

Abstract
Primary cilia are small organelles projecting from the cell surface of various cell types. They play a crucial role in the regulation of various signaling pathway. In this study, we investigated the importance of cilia for heart development by conditionally deleting intraflagellar transport protein Ift88 using the col3.6-cre mouse. Analysis of col3.6;Ift88 offspring showed a wide spectrum of cardiovascular defects including Double Outlet Right Ventricle and Atrioventricular Septal Defects. In addition, we found that in the majority of specimens the pulmonary veins did not properly connect to the developing left atrium. The abnormal connections found resemble those seen in patients with Total Anomalous Pulmonary Venous Return. Analysis of mutant hearts at early stages of development revealed abnormal development of the dorsal mesocardium, a second heart field-derived structure at the venous pole intrinsically related to the development of the pulmonary veins. Data presented support a crucial role for primary cilia in outflow tract development and atrioventricular septation and their significance for the formation of the second heart field derived tissues at the venous pole including the dorsal mesocardium. Furthermore, the results of this study indicate that proper formation of the dorsal mesocardium is critically important for the development of the pulmonary veins.
Keywords: heart, development, cilia, pulmonary veins, congenital herat defect
INTRODUCTION
Primary cilia are small organelles that project from the cell surface of many cell types. The core structure of primary cilia is the axoneme consisting of 9 doublet microtubules that extend from a centriole-derived basal body. Formation, maintenance, and function of cilia are largely dependent on trafficking of particles along the axoneme by the intraflagellar transport machinery.
Ciliopathies are genetic disorders that result in disrupted cilia formation or function. The wide variety of developmental defects associated with dysfunctional cilia reflects the importance of cilia during development. A large number of ciliopathies are associated with congenital heart defects including Alström syndrome, Bardet-Biedl syndrome, Meckel syndrome, Dandy-Walker syndrome, Joubert syndrome, Ellis-van Creveld syndrome, McKusick-Kaufman syndrome, Short-rib polydactyly syndrome, Sensenbrenner syndrome, and nephronophthisis (Koefoed et al., 2014).
Primary cilia are important in the regulation of a number of signaling pathways including the non-canonical Wnt pathway(Wallingford and Mitchell, 2011; May-Simera and Kelley, 2012), the PDGF pathway (Schneider et al., 2005), and the Notch pathway (Ezratty et al., 2011). The most-frequently studied cilia-associated signaling pathway is, however, the Hedgehog (Hh) signaling pathway (Huangfu et al., 2003; Qin et al., 2011). Hh signaling is initiated when a member of the Hh family (typically Sonic Hedgehog, Shh) binds to the membrane receptor patched (Ptc1) localized on the distal end of the axoneme. The interaction between Hh and Ptc1 alleviates inhibition of the transmembrane protein Smoothened (Smo) by Ptc1 (Corbit et al., 2005; Rohatgi et al., 2007). Smo then activates the transcriptional activator Gli2 thereby transmitting the Hh signal to the nucleus. The importance of Hh signaling in heart development has been demonstrated in a number of recently published papers (Goddeeris et al., 2007; Goddeeris et al., 2008; Hoffmann et al., 2009; Briggs et al., 2016). Amongst others, it has been shown that deletion of Smo from the Second Heart Field (SHF) using a SHF-specific cre-mouse model (Mef2c-AHF-cre) leads to the partial (or incomplete) form of Atrioventricular Septal Defect (AVSD) (Briggs et al., 2016), (Goddeeris et al., 2008). In our most recent publication we showed that the pathogenesis of the AVSD in the Mef2c-AHF-cre;Smofl/fl model resulted from the reduction of proliferation in the posterior part of the SHF (pSHF) (Briggs et al., 2016). This led to perturbed development of the pSHF-derived Dorsal Mesenchymal Protrusion (DMP). As a result, the DMP failed to protrude into the common atrium, leading to a primary atrial septal defect (pASD) and common atrioventricular valve (cAVV), hallmarks of all forms of AVSDs. It was also observed that the deletion of Smo from the SHF resulted in suppression of the Wnt/β-catenin pathway. When mice carrying Mef2c-AHF-cre;Smofl/fl embryos were treated with LiCl, an activator of the Wnt/β-catenin pathway, the penetrance of the pAVSD phenotype was significantly reduced, suggesting that the Wnt/β-catenin pathway is acting downstream of the Hh pathway in the development of the DMP (Briggs et al., 2016).
One way of interfering with the development and function of primary cilia is by deleting specific components of the intraflagellar transport (IFT) machinery from specific cell populations. For instance, to determine the importance of cilia in the endocardium and endocardially-derived mesenchyme, Toomer and et al. deleted the IFT protein Ift88 from this cell population using the Nfactc1-cre enhancer mouse and reported that this Nfactc1-cre mediated deletion of Ift88 leads to bicuspid aortic valves (Toomer et al., 2017).
In the current study, we report the outcomes of a series of experiments we have conducted to determine the role of cilia in cardiovascular development by conditionally deleting Ift88 in a wide variety of tissues using a the col3.6-cre mouse (Liu et al., 2004). Whereas this cre mouse model had previously been described as an “osteoblast-targeted cre” (Liu et al., 2004), we found in unrelated studies that it drives cre expression throughout the embryo in a multitude of tissues, including all cell populations contributing to the developing heart. Col3.6-cre mediated deletion of Ift88 resulted in a spectrum of cardiac malformations, including complete atrioventricular septal defect (cAVSD) and a high percentage (>75% penetrance) of total anomalous pulmonary venous return (TAPVR).
To the best of our knowledge, the Col3.6-cre;Ift88fl/- model has the highest penetrance of TAPVR of any previously reported mouse model and therefore represents a unique model for studying the pathogenesis of this phenotype and potentially will lead to the discovery of candidate pathways involved in the etiology of this congenital malformation. Overall, this study contributes to the growing body of evidence that cilia play a very important role in cardiovascular development and that interupting ciliogenesis early in development may result in dysregulation of genes and signaling pathways that collectively result in a spectrum of cardiac defects.
MATERIALS and METHODS
Mice:
The following cre mouse models were used in this study: mWt1-cre (provided by Dr. John Burch), Mef2c-AHF-cre (provided by Dr. Brian Black), Tie2-cre (Kisanuki et al., 2001), and col3.6-cre (Liu et al., 2004). The cre models were used in combination with mice carrying floxed Ift88 alleles (Ift88fl) (Haycraft et al., 2007) and mice with a germline deletion of Ift88 (Ift88∆) (Haycraft et al., 2007). In all our mating protocols, we use male mice to introduce the cre-allele. In some cases, the respective cre mice, as well compound transgenes, were crossed with the B6.129(Cg)-Gt(ROSA)26Sortm4(ACTB-tdTomato,-EGFP)Luo/J reporter mouse (R26mT/mG; Jackson Laboratory; stock no 007576) to allow for determination of cre-driven membrane-targeted green fluorescence (mGFP) after cre-recombinase exposure (Muzumdar et al., 2007). All experiments using animals were approved by the MUSC Institutional Animal Care and Use Committee and complied with federal and institutional guidelines.
Histology:
Following the sacrifice of time-pregnant dams, embryos were isolated in phosphate-buffered saline (PBS), inspected using a dissecting microscope, and staged as described before (Wirrig et al., 2007). Embryos were then fixed in freshly dissolved paraformaldehyde (4% w/v in PBS), processed through a graded series of ethanol, cleared in toluene, embedded in Paraplast Plus (Fisher Brand, catalogue#: 23–021-400), serially sectioned (5 μm), mounted on Superfrost/Plus microscope slides (Fisherbrand catalogue#: 12–550-15) and stored at room temperature. Hematoxylin/eosin staining was performed as previously described (Waller and Wessels, 2000; Snarr et al., 2007a; Snarr et al., 2007b). For immunohistochemistry, sections were deparaffinized in xylenes and rehydrated in graded series of ethanols. Antigen retrieval was typically performed by immersing slides in boiling antigen-unmasking solution (Vector Laboratories, #H-3300) in a pressure cooker for five minutes. To prevent non-specific antibody binding, sections were pretreated with 1% Bovine Serum Albumine (BSA) in PBS for 1 hour before the addition of primary antibodies. Antibodies used in this study included: anti-eGFP (Abcam, ab13970), anti-Acetylated Alpha Tubulin (Sigma, T6793), Gamma Tubulin (Abcam, ab11317), anti-sarcomeric actin (Sigma, A2172), anti-smooth muscle actin (Sigma, A-2547), and anti-Cartilage Link Protein 1 (Crtl1 aka HAPLN1; Developmental Studies Hybridoma Bank, 9/30/8-A-4). Nuclei were visualized using DAPI (Slowfade Gold Antifade Reagent with DAPI, Invitrogen S36938). Fluorescence was visualized using a Zeiss AxioImager II microscope.
RESULTS
Expression domain of cre-recombinase in the Col3.6-cre mouse
The col3.6cre mouse is frequently used for studies on bone formation and has previously been described as an “osteoblast-targeted cre” (Liu et al., 2004). In the context of an unrelated study, we had observed that cre-recombinase activity was actually conveyed to a wide variety of other cell types. To assess the extent of the col3.6cre expression domain, and to establish in which cell populations involved in cardiovascular development cre-activity could be detected, we collected col3.6cre;ROSA26mT/mG offspring between ED9.5 and ED14.5. Cre activity, as determined by eGFP expression, was found throughout the embryo including tissues such as the neural tube, foregut endoderm, and the walls of the dorsal aortae. In the heart, eGFP expression was observed in the myocardium, the endocardium, the epicardium, and the posterior second heart field (pSHF) adjacent and ventral to the pulmonary endoderm. It is important to note that the extent of eGFP expression varied from specimen to specimen (Fig.1).
Figure 1. Col3.6cre-driven eGFP expression.
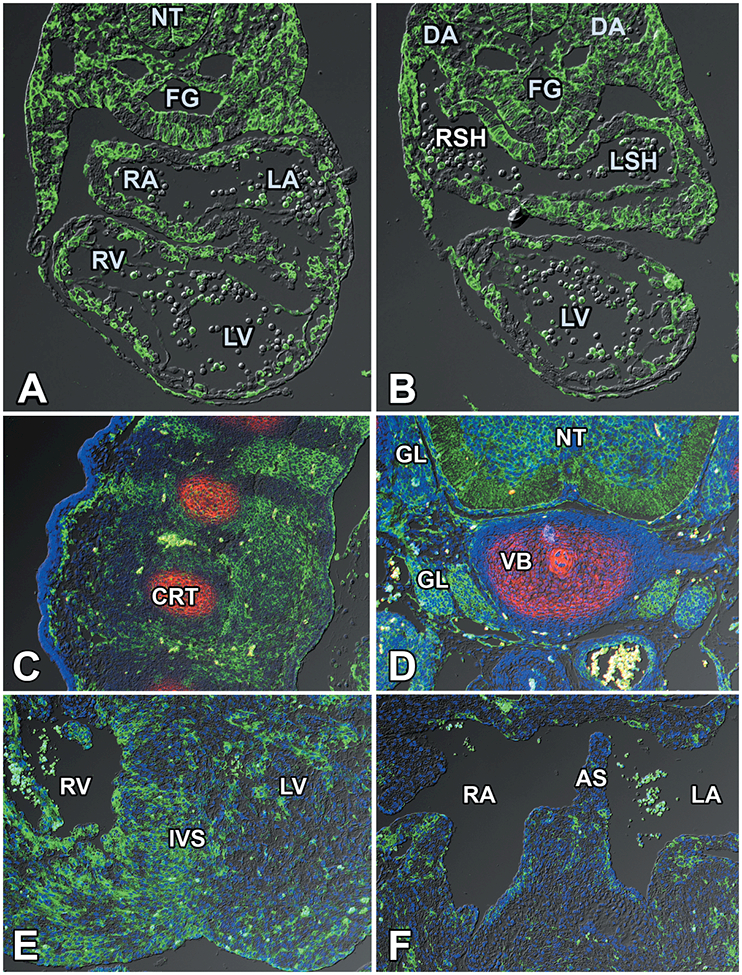
Sections of a col3.6cre;ROSA26mT/mG mouse embryo at ED9.5 showing the cre-mediated expression of eGFP throughout the embryo, including the tissues of the heart. DA=dorsal aorta; FG=foregut; LA=left atrium; LSH=left sinus horn; LV=left ventricle; RA=right atrium; RSH=right sinus horn; RV=right ventricle
Cilia in the heart and surrounding tissues
Cilia are widely expressed in embryonic tissues including the heart (Slough et al., 2008; Willaredt et al., 2008; Willaredt et al., 2012; Klena et al., 2017; Toomer et al., 2017). To determine where in the developing heart cilia are located, immunofluorescent analysis was conducted at stages ED10.5, ED12.5, and ED14.5. Cilia were found on the myocardium (Fig.2A,C), the endocardial cushion mesenchyme (Fig.2B), and the epicardium (Fig.2A,D). We have previously demonstrated the importance of the pSHF and the pSHF-derived Dorsal Mesenchymal Protrusion (DMP) for atrioventricular septation (Snarr et al., 2007a; Snarr et al., 2007b; Briggs et al., 2013; Briggs et al., 2016). To determine the presence of cilia in the pSHF, immunofluorescent analysis was performed in Mef2c-AHF-cre;R26mT/mG specimens at ED10.5 in which SHF cells can be identified by eGFP expression. This analysis revealed an abundance of cilia within the pSHF (Fig.2E,F).
Figure 2. Cilia in the developing mouse heart.
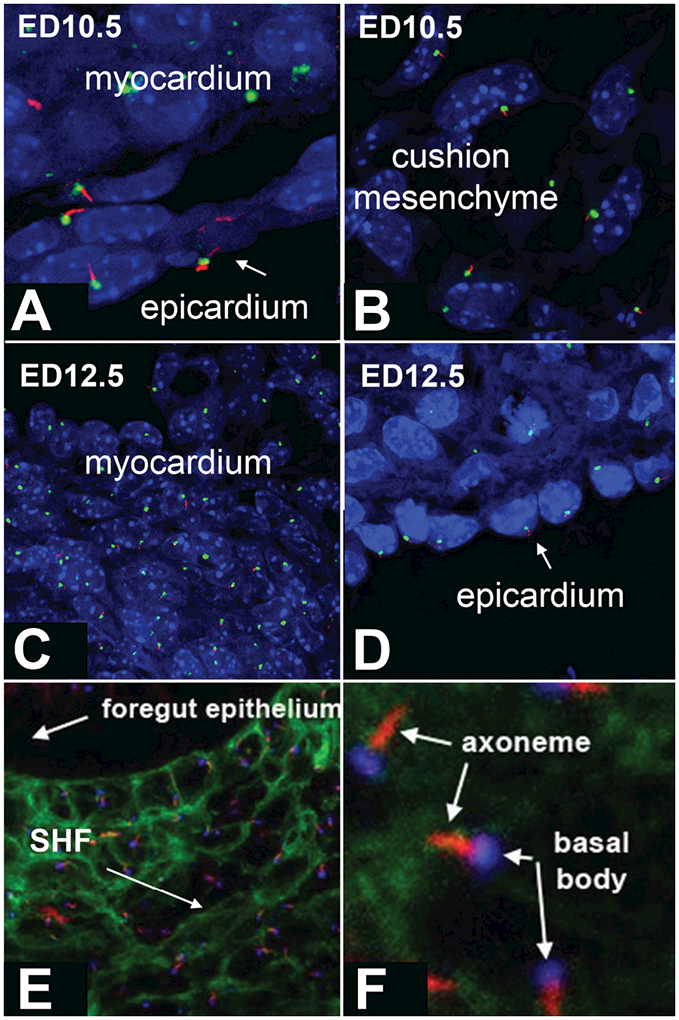
Sections of mouse embryos at ED10.5 (A,B,E,F) and ED12.5 (C,D) were stained with antibodies recognizing acetylated alpha tubulin to visualize the ciliary axoneme (red in A-F), and gamma tubulin to visualize the basal bodies (green in A-D, blue in E-F). The sections in E and F are from a Mef2c-AHF-cre;R26mT/mG mouse embryo at ED10.5, in which second heart field (SHF) cells are identified by the expression of eGFP.
Conditional cre-mediated deletion of Ift88 from the endocardium and second heart field.
To investigate whether and where in the heart intact cilia are necessary for proper heart development, we started with conditionally deleting Ift88 by cre-lox strategy using a variety of establised “tissue-specific” cre mouse models in combination with two mouse models that can be used for determining the relevance of Ift88 in development and/or ciliary function, i.e. the Ift88fl carrying a floxed Ift88 allele (Haycraft et al., 2007) and the Ift88∆ mouse, a mouse with a germline deletion of Ift88 (Haycraft et al., 2007).
First, to test the importance of cilia in the pSHF, we crossed the above mentioned Ift88 models with the Mef2c-AHF-cre mouse (Verzi et al., 2005) creating Mef2c-AHF-cre;Ift88fl/fl and Mef2c-AHF-cre;Ift88∆/fl offspring, following a strategy used in prior studies in which we deleted the BMP receptor Alk3 and the Hh co-receptor Smo respectively from the SHF using the Mef2c-AHF-cre mouse(Briggs et al., 2013; Briggs et al., 2016). Following this protocol, we collected a total of 8 Mef2c-AHF-cre;Ift88fl/fl and 3 Mef2c-AHF-cre;Ift88∆/fl specimens between ED10.5 and ED14.5. Histological analysis did, against expectations, not reveal major cardiac abnormalities with the exception of one Mef2c-AHF-cre;Ift88∆/fl specimen at ED14.5 which presented with a ventricular septal defect (VSD).
To further explore the importance of Ift88, and hence primary cilia, for cardiovascular development, we then turned to the col3.6cre mouse. We predicted, based on the expression of col3.6-cre mediated eGFP expression, that this more robust approach would lead to a more rigorous perturbation of cilia assembly and/or function throughout the embryo. Using the col3.6cre mouse we generated a total of 15 litters containing col3.6-cre;Ift88fl/fl and col3.6-cre;Ift88∆/fl offspring and littermate controls at stages ED9.5, ED10.5, ED14.5, and ED15.5. While this approach did have consequences for the development of other tissues as well, in this contribution we will focus on the impact on cardiac development.
Valvuloseptal Defects in the col3.6-cre;Ift88∆/fl mouse.
During normal development in the mouse, septation of the cardiac chambers and outflow tract should be completed by ED14.5 (Waller and Wessels, 2000; Wessels and Markwald, 2000). Morphological assessment of col3.6-cre;Ift88fl/fl and col3.6-cre;Ift88∆/fl specimens at ED14.5-ED15.5, revealed several structural valvuloseptal defects. At the arterial outflow, 8/14 (57%) of the mutants displayed double outlet right ventricle (DORV), a condition in which both the pulmonary trunk as well as the aorta arise from the right ventricle (Table 1 and Fig.3). In addition, one of the col3.6-cre;Ift88∆/fl specimens had persistent truncus arteriosis (PTA), a situation where the aorta and pulmonary trunk are not divided downstream of the arterial valves. Furthermore, 13 out of the 14 inspected col3.6-cre;Ift88fl/fl and col3.6-cre;Ift88∆/fl embryos (93%) presented with an atrioventricular septal defect (AVSD), characterized by the presence of a primary atrial septum defect (pASD), common AV valve (cAVV), and, in the case of the complete form of AVSD, a ventricular septal defect (VSD) (Table 1 and Fig.4).
Table 1. Cardiovascular defects in col3.6-cre:Ift88 mutants.
| Specimen | Stage | Genotype | DORV | PTA | AVSD | TAPVR |
|---|---|---|---|---|---|---|
| 1 | ED14.5 | col3.6-cre;Ift88∆/fl | no | no | yes | yes |
| 2 | ED14.5 | col3.6-cre;Ift88∆/fl | yes | no | yes | yes |
| 3 | ED14.5 | col3.6-cre;Ift88∆/fl | no | no | yes | ambiguous |
| 4 | ED14.5 | col3.6-cre;Ift88∆/fl | yes | no | yes | yes |
| 5 | ED14.5 | col3.6-cre;Ift88∆/fl | yes | no | yes | yes |
| 6 | ED14.5 | col3.6-cre;Ift88∆/fl | yes | yes | yes | yes |
| 7 | ED14.5 | col3.6-cre;Ift88∆/fl | yes | no | yes | yes |
| 8 | ED14.5 | col3.6-cre;Ift88∆/fl | yes | no | yes | ambiguous |
| 9 | ED14.5 | col3.6-cre;Ift88∆/fl | no | no | yes | yes |
| 10 | ED14.5 | col3.6-cre;Ift88∆/fl | no | no | no | no |
| 11 | ED14.5 | col3.6-cre;Ift88∆/fl | yes | no | yes | yes |
| 12 | ED14.5 | col3.6-cre;Ift88∆/fl | no | no | yes | yes |
| 13 | ED15.5 | col3.6-cre;Ift88fl/fl | yes | no | yes | yes |
| 14 | ED15.5 | col3.6-cre;Ift88fl/fl | no | no | yes | yes |
DORV=double outlet right ventricle, PTA=persistent truncus arteriosus, AVSD=atrioventricular septal defect, TAPVR=total anomalous pulmonary venous return
Figure 3. Double outlet right ventricle (DORV) in the col3.6-cre;Ift88∆/fl mouse.

Serial sections of the arterial pole of a control (A,C) and a col3.6-cre;Ift88∆/fl (B,D) embryo at14.5ED. Note that in both embryos the pulmonary trunk arises from the right ventricle (A,B) but that while the aorta originates from the left ventricle in the control heart (C), in the col3.6-cre;Ift88∆/fl embryo the aorta also comes off the right ventricle. Ao=aorta; LA=left atrium; LV=left ventricle; Pu=pulmonary trunk; RA=right atrium; RV=right ventricle.
Figure 4. Complete atrioventricular septal defects (cAVSD) in the col3.6-cre;Ift88∆/fl mouse.
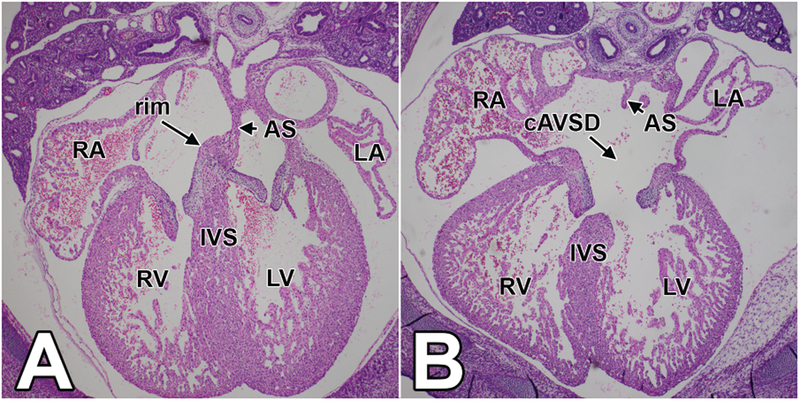
This figure shows 4-chamber views of sections of a control (A) and a col3.6-cre;Ift88∆/fl (B) embryo at 14.5ED at the level of the atrioventricular junction. The four chambers in the control heart are completely septated by a fully formed atrial and ventricular septum. The heart of the col3.6-cre;Ift88∆/fl specimen has a complete atrioventricular septal defect characterized by a primary atrial septal defect as well as a ventricular septal defect. AS=atrial septum; cAVSD=complete atrioventricular septal defect; IVS=interventricular septum; LA=left atrium; LV=left ventricle; RA=right atrium; RV=right ventricle.
Total Anomalous Pulmonary Venous Return (TAPVR) in the col3.6-cre;Ift88∆/fl mouse.
In the mouse, as in the human, the pulmonary veins return the oxygenated blood from the lungs to the heart. However, while in the human the left and right pulmonary veins empty directly into the left atrium through four separate pulmonary orifices, in the mouse, the pulmonary veins come together behind the left atrium and drain through a common orifice into the left atrium (Fig.5). We were able to trace the pulmonary veins, and locate the common pulmonary orifice, in 10 of the 12 col3.6-cre;Ift88∆/fl embryos at ED14.5 and in 2 col3.6-cre;Ift88fl/fl at ED15.5. In 3 col3.6-cre;Ift88∆/fl specimens we were, for various reasons, not able to trace the pulmonary veins. Our analysis revealed that in the majority of the col3.6-cre;Ift88∆/fl hearts (>75%) at ED14.5–15.5 the common pulmonary orifice was not properly positioned in the left atrium (Table 1). The location of the pulmonary venous return in the hearts varied. Whereas in control hearts, the pulmonary veins drain to the left of the atrial septum into the left atrium (Fig.5A), in two ED14.5 col3.6-cre;Ift88∆/fl specimens the pulmonary venous orifice was found to the right of the rudimentary primary atrial septum in the posterior right atrial wall (Fig.6B). In two ED14.5 col3.6-cre;Ift88∆/fl specimens, we found a pulmonary vein connected to the junctional area between the future right superior caval vein (RSCV) and the right atrium Fig.6C). In three ED14.5 col3.6-cre;Ift88∆/fl specimens the common pulmonary vein was seen to drain very low in the right atrium (Fig.6D,E), close to where the left sinus horn (LSH) connects to the right atrium. In one of the ED14.5 col3.6-cre;Ift88∆/fl hearts, the LSH entered the left atrium, a condition often seen in hearts with right atrial isomerism. In this heart the pulmonary vein was found at the junction between the LSH and the left atrial wall. A similar situation was observed in the two col3.6-cre;Ift88fl/fl specimens at ED15.5 where the overall morphology suggested disturbed laterality with the orifice of the pulmonary vein being located in between the bilateral RSVC and LSH.
Figure 5. 3D AMIRA reconstruction of the arrangement of the pulmonary veins in a normal embryonic mouse heart at 14.5ED.

The reconstruction in panel A was prepared using serial sections of a Tie2-cre;ROSA26mT/mG mouse embryo at 14.5ED. All sections were stained with a combination of antibodies recognizing both sarcomeric actin and smooth muscle actin (red in B), a combination of antibodies useful for recognizing cardiomyocytes and smooth muscle cells. Endothelial and endothelial-derived cells in the Tie2-cre;ROSA26mT/mG mouse express eGFP (green in B). C shows the 3D reconstruction “sliced” at the level of the tissue section shown in B. The pulmonary veins in panel A are pseudocolored in yellow, the left sinus horn in blue. AS=atrial septum; CPV=common pulmonary vein; LA=left atrium; LPV=left pulmonary veins; LSH=left sinus horn; LV=left ventricle; RA=right atrium; RPV=right pulmonary vein; RV=right ventricle
Figure 6. Total Anomalous Pulmonary Venous Return (TAPVR) in the col3.6-cre;Ift88∆/fl mouse.

Serial H/E stained section of a control heart (A) and four col3.6-cre;Ift88∆/fl mouse embryos (B-E) at ED14.5. Panel A shows the normal anatomy of the right and left pulmonary veins entering the left atrium through the common pulmonary orifice. In the mutant in B the pulmonary veins enter to the right of the rudimentary primary atrial septum in the posterior right atrial wall. In C a single pulmonary vein connects to the heart in the junctional area of the right superior caval vein and right atrium. In D and E the common pulmonary vein was found to connect very low in the right atrium close to where the left sinus horn enters the right atrial chamber. AS=atrial septum; LA=left atrium; leftPuV=left pulmonary veins; LSH=left sinus horn; LV=left ventricle; RA=right atrium; rightPuV=right pulmonary vein; RV=right ventricle; RVV=right venous valve
The abnormal connections of the pulmonary veins at the venous pole as observed in the col3.6-cre;Ift88∆/fl mouse resemble a cyanotic congenital heart defect in the human known as Total Anomalous Pulmonary Venous Return (TAPVR).
Perturbed development of the dorsal mesocardium in the col3.6-cre;Ift88∆/fl mouse
The development of the pulmonary veins and the final location of the orifice of the common vein in the dorsal wall of the left atrium are intrinsically associated with the development of the dorsal mesocardium and the SHF-derived dorsal mesocardial protrusion (DMP) (Snarr et al., 2008; Burns et al., 2016). During normal development, the DMP penetrates the dorsal mesocardium between ED9.5 and ED10.5 to the right of the primitive pulmonary vein (pPuV) which is initially a midline structure (Briggs et al., 2012). As a result, the future orifice of the common pulmonary vein shifts to the left. The subsequent development of the primary atrial septum eventually determines the final location of the pulmonary orifice in the dorsal atrial wall of the left atrium. Under the hypothesis that the abnormal location of the pulmonary venous return in the col3.6-cre;Ift88 mutants could be linked to abnormal development of the dorsal mesocardium and DMP, we analyzed the venous pole of a small sample of col3.6-cre;Ift88∆/fl specimens at ED9.5. It was observed that in a number of col3.6-cre;Ift88∆/fl specimens at ED9.5 the dorsal mesocardium had an abnormal appearance when compared to control littermates (Fig.7). Specifically, the dorsal mesocardium was found to be a lot wider containing what appears excessive extracellular matrix and mesenchyme. In addition, the pulmonary pit, i.e. the invagination of the endocardial lining that is in contiguity with the pPuV, was typically not well defined (Fig. 7C,C’,D,D’)
Figure 7. The dorsal mesocardium in the col3.6-cre;Ift88∆/fl mouse.
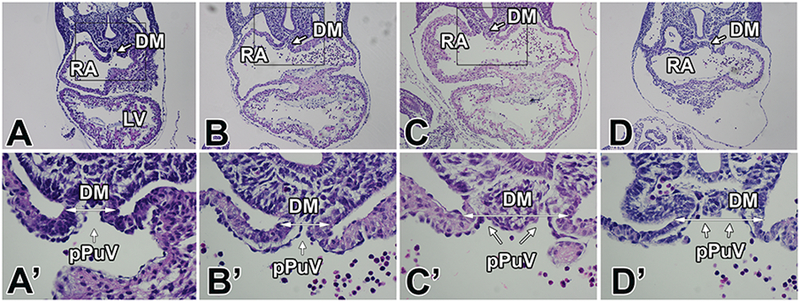
Serial sections of one control embryo (A,A’) and three col3.6-cre;Ift88∆/fl embryos (B,B’,C,C’,D,D’) at the level of the dorsal mesocardium. The higher magnifications in A’-D’ show the normal dorsal mesocardium in the control heart (A’) as well as in one of the col3.6-cre;Ift88∆/fl embryos (B’). Panels C’ and D’ demonstrate that in these specimens the dorsal mesocardium is abnormally wide (arrows) and dysmorphic. The location of the connection of the primitive pulmonary vein(s) to the common atrium is not well-defined. DM=dorsal mesocardium; LV=left ventricle; pPuV=primitive pulmonary vein; RA=right atrium.
DISCUSSION
Relationship of cilia, TAPVR, and other congenital heart defects
Over the last few years, a number of mouse models have been reported in which reduced global expression of IFT proteins results in congenital heart defects. Cardiac abnormalities, including AVSDs have been reported in mice with hypomorphic alleles for Ift88 and Ift172 (Friedland-Little et al., 2011; Willaredt et al., 2012). More recently, the Norris lab has demonstrated that endocardially-expressed Ift88 is of critical importance for proper aortic valve development (Toomer et al., 2017). Combined, these and other studies support the importance of primary cilia for valvuloseptal development. However, the inclusion of TAPVR as part of the spectrum of cardiac abnormalities in mouse models with perturbation of cilia function is novel.
In the human, TAPVR is the more severe form of Anomalous Pulmonary Venous Return (APVR) as all the pulmonary veins connect to the right side of the venous pole (e.g. right atrium, superior caval vein, azygos vein, portal vein, or coronary sinus). TAPVR comprises ~1% of all congenital heart malformations, has a low recurrence risk (3–5%), and is usually found without a family history. Without surgical intervention, TAPVR has a high mortality rate in the first year of life. TAPVR is often found in association with other congenital malformations such as atrial septal defect (ASD), atrioventricular septal defect (AVSD) and double-outlet right ventricle (DORV). TAPVR is frequently seen in syndromic conditions, such as Heterotaxy (Anagnostopoulos et al., 2009), Cat Eye Syndrome (Fahed et al., 2013), Fryns Syndrome (Neville et al., 2002), Turner Syndrome (Gutmark-Little et al., 2012), Williams Syndrome (Park et al., 2012), Scimitar Syndrome (Gudjonsson and Brown, 2006), Smith-Lemli-Opitz Syndrome (Digilio et al., 2003), and Smith-Magenis syndrome (Myers and Challman, 2004). TAPVR is also found in patients with ciliopathies such as Bardet-Biedl (Digilio et al., 2006) (Stamm, 1998) and Ellis-van Creveld Syndrome (Hills et al., 2011).
Etiology of TAPVR
Despite the low recurrence risk for TAPVR, a number of cases with familial TAPVR have been reported which has eventually led to the identification of candidate genes associated with TAPVR. Bleyl and colleagues studied two Utah/Idaho families with non-syndromic TAPVR as an autosomal dominant trait with incomplete penetrance and variable expression and identified PDGFRA as a putative TAPVR gene (Bleyl et al., 1994) (Bleyl et al., 2006). In another study Semaphorin 3d (SEMA3d) was found to be a TAPVR candidate gene (Degenhardt et al., 2013). Other candidate genes include Cardiac Ankyrin Repeat Protein (ANKRD1/CARP) (Cinquetti et al., 2008), Connexin 43 (GJA1) (Fahed et al., 2013), Zinc finger protein 3 (ZIC3) (Fahed et al., 2013), and genes associated with retinoic acid signaling, including Retinol Binding Protein 5 (RBP5), Nodal, and Retinol Dehydrogenase10 (Nash et al., 2015).
Existing hypotheses regarding the pathogenesis of TAPVR–animal studies
Thus far, published studies on the pathogenesis of TAPVR have resulted in two prevailing hypotheses. Based on their findings in the Utah/Idaho families with non-syndromic TAPVR (see above), Bleyl and colleagues investigated the potential role of Pdgfra in the pathogenesis of TAVPR in mice, including the Pdgfra knockout mouse and mouse models in which Pdgfra was conditionally deleted. They found a 7% overall penetrance of TAPVR in these models with only 1 out of 29 Pdgfra knockouts presenting with TAPVR (Bleyl et al., 2010). The authors furthermore stated that Pdgfra is expressed in the DMP and that Pdgfra-deficient embryos have a hypoplastic DMP. Based on these combined, albeit not very convincing, observations and results, it was suggested that hypoplasia of the DMP could be responsible for the presence of persistent pulmonary venous connections to the structures derived from the embryonic sinus venosus. Even though the very low penetrance of TAPVR in these Pdgfra mutants does not make a strong case for a significant role of Pdgfra in the pathogenesis of the development of the pulmonary veins, the Pdgf pathway is of significant interest as Pdgfra, a receptor tyrosine kinase, is the only TAPVR candidate directly associated with primary cilia (Schneider et al., 2005; Plotnikova et al., 2009). It is therefore attempting to speculate that while Pdgfra in itself may not be critically important for proper development of the pulmonary veins, perturbation of Pdgf signaling through cilia-associated Pdgfra may be a contributing factor in the pathogenesis of TAPVR when cilia formation and maintenance is perturbed.
As mentioned, SEMA3D has also been identified as a candidate gene for TAPVR in humans. Studies on the Sema3d knockout mice revealed a higher penetrance of TAPVR (35%) (Degenhardt et al., 2013). Histological analysis of the Sema3d knockout mice led to the suggestion that failure of the developing pulmonary venous plexus to connect properly to the precursor of the common pulmonary vein could be the underlying mechanism responsible for TAPVR. A more or less similar model was proposed by van den Berg and Moorman (2011) who suggested that TAPVR in hearts with normal atrial arrangements may be the result of an error in separation of the splanchnic plexus from which the pulmonary veins derive (van den Berg and Moorman, 2011).
Based on our insights into the development of the DMP and the early stages of pulmonary vein development (Webb et al., 1998; Wessels et al., 2000; Wessels and Markwald, 2000; Snarr et al., 2007a; Snarr et al., 2007b; Snarr et al., 2008; Wu et al., 2011; Briggs et al., 2012; Briggs et al., 2013), we do not believe that either of the two prevailing models sufficiently explains TAPVR pathogenesis. Specifically, neither model provides an explanation of why the point of entry of the pulmonary veins (the pulmonary orifice) finds its way into the right atrium (or its venous tributaries) as observed in the col3.6cre;Ift88fl/Δ mouse. While abnormal anastomosis of the pulmonary venous plexus to the precursor of the common pulmonary vein certainly can be involved in the generation of ectopic “extracardiac” vasculature as proposed by Degenhardt et al. (2013), it would not explain the abnormal location of the pulmonary venous orifice in the models of TAPVR. So, what might cause altering the location of the pulmonary venous orifice in the context of TAPVR?
An alternative model for TAPVR pathogenesis based on observations in the col3.6-cre;Ift88∆/fl mouse.
During the early stages of heart development, the primitive pulmonary vein is a midline structure (Snarr et al., 2008; Briggs et al., 2012; Burns et al., 2016). As the DMP, using the dorsal mesocardium as its “portal of entry”, wedges itself in between the primitive pulmonary vein (PuV) and the right-sided mesocardial reflection into the common atrium, the common orifice of the developing pulmonary vein shifts to the left. In the mouse, this process takes place between ED9.5 and ED10.5 (Briggs et al., 2012; Briggs et al., 2013; Briggs et al., 2016). Completion of the atrial septation process, in combination with myocardialization of the DMP (Wessels et al., 2000), eventually results in the fact that the pulmonary veins drain into the left atrium. In the col3.6cre;Ift88fl/Δ embryos analyzed, we found several specimens in which the dorsal mesocardium was exceptionally wide and in which the location of the mesenchymal tissues that were found within the dorsal mesocardium led to ambiguous location of the pPuV.
An important aspect heart development is the establishment of the dorsal mesocardium at the venous pole of the heart at the completion of cardiac looping, which we will call here the “definitive” dorsal mesocardium. The first step in creating the definitive dorsal mesocardium is the formation of the “primary” dorsal mesocardium. This primary dorsal mesocardium is formed when, in the first stages of cardiac development, the two bilateral heart fields come together and form the straight primary heart tube. After the establishment of the primary heart tube it hangs suspended over its entire length by the primary dorsal mesocardium from the rest of the embryonic foregut. The fusion of the two heart fields also results in the creation of the midpharyngeal endothelial strand which is the precursor of the pPuV and is located within the dorsal mesocardium (DeRuiter et al., 1995). As the heart is undergoing looping, a significant portion of the primary dorsal mesocardium disintegrates, the only persisting dorsal mesocardial connection being the definitive dorsal mesocardium harboring the pPuV (Snarr et al., 2008).
Several years ago, we published a paper on the effect of elevated levels of vascular endothelial growth factor (VEGF) on heart development in the quail (Drake et al., 2006). Amongst other observations, we found that injection of VEGF in early quail embryos in an in vitro setting, resulted in an increase of the number of endothelial cells within the primary dorsal mesocardium. This accumulation of endothelial cells led to the widening of the dorsal mesocardium and to the inhibition of disintegration of the primary dorsal mesocardium. As a result, the primary dorsal mesocardium persisted over a considerable length and inhibited cardiac looping.
The widening of the dorsal mesocardium in the VEGF treated quail embryos is reminiscent of the widening of the dorsal mesocardium observed in the col3.6cre;Ift88fl/Δ mouse. Thus far, we have not determined how deletion of Ift88 in the col3.6cre;Ift88fl/Δ mouse interferes with the formation of the splanchnic plexus and/or formation of endothelial precursors in the definitive dorsal mesocardium. We have also not yet been able to establish whether interference with Ift88 expression has an impact on the disintegration process of the primary dorsal mesocardium over the length of the primary heart tube, a process which potentially can have an effect on the development and location of the endothelial structures involved in the development of the pulmonary veins. In ongoing studies in the lab, we are focusing on elucidating the cellular and molecular mechanisms that underlie the pathogenesis of TAPVR in our model.
The development of the pulmonary vein is a topic of continuing debate. In particular the question of whether the point of entry of the pPuV is completely situated within the confines of the future atrial chambers or whether the pPuV is (also) partly committed to the sinus venosus, has been a contentious subject (DeRuiter et al., 1995; Webb et al., 1998; Wessels et al., 2000; Anderson et al., 2006; Sizarov et al., 2010; Douglas et al., 2011; van den Berg and Moorman, 2011). Where one draws the boundaries “separating” the tissues of the sinus venosus and the developing atrial chambers largely depends on the morphological and molecular criteria used to describe the respective components of the developing heart. This is not the place to rehash this issue. It is beyond doubt, however, that even in the normally developing heart, the “pulmonary pit” is in close proximity to the junction between the tissues of the common atrium and the sinus venosus. We believe that it is conceivable that if the primary dorsal mesocardium does not disintegrate properly in the midsection of the primary heart tube, and that, as a consequence, the dorsal mesocardium does not properly develop, the relative position of the pulmonary pit may change within this crucial area of the venous pole. This, combined with an abnormally developing pSHF and pSHF-derived DMP, and possibly the failure of the pulmonary plexus to properly form, may lead to the various forms of TAPVR encountered in the col3.6-cre;Ift88∆/fl mouse and potentially the spectrum of congenital defects as seen in patients with TAPVR.
Acknowledgments
Sources of Funding
The authors would like to acknowledge the financial support by the following grants: the “South Carolina COBRE for Developmentally Based Cardiovascular Diseases”, P30 GM103342 (AW, RND), the “South Carolina COBRE for Bioengineering Center of Regeneration and Formation of Tissues”, P20GM103444 (RAN), R01HL033756–30 (AW,RND,TAB,EH,RAN), R01HL131546 (RAN, AW), NIH 5T32HL007260 (TAB, KAT), American Heart Association: 17CSA33590067 (RAN), The Foundation Leducq (Paris, France) Transatlantic Mitral Network of Excellence grant 07CVD04 (RAN). The contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH, the AHA, and The Foundation Leducq.
LITERATURE CITED
- Anagnostopoulos PV, Pearl JM, Octave C, Cohen M, Gruessner A, Wintering E, Teodori MF. 2009. Improved current era outcomes in patients with heterotaxy syndromes. Eur J Cardiothorac Surg 35:871–877; discussion 877–878. [DOI] [PubMed] [Google Scholar]
- Anderson RH, Brown NA, Moorman AF. 2006. Development and structures of the venous pole of the heart. Dev Dyn 235:2–9. [DOI] [PubMed] [Google Scholar]
- Bleyl S, Ruttenberg HD, Carey JC, Ward K. 1994. Familial total anomalous pulmonary venous return: a large Utah-Idaho family. American journal of medical genetics 52:462–466. [DOI] [PubMed] [Google Scholar]
- Bleyl SB, Botto LD, Carey JC, Young LT, Bamshad MJ, Leppert MF, Ward K. 2006. Analysis of a Scottish founder effect narrows the TAPVR-1 gene interval to chromosome 4q12. Am J Med Genet A 140:2368–2373. [DOI] [PubMed] [Google Scholar]
- Bleyl SB, Saijoh Y, Bax NA, Gittenberger-de Groot AC, Wisse LJ, Chapman SC, Hunter J, Shiratori H, Hamada H, Yamada S, Shiota K, Klewer SE, Leppert MF, Schoenwolf GC. 2010. Dysregulation of the PDGFRA gene causes inflow tract anomalies including TAPVR: integrating evidence from human genetics and model organisms. Human molecular genetics 19:1286–1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briggs LE, Burns TA, Lockhart MM, Phelps AL, Van den Hoff MJ, Wessels A. 2016. Wnt/beta-catenin and sonic hedgehog pathways interact in the regulation of the development of the dorsal mesenchymal protrusion. Dev Dyn 245:103–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briggs LE, Kakarla J, Wessels A. 2012. The pathogenesis of atrial and atrioventricular septal defects with special emphasis on the role of the dorsal mesenchymal protrusion. Differentiation 84:117–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briggs LE, Phelps AL, Brown E, Kakarla J, Anderson RH, van den Hoff MJ, Wessels A. 2013. Expression of the BMP Receptor Alk3 in the Second Heart Field Is Essential for Development of the Dorsal Mesenchymal Protrusion and Atrioventricular Septation. Circ Res 112:1420–1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns T, Yang Y, Hiriart E, Wessels A. 2016. The Dorsal Mesenchymal Protrusion and the Pathogenesis of Atrioventricular Septal Defects. J Cardiovasc Dev Dis 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cinquetti R, Badi I, Campione M, Bortoletto E, Chiesa G, Parolini C, Camesasca C, Russo A, Taramelli R, Acquati F. 2008. Transcriptional deregulation and a missense mutation define ANKRD1 as a candidate gene for total anomalous pulmonary venous return. Hum Mutat 29:468–474. [DOI] [PubMed] [Google Scholar]
- Corbit KC, Aanstad P, Singla V, Norman AR, Stainier DY, Reiter JF. 2005. Vertebrate Smoothened functions at the primary cilium. Nature 437:1018–1021. [DOI] [PubMed] [Google Scholar]
- Degenhardt K, Singh MK, Aghajanian H, Massera D, Wang Q, Li J, Li L, Choi C, Yzaguirre AD, Francey LJ, Gallant E, Krantz ID, Gruber PJ, Epstein JA. 2013. Semaphorin 3d signaling defects are associated with anomalous pulmonary venous connections. Nat Med 19:760–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeRuiter MC, Gittenberger-De Groot AC, Wenink AC, Poelmann RE, Mentink MM. 1995. In normal development pulmonary veins are connected to the sinus venosus segment in the left atrium. Anat Rec 243:84–92. [DOI] [PubMed] [Google Scholar]
- Digilio MC, Dallapiccola B, Marino B. 2006. Atrioventricular canal defect in Bardet-Biedl syndrome: clinical evidence supporting the link between atrioventricular canal defect and polydactyly syndromes with ciliary dysfunction536. Genet Med 8:536–538. [DOI] [PubMed] [Google Scholar]
- Digilio MC, Marino B, Giannotti A, Dallapiccola B, Opitz JM. 2003. Specific congenital heart defects in RSH/Smith-Lemli-Opitz syndrome: postulated involvement of the sonic hedgehog pathway in syndromes with postaxial polydactyly or heterotaxia. Birth Defects Res A Clin Mol Teratol 67:149–153. [DOI] [PubMed] [Google Scholar]
- Douglas YL, Jongbloed MR, Deruiter MC, Gittenberger-de Groot AC. 2011. Normal and abnormal development of pulmonary veins: state of the art and correlation with clinical entities. Int J Cardiol 147:13–24. [DOI] [PubMed] [Google Scholar]
- Drake CJ, Wessels A, Trusk T, Little CD. 2006. Elevated vascular endothelial cell growth factor affects mesocardial morphogenesis and inhibits normal heart bending. Dev Dyn 235:10–18. [DOI] [PubMed] [Google Scholar]
- Ezratty EJ, Stokes N, Chai S, Shah AS, Williams SE, Fuchs E. 2011. A role for the primary cilium in Notch signaling and epidermal differentiation during skin development. Cell 145:1129–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahed AC, Gelb BD, Seidman JG, Seidman CE. 2013. Genetics of congenital heart disease: the glass half empty. Circ Res 112:707–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedland-Little JM, Hoffmann AD, Ocbina PJ, Peterson MA, Bosman JD, Chen Y, Cheng SY, Anderson KV, Moskowitz IP. 2011. A novel murine allele of Intraflagellar Transport Protein 172 causes a syndrome including VACTERL-like features with hydrocephalus. Hum Mol Genet 20:3725–3737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goddeeris MM, Rho S, Petiet A, Davenport CL, Johnson GA, Meyers EN, Klingensmith J. 2008. Intracardiac septation requires hedgehog-dependent cellular contributions from outside the heart. Development 135:1887–1895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goddeeris MM, Schwartz R, Klingensmith J, Meyers EN. 2007. Independent requirements for Hedgehog signaling by both the anterior heart field and neural crest cells for outflow tract development. Development 134:1593–1604. [DOI] [PubMed] [Google Scholar]
- Gudjonsson U, Brown JW. 2006. Scimitar syndrome. Semin Thorac Cardiovasc Surg Pediatr Card Surg Annu:56–62. [DOI] [PubMed] [Google Scholar]
- Gutmark-Little I, Hor KN, Cnota J, Gottliebson WM, Backeljauw PF. 2012. Partial anomalous pulmonary venous return is common in Turner syndrome. J Pediatr Endocrinol Metab 25:435–440. [DOI] [PubMed] [Google Scholar]
- Haycraft CJ, Zhang Q, Song B, Jackson WS, Detloff PJ, Serra R, Yoder BK. 2007. Intraflagellar transport is essential for endochondral bone formation. Development 134:307–316. [DOI] [PubMed] [Google Scholar]
- Hills CB, Kochilas L, Schimmenti LA, Moller JH. 2011. Ellis-van Creveld syndrome and congenital heart defects: presentation of an additional 32 cases. Pediatr Cardiol 32:977–982. [DOI] [PubMed] [Google Scholar]
- Hoffmann AD, Peterson MA, Friedland-Little JM, Anderson SA, Moskowitz IP. 2009. sonic hedgehog is required in pulmonary endoderm for atrial septation. Development 136:1761–1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huangfu D, Liu A, Rakeman AS, Murcia NS, Niswander L, Anderson KV. 2003. Hedgehog signalling in the mouse requires intraflagellar transport proteins. Nature 426:83–87. [DOI] [PubMed] [Google Scholar]
- Kisanuki YY, Hammer RE, Miyazaki J, Williams SC, Richardson JA, Yanagisawa M. 2001. Tie2-Cre transgenic mice: a new model for endothelial cell-lineage analysis in vivo. Developmental biology 230:230–242. [DOI] [PubMed] [Google Scholar]
- Klena NT, Gibbs BC, Lo CW. 2017. Cilia and Ciliopathies in Congenital Heart Disease. Cold Spring Harb Perspect Biol 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koefoed K, Veland IR, Pedersen LB, Larsen LA, Christensen ST. 2014. Cilia and coordination of signaling networks during heart development. Organogenesis 10:108–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, Woitge HW, Braut A, Kronenberg MS, Lichtler AC, Mina M, Kream BE. 2004. Expression and activity of osteoblast-targeted Cre recombinase transgenes in murine skeletal tissues. Int J Dev Biol 48:645–653. [DOI] [PubMed] [Google Scholar]
- May-Simera HL, Kelley MW. 2012. Cilia, Wnt signaling, and the cytoskeleton. Cilia 1:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muzumdar MD, Tasic B, Miyamichi K, Li L, Luo L. 2007. A global double-fluorescent Cre reporter mouse. Genesis 45:593–605. [DOI] [PubMed] [Google Scholar]
- Myers SM, Challman TD. 2004. Congenital heart defects associated with Smith-Magenis syndrome: two cases of total anomalous pulmonary venous return. Am J Med Genet A 131:99–100. [DOI] [PubMed] [Google Scholar]
- Nash D, Arrington CB, Kennedy BJ, Yandell M, Wu W, Zhang W, Ware S, Jorde LB, Gruber PJ, Yost HJ, Bowles NE, Bleyl SB. 2015. Shared Segment Analysis and Next-Generation Sequencing Implicates the Retinoic Acid Signaling Pathway in Total Anomalous Pulmonary Venous Return (TAPVR). PLoS One 10:e0131514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neville HL, Jaksic T, Wilson JM, Lally PA, Hardin WD Jr., Hirschl RB, Langham MR Jr., Lally KP, Congenital Diaphragmatic Hernia Study G. 2002. Fryns syndrome in children with congenital diaphragmatic hernia. J Pediatr Surg 37:1685–1687. [DOI] [PubMed] [Google Scholar]
- Park HK, Heinle JS, Morales DL. 2012. Williams syndrome and obstructed total anomalous pulmonary venous return: a previously unreported association. The Annals of thoracic surgery 94:289–291. [DOI] [PubMed] [Google Scholar]
- Plotnikova OV, Pugacheva EN, Golemis EA. 2009. Primary cilia and the cell cycle. Methods Cell Biol 94:137–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin J, Lin Y, Norman RX, Ko HW, Eggenschwiler JT. 2011. Intraflagellar transport protein 122 antagonizes Sonic Hedgehog signaling and controls ciliary localization of pathway components. Proceedings of the National Academy of Sciences of the United States of America 108:1456–1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohatgi R, Milenkovic L, Scott MP. 2007. Patched1 regulates hedgehog signaling at the primary cilium Science (New York, NY: 317:372–376. [DOI] [PubMed] [Google Scholar]
- Schneider L, Clement CA, Teilmann SC, Pazour GJ, Hoffmann EK, Satir P, Christensen ST. 2005. PDGFRalphaalpha signaling is regulated through the primary cilium in fibroblasts. Curr Biol 15:1861–1866. [DOI] [PubMed] [Google Scholar]
- Sizarov A, Anderson RH, Christoffels VM, Moorman AF. 2010. Three-dimensional and molecular analysis of the venous pole of the developing human heart. Circulation 122:798–807. [DOI] [PubMed] [Google Scholar]
- Slough J, Cooney L, Brueckner M. 2008. Monocilia in the embryonic mouse heart suggest a direct role for cilia in cardiac morphogenesis. Dev Dyn 237:2304–2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snarr BS, Kern CB, Wessels A. 2008. Origin and fate of cardiac mesenchyme. Dev Dyn 237:2804–2819. [DOI] [PubMed] [Google Scholar]
- Snarr BS, O’Neal JL, Chintalapudi MR, Wirrig EE, Phelps AL, Kubalak SW, Wessels A. 2007a. Isl1 expression at the venous pole identifies a novel role for the second heart field in cardiac development. Circulation research 101:971–974. [DOI] [PubMed] [Google Scholar]
- Snarr BS, Wirrig EE, Phelps AL, Trusk TC, Wessels A. 2007b. A spatiotemporal evaluation of the contribution of the dorsal mesenchymal protrusion to cardiac development. Developmental dynamics: an official publication of the American Association of Anatomists 236:1287–1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamm ER. 1998. Total anomalous pulmonary venous connection. In: Drose JA, editor. Fetal echocardiography: WB Saunders; p 215–225. [Google Scholar]
- Toomer KA, Fulmer D, Guo L, Drohan A, Peterson N, Swanson P, Brooks B, Mukherjee R, Body S, Lipschutz JH, Wessels A, Norris RA. 2017. A role for primary cilia in aortic valve development and disease. Dev Dyn 246:625–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Berg G, Moorman AF. 2011. Development of the pulmonary vein and the systemic venous sinus: an interactive 3D overview. PLoS One 6:e22055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verzi MP, McCulley DJ, De Val S, Dodou E, Black BL. 2005. The right ventricle, outflow tract, and ventricular septum comprise a restricted expression domain within the secondary/anterior heart field. Dev Biol 287:134–145. [DOI] [PubMed] [Google Scholar]
- Waller BR, 3rd, Wessels A 2000. Cardiac morphogenesis and dysmorphogenesis. An immunohistochemical approach Methods Mol Biol 135:151–161. [DOI] [PubMed] [Google Scholar]
- Wallingford JB, Mitchell B. 2011. Strange as it may seem: the many links between Wnt signaling, planar cell polarity, and cilia. Genes & development 25:201–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb S, Brown NA, Wessels A, Anderson RH. 1998. Development of the murine pulmonary vein and its relationship to the embryonic venous sinus. Anat Rec 250:325–334. [DOI] [PubMed] [Google Scholar]
- Wessels A, Anderson RH, Markwald RR, Webb S, Brown NA, Viragh S, Moorman AF, Lamers WH. 2000. Atrial development in the human heart: an immunohistochemical study with emphasis on the role of mesenchymal tissues. Anat Rec 259:288–300. [DOI] [PubMed] [Google Scholar]
- Wessels A, Markwald R. 2000. Cardiac morphogenesis and dysmorphogenesis. I. Normal development. Methods Mol Biol 136:239–259. [DOI] [PubMed] [Google Scholar]
- Willaredt MA, Gorgas K, Gardner HA, Tucker KL. 2012. Multiple essential roles for primary cilia in heart development. Cilia 1:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willaredt MA, Hasenpusch-Theil K, Gardner HA, Kitanovic I, Hirschfeld-Warneken VC, Gojak CP, Gorgas K, Bradford CL, Spatz J, Wolfl S, Theil T, Tucker KL. 2008. A crucial role for primary cilia in cortical morphogenesis. J Neurosci 28:12887–12900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirrig EE, Snarr BS, Chintalapudi MR, O’Neal JL, Phelps AL, Barth JL, Fresco VM, Kern CB, Mjaatvedt CH, Toole BP, Hoffman S, Trusk TC, Argraves WS, Wessels A. 2007. Cartilage link protein 1 (Crtl1), an extracellular matrix component playing an important role in heart development. Developmental biology 310:291–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu B, Wang Y, Lui W, Langworthy M, Tompkins KL, Hatzopoulos AK, Baldwin HS, Zhou B. 2011. Nfatc1 Coordinates Valve Endocardial Cell Lineage Development Required for Heart Valve Formation. Circ Res 109:183–192. [DOI] [PMC free article] [PubMed] [Google Scholar]