1+/+ (C, D) and mutant Fbn1C1039G/+ (H, I) mice. Panels C’, D’, H’, H’’, I’ represent magnified images of corresponding areas of the valve leaflets. Note that αSMA expression is occasionally detected subjacent to but not in endothelial cells in overlays of red and green channel images (arrows; H’’’). E: Fluorescent detection of αSMA (red) staining using the aorta as an internal positive control. E’: Note that αSMA+ smooth muscle cells are present adjacent to GFP+ endothelial cells in the coronary vessel (Co). J: qPCR analysis using RNA isolated from Fbn1C1039G/+ mitral valves demonstrates no significant changes in mRNA expression of mesenchymal transcription factors (Snai1, Snai2, Twist1) and endothelial marker (Cdh5) at 1, 2, and 4 months-of-age versus wildtype Fbn1+/+ controls (n=9 per genotype per time-point). Fold changes in gene expression were calculated using the ΔΔCT method normalized to B2M. Mann-Whitney U Test was used with statistical significance defined as *p<0.05. Data are reported as mean (+/− SD). Ao = aorta; AoV = aortic valve; Co = coronary vessel; MV = mitral valve. Scale Bar = 50μm.
Endothelial cell expression of the mesenchymal marker αSMA has been used previously to indicate endothelial cells undergoing EMT, and co-staining of αSMA and CD31 has been used as a marker of EMT in diseased valves (Dal-Bianco et al., 2009; Mahler et al., 2013; Wylie-Sears et al., 2014; Shapero et al., 2015). Co-labeling of Cdh5 lineage traced GFP-labeled cells with αSMA expression was examined as an additional potential indicator of EMT. As expected, αSMA was detected in the wall of the aorta (Ao; Fig. 5E) and smooth muscle cells surrounding coronary vessels (Co; arrows; Fig. 5E, E’). In contrast, αSMA expression was not detected in GFP-expressing cells in mitral valves from Fbn1+/+ wildtype controls (Fig. 5C, C’, D, D’) and Fbn1C1039G/+ mutant animals (Fig. 5H, H’’, I, I’). On rare occasions, αSMA was detected in interstitial cells subjacent to endothelial cells of the MV atrialis layer (arrows; Fig. 5H’, H’’’), but expression was not detected within GFP-expressing endothelial lineage cells.
To supplement histological analyses, mRNA expression of well-characterized EMT transcription factors expressed in mesenchymal cells Snai1, Snai2, and Twist1 (Peinado et al., 2007) and the endothelial marker Cdh5 were evaluated using RNA isolated from Fbn1+/+ and Fbn1C1039G/+ mitral valves at 1, 2, and 4 months of age (n=9 per genotype per time-point). Consistent with the results from Cdh5-based lineage tracing, no significant changes in mRNA expression of the EMT transcription factors or endothelial marker were observed (Fig. 5J). These data provide further evidence that EMT is not a pathogenic feature of myxomatous valve disease in Marfan syndrome mice.
Hematopoietic cells do not arise from valvular endothelial cells during disease
Immune cells populate normal and diseased heart valves and are mostly of myeloid origin (Hulin et al., 2018). In addition, there has been speculation that CD45+ cells arise from VECs in myxomatous valve disease (Bischoff et al., 2016). In order to examine potential contributions of endothelial cells to CD45+ cells during disease, Cdh5(PAC)-CreERT2;RosamTmG;Fbn1C1039G/+ mice were administered intragastric tamoxifen postnatally and harvested at 2 months of age. Qualitative examination of sections suggest that mutant mitral valves exhibit more CD45+ cells (Fig. 6A-D). Based on lineage tracing, however, CD45+ cells are negative for Cdh5-lineage GFP and the endothelial marker CD31, suggesting that hematopoietic cells do not arise from endothelial cells (Fig. 6A’-D’, A’’-D’’, A’’’-D’’’). Instead, hematopoietic cells are likely to have infiltrated from a circulating population, given that expression of Ccl2, a major pro-inflammatory chemoattractant of circulating monocytes, is increased in Fbn1C1039G/+ heart valves, as detected by qPCR (Fig. 6E). Co-expression of CD45+ and the Tie2-GFP lineage marker in Fbn1C1039G/+ mitral valves (Fig. 3E-E’’) is further evidence for the presence of hematopoietic cells derived from circulating monocytes in myxomatous disease. Taken together, these cell lineage data suggest that immune cell infiltration, rather than EMT, occurs in the pathogenesis of myxomatous valve disease.
FIGURE 6: Hematopoietic cells do not arise from valve endothelial cells in adult Marfan syndrome mice.
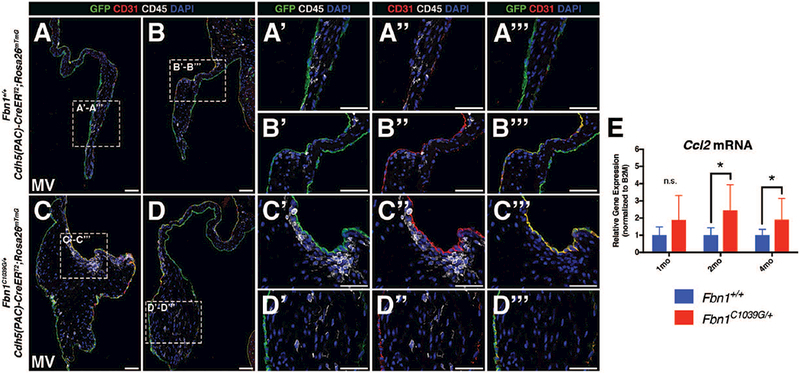
A-B: Fluorescent detection of endothelial cells (CD31+, GFP) and hematopoietic cells (CD45+) in 2 month-old Fbn1+/+ mitral valves. CD45+ cells (white) are a distinct population from VECs (green) (Panels A’-A’’’ and B’-B’’’). C-D: 2 month-old Fbn1C1039G/+ Marfan syndrome mitral valves are thickened and exhibit more CD45+ cells versus control. CD45+ cells do not express the Cdh5-CreERT2-derived GFP lineage marker (Panels C’-C’’’ and D’-D’’’). E: qPCR analysis using RNA isolated from Fbn1C1039G/+ mitral valves demonstrates increased mRNA expression of Ccl2 at 2 and 4 months-of-age, but not at 1 month, versus Fbn1+/+ controls (n=9 per genotype per time-point). Mann-Whitney U Test was used with statistical significance defined as *p<0.05. Data are reported with error bars indicating SD. Scale Bar = 50μm.
DISCUSSION
In this study, we used inducible Cre-mediated cell lineage analysis, together with analysis of cell-type specific markers and gene expression, to examine whether EMT occurs during adult valve homeostasis or the pathogenesis of myxomatous valve disease. Two tamoxifen-inducible CreER lines, Tie2-CreERT2 and Cdh5(PAC)-CreERT2, were used to permanently label VECs and determine whether they contribute to valve interstitial cells (VICs) under homeostatic or MVD conditions. Comparison of the two Cre lines demonstrates that Tie2-CreERT2 labels endothelial and hematopoietic lineages, whereas Cdh5(PAC)-CreERT2 is endothelial-specific in heart valves. Assessment of cell lineages in adult valves showed no evidence of EMT-derived interstitial cells from VECs in mice with mutations in Col1a2, that model OI, or in Fbn1, that model MFS. Additionally, mutant valves did not exhibit changes in EMT transcription factor mRNA expression levels or immunofluorescent detection of double-positive CD31/αSMA cells. Furthermore, EMT was not detected during normal valve maturation and homeostasis after birth. Interestingly, hematopoietic cells were detected by both histology and lineage tracing, suggesting that they may play a more significant role than EMT in myxomatous valve disease.
Tie2-Cre has been used effectively in visualizing EMT during valve development (de Lange et al., 2004; Lincoln et al., 2004) and has also been used to examine EMT in multiple disease states in other organs (Zeisberg et al., 2008). Here, we show that recombination using Tie2-CreERT2 is not specific to adult valve endothelial cells and that Tie2-CreERT2 labeled cells include additional CD45+ hematopoietic lineages. The observed recombination in a subset of adult endothelial cells is likely due to declining Tie2 promoter activity with age (Anstine et al., 2016). More importantly, Tie2 is expressed in both endothelial and hematopoietic cells, therefore leading to ambiguity regarding EMT, and the use of Tie2 as an endothelial cell lineage marker to evaluate EMT has already been discredited in an endocardial fibrosis model (Zhang et al., 2017). In contrast, the Cdh5(PAC)-CreERT2 line more completely and specifically labels valve endothelial cells at postnatal and adult stages, making it an optimal driver for studies of EMT in a variety of contexts. Here, we show that comparison of Tie2-CreERT2 cell lineages with the more endothelial-specific Cdh5(PAC)-CreERT2 lineage driver clearly demonstrates the lack of endothelial-derived interstitial cells in normal or diseased valve interstitium. Together, these data provide no evidence of EMT in normal or myxomatous OI and MFS adult mouse valves.
EMT has been reported previously in sheep models of surgically-induced ischemic mitral regurgitation and leaflet tethering based on the identification of cells double-positive for CD31 and αSMA. This model has been characterized as secondary MVD resulting from ischemic injury in contrast to the primary MVD of genetic connective tissue disorders in the current study. In sheep models, longitudinal cell lineage analysis is problematic due to the lack of genetically modified animals. In addition, the identification of EMT by static histological analysis is limited since the derivation of mesenchymal cells from endothelial lineages cannot be measured directly or longitudinally. The use of CD31 and αSMA+ co-expression as an indicator of EMT is controversial because αSMA+ can also be expressed by resident VICs, and smooth muscle cells are located in close proximity to surface VECs of mitral valves (Nordrum and Skallerud, 2012). Additional reports of adult valve EMT have been based on CD31 and αSMA+ co-labeling in wound-healing scratch assays of isolated VEC/VIC cell cultures (Hjortnaes et al., 2015; Shapero et al., 2015; Bischoff et al., 2016). Cultured VECs exhibit mesenchymal characteristics under these conditions, but it is not clear how the in vitro culture environment, notably media and substrate stiffness of tissue culture, influences cell behavior and phenotype (Quinlan and Billiar, 2012; Balaoing et al., 2015; Pinto et al., 2016). Additional studies are needed to definitively determine if EMT is a feature of MVD in large animal models or human patients. With the advent of gene-editing techniques, it is now possible to generate fluorescent reporter lines in larger animals (Li et al., 2014), so it would be interesting to perform similar lineage tracing studies of valve endothelial cells in these animals.
In mouse heart valves, the presence of Tie2-CreERT2-expressing hematopoietic cells adds to accumulating evidence that immune cells populate valves postnatally and comprise a significant portion of cells in normal and diseased adult valves (Hulin et al., 2018). This is the case for mice with MVD resulting from ECM mutations related to human disease, such as Col1a2 and Fbn1 shown here, and also Axin2 deficiency leading to increased Wnt signaling (Hulin et al., 2017). Cell sorting and lineage analysis demonstrate that these CD45+ cells are primarily myeloid and express markers of macrophage and dendritic lineages (Hulin et al., 2018). Moreover, increased numbers of valvular CD45+ cells have been reported in mouse (Sauls et al., 2015), sheep (Dal-Bianco et al., 2016), and human valve disease (Geirsson et al., 2012; Cote et al., 2013). The current Cdh5-based lineage tracing shows that CD45+ cells are not derived from VECs, contrary to prior speculation (Bischoff et al., 2016), but instead arise from circulating hematopoietic cells. This is reinforced by the observation that Tie2+ lineage cells are also CD45+, representing a subset of circulating monocytes (De Palma et al., 2003; De Palma et al., 2005; Lewis et al., 2007). Furthermore, MFS mitral valves exhibit increased expression for Ccl2, a pro-inflammatory chemokine known to recruit blood monocytes. Altogether, it is likely that infiltrating CD45+ cells, including macrophages, are a feature of valve pathogenesis or repair independent of EMT. Additional studies, both in mice and larger animals, are needed to determine the contributions of immune cells to valve disease.
ACKNOWLEDGMENTS
We would like to thank Alexia Hulin and Jonathan Cheek for their technical advice and support, Elaine Wirrig-Schwendeman for assistance with initial maintenance of mouse lines, and M. Victoria Gomez-Stallons for proof-reading this manuscript.
Funding Sources:
Grant Sponsor: American Heart Association
Grant Number: 16PRE30180000 (A.J. Kim)
Grant Sponsor: National Institutes of Health
Grant Number: R01HL094319 (K.E. Yutzey)
LITERATURE CITED
- Anstine LJ, Bobba C, Ghadiali S, Lincoln J. 2016. Growth and maturation of heart valves leads to changes in endothelial cell distribution, impaired function, decreased metabolism and reduced cell proliferation. J Mol Cell Cardiol 100:72–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balaoing LR, Post AD, Lin AY, Tseng H, Moake JL, Grande-Allen KJ. 2015. Laminin Peptide-Immobilized Hydrogels Modulate Valve Endothelial Cell Hemostatic Regulation. PLoS One 10:e0130749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernanke DH, Markwald RR. 1982. Migratory behavior of cardiac cushion tissue cells in a collagen-lattice culture system. Dev Biol 91:235–245. [DOI] [PubMed] [Google Scholar]
- Bischoff J, Casanovas G, Wylie-Sears J, Kim DH, Bartko PE, Guerrero JL, Dal-Bianco JP, Beaudoin J, Garcia ML, Sullivan SM, Seybolt MM, Morris BA, Keegan J, Irvin WS, Aikawa E, Levine RA. 2016. CD45 Expression in Mitral Valve Endothelial Cells After Myocardial Infarction. Circ Res 119:1215–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheek JD, Wirrig EE, Alfieri CM, James JF, Yutzey KE. 2012. Differential activation of valvulogenic, chondrogenic, and osteogenic pathways in mouse models of myxomatous and calcific aortic valve disease. J Mol Cell Cardiol 52:689–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Combs MD, Yutzey KE. 2009. Heart valve development: regulatory networks in development and disease. Circ Res 105:408–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cote N, Mahmut A, Bosse Y, Couture C, Page S, Trahan S, Boulanger MC, Fournier D, Pibarot P, Mathieu P. 2013. Inflammation is associated with the remodeling of calcific aortic valve disease. Inflammation 36:573–581. [DOI] [PubMed] [Google Scholar]
- Dal-Bianco JP, Aikawa E, Bischoff J, Guerrero JL, Handschumacher MD, Sullivan S, Johnson B, Titus JS, Iwamoto Y, Wylie-Sears J, Levine RA, Carpentier A. 2009. Active adaptation of the tethered mitral valve: insights into a compensatory mechanism for functional mitral regurgitation. Circulation 120:334–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dal-Bianco JP, Aikawa E, Bischoff J, Guerrero JL, Hjortnaes J, Beaudoin J, Szymanski C, Bartko PE, Seybolt MM, Handschumacher MD, Sullivan S, Garcia ML, Mauskapf A, Titus JS, Wylie-Sears J, Irvin WS, Chaput M, Messas E, Hagege AA, Carpentier A, Levine RA, Leducq Transatlantic Mitral N. 2016. Myocardial Infarction Alters Adaptation of the Tethered Mitral Valve. J Am Coll Cardiol 67:275–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lange FJ, Moorman AF, Anderson RH, Manner J, Soufan AT, de Gier-de Vries C, Schneider MD, Webb S, van den Hoff MJ, Christoffels VM. 2004. Lineage and morphogenetic analysis of the cardiac valves. Circ Res 95:645–654. [DOI] [PubMed] [Google Scholar]
- De Palma M, Venneri MA, Galli R, Sergi Sergi L, Politi LS, Sampaolesi M, Naldini L. 2005. Tie2 identifies a hematopoietic lineage of proangiogenic monocytes required for tumor vessel formation and a mesenchymal population of pericyte progenitors. Cancer Cell 8:211–226. [DOI] [PubMed] [Google Scholar]
- De Palma M, Venneri MA, Roca C, Naldini L. 2003. Targeting exogenous genes to tumor angiogenesis by transplantation of genetically modified hematopoietic stem cells. Nat Med 9:789–795. [DOI] [PubMed] [Google Scholar]
- Eisenberg LM, Markwald RR. 1995. Molecular regulation of atrioventricular valvuloseptal morphogenesis. Circ Res 77:1–6. [DOI] [PubMed] [Google Scholar]
- Folkestad L, Hald JD, Gram J, Langdahl BL, Hermann AP, Diederichsen AC, Abrahamsen B, Brixen K. 2016. Cardiovascular disease in patients with osteogenesis imperfecta - a nationwide, register-based cohort study. Int J Cardiol 225:250–257. [DOI] [PubMed] [Google Scholar]
- Forde A, Constien R, Grone HJ, Hammerling G, Arnold B. 2002. Temporal Cre-mediated recombination exclusively in endothelial cells using Tie2 regulatory elements. Genesis 33:191–197. [DOI] [PubMed] [Google Scholar]
- Geirsson A, Singh M, Ali R, Abbas H, Li W, Sanchez JA, Hashim S, Tellides G. 2012. Modulation of transforming growth factor-beta signaling and extracellular matrix production in myxomatous mitral valves by angiotensin II receptor blockers. Circulation 126:S189–197. [DOI] [PubMed] [Google Scholar]
- Gomez-Stallons MV, Wirrig-Schwendeman EE, Hassel KR, Conway SJ, Yutzey KE. 2016. Bone Morphogenetic Protein Signaling Is Required for Aortic Valve Calcification. Arterioscler Thromb Vasc Biol 36:1398–1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinton RB, Yutzey KE. 2011. Heart valve structure and function in development and disease. Annu Rev Physiol 73:29–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hjortnaes J, Shapero K, Goettsch C, Hutcheson JD, Keegan J, Kluin J, Mayer JE, Bischoff J, Aikawa E. 2015. Valvular interstitial cells suppress calcification of valvular endothelial cells. Atherosclerosis 242:251–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopper RK, Moonen JR, Diebold I, Cao A, Rhodes CJ, Tojais NF, Hennigs JK, Gu M, Wang L, Rabinovitch M. 2016. In Pulmonary Arterial Hypertension, Reduced BMPR2 Promotes Endothelial-to-Mesenchymal Transition via HMGA1 and Its Target Slug. Circulation 133:1783–1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulin A, Anstine LJ, Kim AJ, Potter SJ, DeFalco T, Lincoln J, Yutzey KE. 2018. Macrophage Transitions in Heart Valve Development and Myxomatous Valve Disease. Arterioscler Thromb Vasc Biol 38:636–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulin A, Moore V, James JM, Yutzey KE. 2017. Loss of Axin2 results in impaired heart valve maturation and subsequent myxomatous valve disease. Cardiovasc Res 113:40–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- I-Ida T, Tamura K, Tanaka S, Asano G. 2001. [Blood vessels in normal and abnormal mitral valve leaflets]. J Nippon Med Sch 68:171–180. [DOI] [PubMed] [Google Scholar]
- Levine RA, Hagege AA, Judge DP, Padala M, Dal-Bianco JP, Aikawa E, Beaudoin J, Bischoff J, Bouatia-Naji N, Bruneval P, Butcher JT, Carpentier A, Chaput M, Chester AH, Clusel C, Delling FN, Dietz HC, Dina C, Durst R, Fernandez-Friera L, Handschumacher MD, Jensen MO, Jeunemaitre XP, Le Marec H, Le Tourneau T, Markwald RR, Merot J, Messas E, Milan DP, Neri T, Norris RA, Peal D, Perrocheau M, Probst V, Puceat M, Rosenthal N, Solis J, Schott JJ, Schwammenthal E, Slaugenhaupt SA, Song JK, Yacoub MH, Leducq Mitral Transatlantic N. 2015. Mitral valve disease--morphology and mechanisms. Nat Rev Cardiol 12:689–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis CE, De Palma M, Naldini L. 2007. Tie2-expressing monocytes and tumor angiogenesis: regulation by hypoxia and angiopoietin-2. Cancer Res 67:8429–8432. [DOI] [PubMed] [Google Scholar]
- Li S, Flisikowska T, Kurome M, Zakhartchenko V, Kessler B, Saur D, Kind A, Wolf E, Flisikowski K, Schnieke A. 2014. Dual fluorescent reporter pig for Cre recombination: transgene placement at the ROSA26 locus. PLoS One 9:e102455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lincoln J, Alfieri CM, Yutzey KE. 2004. Development of heart valve leaflets and supporting apparatus in chicken and mouse embryos. Dev Dyn 230:239–250. [DOI] [PubMed] [Google Scholar]
- Mahler GJ, Farrar EJ, Butcher JT. 2013. Inflammatory cytokines promote mesenchymal transformation in embryonic and adult valve endothelial cells. Arterioscler Thromb Vasc Biol 33:121–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markwald RR, Fitzharris TP, Manasek FJ. 1977. Structural development of endocardial cushions. Am J Anat 148:85–119. [DOI] [PubMed] [Google Scholar]
- Ng CM, Cheng A, Myers LA, Martinez-Murillo F, Jie C, Bedja D, Gabrielson KL, Hausladen JM, Mecham RP, Judge DP, Dietz HC. 2004. TGF-beta-dependent pathogenesis of mitral valve prolapse in a mouse model of Marfan syndrome. J Clin Invest 114:1586–1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieto MA. 2013. Epithelial plasticity: a common theme in embryonic and cancer cells. Science 342:1234850. [DOI] [PubMed] [Google Scholar]
- Nieto MA, Huang RY, Jackson RA, Thiery JP. 2016. Emt: 2016. Cell 166:21–45. [DOI] [PubMed] [Google Scholar]
- Nordrum IS, Skallerud B. 2012. Smooth muscle in the human mitral valve: extent and implications for dynamic modelling. APMIS 120:484–494. [DOI] [PubMed] [Google Scholar]
- Ocana OH, Coskun H, Minguillon C, Murawala P, Tanaka EM, Galceran J, Munoz-Chapuli R, Nieto MA. 2017. A right-handed signalling pathway drives heart looping in vertebrates. Nature 549:86–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peinado H, Olmeda D, Cano A. 2007. Snail, Zeb and bHLH factors in tumour progression: an alliance against the epithelial phenotype? Nat Rev Cancer 7:415–428. [DOI] [PubMed] [Google Scholar]
- Pinto MT, Covas DT, Kashima S, Rodrigues CO. 2016. Endothelial Mesenchymal Transition: Comparative Analysis of Different Induction Methods. Biol Proced Online 18:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitulescu ME, Schmidt I, Benedito R, Adams RH. 2010. Inducible gene targeting in the neonatal vasculature and analysis of retinal angiogenesis in mice. Nat Protoc 5:1518–1534. [DOI] [PubMed] [Google Scholar]
- Quinlan AM, Billiar KL. 2012. Investigating the role of substrate stiffness in the persistence of valvular interstitial cell activation. J Biomed Mater Res A 100:2474–2482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Runyan RB, Markwald RR. 1983. Invasion of mesenchyme into three-dimensional collagen gels: a regional and temporal analysis of interaction in embryonic heart tissue. Dev Biol 95:108–114. [DOI] [PubMed] [Google Scholar]
- Sauls K, Toomer K, Williams K, Johnson AJ, Markwald RR, Hajdu Z, Norris RA. 2015. Increased Infiltration of Extra-Cardiac Cells in Myxomatous Valve Disease. J Cardiovasc Dev Dis 2:200–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Searcy RD, Vincent EB, Liberatore CM, Yutzey KE. 1998. A GATA-dependent nkx-2.5 regulatory element activates early cardiac gene expression in transgenic mice. Development 125:4461–4470. [DOI] [PubMed] [Google Scholar]
- Shapero K, Wylie-Sears J, Levine RA, Mayer JE Jr., Bischoff J. 2015. Reciprocal interactions between mitral valve endothelial and interstitial cells reduce endothelial-to-mesenchymal transition and myofibroblastic activation. J Mol Cell Cardiol 80:175–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soriano P. 1999. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet 21:70–71. [DOI] [PubMed] [Google Scholar]
- Swanson JC, Davis LR, Arata K, Briones EP, Bothe W, Itoh A, Ingels NB, Miller DC. 2009. Characterization of mitral valve anterior leaflet perfusion patterns. J Heart Valve Dis 18:488–495. [PMC free article] [PubMed] [Google Scholar]
- Tae HJ, Petrashevskaya N, Marshall S, Krawczyk M, Talan M. 2016. Cardiac remodeling in the mouse model of Marfan syndrome develops into two distinctive phenotypes. Am J Physiol Heart Circ Physiol 310:H290–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Nakayama M, Pitulescu ME, Schmidt TS, Bochenek ML, Sakakibara A, Adams S, Davy A, Deutsch U, Luthi U, Barberis A, Benjamin LE, Makinen T, Nobes CD, Adams RH. 2010. Ephrin-B2 controls VEGF-induced angiogenesis and lymphangiogenesis. Nature 465:483–486. [DOI] [PubMed] [Google Scholar]
- Weind KL, Ellis CG, Boughner DR. 2000. The aortic valve blood supply. J Heart Valve Dis 9:1–7; discussion 7–8. [PubMed] [Google Scholar]
- Weind KL, Ellis CG, Boughner DR. 2002. Aortic valve cusp vessel density: relationship with tissue thickness. J Thorac Cardiovasc Surg 123:333–340. [DOI] [PubMed] [Google Scholar]
- Wirrig EE, Gomez MV, Hinton RB, Yutzey KE. 2015. COX2 inhibition reduces aortic valve calcification in vivo. Arterioscler Thromb Vasc Biol 35:938–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wylie-Sears J, Levine RA, Bischoff J. 2014. Losartan inhibits endothelial-to-mesenchymal transformation in mitral valve endothelial cells by blocking transforming growth factor-beta-induced phosphorylation of ERK. Biochem Biophys Res Commun 446:870–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeisberg EM, Potenta SE, Sugimoto H, Zeisberg M, Kalluri R. 2008. Fibroblasts in kidney fibrosis emerge via endothelial-to-mesenchymal transition. J Am Soc Nephrol 19:2282–2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Huang X, Liu K, Tang J, He L, Pu W, Liu Q, Li Y, Tian X, Wang Y, Zhang L, Yu Y, Wang H, Hu R, Wang F, Chen T, Wang QD, Qiao Z, Zhang L, Lui KO, Zhou B. 2017. Fibroblasts in an endocardial fibroelastosis disease model mainly originate from mesenchymal derivatives of epicardium. Cell Res 27:1157–1177. [DOI] [PMC free article] [PubMed] [Google Scholar]