

ABSTRACT
Background
Shark new antigen receptor variable domain (VNAR) antibodies can bind restricted epitopes that may be inaccessible to conventional antibodies.
Methods
Here, we developed a library construction method based on polymerase chain reaction (PCR)-Extension Assembly and Self-Ligation (named “EASeL”) to construct a large VNAR antibody library with a size of 1.2 × 1010 from six naïve adult nurse sharks (Ginglymostoma cirratum).
Results
The next-generation sequencing analysis of 1.19 million full-length VNARs revealed that this library is highly diversified because it covers all four classical VNAR types (Types I–IV) including 11% of classical Type I and 57% of classical Type II. About 30% of the total VNARs could not be categorized as any of the classical types. The high variability of complementarity determining region (CDR) 3 length and cysteine numbers are important for the diversity of VNARs. To validate the use of the shark VNAR library for antibody discovery, we isolated a panel of VNAR phage binders to cancer therapy-related antigens, including glypican-3, human epidermal growth factor receptor 2 (HER2), and programmed cell death-1 (PD1). Additionally, we identified binders to viral antigens that included the Middle East respiratory syndrome (MERS) and severe acute respiratory syndrome (SARS) spike proteins. The isolated shark single-domain antibodies including Type I and Type II VNARs were produced in Escherichia coli and validated for their antigen binding. A Type II VNAR (PE38-B6) has a high affinity (Kd = 10.1 nM) for its antigen.
Conclusions
The naïve nurse shark VNAR library is a useful source for isolating single-domain antibodies to a wide range of antigens. The EASeL method may be applicable to the construction of other large diversity gene expression libraries.
Keywords: shark VNAR, phage display, single-domain antibody, gene library construction, next-generation sequencing
Statement of Significance
A method called “EASeL” for overlap extension PCR combined with self-ligation has been established for constructing a large phage-displayed VNAR single-domain antibody library from six nurse sharks. The shark single-domain library provides an alternative platform for selecting therapeutic antibodies for treating cancer and other human diseases.
INTRODUCTION
Classical immunoglobin G (IgG) is widely used in many biotechnologies and therapeutics [1]. IgG is best described as a heterodimeric homodimer, consisting of two copies of disulfide-bonded heavy (H) and light (L) chains. The H and L chain variable (V) domains (VH and VL, respectively) combine to form the antigen-binding region. When these two V domains are synthesized as a dual-domain single-chain V fragment (scFv), the minimum size of the fragment is 25–30 kDa. In recent years, single-domain immunoglobulins such as the shark VNAR (new antigen receptor variable domain) and the camelid heavy-chain variable domain (VHH) antibodies have been explored, both of which can be isolated as soluble, stable, monomeric binding domains [2–4]. These VNAR and VHH domain antibodies range from 12 to 15 kDa in size, roughly half the size of a scFv binding domain.
Cartilaginous fish (sharks, rays, skates, and chimaeras) are phylogenetically the oldest living organisms that use antibodies as part of their adaptive immune system [3, 4]. They produce three different antibody isotypes that function in their humoral immune responses, immunoglobulin M (IgM), immunoglobulin W (IgW), and immunoglobulin new antigen receptor (IgNAR) [3–6]. IgNAR antibodies are homodimeric proteins composed of heavy chains with an antigen-binding region at the end of each chain. They serve as a major component in humoral responses [4, 7]. A number of camelid VHH domains are being evaluated in phase I, II, and III clinical trials [8, 9]. Even though the shark VNAR is less known, it has the potential to be used in biological therapeutics based on (i) their small size and ability to penetrate dense tissues inaccessible to IgG [10], (ii) their ability to bind in protein clefts and buried functional sites (e.g. enzyme pocket sites for substrate) [11], (iii) their solubility and robustness in harsh conditions [12], and (iv) their ability for high-affinity (including sub-nanomolar) binding. Additionally, these antibodies have the potential to bind a wide range of antigens, despite the nature of their single-domain architecture [13].
The VNAR domain is an Ig superfamily domain with two β sheets held together by two canonical cysteine residues in framework regions (FRs) 1 and 3. In addition to these canonical cysteines, complementarity determining region 3 (CDR3) can have one or two additional cysteines forming additional disulfide bonds within the V domain. IgNARs are classified into four types based on the number and position of non-canonical cysteines in the VNAR domain [14]. Type I VNAR contains two non-canonical cysteine residues in CDR3 encoded by the diversity region or by N-nucleotide additions that form two disulfide bonds with FR2 and 4. Interestingly, Type I has only been reported in nurse sharks, Ginglymostoma cirratum [14]. Type II VNAR domains form disulfide bonds between one D-encoded non-canonical cysteine in CDR3 and another non-canonical cysteine in CDR1. Type III is similar to Type II except there is a highly conserved tryptophan residue in CDR1 positioned adjacent to the disulfide bond. Type IV has no non-canonical disulfide bonds as found in other three VNAR types. Both Type I and Type II VNARs have protruding CDR3s that enable binding to pockets and grooves [15, 16]. Classical IgG and camelid VHH contain a CDR2 loop that is not present in shark VNARs and are replaced with highly diverse amino acids, termed hypervariable region 2 (HV2) [11]. Additionally, there is a second hypervariable region, named HV4, which is inserted in the middle of FR3, therefore separating FR3 into FR3a and FR3b.
Shark VNAR domains may have advantageous properties over conventional IgG. First, sharks are evolutionarily distant from mammals on the phylogenetic tree, therefore can generate high-affinity binders to structurally conservative mammalian drug targets. These may include highly conserved heparan sulfate proteoglycans, G-protein coupled receptors, and ion channels that may exhibit poor immunogenicity in mice and rabbits [15, 17]. Second, the elongated CDR3 in shark and camel antibodies has the ability to seek out buried epitopes and enzyme functional sites [2, 15, 17, 18]. The shark IgNAR CDR3 regions are relatively longer (ranging from 9 to 34 a.a. and including various numbers of cysteine residues) compared to mouse or human counterparts. This can potentially lead to a larger diversity of structures that can interact with more diversified antigens [16]. Similarly, the longer CDR3 region in shark antibodies possesses the extraordinary capacity to form long finger-like extensions that can probe proteins for hidden epitopes [2]. Third, conventional antibodies may have poor tissue penetration ability due to their large size [19]. Whole IgG is 150 kDa and scFv fragments 25–30 kDa. VNAR domains can be as small as 12–15 kDa. Finally, shark VNAR domain antibodies have structural advantages and are easily expressed in Escherichia coli systems [2]. Sharks enrich their blood with urea to maintain osmotic balance in the marine environment, so shark antibody structure has evolved to become particularly stable [4].
Phage display technology has been used to isolate shark VNAR antibodies. In one study, two shark VNAR libraries with a size of 107 clones were constructed from both naïve spiny dogfish (Squalus acanthias) and smooth dogfish (Mustelus canis) sharks [20]. Phage antibodies were isolated from immunized sharks for specific antigens such as hen egg white lysozyme [21–24]. Synthetic shark VNAR single-domain libraries were also pursued [25–29]. However, none of these approaches has generated a large shark VNAR single-domain library that covers the wide range of diversity required (>1010) commonly for therapeutic antibody discovery. In this study, we developed a PCR-Extension Assembly and Self-Ligation-based method (named EASeL) to make a large phage-displayed VNAR single-domain library from six nurse sharks. Nurse sharks were chosen to maximize the diversity of the VNAR library because they exclusively have been reported to contain Type I VNAR domains. To assess its diversity and analyze the VNAR sequences on the largest scale in the field, we conducted next-generation sequencing (NGS) analysis on 1.19 million unique full-length VNARs. The unique sequences were then analyzed using unbiased methods to investigate their cysteine numbers and CDR3 lengths for all VNAR sequences together as well as Type I and Type II/III VNARs separately. To validate the potential of this library as a new platform for therapeutic antibody discovery, we conducted phage panning to identify shark VNAR binders to a panel of tumor and viral antigens. These included antigens associated with liver and breast cancers, as well as antigens against viral proteins associated with SARS and MERS. These selected binders including Type I and II VNARs were produced successfully in E. coli as soluble proteins for antigen-binding validation. This work validated the large diversity of the nurse shark VNAR library and the utility of the shark VNAR library as a platform for therapeutic antibody discovery.
RESULTS
Construction of a nurse shark VNAR phage library
To construct a large shark VNAR library, we developed a method called EASeL. As illustrated in Figure 1A, we isolated peripheral blood leukocytes from six adult nurse sharks (G. cirratum) and amplified shark VNAR sequences with PCR primers. The reverse primers contain a 19-nucleotide sequence on the pComb3x backbone. Extension overlap PCR was conducted to combine the VNAR and phagemid backbone (Fig. 1B). Finally, after gel purification we performed a self-ligation with T4 ligase to circularize the assembled VNAR pComb3x plasmids. Our shark VNAR library size (~1.2 × 1010) was determined by titration based on the number of individual TG1 bacteria colonies on agar plates. This high-efficiency method is a significant improvement over the conventional phage library construction method, which usually requires many rounds of restriction enzyme digestion and ligation, making the previous method labor intensive and time consuming. Moreover, the sequence diversity of the shark VNAR is preserved and represented in our method to a maximum degree due to highly efficient PCR-based amplification and ligation.
Figure 1.
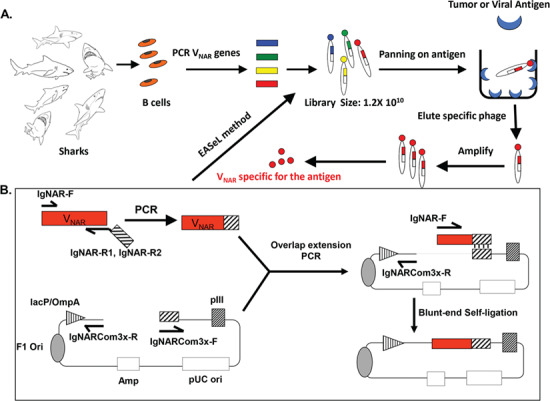
Strategy of the library construction and panning. (A) Flow of the project, starting from B-cell collection, PCR amplification of the VNAR sequences, assemblage of VNAR fragments to vector backbone, electroporation of TG1 cells to make the library (containing 1.2 × 1010 individual clones), and panning on plastic plates coated with different antigens. (B) Diagramed protocol for the assemblage of VNAR fragments with vector backbone by PCR-EASeL.
To analyze the diversity of the library, we performed deep sequencing of the whole library using NGS. Each sequencing was done twice in both forward and reverse direction for paired-end reads. The merging of paired-end reads was then performed with high stringency by combining the forward and reverse sequencing results to ensure accuracy for each unique sequence. Nearly 1.2 million full-length VNAR sequences with in-frame translation were used for further analysis with a focus on cysteine numbers, CDR3 length, amino acid variability, and VNAR type counts. This is the largest scale shark VNAR sequence analysis reported thus far. As shown in Figure 2A, the presence of two canonical cysteines located at both amino acid 21 and 82 are used as a key criterion to characterize Type I–IV VNARs. The sequences that do not contain one or both of these cysteines are considered as other types (n = 56 508; ~5% of the total VNARs) because they do not fit in with the four known VNAR type families. The sequences that have both 21C and 82C (n = 1 138 843) are further categorized based on their placement of additional cysteines. Type IV VNARs contain only two canonical cysteines found at position 21 and 82 (n = 19 494). Type I VNARs contain an extra cysteine at position 34 (n = 281 361; ~24% of the total VNARs). This group can be further divided into subtypes based on how many additional cysteines they contain. The classical definition of a Type I VNAR describes an antibody that has a total of six cysteines. About 11% of the total VNARs are classical Type I. Type II and III VNARs have at least one extra cysteine at amino acid 28 (n = 837 988; 70% of the total VNARs). Among them, ~57% of the total VNARs are classical Type II. Unique full-length VNARs are defined as having one differing amino acid in sequence. As shown in Figure 2B, 85% (1 022 715) of the 1.2 million sequences only appeared once in NGS results, and 8.5% of the 1.2 million sequences appeared twice. Only 1% of the 1.2 million sequences appeared more than 10 times. The most frequently repeated clone appeared 2832 times in NGS results. The percentage of the different types of VNAR is plotted in the pie chart in Figure 2C. Based on the number of extra cysteines in these sequences, the Type I (six cysteines in total) and Type II (four cysteines in total) VNARs are considered classical to others (other numbers of cysteines) as shown in Figure 2D and E, respectively. Representative sequences of Type I–IV shark VNARs with different numbers of cysteines were randomly picked from NGS data and shown here as examples (Fig. 3). The FRs and CDR1, CDR3, HV2, and HV4 are marked based on Stanfield et al. [11] and Fennell et al. [13] in Figure 3. These sequences were aligned to sequences on IMGT database for CDR determination. As shown in Figure 3, part of the HV2 sequence was identified as “CDR2” in the IMGT database.
Figure 2.
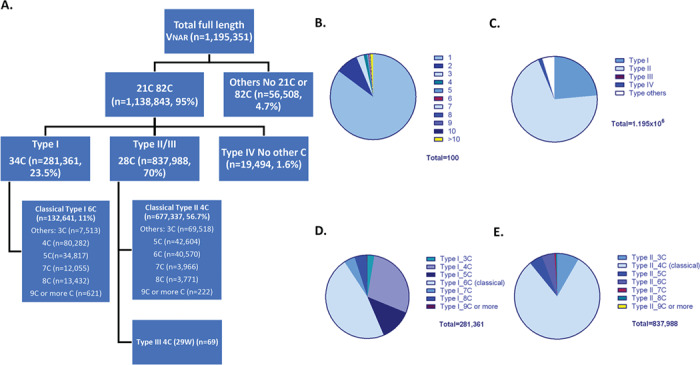
NGS analysis of the shark VNAR library. (A) Flow chart of the criteria used to categorize the VNAR types for 1.2 million validated full-length shark VNAR sequences. (B) Pie chart of the percentage of repeated sequences. Numbers represent the times each unique sequences are repeated in NGS results. (C) Pie chart of the percentage of four types of VNARs and other type of VNARs. (D) Pie chart of the percentage of classical Type I VNARs with six cysteines and the other non-canonical Type I VNARs. (E) Pie chart of the percentage of classical Type II VNARs with four cysteines and the other non-canonical Type II VNARs.
Figure 3.
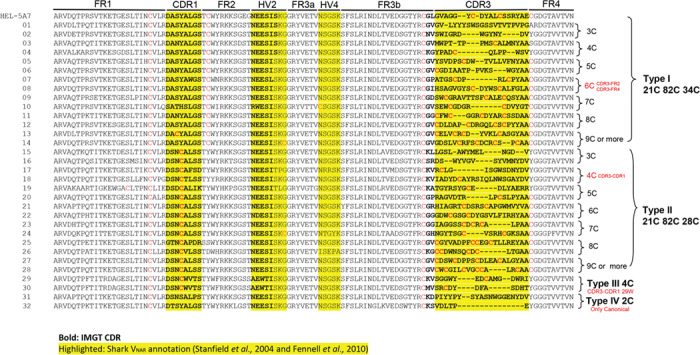
Sequence alignment of the randomly picked clones from the shark VNAR library that represent sequences have different numbers of cysteines. The bold areas represent CDRs determined by IMGT database. The areas highlighted by yellow are the CDRs determined by Stanfield et al. [11] and Fennell et al. [13].
These VNARs may have multiple cysteine residues in CDR3 based on the sequences from NGS data. We analyzed the total number of cysteines, the number of cysteines found in CDR3, the length of CDR3, and the amino acid sequence variability. Due to the high variability of CDR3 lengths, we defined CDR3 to be the sequence between conserved framework sequences TYRC (end of FR3b) and XXXGTXXTVN (FR4). The total cysteines in VNAR sequences can vary from 0 to 11 (Fig. 4A) and the CDR3 can have 0–6 cysteines (Fig. 4B). The CDR3 length varies greatly as well, and it can be between 0–40 amino acids according to the NGS data (Fig. 3C). The separate analysis of Type I and Type II/III VNARs showed Type I VNARs (shown as blue lines) have more total cysteines and in CDR3 than Type II/III (shown as red lines) (Fig. 4A and B). The CDR3 lengths for Type I VNARs are also slightly longer compared to Type II/III (Fig. 4C). These findings are consistent with published small-scale sequence analysis: Type I VNARs have more even numbers of cysteines in CDR3 (0, 2, or 4) (Fig. 4B) [7, 16]. The high variability of CDR3 length and cysteine numbers are crucial to the diversity of VNARs since binding diversity is dependent on the CDR3 structure diversity. Amino acid sequence variation analysis showed the sequence diversity is mainly contributed to CDR3 with minimal variation in CDR1, HV2, and HV4 (Fig. 5). Taken together, we designed a PCR-based method to establish a large shark VNAR single-domain antibody library with the size of 1010. The library contains all types (I–IV) of shark VNAR sequences as well as many other previously undefined types.
Figure 4.
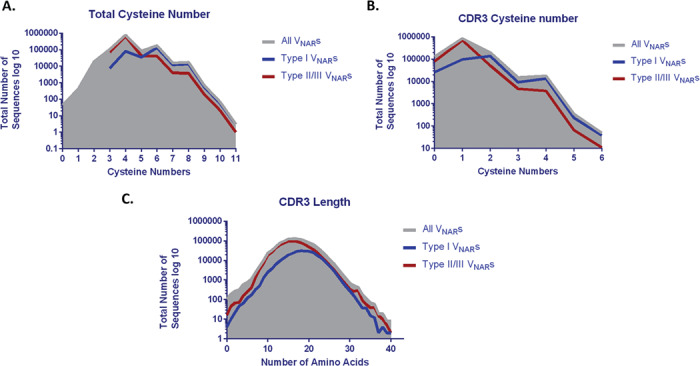
Sequence analysis of the unique full-length VNARs. (A) Distribution plots of total cysteine numbers in VNAR full-length sequences. (B) Distribution plots of the cysteine numbers in CDR3. (C) Distribution plots of CDR3 length.
Figure 5.
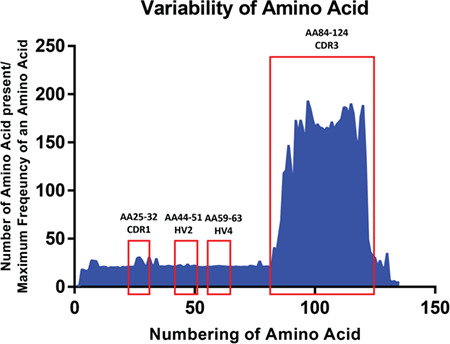
Amino acid sequence variability analysis for full-length VNAR sequences.
Isolation of VNAR binders to various tumor and viral targets
To evaluate the library’s potential for therapeutic development, we chose a variety of human tumor biomarkers and virus antigen proteins as selection targets. These include glypican-3 (GPC3), HER2 and PD1, the spike proteins of the MERS and SARS viruses, and Pseudomonas exotoxin (PE38). After four rounds of panning, specific binders to the listed targets were identified by monoclonal phage ELISA (Fig. 6A). The binders were assigned the following names based on their targets and well numbers, which include GPC3-F1, HER2-A6, PD1-A1, MERS-A3, MERS-A7, MERS-A8, MERS-B4, MERS-B5, SARS-01, and PE38-B6. Only one binder was isolated for all antigens, except MERS spike protein. Five binders were isolated for MERS spike protein. Sequence analysis showed that most of these binders were Type II VNAR except for one Type I and two undefined types. One Type II binder (PE38-B6) targeting PE38 fragment was produced in E. coli HB2151 strain as a single-domain protein. It had 10.1 nM Kd binding affinity for its antigen as measured by Octet kinetic assay (Fig. 6B and C). The affinity is high as a monomeric single-domain soluble protein isolated from a naïve shark library without immunization. We also produced the Type I binder (MERS A8) and Type II binder MERS A7 in E. coli. The protein yield varied based on protein sequences. PE38-B6 has a relatively low yield (3 mg from 2 L E. coli culture). MERS A8 binder yielded 3.1 mg and MERS A7 yielded 8.7 mg of single-domain soluble protein from 600 ml E. coli culture. Nickel-charged HisTrap columns (GE Healthcare, Chicago, IL) were used to purify these proteins from the polymyxin B lysed bacteria pellet supernatants. The elution profile showed the protein elution in Figure 7A and D. sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) of the elution peaks in Figure 7B and E showed the later peak eluted by higher imidazole concentrations had over 90% purity of the target soluble VNARs. Both MERS A8 and A7 had specific binding to the phage panning antigen MERS spike protein in Figure 7C and F. The Type I binder (MERS A8) had higher signals. TSK size exclusion column purification for the Type II binder PE38-B6-his soluble single-domain protein showed this protein is monomeric. The 16.8 kDa protein was eluted with one peak that is slightly earlier than 13.7 kDa control protein peak (Fig. 7G and H). Taken together, binders to tumor and viral antigens, including Type I and Type II single domains containing multiple cysteine residues, can be selected from the library and produced as functional single-domain proteins in E. coli. Our data indicate that the new shark phage library is a valuable platform for discovery of VNAR binders.
Figure 6.
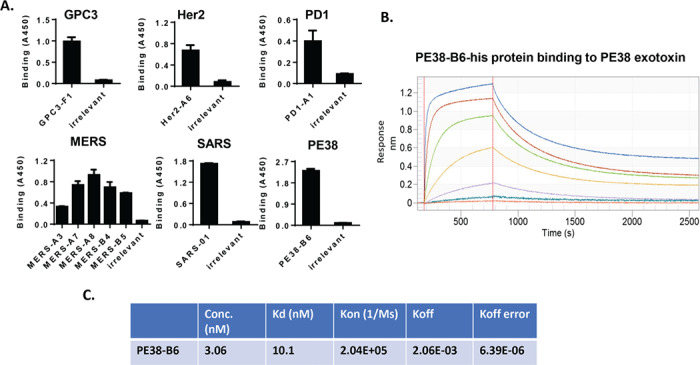
Identification of the binders by phage ELISA. (A) Monoclonal phage ELISA was carried out to identify the binders to GPC3, Her2, PD1, MERS S-protein, SARS S-protein, PE38, and hFc. A random phage that had no binding to all tested antigens was used as irrelevant control in the phage ELISA. (B) Octet kinetic assay for PE38-B6-his single-domain soluble protein. Seven concentrations of PE38-B6-his protein used from high to low are 780 nM, 195 nM, 48.8 nM, 12.2 nM, 3.06 nM, 0.76 nM, and 0.19 nM. (C) The representative data were shown for PE38-B6 concentration 3.06 nM. The Kd, kon, koff, and koff standard error was summarized in this table.
Figure 7.
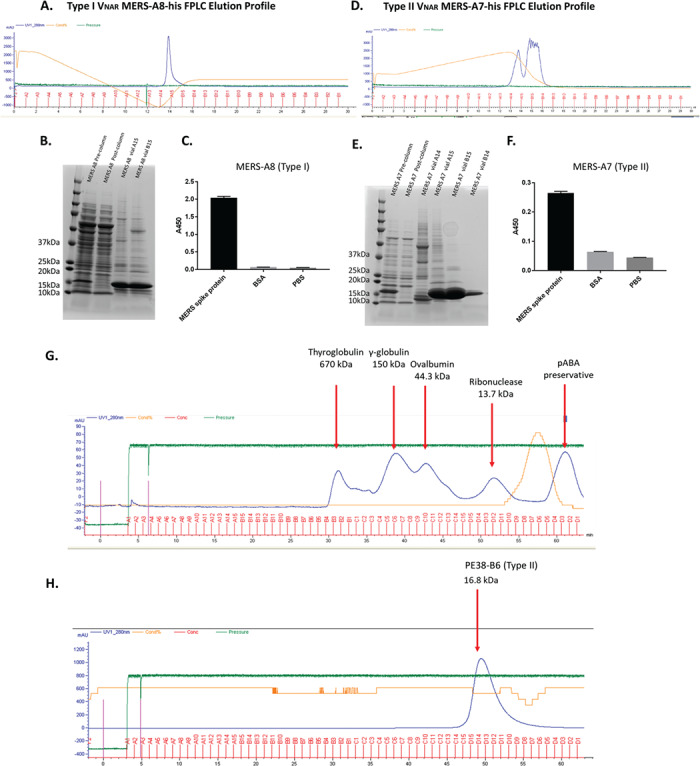
Soluble VNAR binder protein production and purification. (A) Type I VNAR MERS A8 AKTA His-Trap column elution profile. (B) SDS-PAGE of the MERS A8 eluted protein peaks and the pre- and post-column supernatant controls. (C) Soluble protein ELISA for MERS A8-his protein binding to MERS spike protein. (D) Type II VNAR MERS A7 AKTA His-Trap column elution profile. (E) SDS-PAGE of the MERS A7 eluted protein peaks and the pre- and post-column supernatant controls. (F) Soluble protein ELISA for MERS A7-his protein binding to MERS spike protein. (G) TSK size exclusion column purification of a mixture of protein standards containing four control proteins of different sizes. (H) TSK size exclusion column purification of Type II shark binder PE38-B6-his soluble protein.
Discussion
In the present study, we developed the EASeL method to construct phage display libraries based on PCR extension assembly followed by self-ligation. By using this EASeL method, we successfully constructed a large phage-displayed VNAR library with a size of 1010 from six antigen-naïve nurse sharks. The NGS analysis data show that our phage display library has a large diversity. Finally, using a panel of tumor and viral antigen proteins as targets, we isolated shark binders to all of the antigens screened. We also demonstrated that Type I and Type II shark VNAR binders can be produced from E. coli as functional single-domain proteins. The large nurse shark VNAR single-domain phage library described here provides an attractive single-domain antibody platform for drug discovery.
In this study, we use two canonical cysteines located at both amino acid 21 and 82 as a key criterion to characterize Type I–IV VNAR families. About 5% of the total VNARs do not fit in any one of the four known VNAR type families. We have identified ~11% of the total VNARs are classical Type I and 57% classical Type II. While a majority of the VNARs are either classical Type I or Type II, ~30% of the VNARs do not fit any of the four classical types. Most of these non-classical VNARs have various numbers of cysteines (Fig. 2). Future structural and functional analysis of the binders isolated from the library for various antigens will be needed to understand the role of these non-classical VNARs in therapeutic antibody discovery and engineering.
The key for a successful isolation of antibodies is the phage library used for the selection [30]. Various methods have been employed to make antibody phage display libraries. Most widely used methods consist of PCR amplification of antibody fragments, followed by enzymatic digestion and ligation with the vector. These methods are time consuming and have a low efficiency in the ligation and transformation step. In the present study, we developed the EASeL method to construct a large shark VNAR phage library. We used a labeling PCR to add homologue regions to the VNAR pool. Then an overlap extension PCR was used to assemble VNAR and phagemid DNA, followed by self-ligation of the whole library DNA. It only took several weeks to make the shark VNAR library while conventional approaches would require several months. Our protocol has significantly shortened the time required to construct a large gene library. Another contemporary method used for gene cloning is “Gibson Assembly”. This method is based on enzymatic assembly for joining multiple overlapping DNA fragments into a single reaction system [31]. Our method may be more efficient than Gibson Assembly. Gibson Assembly requires 5-exonuclease to nick the 5-terminals of the DNA fragments to make them complementary followed by annealing together. DNA polymerase was then used to fill the gap, and DNA ligase was used to seal the gap. Therefore, the two termini that are being ligated have to be identical for each other for ~20 nucleotides. The Gibson Assembly reaction may fail due to complications related to total repeat density (direct, inverse, and palindromic repeat elements), extremes in G/C content, and secondary structures near the 3’ and 5’ termini of the sequence. To our best knowledge, there is no report using the Gibson Assembly for the development of a large phage-displayed antibody library. In our method, we only need the conventional T4 DNA ligase to run a standard ligation at 16°C for 12–24 h. The two ends of the DNA are blunt and do not need to be identical for blunt-end ligation. Therefore, our method is similar to traditional blunt-end ligation, and the terminal part of the DNA fragment sequence can be varied. During the preparation of this manuscript, our laboratory has applied the EASeL method to successfully make 20 large camel VHH single-domain phage libraries. Taken together, the new EASeL library construction method is robust, comprehensive, and quick.
The new EASeL method also maximized the sequence diversity of CDR3 represented in the phage library to increase the selection of high-affinity binders. A previous study produced a shark phage library with 107 diversity using the conventional cloning method. Our library has 1010 diversity and contains all known four types of VNAR single domains. The majority of the sequences are Type II and Type I VNAR as shown in Figure 2C. Interestingly, Type IV VNAR sequences are significantly under-represented as shown in Figure 2C. The shark VNAR sequences in the NCBI database are mostly derived from bamboo shark, dogfish shark, wobbegong shark, and other types of smaller sharks [12–17]. The percentage of VNAR types can be significantly different between various species of sharks. Interestingly, our deep sequencing analysis showed that 56 174 of the nurse shark VNAR sequences in our library were not categorized in any one of the known VNAR types (Types I–IV). Whether these novel types of VNARs possess unique confirmations and biophysical properties should be analyzed structurally and functionally when binders for cancer or viral antigens are found from these novel types of VNARs in the future. Since the method we used to assemble the unique VNAR sequences for analysis is highly accurate and the number of the unique sequences analyzed is large, we used unbiased methods to analyze all the sequences for cysteine number and CDR3 length without further subdividing them into the four known VNAR types. A significant percentage of the sequences are not classical VNAR as defined in the known four types. Analyzing the data sequences altogether would provide more comprehensive picture of the sequence patterns in the naïve nurse shark without having to fit the sequences into a defined VNAR type. The extra cysteines in both Type I and Type II VNARs are important for stabilizing the antigen-binding regions [11]. Extra disulfide bonds formed by these cysteines are essential for forming diverse antigen-binding surfaces. It may be one strategy for increasing antigen-binding region diversity in heavy chain-only antibodies.
Interestingly, the sequence variation mostly lies in CDR3 with minimal variation in CDR1, HV2, and HV4. Previous literature suggested there are sequence variability in HV2 and HV4 during in vivo affinity maturation [24]. It was also shown that somatic mutations within HV2/4 can contribute to antigen binding [21]. However, NGS results in the present study showed there is minimal sequence variability in CDR1, HV2, and HV4 in our naïve shark library. It is possible that these naïve sharks used in our study have been kept in captivity for a long time, therefore do not have as much sequences diversity in CDR1, HV2, and HV4. Another major type of heavy chain-only domain antibody VHH from camels also has highly diversified CDR3. The lengths of VHH CDR3 are much longer than conventional VH from camel IgG. The median CDR3 length of VHH is 20 amino acids (similar with shark VNAR), whereas median length for camel VH CDR3 is only 15 amino acids [32]. However, there is no comprehensive study on the cysteine numbers and locations in camel VHH sequences for comparison with shark VNAR.
We hypothesize that the diversity of this large naïve shark library can be further increased significantly by randomizing the CDR1 sequences and keeping the highly diverse CDR3 [33, 34]. For example, the eight amino acids in CDR1 can be completely randomized to further diversify our VNAR library. Furthermore, affinity maturation of the binders in Figure 6 can be performed by randomizing CDR1. In summary, we believe this shark library has a suitable size and diversity for antibody discovery and can potentially be used as a large single-domain antibody discovery platform.
The potential immunogenicity of shark antibodies could be a concern in clinical applications. These concerns could be addressed by identifying the B-cell and T-cell immunogenic epitopes and silence them by specific site mutations [35, 36]. Furthermore, durable immune tolerance to highly immunogenic proteins (e.g. bacterial toxins) can be induced by nanoparticles containing rapamycin in vivo [37].
In conclusion, we have built a large, highly diverse phage-displayed VNAR library using B cells isolated from nurse sharks. It provides a new platform to discover single-domain antibodies for therapeutic and diagnostic applications. The EASeL library construction method described here may be applicable to the construction of other large gene expression libraries including antibody libraries derived from other species and T-cell receptors.
MATERIALS AND METHODS
Cell lines
The GPC3-positive human hepatocellular carcinoma cell line HepG2 was maintained as adherent monolayer cultures in DMEM medium (Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum (HyClone, Logan, UT), 1% L-glutamine, and 1% penicillin-streptomycin (Invitrogen) in a 5% CO2 incubator at 37°C. GPC3-negative A431 cells (Human epithelial carcinoma cell line) were engineered in our laboratory to express high levels of human GPC3 by transfection with a plasmid encoding full-length GPC3 [38, 39]. Both A431 and the stably transfected cells (G1) [39] were maintained in DMEM with supplements.
Protein reagents
The GPC3 peptide (a.a. 510–560) was synthesized. The recombinant extracellular domain of Her2 and PD1, the S-protein of MERS and SRS were purchased from Sino Biological (Beijing, China). The recombinant PE38 and mPE24 were made in our laboratory according to the published methods [40]. The recombinant GPC3-hFc was generated as previously published [41].
Construction of a nurse shark VNAR phage library
Six naïve nurse sharks (three males and three females, ranging from 2.5 to 6 ft long) were bled for 10 ml of blood in phosphate buffered saline (PBS)/1000 IU/ml heparin. The buffy coat was collected, spun, and resuspended in 3.5 ml of Trizol for RNA preparation. Total RNA was isolated using the TRIzol reagent (Thermo Fisher Scientific, Grand Island, NY) according to the manufacturer’s instruction. Five micrograms of total RNA were reverse-transcribed into cDNA in a total of 20 μl volume using the SuperScript III First-Strand Synthesis System (Thermo Fisher Scientific) according to the manufacturer’s instruction. One forward primer and two reverse primers were synthesized to PCR amplify the VNAR sequence from the cDNA product. Forward primer IgNAR-F: GCTCGAGTGACCAAACACCG, reverse primer IgNAR-R1: GGTGGCCGGCCTGGCCACTATTCACAGTCACGGCAGTGCCAT, reverse primer IgNAR-R2: GGTGGCCGGCCTGGCCACTATTCACAGTCACGACAGTGCCACC. Primer pairs of IgNAR-F/IgNAR-R1, IgNAR-F/IgNAR-R1 were used to amplify the VNAR fragment in a 50 μl of PCR volume that contains 1 μl of cDNA product. The PCR cycling parameters were the following: initial denaturation at 94°C for 3 min, 40 cycles of denaturation at 98°C for 10 s, annealing at 60°C for 15 s, and elongation at 72°C for 45 s using PrimeStar (CloneTech). In the meantime, the linear vector backbone fragment was prepared by PCR using forward primer IgNARCom3x-F: AGTGGCCAGGCCGGCCACC, and reverse primer IgNARCom3x-R: GGCCGCCTGGGCCACGGTA. Five nanograms of the plasmid pComb3X was used as the template in a total of 50 μl of PCR reaction volume. The primers to amplify the vector backbone were forward IgNARCom3x-F: AGTGGCCAGGCCGGCCACC and reverse IgNARCom3x-R: GGCCGCCTGGGCCACGGTA. The PCR cycling parameters were the following: initial denaturation at 94°C for 3 min, then 25 cycles of denaturation at 98°C for 10 s, annealing at 60°C for 15 s, and elongation at 72°C for 3 min using PrimeStar (Takara, Shiga, Japan). To assemble the VNAR and the amplified vector backbone, 100 ng of vector backbone was mixed with 30 ng of VNAR PCR products in a 50 μl of PCR reaction volume, the overlapping extension PCR was primed by primers of IgNAR-F/IgNARCom3x-R using PrimeStar. Twenty micrograms of the assembled PCR product were circularized by intra-molecular self-ligation in a 1 ml of ligation buffer using T4 DNA ligase (New England Biolabs, Ipswich, MA). The ligation products were cleaned up by removing the enzymes and transformed into 500 μl of electroporation competent TG1 cells (Lucigen, Middleton, WI) to make the library. This method is referred to as EASeL method in this study.
NGS and library composition analysis of the shark library
NGS of the library was generated from shark VNAR insert DNA digested out of plasmid library using XhoI and MscI restriction enzymes (New England Biolabs). The shark VNAR library was excised out from the vector with restriction enzyme (XhoI and MscI) and gel purified. The insert fragments were ligated to Illumina adaptor and sequenced on Illumina MiSeq with 2 × 250 bp paired-end reads using both NEBNext Ultra II DNA library preparation kit and Kappa Hyper Prep library preparation kit. Paired-end reads were merged to cover the full length of the insert using FLASH [42]. Merged sequence reads were then analyzed with custom Perl Scripts. All DNA sequences were oriented and translated to protein sequences. Sequences with stop codons were removed from further analysis. We also required the amino acid sequences to start with “RV” at the N-terminus and end with “XXXGTXXTVNS” at the C-terminus to be considered as full-length VNARs. After the selection with these criteria, there are almost 2 million VNAR amino acid sequences from this experiment, with more than 1 million unique VNAR sequences. We aligned all VNAR sequences by anchoring the constant regions and allow CDR3 regions to have variable lengths from 0 to 40 amino acids. We then calculated the variability according to methods described in Wu and Kabat [43]. We further classified VNAR sequences into different subtypes based on residue numbers and positions. Type I and Type II/III sequences were also analyzed separately for the CDR3 lengths, total cysteine numbers, and CDR3 cysteine numbers.
Phage display and panning method
Library TG1 bacterial stock was inoculated into 2.5 L of 2YT media containing 2% glucose, 100 μg/ml ampicillin, and cultured at 37°C with shaking (250 rpm). When the cells reached mid-log phase (OD600 between 0.4–0.8), super-infection was performed by adding helper phage M13KO7 at 5 × 109 pfu/ml. After 1 h of continued growth, the TG1 cells were pelleted and resuspended in 2.5 L of 2YT media containing 100 μg/ml ampicillin and 50 μg/ml kanamycin, and incubated at 25°C overnight. After the cells were centrifuged and filtered with a 0.22 μm membrane, the supernatant was stored at 4°C for panning.
The phage panning protocol has been described previously [44, 45]. Briefly, a 96 well Maxisorp ELISA plate (Nunc/Thermo Fisher Scientific, Rochester, NY) was used to capture various antigens (100 μg/ml) in PBS buffer at 4°C overnight. After the coating buffer was decanted, the plate was treated with blocking buffer (2% bovine serum albumin in PBS) at room temperature for 1 h. Then 30 μl pre-blocked phage supernatant (typically contained 1010–1011 cfu) in 30 μl blocking buffer was added per well for 1 h at room temperature to allow binding. After four washes with PBS containing 0.05% Tween-20, bound phages were eluted with 100 mM triethylamine. After four rounds of panning, single colonies were picked and identified by using phage ELISA.
Phage ELISA
The antigenic proteins were used to coat a 96 well plate at 5 μg/ml in PBS buffer, 50 μl/well, at 4°C overnight. The irrelevant antigen used was 5 μg/ml BSA in PBS. After the plate was blocked with 2% BSA in PBS buffer, 25 μl pre-blocked phage supernatant (typically 1010–1011 cfu) were added to the plate. Binding was detected by HRP conjugated mouse anti-M13 antibody (GE Healthcare Life Sciences, Pittsburg, PA). The cut-off value for positive binder was set as 3× higher signal of antigen binding compared to background noise.
Soluble protein production and purification
The pComb3x phagemids containing the VNAR binders were transformed into HB2151 E. coli cells. The formed colonies were pooled for culture in 2 L 2YT media containing 2% glucose, 100 μg/ml ampicillin at 37°C until the OD600 reaches 0.8–1. Culture media was then replaced with 2YT media containing 1 mM IPTG (Sigma), 100 μg/ml ampicillin, and shook at 30°C overnight for soluble protein production. Bacteria pellet was spun down and lysed with polymyxin B (Sigma) for 1 h at 37°C to release the soluble protein. The supernatant was harvested after lysis and purified using HisTrap column (GE Healthcare) using AKTA.
Biophysical analysis
The shark single-domain soluble protein PE38-B6-His was buffer exchanged in PBS buffer after purification. The binding kinetics of PE38-B6-His was measured with ForteBio Octet RED96 located at the Biophysics Core in National Heart, Lung and Blood Institute. PE38-B6-His diluted in assay buffer PBS supplemented with 0.1% Tween-20 and 1% (w/v) BSA. The Octet RED96 program was as follows: 10 min presoak, 120 s baseline establishment, 300 s antigen loading, 60 s baseline re-establishment after antigen loading, 600 s PE38-B6-His association, 30 min dissociation. A total of 1 μg/ml PE38-B6-his was used to load the Ni-NTA biosensor, and serial diluted antigen protein PE38 was used for binding assay. The binding kinetics was calculated with forteBio Octet RED96 software.
Statistical analysis
All statistical analyses were conducted using GraphPad Prism 5 (GraphPad Software, Inc., La Jolla, CA). Differences between groups were analyzed using the two-tailed Student’s t-test of means.
FUNDING
National Cancer Institute Center for Cancer Research FLEX Program Technology Development Award [to M.H.]. Intramural Research Program of the National Institutes of Health, National Cancer Institute, Center for Cancer Research (Z01 BC 010891 and ZIA BC 010891 to M.H.). National Cancer Institute, National Institutes of Health, under Contract No. HHSN261200800001E (to X.W.).
ACKNOWLEDGMENTS
We thank Dr Gregory Piszczek from Biophysics Core Facility in National Heart, Lung, and Blood institute for Octet assay technical support.
REFERENCES
- 1. Weiner, LM, Murray, JC, Shuptrine, CW. Antibody-based immunotherapy of cancer. Cell 2012; 148: 1081–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wesolowski, J, Alzogaray, V, Reyelt, J et al. Single domain antibodies: promising experimental and therapeutic tools in infection and immunity. Med Microbiol Immunol 2009; 198: 157–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Dooley, H, Flajnik, MF. Antibody repertoire development in cartilaginous fish. Dev Comp Immunol 2006; 30: 43–56. [DOI] [PubMed] [Google Scholar]
- 4. Feige, MJ, Grawert, MA, Marcinowski, M et al. The structural analysis of shark IgNAR antibodies reveals evolutionary principles of immunoglobulins. Proc Natl Acad Sci U S A 2014; 111: 8155–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rumfelt, LL, Lohr, RL, Dooley, H et al. Diversity and repertoire of IgW and IgM VH families in the newborn nurse shark. BMC Immunol 2004; 5: 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Rumfelt, LL, Diaz, M, Lohr, RL et al. Unprecedented multiplicity of Ig transmembrane and secretory mRNA forms in the cartilaginous fish. J Immunol 2004; 173: 1129–39. [DOI] [PubMed] [Google Scholar]
- 7. Roux, KH, Greenberg, AS, Greene, L et al. Structural analysis of the nurse shark (new) antigen receptor (NAR): molecular convergence of NAR and unusual mammalian immunoglobulins. Proc Natl Acad Sci U S A 1998; 95: 11804–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Van Bockstaele, F, Holz, JB, Revets, H. The development of nanobodies for therapeutic applications. Curr Opin Investig Drugs 2009; 10: 1212–24. [PubMed] [Google Scholar]
- 9. Van Audenhove, I, Gettemans, J. Nanobodies as versatile tools to understand, diagnose, visualize and treat cancer. EBioMedicine 2016; 8: 40–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Irving, RA, Coia, G, Roberts, A et al. Ribosome display and affinity maturation: from antibodies to single V-domains and steps towards cancer therapeutics. J Immunol Methods 2001; 248: 31–45. [DOI] [PubMed] [Google Scholar]
- 11. Stanfield, RL, Dooley, H, Flajnik, MF et al. Crystal structure of a shark single-domain antibody V region in complex with lysozyme. Science 2004; 305: 1770–3. [DOI] [PubMed] [Google Scholar]
- 12. Simmons, DP, Abregu, FA, Krishnan, UV et al. Dimerisation strategies for shark IgNAR single domain antibody fragments. J Immunol Methods 2006; 315: 171–84. [DOI] [PubMed] [Google Scholar]
- 13. Fennell, BJ, Darmanin-Sheehan, A, Hufton, SE et al. Dissection of the IgNAR V domain: molecular scanning and orthologue database mining define novel IgNAR hallmarks and affinity maturation mechanisms. J Mol Biol 2010; 400: 155–70. [DOI] [PubMed] [Google Scholar]
- 14. Zielonka, S, Empting, M, Grzeschik, J et al. Structural insights and biomedical potential of IgNAR scaffolds from sharks. MAbs 2015; 7: 15–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Barelle, C, Gill, DS, Charlton, K. Shark novel antigen receptors––the next generation of biologic therapeutics? Adv Exp Med Biol 2009; 655: 49–62. [DOI] [PubMed] [Google Scholar]
- 16. Diaz, M, Stanfield, RL, Greenberg, AS et al. Structural analysis, selection, and ontogeny of the shark new antigen receptor (IgNAR): identification of a new locus preferentially expressed in early development. Immunogenetics 2002; 54: 501–12. [DOI] [PubMed] [Google Scholar]
- 17. Nuttall, SD, Walsh, RB. Display scaffolds: protein engineering for novel therapeutics. Curr Opin Pharmacol 2008; 8: 609–15. [DOI] [PubMed] [Google Scholar]
- 18. Ho, M. Inaugural editorial: searching for magic bullets. Antib Ther 2018; 1: 1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mordenti, J, Cuthbertson, RA, Ferrara, N et al. Comparisons of the intraocular tissue distribution, pharmacokinetics, and safety of 125I-labeled full-length and Fab antibodies in rhesus monkeys following intravitreal administration. Toxicol Pathol 1999; 27: 536–44. [DOI] [PubMed] [Google Scholar]
- 20. Liu, JL, Anderson, GP, Delehanty, JB et al. Selection of cholera toxin specific IgNAR single-domain antibodies from a naive shark library. Mol Immunol 2007; 44: 1775–83. [DOI] [PubMed] [Google Scholar]
- 21. Stanfield, RL, Dooley, H, Verdino, P et al. Maturation of shark single-domain (IgNAR) antibodies: evidence for induced-fit binding. J Mol Biol 2007; 367: 358–72. [DOI] [PubMed] [Google Scholar]
- 22. Dooley, H, Flajnik, MF, Porter, AJ. Selection and characterization of naturally occurring single-domain (IgNAR) antibody fragments from immunized sharks by phage display. Mol Immunol 2003; 40: 25–33. [DOI] [PubMed] [Google Scholar]
- 23. Dooley, H, Flajnik, MF. Shark immunity bites back: affinity maturation and memory response in the nurse shark, Ginglymostoma cirratum. Eur J Immunol 2005; 35: 936–45. [DOI] [PubMed] [Google Scholar]
- 24. Dooley, H, Stanfield, RL, Brady, RA et al. First molecular and biochemical analysis of in vivo affinity maturation in an ectothermic vertebrate. Proc Natl Acad Sci U S A 2006; 103: 1846–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Shao, CY, Secombes, CJ, Porter, AJ. Rapid isolation of IgNAR variable single-domain antibody fragments from a shark synthetic library. Mol Immunol 2007; 44: 656–65. [DOI] [PubMed] [Google Scholar]
- 26. Liu, JL, Anderson, GP, Goldman, ER. Isolation of anti-toxin single domain antibodies from a semi-synthetic spiny dogfish shark display library. BMC Biotechnol 2007; 7: 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Nuttall, SD, Krishnan, UV, Hattarki, M et al. Isolation of the new antigen receptor from wobbegong sharks, and use as a scaffold for the display of protein loop libraries. Mol Immunol 2001; 38: 313–26. [DOI] [PubMed] [Google Scholar]
- 28. Nuttall, SD, Krishnan, UV, Doughty, L et al. Isolation and characterization of an IgNAR variable domain specific for the human mitochondrial translocase receptor Tom70. Eur J Biochem 2003; 270: 3543–54. [DOI] [PubMed] [Google Scholar]
- 29. Nuttall, SD, Humberstone, KS, Krishnan, UV et al. Selection and affinity maturation of IgNAR variable domains targeting Plasmodium falciparum AMA1. Proteins 2004; 55: 187–97. [DOI] [PubMed] [Google Scholar]
- 30. Hust, M, Frenzel, A, Meyer, T et al. Construction of human naive antibody gene libraries. Methods Mol Biol 2012; 907: 85–107. [DOI] [PubMed] [Google Scholar]
- 31. Gibson, DG, Young, L, Chuang, RY et al. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods 2009; 6: 343–5. [DOI] [PubMed] [Google Scholar]
- 32. Li, X, Duan, X, Yang, K et al. Comparative analysis of immune repertoires between Bactrian camel’s conventional and heavy-chain antibodies. PLoS One 2016; 11: e0161801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Grzeschik, J, Konning, D, Hinz, SC et al. Generation of semi-synthetic shark IgNAR single-domain antibody libraries. Methods Mol Biol 2018; 1701: 147–67. [DOI] [PubMed] [Google Scholar]
- 34. Konning, D, Hinz, S, Grzeschik, J et al. Construction of histidine-enriched shark IgNAR variable domain antibody libraries for the isolation of pH-sensitive vNAR fragments. Methods Mol Biol 2018; 1827: 109–27. [DOI] [PubMed] [Google Scholar]
- 35. Mazor, R, Zhang, J, Xiang, L et al. Recombinant immunotoxin with T-cell epitope mutations that greatly reduce immunogenicity for treatment of mesothelin-expressing tumors. Mol Cancer Ther 2015; 14: 2789–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Weldon, JE, Xiang, L, Zhang, J et al. A recombinant immunotoxin against the tumor-associated antigen mesothelin reengineered for high activity, low off-target toxicity, and reduced antigenicity. Mol Cancer Ther 2013; 12: 48–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mazor, R, King, EM, Onda, M et al. Tolerogenic nanoparticles restore the antitumor activity of recombinant immunotoxins by mitigating immunogenicity. Proc Natl Acad Sci U S A 2018; 115: E733–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gao, W, Kim, H, Feng, M et al. Inactivation of Wnt signaling by a human antibody that recognizes the heparan sulfate chains of glypican-3 for liver cancer therapy. Hepatology 2014; 60: 576–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Phung, Y, Gao, W, Man, Y-G et al. High-affinity monoclonal antibodies to cell surface tumor antigen glypican-3 generated through a combination of peptide immunization and flow cytometry screening. MAbs 2012; 4: 592–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gao, W, Tang, Z, Zhang, YF et al. Immunotoxin targeting glypican-3 regresses liver cancer via dual inhibition of Wnt signalling and protein synthesis. Nat Commun 2015; 6: 6536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Feng, M, Gao, W, Wang, R et al. Therapeutically targeting glypican-3 via a conformation-specific single-domain antibody in hepatocellular carcinoma. Proc Natl Acad Sci U S A 2013; 110: E1083–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Magoč, T, Salzberg, SL. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011; 27: 2957–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wu, TT, Kabat, EA. An analysis of the sequences of the variable regions of Bence Jones proteins and myeloma light chains and their implications for antibody complementarity. J Exp Med 1970; 132: 211–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ho, M, Pastan, I. In vitro antibody affinity maturation targeting germline hotspots. Methods Mol Biol 2009; 525: 293–308 xiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ho, M, Kreitman, RJ, Onda, M et al. In vitro antibody evolution targeting germline hot spots to increase activity of an anti-CD22 immunotoxin. J Biol Chem 2005; 280: 607–17. [DOI] [PubMed] [Google Scholar]