

Abstract
Alcoholic liver disease (ALD) is characterized by macrophage and neutrophil leukocyte recruitment and activation in the liver. Damage‐ and pathogen‐associated molecular patterns contribute to a self‐perpetuating proinflammatory state in ALD. Triggering receptor expressed on myeloid cells 1 (TREM‐1) is a surface receptor that amplifies inflammation induced by toll‐like receptors (TLRs) and is expressed on neutrophils and monocytes/macrophages. We hypothesized that TREM‐1 signaling contributes to proinflammatory pathway activation in ALD. Using an in vivo ALD model in mice, we tested the effects of ligand‐independent TREM‐1 inhibitory peptides that were formulated into human high‐density lipoprotein (HDL)‐mimicking complexes GF9‐HDL and GA/E31‐HDL. As revealed in vitro, macrophages endocytosed these rationally designed complexes through scavenger receptors. A 5‐week alcohol feeding with the Lieber‐DeCarli diet in mice resulted in increased serum alanine aminotransferase (ALT), liver steatosis, and increased proinflammatory cytokines in the liver. TREM‐1 messenger RNA (mRNA) expression was significantly increased in alcohol‐fed mice, and TREM‐1 inhibitors significantly reduced this increase. TREM‐1 inhibition significantly attenuated alcohol‐induced spleen tyrosine kinase (SYK) activation, an early event in both TLR4 and TREM‐1 signaling. The TREM‐1 inhibitors significantly inhibited macrophage (epidermal growth factor‐like module‐containing mucin‐like hormone receptor‐like 1 [F4/80], clusters of differentiation [CD]68) and neutrophil (lymphocyte antigen 6 complex, locus G [Ly6G] and myeloperoxidase [MPO]) markers and proinflammatory cytokines (monocyte chemoattractant protein 1 [MCP‐1], tumor necrosis factor α [TNF‐α], interleukin‐1β [IL‐1β], macrophage inflammatory protein 1α [MIP‐1α]) at the mRNA level compared to the HDL vehicle. Administration of TREM‐1 inhibitors ameliorated liver steatosis and early fibrosis markers (α‐smooth muscle actin [αSMA] and procollagen1α [Pro‐Col1α]) at the mRNA level in alcohol‐fed mice. However, the HDL vehicle also reduced serum ALT and some cytokine protein levels in alcohol‐fed mice, indicating HDL‐related effects. Conclusion: HDL‐delivered novel TREM‐1 peptide inhibitors ameliorate early phases of inflammation and neutrophil and macrophage recruitment and activation in the liver and attenuate hepatocyte damage and liver steatosis. TREM‐1 inhibition represents a promising therapeutic approach for further investigations in ALD.
Abbreviations
- ACC1
acetyl‐coenzyme A carboxylase 1
- ADRP
perilipin‐2
- ALD
alcoholic liver disease
- ALT
alanine aminotransferase
- Apo A‐1
apolipoprotein A‐I
- BLT‐1
blocker of lipid transport 1
- CD
clusters of differentiation
- CPT1A
carnitine palmitoyltransferase 1A
- DAMP
damage‐associated molecular pattern
- ELISA
enzyme‐linked immunosorbent assay
- EtOH
ethanol
- F4/80
epidermal growth factor‐like module‐containing mucin‐like hormone receptor‐like 1
- H&E
hematoxylin and eosin
- HDL
high‐density lipoprotein
- IHC
immunohistochemistry
- IL
interleukin
- LPS
lipopolysaccharide
- Ly6G
lymphocyte antigen 6 complex, locus G
- MCAD
medium‐chain acyl‐coenzyme A dehydrogenase
- MCP‐1
monocyte chemoattractant protein 1
- MIP‐1α
macrophage inflammatory protein 1α
- MPO
myeloperoxidase
- mRNA
messenger RNA
- p‐SYK
activated phosphorylated‐SYK
- PAMP
pathogen‐associated molecular pattern
- PCR
polymerase chain reaction
- PF
pair‐fed
- POPC
1‐palmitoyl‐2‐oleoyl‐sn‐glycero‐3‐phosphocholine
- PPARα
peroxisome proliferator‐activated receptor α
- Pro‐Col1α
procollagen‐1α
- RANTES
regulated on activation, normal T cell expressed, and secreted
- rho B
rhodamine B
- SR
scavenger receptor
- SREBF1
sterol regulatory element binding transcription factor 1
- SYK
spleen tyrosine kinase
- TLR
toll‐like receptor
- TNF‐α
tumor necrosis factor α
- TREM‐1
triggering receptor expressed on myeloid cells 1
- UMMS
University of Massachusetts Medical School
ALD has been a long‐standing public health problem globally, yet the pathomechanisms leading to alcoholic hepatitis, a severe and often deadly form of the disease, are only partially understood. The direct effects of alcohol and its metabolites on hepatocytes result in steatosis and endoplasmic reticulum stress and trigger release of damage‐associated molecular patterns (DAMPs). In addition, excessive and/or chronic alcohol consumption disrupts the gut barrier function and changes the gut microbiome, leading to increased levels of microbial pathogen‐associated molecular patterns (PAMPs) in the circulation. PAMPs from portal circulation and locally released DAMPs provide proinflammatory signals in the liver for activation of resident Kupffer cells and recruitment of activated macrophages and neutrophil leukocytes from the bone marrow.1, 2 Increased lipopolysaccharide (LPS) levels in animal models of ALD and in patients with alcoholic hepatitis contribute to macrophage and neutrophil activation and proinflammatory cascade activation by theTLR4 receptor complex. Cytokines induced by PAMPs and DAMPs contribute to a self‐perpetuating proinflammatory state that characterizes alcoholic hepatitis.3
TREM‐1 is an activating receptor complex that is expressed on neutrophils and monocytes/macrophages and amplifies TLR signaling.4 It has been shown that TREM‐1 can amplify TLR4‐mediated as well as TLR2‐mediated proinflammatory signals.5, 6 The expression and function of TREM‐1 has been indicated in various inflammatory diseases, including sepsis, cancer, retinopathy of prematurity (ROP), atherosclerosis, and experimental colitis.7, 8, 9 Expression of TREM‐1 in hepatic stellate cells was proposed as a prognostic factor in hepatitis B‐related hepatocellular carcinoma (HCC), and TREM‐1‐mediated Kupffer cell activation was found in HCC.10, 11 Little is known about the role of TREM‐1 in ALD. Here, we hypothesized that inhibition of TREM‐1 will ameliorate inflammation and liver damage in ALD, given the known role of TREM‐1 in myeloid cell activation and amplification of proinflammatory signaling.
Despite some recent evidence that peptidoglycan recognition protein 1 (PGLYRP1) may potentially act as a ligand for TREM‐1,12 the actual TREM‐1 ligand(s) and molecular mechanisms of TREM‐1 signaling are not well understood, impeding the development of clinically relevant inhibitors of TREM‐1. Previously, we used our signaling chain homo‐oligomerization (SCHOOL) model of TREM‐1 signaling13, 14 to rationally design a novel, first‐in‐class, TREM‐1 inhibitory peptide (GF9) and demonstrated that GF9 suppresses inflammation in vitro and in vivo and exhibits therapeutic effects in animal models of sepsis, cancer, rheumatoid arthritis, and ROP.15, 16, 17, 18 We also significantly improved peptide half‐life and its targeted delivery to macrophages in vitro and in vivo by formulation of the GF9 peptide sequence into self‐assembling lipopeptide complexes that mimic human HDLs but in contrast to native HDLs are endocytosed by macrophages.15, 16, 17, 18, 19, 20 These complexes have either GF9 and two modified peptides with sequences corresponding to those of helices 4 (H4 [or PE22]) and 6 (H6 [or PA22]) of the major HDL protein apolipoprotein (apo) A‐I (GF9‐HDL) or an equimolar mixture of 31 amino acid‐long peptides GA31 and GE31, with sequences corresponding to those of GF9 and either PA22 or PE22, respectively (GA/E31‐HDL). By combining these sequences, GA31 and GE31 were shown to assist in the self‐assembly of HDLs, target HDLs to macrophages, and inhibit TREM‐1 in vivo. 15, 16, 17
In the present study, we investigated the role of TREM‐1 in ALD and the potential therapeutic effect of the TREM‐1 inhibitory GF9‐HDL and GA/E31‐HDL formulations in the Lieber‐DeCarli ALD mouse model.21
Materials and Methods
Reagents And Cells
The murine macrophage J774A.1 cell line was purchased from ATCC (Manassas, VA). Cytochalasin D was purchased from MP Biomedicals (Solon, OH). Blocker of lipid transport 1 (BLT‐1) was purchased from Calbiochem (Torrey Pines, CA). Sodium cholate, cholesteryl oleate, fucoidan, and other chemicals were purchased from Sigma‐Aldrich (St. Louis, MO). 1‐Palmitoyl‐2‐oleoyl‐sn‐glycero‐3‐phosphocholine (POPC), 1,2‐dimyristoyl‐sn‐glycero‐3‐phosphoethanolamine‐N‐(lissamine rhodamine B sulfonyl) (rho B), and cholesterol were purchased from Avanti Polar Lipids (Alabaster, AL).
Peptide Synthesis
The following synthetic peptides were ordered from Bachem (Torrance, CA): one 9‐mer peptide, GFLSKSLVF (human TREM‐1213‐221, GF9); two 22‐mer methionine sulfoxidized peptides, PYLDDFQKKWQEEM(O)ELYRQKVE (H4) and PLGEEM(O)RDRARAHVDALRTHLA (H6), which correspond to human apo A‐I helices 4 (apo A‐I123‐144) and 6 (apo A‐I167‐188), respectively; and two 31‐mer methionine sulfoxidized peptides, GFLSKSLVFPYLDDFQKKWQEEM(O)ELYRQKVE (GE31) and GFLSKSLVFPLGEEM(O)RDRARAHVDALRTHLA (GA31).
Lipopeptide Complexes
HDL‐mimicking lipopeptide complexes of spherical morphology that contained either GF9 and an equimolar mixture of PE22 and PA22 (GF9‐HDL) or an equimolar mixture of GA31 and GE31 (GA/E31‐HDL) were synthesized using the sodium cholate dialysis procedure, purified, and characterized as described.16, 17, 18, 22 For GF9‐HDL, the initial molar ratio was 125:6:2:3:1:210, corresponding to POPC:cholesterol:cholesteryl oleate:GF9:apo A‐I:sodium cholate, respectively, where apo A‐I was an equimolar mixture of PE22 and PA22. For GA/E31‐HDL, the initial molar ratio was 125:6:2:1:210, corresponding to POPC:cholesterol:cholesteryl oleate:GA/E31:sodium cholate, respectively, where GA/E31 was an equimolar mixture of GA31 and GE31.
In Vitro Macrophage Uptake Of GF9‐HDL And GA/E31‐HDL
A quantitative in vitro macrophage assay of endocytosis of rho B‐labeled HDL‐mimicking lipopeptide complexes by J774 macrophage was performed as described.18, 19, 20 Briefly, BALB/c murine macrophage J774A.1 cells (ATCC) were cultured at 37°C with 5% CO2 in Dulbecco’s modified Eagle’s medium (Cellgro Mediatech, Manassas, VA) with 2 mM glutamine, 100 U/mL penicillin, 0.1 mg/mL streptomycin, and 10% heat‐inactivated fetal bovine serum (Cellgro Mediatech) and grown to approximately 90% confluency in 12‐well tissue culture plates (Corning Costar, Corning, NY). After reaching target confluency, cells were incubated for 1 hour in medium with or without fucoidan (400 μg/mL), BLT‐1 (10 μM), or cytochalasin D (40 μM). Cells were subsequently incubated for 4 hours and 22 hours at 37°C in medium containing 2 μM of rho B‐labeled GF9‐HDL or GA/E31‐HDL (as calculated for rho B). Cells were washed twice using phosphate‐buffered saline and lysed using Passive Lysis Buffer (Promega, Madison, WI). Rho B fluorescence was measured in the lysates with 544‐nm excitation and 590‐nm emission filters, using a Fluoroscan Ascent CF fluorescence microplate reader (Thermo Labsystems, Vantaa, Finland). Protein concentrations in the lysates were measured using Bradford reagent (Sigma‐Aldrich) and an MRX microplate reader (Dynex Technologies, Chantilly, VA) according to the manufacturer’s recommended protocol.
Animals
C57BL/6 female mice (10‐ to 12‐week‐old) were purchased from the Jackson Laboratory (Bar Harbor, ME) and housed at the University of Massachusetts Medical School (UMMS) animal facility. All animals received humane care in accordance with protocols approved by the UMMS Institutional Animal Use and Care Committee. Mice (n = 6‐9/group) were acclimated to a Lieber‐DeCarli liquid diet of 5% ethanol (EtOH) (volume [vol]/vol) over a period of 1 week, then maintained on the 5% diet for 4 weeks. Pair‐fed (PF) control mice were fed a calorie‐matched dextran‐maltose diet. All animals had unrestricted access to water throughout the entire experimental period. In treated groups, mice were intraperitoneally treated 5 days/week with vehicle (empty HDL) or the TREM‐1 inhibitory formulations GF9‐HDL (2.5 mg of GF9/kg) or GA/E31‐HDL (4 mg equivalent of GF9/kg) (SignaBlok, Shrewsbury, MA) from the first day on a 5% EtOH diet. At the end of all animal experiments, cheek blood samples were collected in serum collection tubes (BD Biosciences, San Jose, CA) and processed within an hour. After blood collections, mice were euthanized and liver samples were harvested and stored at −80°C until further analysis.
Total Protein Isolation From Liver
Total protein was extracted from liver samples using radio immunoprecipitation assay buffer (BP‐115; Boston BioProducts) supplemented with protease inhibitor cocktail tablets (11836153001; Roche) and Phospho Stop phosphatase inhibitor (04906837001; Roche). Cell debris was removed from cell lysates by 10 minutes centrifugation at 12,000 rpm.
Biochemical Assays And Cytokines
Serum ALT levels were determined by the kinetic method using commercially available reagents from Teco Diagnostics (Anaheim, CA). Cytokine levels were measured in serum samples, and whole‐liver lysates were diluted in assay diluent following the manufacturer’s instructions. Specific anti‐mouse enzyme‐linked immunosorbent assay (ELISA) kits were used for the quantification of MCP‐1, TNF‐α (BioLegend Inc., San Diego, CA), and IL‐1β (R&D Systems, Minneapolis, MN) levels. For normalization, the total protein concentration of the whole‐liver lysate was determined using the Pierce bicinchoninic acid protein assay.
Western Blot Analysis
Whole‐liver proteins were boiled in Laemmli’s buffer. Samples were resolved in 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis gel under reducing conditions, using a Tris‐glycine buffer system; resolved proteins were transferred onto a nitrocellulose membrane. SYK proteins were detected by specific primary antibodies (SYK, 2712 [Cell Signaling]; phospho‐SYKY525/526, ab58575 [Abcam]) followed by an appropriate secondary horseradish peroxidase‐conjugated immunoglobulin G antibody from Santa Cruz Biotechnology. β‐actin, detected by an ab49900 antibody (Abcam), was used as a loading control. The specific immunoreactive bands of interest were visualized by chemiluminescence (Bio‐Rad Laboratories) using the Fujifilm LAS‐4000 luminescent image analyzer.
RNA Extraction and Quantitative Real‐Time Polymerase Chain Reaction Analysis
Total RNA was extracted using the Qiagen RNeasy kit (Qiagen) according to the manufacturer’s instructions with on‐column deoxyribonuclease treatment. RNA was quantified using a Nanodrop 2000 spectrophotometer (Thermo Fisher Scientific), and complementary DNA synthesis was performed using the iScript Reverse Transcription Supermix (Bio‐Rad Laboratories) and 1 µg total RNA. Real‐time quantitative polymerase chain reaction (PCR) was performed using Bio‐Rad iTaq Universal SYBR Green Supermix and a CFX96 real‐time detection system (Bio‐Rad Laboratories). Relative gene expression was calculated by the comparative ∆∆Ct method. The expression level of target genes was normalized to the housekeeping gene 18S ribosomal RNA in each sample, and the fold change in the target gene expression among experimental groups was expressed as a ratio. Primers were synthesized by IDT, Inc.; the sequences are listed in Table 1.
Table 1.
Sequences of Primers Used in This Study
| Mouse Primers | Primers | |
|---|---|---|
| Forward Sequence 5′ to 3′ | Reverse Sequence 5′ to 3′ | |
| 18s | GTA ACCCGTTGAACCCCATT | CCATCCAATCGGTAGTAGCG |
| TREM‐1 | TCCTATTACAAGGCTGACAGAGCGTC | AAGACCAGGAGAGGAAACAACCGC |
| TNF‐α | CACCAC CATCAA GGACTC AA | AGGCAACCTGACCAC TCTCC |
| MCP‐1 | CAGGTCCCT GTCATGCTTCT | CAGGTCCCTGTC ATGCTTCT |
| IL‐1β | CTTTGAAGTTGACGGACCC | TGAGTGATACTGCCTGCCTG |
| MPO | CATCCAACCCTTCATGTTCC | CTGGCGATTCAGTTTGG |
| LY6G | TGCGTTGCTCTGCTGGAGATAGA | CAGAGTAGTGGGGCAGATGG |
| F4/80 | TGCATCTAGCAATGGACAGC | GCCTTCTGGATCCATTTGAA |
| CD68 | TGTCTGATCTTGCTAGGACCG | GAGAGTAACGGCCTTTTTGTG |
| Pro‐Col1α | GCTCCTCTTAGGGGCCACT | CCACGTCTCACCATTGGG |
| α‐SMA | GTCCCAGACATCAGGGAGTAA | TCGGATACTTCAGCGTCAGGA |
| ACC1 | AGCAGATCCGCAGCTTG | ACCTCTGCTCGCTGAGTGC |
| MIP‐1α | TTCTCTGTACCATGACACTCTGC | GCATTAGCTTCAGATTTACGGGT |
| RANTES | GCTGCTTTGCCTACCTCTCC | TCGAGTGACAAACACGACTGC |
| ADRP | CTGTCTACCAAGCTCTGCTC | CGATGCTTCTCTTCCACTCC |
| PPARα | AACATCGAGTGTCGAATATGTGG | AGCCGAATAGTTCGCCGAAAG |
| SREBF1 | GCTTCTTACAGCACAGCAACC | TTTCATGCCCTCCATAGACAC |
| CPT1A | CCAGGCTACAGTGGGACATT | GAACTTGCCCATGTCCTTGT |
| MCAD | GATCGCAATGGGTGCTTTTGATAGAA | AGCTGATTGGCAATGTCTCCAGCAAA |
Liver Histopathology
Sections of formalin‐fixed paraffin‐embedded liver specimens from mice were stained with hematoxylin and eosin (H&E) or F4/80 (MF48000; Thermo Fisher Scientific) and MPO (ab9535; Abcam) antibodies for immunohistochemistry (IHC). The fresh‐frozen samples were stained with Oil Red O at the UMMS Diabetes and Endocrinology Research Center histology core facility.
Statistical Analysis
All statistical analyses were performed using GraphPad Prism 7.02 (GraphPad Software Inc.). Significance levels were determined using one‐way analysis of variance followed by a post‐hoc test for multiple comparisons. Data are shown as mean ± SEM, and differences were considered statistically significant when P ≤ 0.05.
Results
Blockade Of TREM‐1 Signaling Reduces The Expression Of Inflammation‐Associated Genes In ALD In Mice
Previous reports showed that TREM‐1 activation leads to the expression and release of proinflammatory cytokines and chemokines through nuclear factor κB activation, which also regulates the expression of TREM‐1, providing a positive feedback loop on the expression of the receptor.4 Proinflammatory cytokine expression is increased in ALD1, 2, 3, 23, 24; therefore, we hypothesized that TREM‐1 signaling contributes to the amplification of proinflammatory pathways in ALD. To evaluate this hypothesis, first we tested whole‐liver mRNAs of EtOH‐fed and PF mice with or without treatment with two different TREM‐1 inhibitory formulations and a vehicle control in a 5‐week alcohol administration model of ALD in mice.25 We found that mRNA levels of TREM‐1 and MCP‐1 were significantly increased in livers of alcohol‐fed mice compared to PF controls (Fig. 1A,B). In contrast, in mice treated with the TREM‐1 inhibitors, both GF9‐HDL and GA/E31‐HDL inhibited alcohol‐related changes in TREM‐1; in addition, MCP‐1 mRNA levels corresponded to those of the PF controls (Fig. 1A,B). Although induction of TNF‐α and IL‐1ß in alcohol‐fed mice did not reach statistical significance compared to PF controls, TREM‐1 blockade by GF9‐HDL resulted in a significant inhibition of TNF‐α mRNA in the alcohol‐fed mice compared to vehicle treatment (Fig. 1C), while IL‐1ß mRNA expression was also significantly attenuated by both the GF9‐HDL and GA/E31‐HDL formulations in the alcohol‐fed as well as in the PF groups (Fig. 1D). MIP‐1α mRNA levels were increased in alcohol‐fed mice, but TREM‐1 blockade with GF9‐HDL or GA/E31‐HDL significantly attenuated this increase compared to the vehicle control (Fig. 1E). Regulated on activation, normal T cell expressed, and secreted (RANTES) mRNA levels did not change regardless of alcohol feeding or TREM‐1 treatment (Fig. 1 F).
Figure 1.
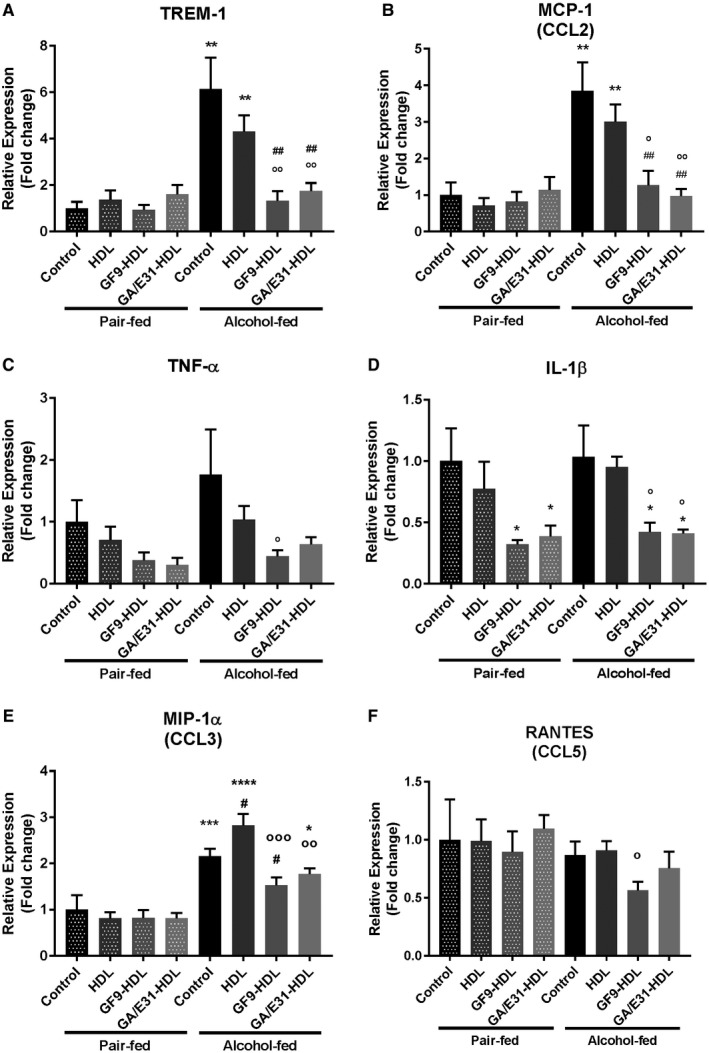
Impact of TREM‐1 pathway inhibition on TREM‐1 and inflammatory cytokine mRNA expression. [Correction made 31 October, 2018. The table title was modified to the correct description.] TREM‐1 pathway inhibition suppresses the expression of (A) TREM‐1 and inflammatory cytokines (B) MCP‐1, (C) TNF‐α, (D) IL‐1β, and (E) MIP‐1α but not (F) RANTES at the mRNA level as measured in whole‐liver lysates by real‐time quantitative PCR. * indicates significance level compared to nontreated PF group; # indicates significance level compared to nontreated alcohol‐fed group; o indicates significance level compared to vehicle‐treated alcohol‐fed group. Significance levels are as follows: */#/o P ≤ 0.05; **/##/oo P ≤ 0.01; ***/ooo P ≤ 0.001; ****P ≤ 0.0001. Abbreviation: CCL, chemokine (C‐C motif) ligand.
Next, we used specific ELISA kits to assess the protein levels of cytokines in the serum and in whole‐liver lysates (Fig. 2). We found a significant increase in MCP‐1 level in the serum and liver and TNF‐α in the liver of alcohol‐fed mice compared to PF controls (Fig. 2A‐D). All these alcohol‐induced increases were prevented both in the serum and liver by administration of either TREM‐1 inhibitor. Interestingly, we found attenuation of alcohol‐induced liver MCP‐1 and TNF‐α induction even in the vehicle‐treated (HDL only) groups (Fig. 2A‐C). The increase in total IL‐1ß levels after alcohol feeding and its attenuation by TREM‐1 inhibition did not reach statistical significance (Fig. 2D).
Figure 2.
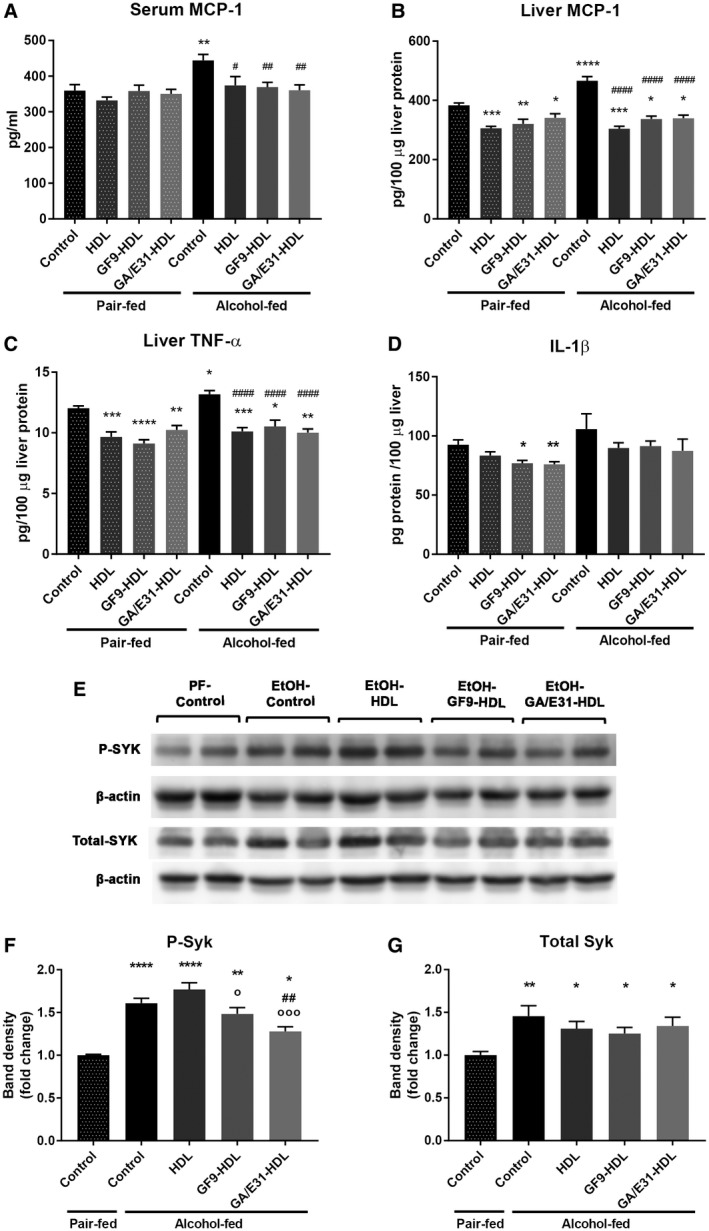
TREM‐1 blockade and protein levels of inflammatory pathway molecules. [Correction made 31 October, 2018. The table title was modified to the correct description.] TREM‐1 blockade reduces inflammatory cytokine levels in (A) serum and (B‐D) whole‐liver lysates as measured with specific ELISA kits. (E‐G) Total liver protein was analyzed for total SYK and activated p‐SYKY525/526 expression by western blotting using β‐actin as a loading control. Statistical analysis was performed by evaluating two blots (n = 4/group).* indicates significance level compared to the nontreated PF group; # indicates significance level compared to the nontreated alcohol‐fed group; o indicates significance level compared to the vehicle‐treated alcohol‐fed group. Significance levels are as follows: */#/o P ≤ 0.05; **/## P ≤ 0.01; ***P ≤ 0.001; ****/#### P ≤ 0.0001.
Because TREM‐1 is a membrane‐associated molecule that triggers SYK activation as one of its proximal signaling molecules and we previously found increased SYK phosphorylation in liver in ALD,24 we tested the levels of total and activated phospho‐SYK (p‐SYKY525/526) in the livers. We found significantly increased total and p‐SYKY525/526 levels after alcohol feeding (Fig. 2E‐G). Treatment with GA/E31‐HDL significantly decreased the p‐SYKY525/526 levels in alcohol‐fed mice compared to the untreated and vehicle‐treated alcohol‐fed groups, while GF9‐HDL decreased p‐SYKY525/526 levels compared to the vehicle‐treated group. (Fig. 2E,F).
Blockade Of TREM‐1 Activation Reduces Expression Of Macrophage And Neutrophil Markers In Liver
In agreement with previous studies indicating that chronic alcohol use causes hepatic macrophage infiltration and activation,1, 3, 26 we found increased expression of the Kupffer cell/macrophage markers F4/80 and CD68 at the mRNA level. Treatment with the TREM‐1 inhibitors significantly attenuated alcohol‐induced expression of both F4/80 and CD68 in the liver, indicating anti‐inflammatory effects (Fig. 3A,B). We also found a significant decrease in F4/80 expression on paraffin‐embedded liver sections by IHC in alcohol‐fed mice treated with either GF9‐HDL or GA/E31‐HDL compared to the EtOH‐fed vehicle‐treated group (Fig. 3C,D).
Figure 3.
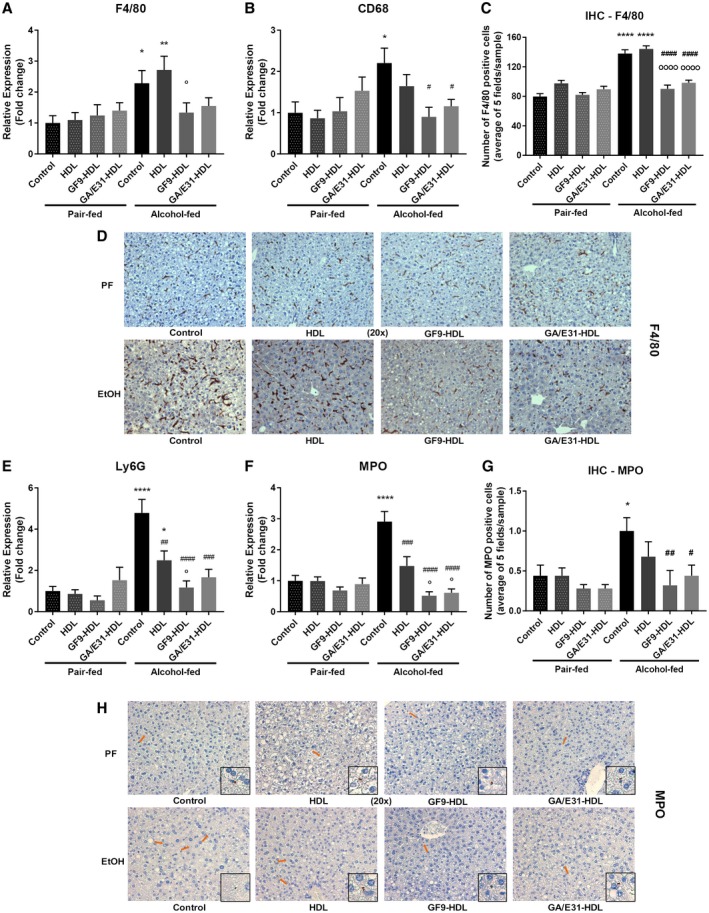
TREM‐1 inhibition and markers of immune cell infiltration. [Correction made 31 October, 2018. The table title was modified to the correct description.] (A,B) TREM‐1 inhibition suppresses the mRNA expression of macrophage cell markers in the liver as measured by real‐time quantitative PCR. (C,D) Both TREM‐1 inhibitors attenuated F4/80 as shown by IHC. (E,F) TREM‐1 inhibition suppresses the mRNA expression of neutrophil cell markers in the liver as measured by real‐time quantitative PCR. (G,H) Both TREM‐1 inhibitors attenuated MPO‐positive cell infiltration as shown by IHC. * indicates significance level compared to the nontreated PF group; # indicates significance level compared to the nontreated alcohol‐fed group; o indicates significance level compared to the vehicle‐treated alcohol‐fed group. Significance levels are as follows: */#/o P ≤ 0.05; **/## P ≤ 0.01; ### P ≤ 0.001; ****/#### P ≤ 0.0001.
Neutrophil infiltration of the liver is a characteristic of alcoholic hepatitis; therefore, we investigated markers associated with this cell population. Expression of the neutrophil markers Ly6G and MPO were significantly increased in livers of alcohol‐fed mice compared to PF controls. This was fully prevented by TREM‐1 blockade (Fig. 3E,F). Interestingly, the HDL vehicle alone also resulted in a decreasing trend of Ly6G and MPO expression in alcohol‐fed mice; however, the GF9‐HDL and GA/E31‐HDL TREM‐1 inhibitors significantly attenuated Ly6G and MPO levels even when compared to the vehicle‐treated alcohol‐fed mice (Fig. 3E,F). MPO staining on IHC confirmed that both TREM‐1 inhibitors significantly reduced MPO‐positive cell numbers compared to the untreated alcohol‐fed control group (Fig. 3G,H).
TREM‐1 Inhibitory Formulations And HDL Ameliorate Chronic Alcohol‐Induced Liver Injury And Steatosis
Next, we evaluated the impact of the TREM‐1 inhibitors on hepatocyte damage and steatosis in liver. Serum ALT levels obtained during week 5 of the alcohol feeding showed significant increases in alcohol‐fed mice compared to PF controls. This ALT increase was attenuated in both TREM‐1 inhibitor‐treated groups, indicating attenuation of liver injury (Fig. 4A). Interestingly, vehicle treatment (HDL) also showed a similar protective effect (Fig. 4A).
Figure 4.
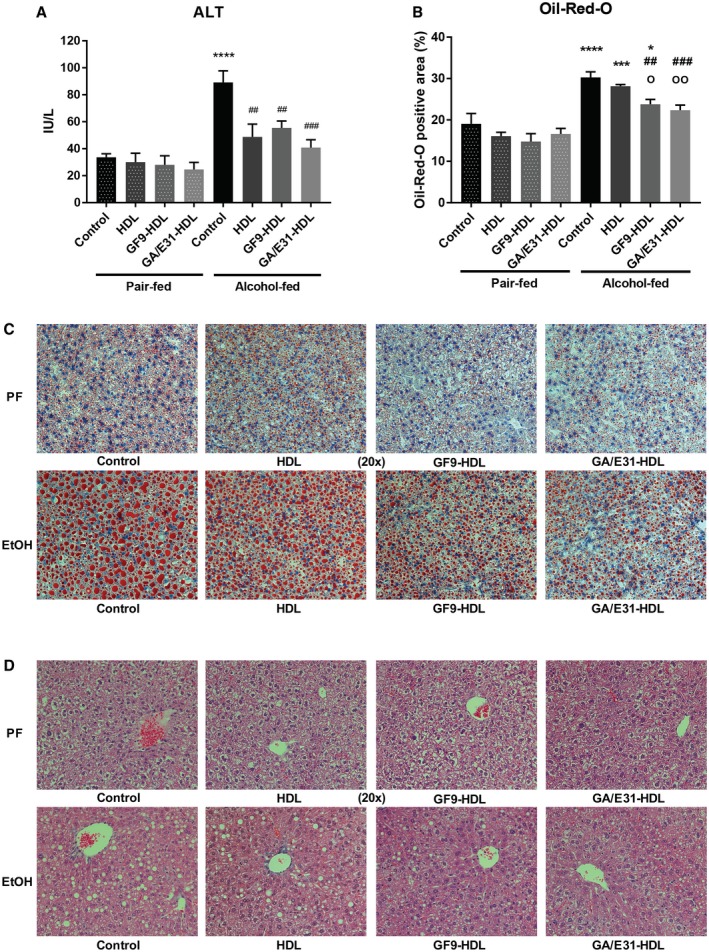
Liver damage after 5 weeks of alcohol feeding and effect of TREM‐1 pathway inhibition in a mouse model of ALD. Cheek blood and livers were harvested at death. (A) Serum ALT levels were measured using a kinetic method. (B‐D) Liver sections were stained with (B,C) Oil Red O and (D) H&E staining, and the lipid content was analyzed by ImageJ (B). * indicates significance level compared to the nontreated PF group; # indicates significance level compared to the nontreated alcohol‐fed group; o indicates significance level compared to the vehicle‐treated alcohol‐fed group. Significance levels are as follows: */o P ≤ 0.05; ##/oo P ≤ 0.01;***/### P ≤ 0.001; ****P ≤ 0.0001.
Consistent with steatosis, we found a significant increase in Oil Red O staining in livers of alcohol‐fed mice compared to PF controls (Fig. 4C). Oil Red O (Fig. 4B‐D) and H&E (Fig. 4D) staining revealed attenuation of steatosis in the alcohol‐fed TREM‐1 inhibitor‐treated mice compared to both untreated and vehicle (HDL)‐treated alcohol‐fed groups (Fig. 4B‐D).
To further assess the effects of the TREM‐1 inhibitors on mechanisms of lipid metabolism, we tested genes involved in lipid synthesis (sterol regulatory element binding transcription factor 1 [SREBF1] and acetyl‐coenzyme A carboxylase 1 [ACC1]) along with the lipid accumulation marker perilipin‐2 (ADRP) (Fig. 5A‐C). Both TREM‐1 inhibitors but not vehicle treatment prevented alcohol‐induced up‐regulation of SREBF1, ACC1, and ADRP at the mRNA level (Fig. 5A‐C). To assess lipid oxidation, we tested peroxisome proliferator‐activated receptor α (PPARα), carnitine palmitoyl transferase 1A (CPT1A), and medium‐chain acyl‐coenzyme A dehydrogenase (MCAD) mRNA levels in whole‐liver samples (Fig. 5D‐F). Alcohol feeding significantly reduced mRNA expression of PPARα and CPT1A, while MCAD had a decreasing trend. Both TREM‐1 inhibitors as well as the vehicle treatment significantly increased PPARα and MCAD levels compared to the untreated alcohol‐fed controls (Fig. 5D‐F).
Figure 5.
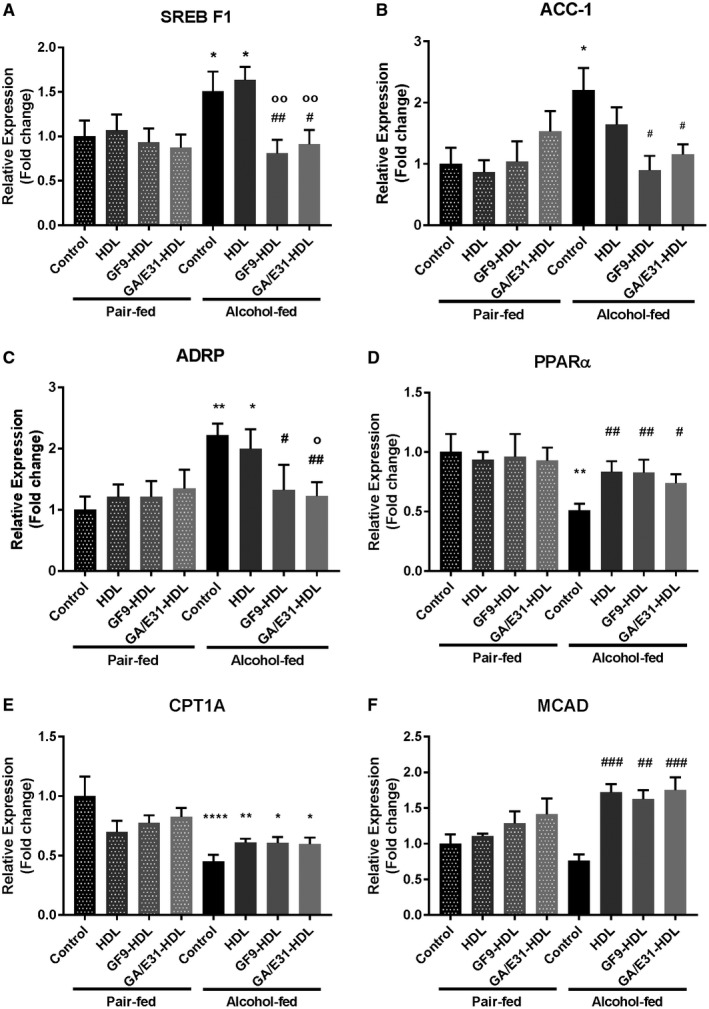
TREM‐1 inhibition alters mRNA expression of enzymes involved in lipid metabolism. mRNA expression of genes involved in (A,B) lipid synthesis (SERBF1, ACC1), (D‐F) lipid oxidation (PPARα, CPT1A, MCAD), and (C) the lipid accumulation marker (ADRP) were measured in whole liver. [Correction made 31 October, 2018. The table title and label descriptions included the wrong information and have been modified.] * indicates significance level compared to the nontreated PF group; # indicates significance level compared to the nontreated alcohol‐fed group; o indicates significance level compared to the vehicle‐treated alcohol‐fed group. Significance levels are as follows: */#/o P ≤ 0.05; **/##/oo P ≤ 0.01; ### P ≤ 0.001; ****P ≤ 0.0001.
TREM‐1 Blockade Ameliorates Expression Of Early Fibrosis Marker Genes Induced By Chronic Alcohol Consumption
The clinical progression of ALD is associated with liver fibrosis.27 Our mouse model of ALD mimics the early phase of the human disease, yet mRNA levels of early fibrosis markers Pro‐Col1α and α‐SMA were significantly increased in alcohol‐fed mice compared to PF controls in the whole‐liver samples (Fig. 6A,B). Induction of these makers was remarkably attenuated in the vehicle‐treated group and, importantly, further decreased by the TREM‐1 inhibitory formulations used (Fig. 6A,B).
Figure 6.
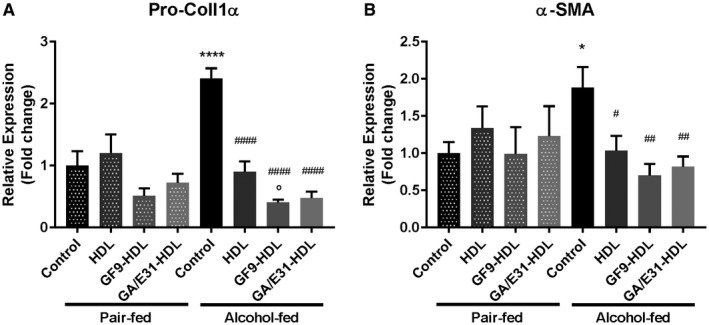
TREM‐1 inhibition suppresses the expression of fibrinogenesis marker molecules. (A) Pro‐Col1α and (B) α‐SMA are suppressed by TREM‐1 blockade at the RNA level as measured in whole‐liver lysates. [Correction made 31 October, 2018. The label descriptions included the wrong description and have been modified.] * indicates significance level compared to the nontreated PF group; # indicates significance level compared to the nontreated alcohol‐fed group; o indicates significance level compared to the vehicle‐treated alcohol‐fed group. Significance levels are as follows: */#/o P ≤ 0.05; ## P ≤ 0.01; ****/#### P ≤ 0.0001. Data are presented as mean ± SEM.
Quantitatively Similar Macrophage Endocytosis Of GF9‐HDL And GA/E31‐HDL Is Mainly Mediated By SR‐A
We studied the uptake of GF9‐HDL and GA/E31‐HDL in vitro in order to evaluate potential mechanisms of targeted delivery of GF9 (GA/E31). Kupffer cells and recruited hepatic macrophages express high levels of SRs, including SR‐A, that are involved in phagocytosis and removal of oxidatively damaged lipoproteins and cells from the blood circulation.28, 29 We previously demonstrated intracellular macrophage delivery of GF9, GA31, and GE31 by macrophage‐targeted GF9‐HDL and GA/E31‐HDL, respectively, and hypothesized that the observed macrophage endocytosis of these complexes is SR mediated.16, 17 To further investigate the molecular mechanisms involved in this process, we used J774 macrophages as a model for Kupffer cells and incubated them with rho B‐labeled GF9‐HDL or GA/E31‐HDL in the presence or absence of cytochalasin D, fucoidan, or BLT‐1, which are known to inhibit all SRs,30 SR‐A,31 or SR‐BI,32 respectively.
In the presence of cytochalasin D, which inhibits both SR‐A and SR‐BI, the macrophage uptake of both TREM‐1 inhibitor complexes was significantly inhibited, suggesting that this uptake is SR mediated. Fucoidan, an SR‐A inhibitor, substantially suppressed endocytosis of TREM‐1 inhibitor complexes at 22 hours but not at 4 hours, indicating time‐dependent mechanisms of SR‐A‐mediated endocytosis (Fig. 7B). In contrast, BLT‐1, which inhibits SR‐BI, similarly inhibited the uptake of the complexes at both time points but to a lesser extent compared with that of fucoidan (Fig. 7C), presumably because of lower expression of SR‐BI on J774 macrophages.33 These findings suggest that SR‐A is the main contributor in SR‐mediated endocytosis of both GF9‐HDL and GA/E31‐HDL.
Figure 7.
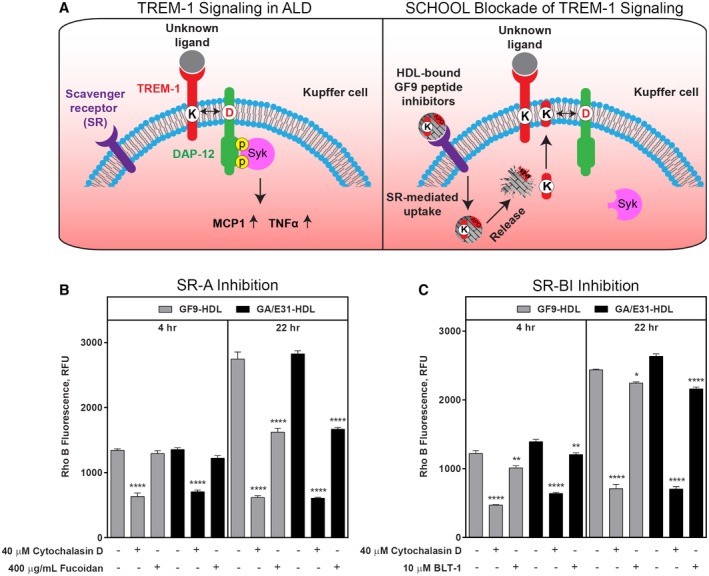
Scavenger receptors SR‐A and SR‐BI mediate the macrophage uptake of GF9‐HDL and GA/E31‐HDL. (A) Schematic representation for the proposed role of TREM‐1 signaling in ALD and the SCHOOL mechanism of TREM‐1 blockade. (A, left panel) Activation of the TREM‐1/DAP12 receptor complex expressed on Kupffer cells leads to phosphorylation of the DAP12 cytoplasmic signaling domain, subsequent SYK recruitment, and the downstream inflammatory cytokine response. (A, right panel) SR‐mediated endocytosis of HDL‐bound GF9 peptide inhibitors by Kupffer cells results in the release of GF9 (GA31 or GE31) into the cytoplasm; GF9 self‐penetrates the cell membrane and blocks intramembrane interactions between TREM‐1 and DAP12, thereby preventing DAP12 phosphorylation and the downstream signaling cascade. (B,C) Macrophage uptake of GF9‐HDL and GA/E31‐HDL in vitro is SR mediated in a time‐dependent manner and is largely driven by SR‐A. As described in the Materials and Methods, J774 macrophages were cultured at 37°C overnight with medium. Prior to uptake of GF9‐HDL and GA/E31‐HDL, cells were treated for 1 hour at 37°C with 40 μM cytochalasin D and either (B) 400 μg/mL fucoidan or (C) 10 μM BLT‐1, as indicated. Cells were then incubated for either 4 hours or 22 hours with medium containing 2 μM rho B‐labeled GF9‐HDL (gray bars) or GA/E31‐HDL (black bars), respectively. Cells were lysed, and rho B fluorescence intensities of lysates were measured and normalized to the protein content. Results are expressed as mean ± SEM (n = 3); *P ≤ 0.05; **P ≤ 0.01; ****P ≤ 0.0001 versus uptake of GF9‐HDL and GA/E31‐HDL in the absence of inhibitor. Abbreviations: D, DAP12; DAP12, DNAX activation protein of 12 kDa; K, Kupffer cell; RFU, relative fluorescence units; SCHOOL, signaling chain homo‐oligomerization.
Interestingly, quantitatively determined macrophage uptake levels in the presence or absence of fucoidan or BLT‐1 were similar for GF9‐HDL and GA/E31‐HDL (Fig. 7). This importantly suggests that the combination of GF9 and apo A‐I peptide sequences in GA31 and GE31 sequences does not change the level and mechanisms of macrophage endocytosis of GA/E31‐HDL compared with those of GF9‐HDL.
Discussion
In this study, we report for the first time an important role for TREM‐1 in ALD. Using a mouse model, we found a significant up‐regulation of TREM‐1 in livers of mice following chronic alcohol feeding. Treatment with novel ligand‐independent TREM‐1 inhibitors reduced the expression of the TREM‐1 molecule itself, attenuated or fully prevented alcohol‐induced increases in proinflammatory cytokines at the mRNA level, and inhibited SYK activation. We demonstrated that TREM‐1 blockade results in reduced macrophage and neutrophil infiltration and activation indicated by reduced F4/80, CD68, Ly6G, and MPO expression in the liver. These findings complement our previous data demonstrating that TREM‐1 blockade using GF9‐HDL and GA/E31‐HDL suppresses macrophage infiltration of the tumor in cancer mice.17 The TREM‐1 inhibitors attenuated alcohol‐induced liver steatosis. HDL and the TREM‐1 inhibitors also attenuated liver injury and markers of early fibrosis in alcohol‐fed mice. Interestingly, the HDL vehicle control showed similar efficiency as the inhibitory formulations at the protein level of the proinflammatory cytokines. We discovered that HDL has some protective effects on ALD at the level of ALT and lipid oxidation.
While the ligand of TREM‐1 is still unknown, it has been shown that TREM‐1 activation amplifies inflammation and synergizes with TLR signaling pathways.34 It was also observed that bacterial infection and challenge with LPS or lipoteichoic acid increase TREM‐1 expression,7 indicating a positive feedback loop among PAMP exposure, TREM‐1 expression, and inflammatory cytokine induction. Different DAMPs, such as high‐mobility group box protein 1 [Correction made 31 October, 2018. The correct protein was noted.] and heat shock protein 70, have been suggested to stimulate TREM‐1,35 while other studies found cell (granulocyte and platelet)‐surface‐associated activators as well.35, 36 Both PAMPs and DAMPs are present in ALD, providing potential mechanisms for TREM‐1 up‐regulation in this disease. Alcohol induces changes in the gut microbiome and disrupts the gut barrier function, resulting in increased levels of endotoxin and microbial PAMPs in circulation.1, 37 Alcohol also causes hepatocyte damage that leads to the release of DAMPs,23 and all these processes contribute to TREM‐1 activation.
TREM‐1 signaling leads to phosphorylation and activation of SYK, which has been indicated as a major regulator in inflammatory processes in ALD.38 TREM‐1 also amplifies TLR4 signaling that involves downstream SYK activation and phosphorylation.38 Indeed, we found increased total and phosphorylated SYK levels in the livers of alcohol‐fed mice that was attenuated by TREM‐1 inhibitor administration. A previous study showed that inhibition of SYK activation attenuates alcohol‐induced liver inflammation, cell death, and steatosis, suggesting that the SYK pathway could be a feasible therapeutic target in ALD.24 SYK is expressed in a wild spectrum of cells, while TREM‐1 inhibition may specifically modulate macrophages, neutrophils, and stellate cells that each play a role in ALD. Another advantage of TREM‐1 inhibition is that it likely attenuates signaling from a broader spectrum of TLRs, not only TLR4.
TREM‐1 activation alone has been shown to increase the production of proinflammatory chemokines and cytokines.39 Furthermore, simultaneous stimulation of TREM‐1 and TLRs by an agonistic anti‐TREM‐1 antibody and different TLR ligands synergized in the induction of these proinflammatory molecules. TREM‐1 and TLR4 costimulated monocytes showed increased production of MCP‐1, IL‐1β, and IL‐8. In contrast, the level of the anti‐inflammatory cytokine IL‐10 decreased when anti‐TREM‐1 antibody and the TLR3 ligand poly(I:C) or the TLR4 ligand LPS simultaneously attached to their receptors.40 Because self‐perpetuating proinflammatory pathways are present in alcoholic hepatitis, interruption of these pathways using TREM‐1 inhibition seems attractive.
By inducing TNF‐α, IL‐6, MCP‐1, IL‐8, and granulocyte‐macrophage colony‐stimulating factor and inhibiting IL‐10 production, TREM‐1 is involved in activation and recruitment of monocytes and modulation of inflammatory responses.40 Furthermore, TREM‐1 expression was highly up‐regulated on the surface of infiltrating monocytes and neutrophils in human tissues infected by bacteria, highlighting the importance of this receptor in these processes.7 In alcoholic hepatitis, neutrophils infiltrate the liver, inducing oxidative stress and cytotoxicity that contributes to the high mortality of the disease.2 We showed that these processes can be attenuated by TREM‐1 inhibitors. Mechanistically, the GF9‐HDL and GA/E31‐HDL formulations target the liver more efficiently than only the peptides alone and release the TREM‐1 inhibitory sequences inside the target cells where these peptides likely inhibit TREM‐1 signaling by disrupting the intramembrane interactions of the TREM‐1 receptor and its signaling adaptor molecule DNAX activation protein of 12kDa [Correction made 31 October, 2018. The correct protein was noted.] (Fig. 7).15, 16, 17
We previously hypothesized that the observed preferential endocytosis of GF9‐HDL and GA/E31‐HDL by macrophages and hepatic clearance of these complexes are mediated by SR recognition of putative epitopes in the modified apo A‐I peptide constituents of GF9‐HDL and GA/E31‐HDL.16, 17, 19 Our current findings indicate that GF9‐HDL and GA/E31‐HDL are largely recognized by SR‐A on macrophages (Fig. 7). We also observed SR‐BI‐mediated uptake, which likely explains the previously observed hepatic clearance for these complexes in another animal model.19 While these data confirm our hypothesis, future studies are needed to determine the clearance properties for GF9‐HDL and GA/E31‐HDL in ALD.
Further, our present study demonstrates that GF9‐HDL and GA/E31‐HDL exhibit not only similar macrophage uptake in vitro largely driven by SR‐A (Fig. 7) but also similar therapeutic effect in a mouse model of ALD (Figs. 1, 2, 3, 4, 5, 6). This is in line with our previous studies where GF9‐HDL and GA/E31‐HDL exhibited similar therapeutic activities in cancer and arthritic mice.16, 17 We suggest that SR‐A epitopes are similarly exposed on GA31 and GE31 in GA/E31‐HDL and on PA22 and PE22 in GF9‐HDL, providing similar uptake of these complexes and as a result delivery of TREM‐1 inhibitory GF9 peptide sequences in vivo. The use of GA/E31‐HDL in the further development of effective and low‐toxicity therapy for ALD is advantageous because it makes the entire manufacturing process easier and less expensive. We also suggest that the in vitro macrophage uptake assay can be potentially used to predict the outcomes for macrophage‐targeted TREM‐1 therapy in vivo.
In addition to attenuating inflammatory processes, the TREM‐1 inhibitory formulations also ameliorated hepatocyte damage and steatosis. Serum ALT and liver triglyceride levels were both decreased in the GF9‐HDL, GA/E31‐HDL, and HDL‐vehicle treated groups. The vehicle also had an inhibitory effect on TNF‐α and MCP‐1 protein levels as well as on mRNA expression of neutrophil and fibrosis markers, indicating that the HDL vehicle formulation can attenuate inflammation to a moderate extent. A previous study found evidence that HDL can protect hepatocytes from endoplasmic reticulum stress,41 while other publications reported a scavenger function of HDL for LPS and lipoteichoic acid42, 43 that could prevent immune cells from being activated by those molecules.42, 43 Further, the observed moderate beneficial effect of HDL treatment alone on fatty acid oxidation markers in alcohol‐exposed mice (Fig. 5A‐C) is in line with data that demonstrate infusion of reconstituted HDL reduces fatty acid oxidation in patients with type 2 diabetes mellitus.44 In human and rat plasma, apo A‐I, the major protein of HDL, has been shown to inhibit lipid peroxidation.45 These data might provide an explanation for our findings of the hepatoprotective effects of HDL.
Our study shows that TREM‐1 inhibitors with HDL formulation exerted significant inhibition on early signaling events of proinflammatory processes at the level of cytokine mRNA and the activated p‐SYK protein levels compared to the HDL vehicle alone in a mouse model of ALD. This effect presumably would be even more obvious at the protein level of cytokines in a more severe liver injury. However, in mice, the most commonly used 5‐week alcohol feeding that we used resulted in moderate liver damage and minimal inflammation,25 which is a limitation of our study. Importantly, as shown on the stained liver sections, the GF9‐HDL and GA/E31‐HDL formulations significantly inhibited immune cell infiltration and steatosis compared to the HDL vehicle only group [Correction made 31 October, 2018. The word group was added.] in mice with ALD. Thus, exploration of TREM‐1 inhibitors in decreasing inflammation deserves further investigation in ALD.
Potential conflict of interest
Dr. Sigalov and Dr. Shen are employed by SignaBlok. Dr. Szabo is the Editor‐in‐Chief of Hepatology Communications and was fully excused from the peer review of this manuscript.
Supported by the National Institute on Alcohol Abuse and Alcoholism, National Institutes of Health (grant Nos. U01 AA021907 to G.S. and R43AA024355 to A.B.S.) and SignaBlok, Inc. (additional funding to Z.T.S. and A.B.S.).
The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Contributor Information
Alexander B. Sigalov, Email: Sigalov@signablok.com
Gyongyi Szabo, Email: Gyongyi.Szabo@umassmed.edu.
References
Author names in bold designate shared co‐first authorship.
- 1. Szabo G, Bala S, Petrasek J, Gattu A. Gut‐liver axis and sensing microbes. Dig Dis 2010;28:737‐744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bautista AP. Neutrophilic infiltration in alcoholic hepatitis. Alcohol 2002;27:17‐21. [DOI] [PubMed] [Google Scholar]
- 3. Szabo G, Petrasek J, Bala S. Innate immunity and alcoholic liver disease. Dig Dis 2012;30(Suppl 1):55‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tessarz AS, Cerwenka A. The TREM‐1/DAP12 pathway. Immunol Lett 2008;116:111‐116. [DOI] [PubMed] [Google Scholar]
- 5. Arts RJ, Joosten LA, Dinarello CA, Kullberg BJ, van der Meer JW, Netea MG. TREM‐1 interaction with the LPS/TLR4 receptor complex. Eur Cytokine Netw 2011;22:11‐14. [DOI] [PubMed] [Google Scholar]
- 6. Campanholle G, Mittelsteadt K, Nakagawa S, Kobayashi A, Lin SL, Gharib SA, et al. TLR‐2/TLR‐4 TREM‐1 signaling pathway is dispensable in inflammatory myeloid cells during sterile kidney injury. PLoS One 2013;8:e68640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bouchon A, Facchetti F, Weigand MA, Colonna M. TREM‐1 amplifies inflammation and is a crucial mediator of septic shock. Nature 2001;410:1103‐1107. [DOI] [PubMed] [Google Scholar]
- 8. Zysset D, Weber B, Rihs S, Brasseit J, Freigang S, Riether C, et al. TREM‐1 links dyslipidemia to inflammation and lipid deposition in atherosclerosis. Nat Commun 2016;7:13151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Schenk M, Bouchon A, Seibold F, Mueller C. TREM‐1—expressing intestinal macrophages crucially amplify chronic inflammation in experimental colitis and inflammatory bowel diseases. J Clin Invest 2007;117:3097‐3106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Liao R, Sun TW, Yi Y, Wu H, Li YW, Wang JX, et al. Expression of TREM‐1 in hepatic stellate cells and prognostic value in hepatitis B‐related hepatocellular carcinoma. Cancer Sci 2012;103:984‐992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wu J, Li J, Salcedo R, Mivechi NF, Trinchieri G, Horuzsko A. The proinflammatory myeloid cell receptor TREM‐1 controls Kupffer cell activation and development of hepatocellular carcinoma. Cancer Res 2012;72:3977‐3986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Read CB, Kuijper JL, Hjorth SA, Heipel MD, Tang X, Fleetwood AJ, et al. Cutting edge: identification of neutrophil PGLYRP1 as a ligand for TREM‐1. J Immunol 2015;194:1417‐1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sigalov AB. Multichain immune recognition receptor signaling: different players, same game? Trends Immunol 2004;25:583‐589. [DOI] [PubMed] [Google Scholar]
- 14. Sigalov AB. Immune cell signaling: a novel mechanistic model reveals new therapeutic targets. Trends Pharmacol Sci 2006;27:518‐524. [DOI] [PubMed] [Google Scholar]
- 15. Rojas MA, Shen ZT, Caldwell RB, Sigalov AB. Blockade of TREM‐1 prevents vitreoretinal neovascularization in mice with oxygen‐induced retinopathy. Biochim Biophys Acta Mol Basis Dis 2018;1864:2761‐2768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Shen ZT, Sigalov AB. Rationally designed ligand‐independent peptide inhibitors of TREM‐1 ameliorate collagen‐induced arthritis. J Cell Mol Med 2017;21:2524‐2534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shen ZT, Sigalov AB. Novel TREM‐1 inhibitors attenuate tumor growth and prolong survival in experimental pancreatic cancer. Mol Pharm 2017;14:4572‐4582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sigalov AB. A novel ligand‐independent peptide inhibitor of TREM‐1 suppresses tumor growth in human lung cancer xenografts and prolongs survival of mice with lipopolysaccharide‐induced septic shock. Int Immunopharmacol 2014;21:208‐219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Shen ZT, Zheng S, Gounis MJ, Sigalov AB. Diagnostic magnetic resonance imaging of atherosclerosis in apolipoprotein E knockout mouse model using macrophage‐targeted gadolinium‐containing synthetic lipopeptide nanoparticles. PLoS One 2015;10:e0143453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sigalov AB. Nature‐inspired nanoformulations for contrast‐enhanced in vivo MR imaging of macrophages. Contrast Media Mol Imaging 2014;9:372‐382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lieber CS, DeCarli LM. The feeding of alcohol in liquid diets: two decades of applications and 1982 update. Alcohol Clin Exp Res 1982;6:523‐531. [DOI] [PubMed] [Google Scholar]
- 22. Shen ZT, Sigalov AB. SARS coronavirus fusion peptide‐derived sequence suppresses collagen‐induced arthritis in DBA/1J mice. Sci Rep 2016;6:28672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Iracheta‐Vellve A, Petrasek J, Satishchandran A, Gyongyosi B, Saha B, Kodys K, et al. Inhibition of sterile danger signals, uric acid and ATP, prevents inflammasome activation and protects from alcoholic steatohepatitis in mice. J Hepatol 2015;63:1147‐1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bukong TN, Iracheta‐Vellve A, Saha B, Ambade A, Satishchandran A, Gyongyosi B, et al. Inhibition of spleen tyrosine kinase activation ameliorates inflammation, cell death, and steatosis in alcoholic liver disease. Hepatology 2016;64:1057‐1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bertola A, Mathews S, Ki SH, Wang H, Gao B. Mouse model of chronic and binge ethanol feeding (the NIAAA model). Nat Protoc 2013;8:627‐637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ju C, Mandrekar P. Macrophages and alcohol‐related liver inflammation. Alcohol Res 2015;37:251‐262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Teli MR, Day CP, Burt AD, Bennett MK, James OF. Determinants of progression to cirrhosis or fibrosis in pure alcoholic fatty liver. Lancet 1995;346:987‐990. [DOI] [PubMed] [Google Scholar]
- 28. Terpstra V, van Berkel TJ. Scavenger receptors on liver Kupffer cells mediate the in vivo uptake of oxidatively damaged red blood cells in mice. Blood 2000;95:2157‐2163. [PubMed] [Google Scholar]
- 29. Zingg JM, Ricciarelli R, Azzi A. Scavenger receptors and modified lipoproteins: fatal attractions? IUBMB Life 2000;49:397‐403. [DOI] [PubMed] [Google Scholar]
- 30. Gu BJ, Saunders BM, Jursik C, Wiley JS. The P2X7‐nonmuscle myosin membrane complex regulates phagocytosis of nonopsonized particles and bacteria by a pathway attenuated by extracellular ATP. Blood 2010;115:1621‐1631. [DOI] [PubMed] [Google Scholar]
- 31. Sigola LB, Fuentes AL, Millis LM, Vapenik J, Murira A. Effects of Toll‐like receptor ligands on RAW 264.7 macrophage morphology and zymosan phagocytosis. Tissue Cell 2016;48:389‐396. [DOI] [PubMed] [Google Scholar]
- 32. Yu M, Romer KA, Nieland TJ, Xu S, Saenz‐Vash V, Penman M, et al. Exoplasmic cysteine Cys384 of the HDL receptor SR‐BI is critical for its sensitivity to a small‐molecule inhibitor and normal lipid transport activity. Proc Natl Acad Sci U S A 2011;108:12243‐12248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Dong P, Xie T, Zhou X, Hu W, Chen Y, Duan Y, et al. Induction of macrophage scavenger receptor type BI expression by tamoxifen and 4‐hydroxytamoxifen. Atherosclerosis 2011;218:435‐442. [DOI] [PubMed] [Google Scholar]
- 34. Tammaro A, Derive M, Gibot S, Leemans JC, Florquin S, Dessing MC. TREM‐1 and its potential ligands in non‐infectious diseases: from biology to clinical perspectives. Pharmacol Ther 2017;177:81‐95. [DOI] [PubMed] [Google Scholar]
- 35. El Mezayen R, El Gazzar M, Seeds MC, McCall CE, Dreskin SC, Nicolls MR. Endogenous signals released from necrotic cells augment inflammatory responses to bacterial endotoxin. Immunol Lett 2007;111:36‐44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gibot S, Buonsanti C, Massin F, Romano M, Kolopp‐Sarda MN, Benigni F, et al. Modulation of the triggering receptor expressed on the myeloid cell type 1 pathway in murine septic shock. Infect Immun 2006;74:2823‐2830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bala S, Marcos M, Gattu A, Catalano D, Szabo G. Acute binge drinking increases serum endotoxin and bacterial DNA levels in healthy individuals. PLoS One 2014;9:e96864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Arts RJ, Joosten LA, van der Meer JW, Netea MG. TREM‐1: intracellular signaling pathways and interaction with pattern recognition receptors. J Leukoc Biol 2013;93:209‐215. [DOI] [PubMed] [Google Scholar]
- 39. Klesney‐Tait J, Turnbull IR, Colonna M. The TREM receptor family and signal integration. Nat Immunol 2006;7:1266‐1273. [DOI] [PubMed] [Google Scholar]
- 40. Bleharski JR, Kiessler V, Buonsanti C, Sieling PA, Stenger S, Colonna M, et al. A role for triggering receptor expressed on myeloid cells‐1 in host defense during the early‐induced and adaptive phases of the immune response. J Immunol 2003;170:3812‐3818. [DOI] [PubMed] [Google Scholar]
- 41. Hong D, Li LF, Gao HC, Wang X, Li CC, Luo Y, et al. High‐density lipoprotein prevents endoplasmic reticulum stress‐induced downregulation of liver LOX‐1 expression. PLoS One 2015;10:e0124285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Feingold KR, Grunfeld C. The role of HDL in innate immunity. J Lipid Res 2011;52:1‐3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tobias PS, Ulevitch RJ. Control of lipopolysaccharide‐high density lipoprotein binding by acute phase protein(s). J Immunol 1983;131:1913‐1916. [PubMed] [Google Scholar]
- 44. Drew BG, Carey AL, Natoli AK, Formosa MF, Vizi D, Reddy‐Luthmoodoo M, et al. Reconstituted high‐density lipoprotein infusion modulates fatty acid metabolism in patients with type 2 diabetes mellitus. J Lipid Res 2011;52:572‐581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Mashima R, Yamamoto Y, Yoshimura S. Reduction of phosphatidylcholine hydroperoxide by apolipoprotein A‐I: purification of the hydroperoxide‐reducing proteins from human blood plasma. J Lipid Res 1998;39:1133‐1140. [PubMed] [Google Scholar]