

Abstract
Chronic hepatitis B virus (HBV) infection remains a major global health burden. Currently, two types of treatment, interferons (IFNs) and nucleos(t)ide analogues (NAs), have been approved. These treatments are effective in suppressing HBV replication and in decreasing the risk of developing cirrhosis, liver failure, hepatocellular carcinoma (HCC), and death. However, these treatments do not eliminate the virus, and the risk of HCC remains. This review article summarizes current knowledge about the safety, efficacy, and clinical indications of hepatitis B treatment. It also discusses limitations of existing treatment, gaps in knowledge, and feasibility of a hepatitis B cure.
Abbreviations
- ALT
alanine aminotransferase
- APASL
Asian Pacific Association for the Study of the Liver
- cccDNA
covalently closed circular DNA
- EASL
European Association for the Study of the Liver
- ETV
entecavir
- HBeAg
hepatitis B e antigen
- HBIG
hepatitis B immune globulin
- HBsAg
hepatitis B surface antigen
- HBV
hepatitis B virus
- HCC
hepatocellular carcinoma
- IFN
interferon
- NA
nucleos(t)ide analogue
- TAF
tenofovir alafenamide
- TDF
tenofovir disoproxil fumarate
Chronic hepatitis B virus (HBV) infection remains a major global health burden affecting 292 million persons worldwide.1 Treatments that are effective in suppressing HBV replication are interferons (IFNs) and nucleos(t)ide analogues (NAs); these have been available for nearly 2 decades but do not eliminate the virus. IFNs and NAs have been demonstrated to prevent cirrhosis, liver failure, and hepatocellular carcinoma (HCC),2 but the risk of HCC remains, even for patients in whom the virus is suppressed. Thus, there is a need to develop “curative” therapies for hepatitis B. Equally important is the need to improve diagnosis and linkage to care. Globally, it is estimated that only 10% of persons chronically infected with HBV have been diagnosed and only 5% of those who are eligible for treatment have received treatment.1
HBV Biology and Barriers to Cure
HBV Replication Cycle
HBV is a hepatotropic DNA virus that replicates by means of reverse transcription of a pregenomic RNA. The life cycle of HBV is depicted in Fig. 1.3 The circulating virion comprises an envelope and a nucleocapsid that contains a partially double‐stranded, relaxed, circular DNA. The entry receptor of HBV has been identified to be sodium taurocholate cotransporting polypeptide, which explains the hepatotropism of HBV. Following entry into the hepatocyte, the envelope is shed and the nucleocapsid is transported into the nucleus where the partially double‐stranded, relaxed, circular DNA is repaired and the covalently closed circular DNA (cccDNA) is bound to chromatin. The cccDNA is transcribed into pregenomic RNA, which is then reverse transcribed into HBV DNA. The pregenomic RNA is also transcribed into viral messenger RNAs and in turn translated into the following viral proteins: surface (large, middle, and small surface proteins), core (hepatitis B core antigen as well as precore/core from which hepatitis B e antigen [HBeAg] is derived), polymerase (which also functions as a reverse transcriptase), and X (unclear functions but may play a role in cccDNA transcription). The nucleocapsid is assembled in the cytoplasm and contains the pregenomic RNA as well as core and polymerase proteins. The pregenomic RNA is reverse transcribed into the (–) strand HBV DNA, which serves as a template for the (+) strand HBV DNA; however, the virion is enveloped and secreted before the (+) strand HBV DNA is completely synthesized. Nucleocapsids can be recycled from the cytoplasm back into the nucleus, and thus cccDNA can be replenished without the need for entry of new virions. cccDNA has a long half‐life, and its elimination in untreated patients appears to be predominantly mediated through loss of infected hepatocytes.
Figure 1.
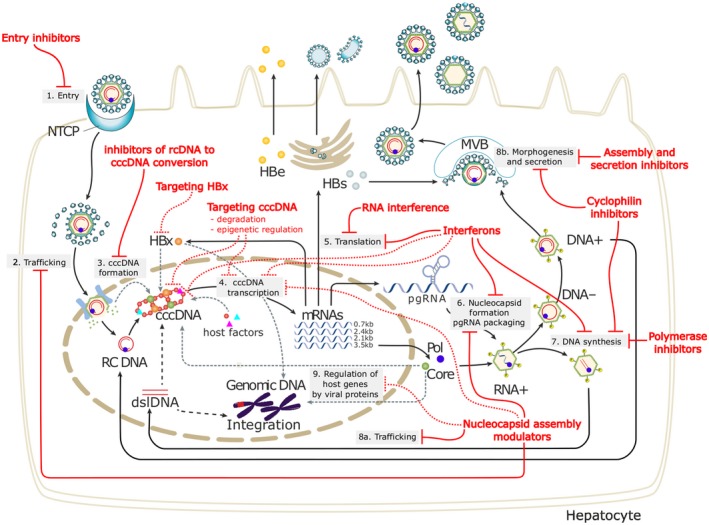
HBV lifecycle and antiviral targets.3 HBV entry: Lipopeptides mimicking pre‐S1 domain competing with a Dane particle for binding to NTCP (e.g., Myrcludex B); other small molecule inhibitors are in development. Targeting cccDNA: Prevention of cccDNA formation; damage and destruction through cytokines or cccDNA sequence‐specific nucleases; functional silencing through modulation of host cellular epigenetic‐modifying enzymes by cytokines or inhibition of viral protein function. HBV mRNAs: Small interfering RNA approaches or anti‐sense oligonucleotides to block viral replication and viral protein expression. HBV polymerase: Reverse transcriptase inhibitors include approved NAs; RNAse H inhibitors are in preclinical evaluation. Nucleocapsid assembly and pgRNA packaging: Capsid assembly modulators can affect nucleocapsid assembly and pgRNA encapsidation and may affect the nuclear functions of HBc (cccDNA regulation and IFN‐stimulated gene expression). Targeting HBsAg: Phosphorothioate oligonucleotides nucleic acid polymer inhibiting HBsAg release and monoclonal antibodies to decrease circulating HBsAg load are under evaluation. Abbreviations: dslDNA, double‐stranded linear DNA; HBc, hepatitis B core protein; HBe, hepatitis B e protein; HBs, hepatitis B surface antigen; HBx, hepatitis B x protein; mRNA, messenger RNA; MVB, multivesicular body; NTCP, sodium taurocholate cotransporting polypeptide; pgRNA, pregenomic RNA; Pol, polymerase; rcDNA, relaxed circular DNA; RNAse H, ribonuclease H.
Mechanisms of Action of NA and IFN
NAs inhibit reverse transcription of pregenomic RNA and synthesis of HBV DNA in the cytoplasm and have no direct effect on cccDNA. IFN has both antiviral and immunomodulatory activities, although the precise mechanisms of action remain unclear. Recent studies suggest that IFN may enhance cccDNA degradation and exert epigenetic modification of cccDNA transcription,4 explaining its greater effect on viral protein production and higher rates of HBeAg and hepatitis B surface antigen (HBsAg) loss compared to NAs.
Barriers to Eliminating HBV
Persistence of cccDNA and its ability to self‐replenish and the lack of direct effects of current therapies on cccDNA account for the difficulty in eliminating cccDNA. There are additional barriers to eliminating HBV. HBV DNA can be integrated into the host genome. Although integrated HBV DNA is often rearranged and/or partially deleted and there is no evidence that it supports the full cycle of HBV replication, recent studies suggest that integrated HBV DNA can be sufficiently intact to support translation of viral proteins, e.g., HBsAg.5 Elimination of integrated HBV DNA will likely require the removal of hepatocytes that harbor these DNA. Control of infections generally requires elimination of the infectious organisms coupled with activation of specific immune responses. Whereas patients who recover from acute HBV infection display rigorous immune responses to multiple HBV epitopes, patients with chronic HBV infection manifest weak immune responses to very few HBV epitopes.6
Persistence of cccDNA, presence of integrated HBV DNA, and impaired innate and adaptive immune responses explain why it would be difficult to eliminate HBV from patients who are chronically infected. Indeed, HBV persists in the livers of persons who have recovered from acute HBV infection with HBsAg to hepatitis B surface antibody seroconversion. Thus, HBV reactivation can occur when “recovered” individuals receive potent immunosuppressive therapy, such as anti‐clusters of differentiation (CD)20, and transmission of HBV infection can occur when livers from these individuals are transplanted into seronegative recipients.
Phases of Chronic HBV Infection
The course of chronic HBV infection is characterized by fluctuations in HBV DNA and alanine aminotransferase (ALT) levels, reflecting variations in the balance between HBV replication and host immune response. Traditionally, three clinical parameters (HBeAg status, HBV DNA level, and ALT level) are used to define the four phases of chronic HBV infection (Fig. 2).
Figure 2.
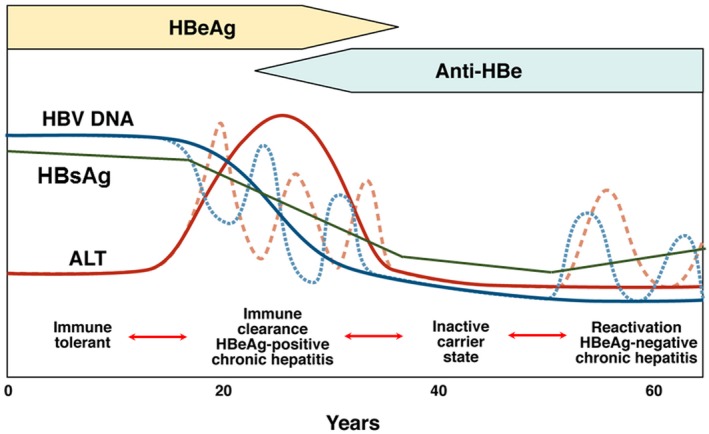
Phases of chronic HBV infection.3 Traditionally, phases of chronic HBV infection are defined by HBeAg status, serum HBV DNA, and ALT levels. Quantitative HBsAg levels are different in each phase and are generally highest in the immune tolerant phase and lowest in the inactive carrier phase. Although most patients progress from one phase to the next, not all patients go through each phase; reversion to an earlier phase can occur. Immune tolerant: HBeAg‐positive, high serum HBV DNA but normal ALT levels. Immune clearance/HBeAg‐positive chronic hepatitis: HBeAg‐positive, high serum HBV DNA, and elevated ALT levels; HBeAg seroconversion to anti‐HBe occurs after varying durations. Inactive carrier: HBeAg‐negative, serum HBV DNA low (generally <2,000 IU/mL) or undetectable. Reactivation/HBeAg‐negative chronic hepatitis: HBeAg‐negative, elevated levels of HBV DNA and ALT in serum, HBV precore and/or basal core promoter variant often present. Abbreviation: HBsAg, hepatitis B surface antigen.
During the first phase, patients are HBeAg‐positive with high HBV DNA (>7 log10 IU/mL) but normal ALT levels. These patients are considered to be in the “immune tolerant” phase, although recent studies showed that HBV‐specific T‐cell responses during this phase are not significantly different from those in active phases and that this phase should be renamed “low inflammatory” phase.7 During the second “HBeAg‐positive chronic hepatitis” phase, ALT becomes elevated. After varying durations, seroconversion from HBeAg to hepatitis B e antibody occurs. In some instances, this may be accompanied by ALT flares and even hepatic decompensation. After HBeAg loss, most patients enter the third “inactive carrier” phase. Patients in this phase are HBeAg‐negative and have low (<2,000 IU/mL) or undetectable HBV DNA and normal ALT. Prognosis is generally favorable if there had not been significant liver injury before entry to this phase and patients remain in this phase. Some patients directly transition from the HBeAg‐positive chronic hepatitis phase to the fourth “HBeAg‐negative chronic hepatitis” phase, whereas most enter this phase after varying durations in the inactive carrier phase. Patients in the HBeAg‐negative chronic hepatitis phase are HBeAg‐negative and have moderate to high HBV DNA (3‐7 log10 IU/mL) levels and elevated ALT. Although most patients progress from one phase to the next sequentially, reversion to the previous phase can occur. Furthermore, depending on the HBV DNA and ALT cut‐off values used to define these phases, as many as 30% to 40% of patients do not fit into any phase. Many of these patients may be in transition from one phase to another, whereas some with low viremia and elevated ALT may have concomitant causes of liver injury, most commonly nonalcoholic fatty liver disease.
Recent studies have shown that quantification of serum HBsAg levels can help in differentiating the phases of HBV infection; levels are highest in the immune tolerant phase and lowest in the inactive carrier phase. Quantitative HBsAg levels are most useful in differentiating whether a patient who is HBeAg‐negative with low HBV DNA is truly in the inactive carrier phase or in the quiescent period of the HBeAg‐negative chronic hepatitis phase and in predicting risk of HCC.8, 9
Eventually, some patients spontaneously clear HBsAg. HBsAg loss is reported to occur at the rate of 0.5% to 1% per year, but the incidence is not linear.10 Risk of adverse clinical outcomes is greatly reduced after HBsAg loss, but risk of HCC remains if HBsAg loss occurs after age 50 years or cirrhosis development or in the presence of hepatitis C or D coinfection.11 Currently, HCC surveillance is recommended for those who lost HBsAg after age 50 or after cirrhosis development, but it is unclear if this needs to be continued for life because the risk may decrease over time to a level when HCC surveillance is no longer cost effective.
Current Treatment
Goals of Treatment
The goals of hepatitis B treatment are to prevent the development of cirrhosis, liver failure, HCC, and HBV‐related deaths. Proving that treatment improves clinical outcomes will require years if not decades of surveillance. Thus, surrogate markers consisting of biochemical (normalization of ALT), virologic (suppression of serum HBV DNA to undetectable using sensitive real‐time polymerase chain reaction assays with detection limits of 10‐20 IU/mL), serologic (HBeAg and/or HBsAg loss with or without seroconversion to hepatitis B e antibody and hepatitis B surface antibody), and histologic (decrease in hepatic inflammation with or without reversal of fibrosis) tests have been used to measure efficacy of treatment.
Indications for Treatment
Chronic HBV infection, if acquired at a young age and if left untreated, is associated with a 20% to 40% lifetime risk of HBV‐related mortality. However, as described above, spontaneous virologic and clinical remissions with HBeAg and even HBsAg loss can occur. Thus, not all patients require treatment. Indications for treatment depend on the activity and severity of liver disease at presentation; for those with no cirrhosis at presentation, indications for treatment depend on the predicted risk of cirrhosis or HCC in the future. Various models have been developed to predict the risk of HCC. Variables included in each model differ but generally include demographics (sex, age), HBV markers (HBeAg status, HBV DNA level, and some also include HBV genotype and quantitative HBsAg), and liver disease parameters (ALT, platelets as a marker of cirrhosis/portal hypertension, cirrhosis status, or liver stiffness measurement as a marker of liver fibrosis). Although some of these models have been validated in external cohorts, prospective validation of these models in determining indications for treatment, intensity of monitoring, and need for HCC surveillance has not been performed.
The American Association for the Study of Liver Diseases (AASLD), the Asian Pacific Association for the Study of the Liver (APASL), and the European Association for the Study of the Liver (EASL) guidelines recommend antiviral treatment for patients with decompensated cirrhosis regardless of HBV DNA or ALT levels.12, 13, 14, 15 Treatment is also recommended for patients with compensated cirrhosis and detectable serum HBV DNA regardless of ALT level. Patients with severe exacerbations of chronic hepatitis B (ALT flares with accompanying increase in bilirubin) and those with acute liver failure are also recommended to receive antiviral treatment. Although high‐quality evidence to support these indications is not available, the potential benefits are undisputed and the possible risks are small. Of note, the landmark double‐blind randomized trial demonstrating a benefit of lamivudine versus placebo in reducing disease progression (defined as increase in Child‐Turcotte‐Pugh score by ≥2 points, clinical decompensation, HCC, or death) in patients with bridging fibrosis or cirrhosis enrolled patients with baseline serum HBV DNA >~140,000 IU/mL.16 Since then, several studies have suggested a benefit of treatment in patients with compensated cirrhosis and lower HBV DNA levels (including those with HBV DNA <2,000 IU/mL).
For patients with no cirrhosis at presentation, guidelines recommend treatment for those with chronic hepatitis, both HBeAg‐positive or HBeAg‐negative. Although HBV DNA levels are highest in the immune tolerant phase, current guidelines do not recommend treatment for patients in this phase because the likelihood of treatment‐induced HBeAg loss is extremely low. In one study comparing tenofovir disoproxil fumarate (TDF) monotherapy versus TDF plus emtricitabine, HBV DNA was suppressed to <69 IU/mL in 65% of patients, HBeAg loss occurred in 3% of patients, and HBsAg loss occurred in none of the patients at the end of 4 years of treatment; relapse occurred in all patients when treatment was discontinued.17
Recognizing that persistently high HBV DNA levels and having a family history of HCC increase the risk of HCC,18, 19 guidelines recommend treatment if patients remain in the immune tolerant phase after the age of 30 to 40 years or if there is a family history of HCC. The AASLD guidelines used age 40 as the cutoff based on data from a study showing the risk of cirrhosis development increased from 1.1% to 4.1% to 28% in patients who lost HBeAg before age 30, between 30 and 39, and after age 40, respectively.20 By contrast, the EASL guidelines selected age 30 as the cutoff. It is unclear which cutoff is more appropriate. Many experts have advocated for treatment early in the course of infection to minimize liver injury, integration of HBV DNA, and risk of HCC. However, it is unclear whether initiating treatment in the immune tolerant phase will prevent integration of HBV DNA because integration can occur early and can be detected in children. Similarly, although there is clear evidence that patients with a family history of HCC are at increased risk of HCC, it is unclear whether the risk is related to the number of family members affected, the biological relationships to the affected family members, and the age of family members when HCC was diagnosed. It is also unclear whether antiviral therapy initiated in the absence of standard indications will prevent HCC in patients who are genetically predisposed, are infected with an unusually virulent strain of HBV, or had been exposed to environmental carcinogens. Answers to these questions may be irrelevant if one day simple, safe, highly effective curative treatments similar to direct‐acting antivirals for hepatitis C become available when universal treatment would be recommended. In the meantime, studies that can provide answers to these nuances would be informative.
Another group of patients in the immune tolerant phase for which antiviral treatment is recommended are women with high viremia (>200,000 IU/mL).13 Several studies showed that administration of antiviral therapy to these mothers in the third trimester of pregnancy further reduced the risk of mother‐to‐child transmission in newborns who received hepatitis B immune globulin (HBIG) and the HBV vaccine at birth.21, 22 This benefit was not observed in a recent study where administration of HBIG and birth dose HBV vaccine occurred immediately after birth (median, 1.2‐1.3 hours) and a total of five versus three doses of HBV vaccine were administered.23 Further studies to determine whether more timely administration of HBIG and birth dose HBV vaccine to newborns might obviate the need for antiviral therapy in highly viremic mothers is warranted. Some experts have indicated that TDF is more readily available and at a lower cost than HBIG in resource‐limited countries; however, HBV DNA testing is also not readily available in these countries, making it difficult to differentiate high versus low viremic carrier mothers.
Antiviral therapy is also recommended as prophylaxis for patients who are HBsAg‐positive as well as patients who are HBsAg‐negative and hepatitis B core antibody‐positive who require treatment with immunosuppressive therapies that are predicted to have a moderate to high risk of HBV reactivation.13 A major challenge is in assessing the risk of HBV reactivation associated with immunosuppressive or cancer therapies because new drugs and biologics come on the market every month, a combination of drugs are frequently used, and treatment may be modified if initial response is unsatisfactory.
Efficacy and Safety of Available Treatments
Currently, two types of treatment, IFNs and NAs, are approved for chronic HBV infection. The virologic responses to these therapies are summarized in Table 1.13, 24, 25 Pegylated IFNs have a more convenient dosing schedule (once versus thrice weekly) and improved efficacy. Among the NAs, entecavir (ETV), TDF, and tenofovir alafenamide (TAF) are preferred because of their potent antiviral activity and high barrier to antiviral resistance. A 1‐year course of pegylated IFN results in higher rates of HBeAg seroconversion (~30% versus 10%‐21%) and HBsAg loss (3% versus <1%‐3%) than the same duration of ETV, TDF, or TAF therapy in patients who are HBeAg‐positive despite lower rates of undetectable HBV DNA (25% versus 61%‐76%). Similarly, in patients who are HBeAg‐negative, a 1‐year course of pegylated IFN results in a higher rate of HBsAg loss (4% versus 0%‐1%) than the same duration of ETV, TDF, or TAF therapy despite a lower rate of undetectable HBV DNA (63% versus 90%‐94%). Response to IFN is more durable, and rates of HBeAg and HBsAg loss continue to increase after cessation of treatment, whereas viral relapse is universal when NA is discontinued after 1 year of therapy.
Table 1.
| ETV | TDF | TAF | Pegylated IFN | |
|---|---|---|---|---|
| HBeAg‐positive patients | ||||
| At week 48 or 52 | ||||
| Undetectable HBV DNA | 67 | 67‐76 | 64 | 25 |
| HBeAg seroconversion | 21 | 12‐21 | 10 | 27 |
| HBsAg loss | 2 | <1‐3 | 1 | 3 |
| During extended treatment or follow‐up † | ||||
| Undetectable HBV DNA | 94 (5) | 97 (5) | 73 (2) | 13 (4.5) |
| HBeAg seroconversion | 41 (5) | 40 (5) | 18 (2) | 37 (4.5) |
| HBsAg loss | 5 (5) | 10 (5) | 1 (2) | 8 (4.5) |
| HBeAg‐negative patients | ||||
| At week 48 or 52 | ||||
| Undetectable HBV DNA | 90 | 93 | 94 | 63 |
| HBsAg loss | <1 | 0 | 0 | 4 |
| During extended treatment or follow‐up † | ||||
| Undetectable HBV DNA | NA | 99 (5) | 90 (2) | 18 (4) |
| HBsAg loss | NA | 0 (5) | <1 (2) | 8 (4) |
Abbreviation: NA, not available.
Responses presented as %.
Time point in which response was assessed in years while on treatment for ETV, TDF, or TAF and during off‐treatment follow‐up for pegylated IFN.
Follow‐up of patients for 3 to 4 years after completion of a 1‐year course of pegylated IFN showed that HBsAg loss increased to 8%. By contrast, continuous treatment with NA for up to 5 years has not been shown to further increase the rate of HBsAg loss except for one study of TDF in patients who were HBeAg‐positive where the rate of HBsAg loss increased to 10%. The low rate of HBsAg loss is related to the lack of direct effect of IFN and NA on cccDNA; thus, despite suppression of HBV DNA replication, cccDNA persists and transcription of pregenomic RNA and translation of viral protein continues. Furthermore, it is now recognized that integrated HBV DNA can also be a source of circulating HBsAg.
IFN has to be administered as injections and is associated with numerous adverse effects. It is contraindicated in patients with decompensated cirrhosis or severe exacerbations of chronic hepatitis and those with autoimmune or psychiatric illnesses and must be used with caution in patients with compensated cirrhosis. By contrast, NAs are administered orally and have negligible adverse effects, and risk of antiviral drug resistance is ≤1% after >5 years of continued treatment with ETV or TDF. NAs can be safely administered in patients with decompensated cirrhosis, HCC, severe exacerbations of chronic hepatitis, or acute liver failure. ETV was reported to be associated with lactic acidosis in one case series of hospitalized patients with decompensated cirrhosis, but other causes, such as sepsis, may have contributed to lactic acidosis in these acutely sick patients. Mitochondrial toxicity and lactic acidosis are potential adverse effects of all NAs, but documented associations with ETV, TDF, or TAF are extremely rare. TDF has been documented to decrease glomerular filtration rate as well as renal tubular function and to decrease bone mineral density. Although population studies suggest that the incidence of adverse events is low,26 given the need for long durations of NA therapy and increasing age of patients with chronic hepatitis B, safer alternatives are preferred. TAF is a new prodrug of tenofovir. TAF in doses of 25 mg has similar antiviral activity as TDF in doses of 300 mg and is associated with smaller decreases in glomerular filtration rate and bone mineral density.25 Whether TAF and ETV have a similar renal and bone safety profile has not been studied. Lamivudine, telbivudine, and TDF have been shown to be safe for the fetus when used in the first trimester of pregnancy.27 TDF is preferred because of its high barrier to resistance.
Resistance to NAs
NA treatment may select for viral variants that confer resistance. Resistance to NAs leads to virologic breakthrough (>1 log10 increase in HBV DNA from nadir while on treatment) that may be accompanied by biochemical breakthrough, hepatitis flares, and hepatic decompensation. Earlier NAs for hepatitis B were associated with very high rates of antiviral drug resistance that reached up to 70% after 5 years of lamivudine treatment. The risk of antiviral drug resistance in patients who are NA naive is ≤1% after 5 years of continued treatment with ETV and 8 years with TDF, but the risk of resistance to ETV is as high as 50% in patients with lamivudine‐resistant HBV.28, 29 For patients with prior NA exposure, TDF or TAF is preferred because mutations conferring resistance to lamivudine or telbivudine (M204V/I) constitute the first step in the two‐hit path to ETV resistance.
Predictors of Response
Among patients who are HBeAg‐positive, high ALT or histologic activity and low HBV DNA pretreatment have been consistently shown to be predictive of higher rates of HBeAg loss in response to IFN as well as NA therapy. HBV genotype is also a strong predictor of IFN‐related HBeAg and HBsAg loss.30 The basis for the genotype effect on IFN‐induced HBeAg and HBsAg loss is unclear. Clinical studies showed that patients with genotype A infection do not have lower baseline HBeAg or HBsAg levels. Understanding the biologic differences in HBV genotypes that contribute to a more favorable response of genotype A to IFN may help in optimizing the design of new antiviral or immunomodulatory therapies. Among patients who are HBeAg‐negative, there are no consistent baseline predictors of response to IFN or NAs. Because of the low event rate, predictors of HBsAg loss have not been identified for NA therapy, whereas genotype A is the only predictor of HBsAg loss for IFN therapy.
In patients who received IFN therapy, lack of or suboptimal decline in quantitative HBsAg but not HBV DNA has a high negative predictive value for response defined as HBeAg loss in patients who are HBeAg‐positive or sustained off‐treatment virus suppression in patients who are HBeAg‐negative. This has led to the development of stop rules such that patients who are predicted not to have a successful response may discontinue IFN after 12 or 24 weeks, sparing them unnecessary adverse effects.31 However, these stop rules are genotype specific and have not been validated in prospective studies.
Choice of Antiviral Drug and Duration of Treatment
For patients who are eligible to receive either IFN or NA therapy, the choice depends on patient preference and predicted likelihood of response. IFN has to be administered as injections and is associated with many side effects, but it is given for a finite duration and has a higher rate of HBeAg and HBsAg loss, particularly for patients with genotype A infection. Patients who are HBeAg‐positive with genotype A infection, high ALT (>2 times normal), and low HBV DNA (<8 log10 IU/mL) with no cirrhosis and no contraindications to the use of IFN are the best candidates for IFN therapy.
NAs are taken orally and have negligible side effects but need to be administered for many years and in some patients for life. Thus, willingness to commit to a long duration of treatment and ability to adhere to the treatment are important deciding factors. ETV, TDF, and TAF are preferred NAs. ETV or TAF is preferred for patients with underlying renal or bone disease. TDF or TAF should be used in patients with prior NA exposure. TDF should be used in pregnant women and women contemplating pregnancy.
De novo combination of NAs has not been shown to be superior to NA monotherapy except for patients with very high baseline HBV DNA (>8 log10 IU/mL) where a more rapid decline in viremia is achieved with combination therapy.32 Similarly, although combination therapy was initially advocated for patients with NA resistance, subsequent studies showed that TDF monotherapy has similar efficacy compared to a combination of TDF plus emtricitabine in patients with lamivudine resistance.33 Several strategies to combine pegylated IFN and NA have been tried, including de novo combination, add‐on, and switch‐to approaches. Although some studies have shown a benefit compared to monotherapy, the results are not consistent and may not be generalized because many studies only enrolled highly selected patients.31 A study comparing de novo combination pegylated IFN plus TDF versus pegylated IFN or TDF monotherapy showed that combination therapy was associated with a higher rate of HBsAg loss, but this benefit was mainly observed in patients with genotype A infection.34 Further studies are needed to identify the subset of patients who are most likely to benefit and to optimize study design of combination therapy with pegylated IFN and NA; however, given the plethora of new antiviral and immunomodulatory therapies in clinical trials, support for these studies might be difficult to garner.
Pegylated IFN is administered for 1 year (48‐52 weeks) in both HBeAg‐positive and HBeAg‐negative patients. A clinical trial comparing 24 versus 48 weeks pegylated IFN in patients who were HBeAg‐positive showed that 24‐week duration was inferior.35 Another trial comparing 48 versus 96 weeks of pegylated IFN in patients who were HBeAg‐negative suggests that a 96‐week duration is superior, but the study included a small number of patients.36 As discussed earlier, the stop rule may be applied at week 12 or 24 to identify patients for whom continued treatment is predicted to be futile.
Guidelines recommend that NA treatment should be continued indefinitely in patients with cirrhosis prior to the start of treatment.12, 13, 14, 15 The APASL guidelines indicate NA may be discontinued in selected patients with cirrhosis provided close monitoring is feasible. All guidelines recommend that NAs should be administered for 12 additional months (consolidation) after HBeAg seroconversion is achieved in patients who are HBeAg‐positive with no cirrhosis. Given that only 40% of patients achieve HBeAg seroconversion after 5 years of ETV or TDF,37, 38 most patients will require more than 6 years of treatment. All guidelines recommend that NAs can be stopped when HBsAg is lost for patients who are HBeAg‐negative with no cirrhosis, but with <1% of patients achieving this outcome after 5 years of treatment, nearly all patients will require lifelong treatment. All patients must be closely monitored after discontinuation of NA such that treatment can be resumed if there is evidence of clinical relapse (HBV DNA and ALT increasing to levels that meet treatment indications).
Recent studies suggest paradoxically that, for patients who are HBeAg‐negative who had been virally suppressed for >2 to 3 years, withdrawal of an NA may be associated with a higher rate of HBsAg loss compared to those who continued the NA.39, 40, 41 This has led the APASL and EASL guidelines to include a provision for such an approach as long as patients agree to close monitoring after treatment is discontinued. However, outcomes, including risks of hepatic decompensation, frequency of monitoring, and criteria for resuming treatment, need to be studied in larger cohorts before this approach is widely adopted.
The reason for a higher rate of HBsAg loss after NA withdrawal is unclear. It has been postulated that viral relapse after a long duration of virus suppression may mimic acute hepatitis B, provoking a rigorous immune response to HBV leading to HBsAg loss. Among patients who experienced clinical relapse, one study found that those who did not resume treatment had a higher rate of HBsAg loss than those who did, suggesting that HBsAg loss is more likely to occur if immune clearance is allowed to proceed and not be curbed too soon. However, overly rigorous or prolonged immune lysis of infected hepatocytes can result in liver failure. Moreover, this same study showed that patients who had no viral relapse and those who had viral but not clinical relapse had higher rates of HBsAg loss than those who had clinical relapse, indicating that hepatitis flares are not required for HBsAg loss.40
The ability to identify which patients will remain in remission, which ones will experience viral relapse only, which ones will experience clinical relapse, and which ones will decompensate when NA is stopped is an important area for research. It is also important to have a standardized definition for viral and clinical relapse so that data from different studies can be compared. Several studies suggest that serum HBsAg, HBV RNA, and hepatitis B core related antigen levels, which are surrogate markers for cccDNA, before stopping treatment may be predictive.42 Validation of these markers is necessary, and if confirmed, standardized assays must be available for clinical use.
Long‐Term Outcomes
Continued follow‐up of patients who completed a course of IFN treatment showed that rates of HBeAg seroconversion increased to 37% after 4 to 5 years and HBsAg loss to 8% after 4 years.43, 44 However, these incremental responses were mainly seen in patients with genotype A infection. Studies have also shown that continued NA treatment is associated with increasing rates of HBeAg seroconversion to 40% after 5 years of ETV or TDF,37, 38 but there is minimal increase in rates of HBsAg loss.
Antiviral treatment has also been shown to improve liver histology. Biopsies performed after 1 year of treatment showed decreased inflammation but not fibrosis, and biopsies performed after 3 to 5 years of continued NA therapy have demonstrated a decrease in both inflammation and fibrosis. A study of 348 patients who had liver biopsies at baseline and after 240 weeks of TDF found that 87% had decreased inflammation and 52% had decreased fibrosis. Importantly, 74% of patients who had cirrhosis at enrollment no longer had cirrhosis.38 It would be important to know whether patients who have regression of cirrhosis have a lower risk of HCC compared to those without regression. Liver biopsies, particularly serial biopsies, are seldom performed in clinical practice. Studies using biomarker panels or elastography are needed to determine what criteria should be used to assess antiviral‐induced fibrosis regression because these noninvasive methods measure not only fibrosis but also inflammation and initial improvement after treatment may be due to decreased inflammation and not fibrosis.
Antiviral therapy has also been shown to improve clinical outcomes. A landmark randomized controlled trial showed that lamivudine decreased the risk of disease progression and HCC in patients with advanced fibrosis or cirrhosis.16 Long‐term follow‐up studies and meta‐analyses indicate that IFN and NA therapy decrease the development of cirrhosis, cirrhosis complications, HCC, and mortality.2, 45, 46 However, the incidence of HCC is not eliminated, and continued surveillance is required. A recent study showed that the incidence of HCC appeared to level off after 5 years of ETV or TDF in patients with cirrhosis,47 suggesting that with a longer duration of suppressed HBV replication and decreased hepatic inflammation, development of new HCC can be prevented. In this study, all new HCC after year 5 occurred in patients >50 years old. These data support the recommendations to initiate treatment as early as possible. Longer follow‐up is needed to determine whether the incidence of HCC will eventually stop in patients with viral suppression on NAs.
Future of HBV Treatment
The availability of a simple, safe, and highly effective cure for hepatitis C has reenergized the search for a cure for hepatitis B. However, as described earlier, a sterilizing cure with elimination of both cccDNA and integrated HBV DNA may not be possible. Instead, experts agree that a functional cure akin to spontaneous HBsAg loss in patients with chronic HBV infection may be a realistic goal.3 The endpoint would be HBsAg loss accompanied by HBeAg loss and undetectable HBV DNA in serum, but HBV DNA may persist in the liver as integrated HBV DNA and as transcriptionally inactive cccDNA. Progression of liver disease would be halted, and over time fibrosis would regress and the incidence of HCC would decrease. Whether seroconversion to hepatitis B surface antibody is necessary to prevent HBsAg seroreversion remains to be determined. This functional cure is currently achievable in a small percentage (<10%) of patients after IFN or NA therapy. The vision for the future is to deploy a combination of antiviral drugs directed against new targets and immunomodulatory therapies to restore innate as well as adaptive immune responses with the goal of achieving HBsAg loss in a higher percentage (>50%) of patients after a finite course (≥2 years) of treatment. These new strategies may or may not include IFN or NAs.
New antiviral drugs in clinical trials include entry receptor inhibitors, capsid assembly modifiers, RNA interference, and nucleic acid polymers.3 To date, capsid assembly modifiers have shown promising results, but the effect on HBsAg levels has been modest and there is a concern for antiviral drug resistance. RNA interference has been shown to result in >1 log decrease in HBsAg levels, but it has to be administered as injections and there is a concern for off‐target effects. Studies of nucleic acid polymers have shown a high rate of HBsAg loss, but the design of those studies is complex, the exact mechanism of action of nucleic acid polymers is unclear, and marked ALT flares were observed in a high percentage of patients; thus, safety needs to be established and efficacy needs to be confirmed. To date, studies of immune modulatory therapies, including therapeutic vaccines to stimulate T‐cell response and toll‐like receptor agonists to enhance innate immune response, have had limited success. Given the recent success of immune therapies in oncology, enthusiasm for immune modulatory therapies, including check‐point inhibitors and engineered T cells, remains high. A major concern of immune modulatory therapies is uncontrolled immune activation leading to fatal hepatitis flares or extrahepatic organ damage.
Major challenges with developing new drugs for hepatitis B include safety (given the excellent safety profile of NAs), ease of administration (NAs are taken as pills once a day), and cost (ETV and TDF are both off patent). However, given the high global burden of hepatitis B, the desire to achieve a “cure” with a finite course of therapy, and the need to effectively control HBV replication at an early stage of chronic HBV infection, it is hoped that there will be sufficient interest and commitment from the pharmaceutical industry and the scientific community to work together to make an HBV cure a reality in the not too distant future.
Potential conflict of interest
Dr. Lok has received research support from Bristol‐Myers Squibb and Gilead Sciences and has served as advisor for Gilead Sciences, Spring Bank, Hoffmann la Roche, and Viravaxx.
References
Author names in bold designate shared co‐first authorship.
- 1. Polaris Observatory Collaborators . Global prevalence, treatment, and prevention of hepatitis B virus infection in 2016: a modelling study. Lancet Gastroenterol Hepatol 2018;3:383‐403. [DOI] [PubMed] [Google Scholar]
- 2. Lok AS, McMahon BJ, Brown RS Jr, Wong JB, Ahmed AT, Farah W, et al. Antiviral therapy for chronic hepatitis B viral infection in adults: a systematic review and meta‐analysis. Hepatology 2016;63:284‐306. [DOI] [PubMed] [Google Scholar]
- 3. Lok AS, Zoulim F, Dusheiko G, Ghany MG. Hepatitis B cure: from discovery to regulatory approval. Hepatology 2017;66:1296‐1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lucifora J, Xia Y, Reisinger F, Zhang K, Stadler D, Cheng X, et al. Specific and nonhepatotoxic degradation of nuclear hepatitis B virus cccDNA. Science 2014;343:1221‐1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wooddell CI, Yuen MF, Chan HL, Gish RG, Locarnini SA, Chavez D, et al. RNAi‐based treatment of chronically infected patients and chimpanzees reveals that integrated hepatitis B virus DNA is a source of HBsAg. Sci Transl Med 2017;9:pii:eaan0241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ferrari C, Penna A, Bertoletti A, Valli A, Antoni AD, Giuberti T, et al. Cellular immune response to hepatitis B virus‐encoded antigens in acute and chronic hepatitis B virus infection. J Immunol 1990;145:3442‐3449. [PubMed] [Google Scholar]
- 7. Bertoletti A, Kennedy PT. The immune tolerant phase of chronic HBV infection: new perspectives on an old concept. Cell Mol Immunol 2015;12:258‐263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tseng TC, Liu CJ, Yang HC, Su TH, Wang CC, Chen CL, et al. High levels of hepatitis B surface antigen increase risk of hepatocellular carcinoma in patients with low HBV load. Gastroenterology 2012;142:1140‐1149.e3. [DOI] [PubMed] [Google Scholar]
- 9. Cornberg M, Wong VW, Locarnini S, Brunetto M, Janssen HLA, Chan HL. The role of quantitative hepatitis B surface antigen revisited. J Hepatol 2017;66:398‐411. [DOI] [PubMed] [Google Scholar]
- 10. Chu CM, Liaw YF. HBsAg seroclearance in asymptomatic carriers of high endemic areas: appreciably high rates during a long‐term follow‐up. Hepatology 2007;45:1187‐1192. [DOI] [PubMed] [Google Scholar]
- 11. Yuen MF, Wong DK, Fung J, Ip P, But D, Hung I, et al. HBsAg Seroclearance in chronic hepatitis B in Asian patients: replicative level and risk of hepatocellular carcinoma. Gastroenterology 2008;135:1192‐1199. [DOI] [PubMed] [Google Scholar]
- 12. Terrault NA, Bzowej NH, Chang KM, Hwang JP, Jonas MM, Murad MH, et al.; American Association for the Study of Liver Diseases . AASLD guidelines for treatment of chronic hepatitis B. Hepatology 2016;63:261‐283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Terrault NA, Lok ASF, McMahon BJ, Chang KM, Hwang JP, Jonas MM, et al. Update on prevention, diagnosis, and treatment of chronic hepatitis B: AASLD 2018 hepatitis B guidance. Hepatology 2018;67:1560‐1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sarin SK, Kumar M, Lau GK, Abbas Z, Chan HL, Chen CJ, et al. Asian‐Pacific clinical practice guidelines on the management of hepatitis B: A 2015 update. Hepatol Int 2016;10:1‐98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. European Association for the Study of the Liver . EASL 2017 clinical practice guidelines on the management of hepatitis B virus infection. J Hepatol 2017;67:370‐398. [DOI] [PubMed] [Google Scholar]
- 16. Liaw YF, Sung JJ, Chow WC, Farrell G, Lee CZ, Yuen H, et al. Cirrhosis Asian Lamivudine Multicentre Study Group. Lamivudine for patients with chronic hepatitis B and advanced liver disease. N Engl J Med 2004;351:1521‐1531. [DOI] [PubMed] [Google Scholar]
- 17. Chan HL, Chan CK, Hui AJ, Chan S, Poordad F, Chang TT, et al. Effects of tenofovir disoproxil fumarate in hepatitis B e antigen‐positive patients with normal levels of alanine aminotransferase and high levels of hepatitis B virus DNA. Gastroenterology 2014;146:1240‐1248. [DOI] [PubMed] [Google Scholar]
- 18. Chen CJ, Yang HI, Su J, Jen CL, You SL, Lu SN, et al.; REVEAL ‐ HBV Study Group . Risk of hepatocellular carcinoma across a biological gradient of serum hepatitis B virus DNA level. JAMA 2006;295:65‐73. [DOI] [PubMed] [Google Scholar]
- 19. Loomba R, Liu J, Yang HI, Lee MH, Lu SN, Wang LY, et al.; REVEAL‐HBV Study Group . Synergistic effects of family history of hepatocellular carcinoma and hepatitis B virus infection on risk for incident hepatocellular carcinoma. Clin Gastroenterol Hepatol 2013;11:1636‐1645.e1‐e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chu CM, Liaw YF. Chronic hepatitis B virus infection acquired in childhood: special emphasis on prognostic and therapeutic implication of delayed HBeAg seroconversion. J Viral Hepat 2007;14:147‐152. [DOI] [PubMed] [Google Scholar]
- 21. Brown RS Jr, McMahon BJ, Lok AS, Wong JB, Ahmed AT, Mouchli MA, et al. Antiviral therapy in chronic hepatitis B viral infection during pregnancy: a systematic review and meta‐analysis. Hepatology 2016;63:319‐333. [DOI] [PubMed] [Google Scholar]
- 22. Pan CQ, Duan Z, Dai E, Zhang S, Han G, Wang Y, et al.; China Study Group for the Mother‐to‐Child Transmission of Hepatitis B . Tenofovir to prevent hepatitis B transmission in mothers with high viral load. N Engl J Med 2016;374:2324‐2334. [DOI] [PubMed] [Google Scholar]
- 23. Jourdain G, Ngo‐Giang‐Huong N, Harrison L, Decker L, Khamduang W, Tierney C, et al. Tenofovir versus placebo to prevent perinatal transmission of hepatitis B. N Engl J Med 2018;378:911‐923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yapali S, Talaat N, Lok AS. Management of hepatitis B: our practice and how it relates to the guidelines. Clin Gastroenterol Hepatol 2014;12:16‐26. [DOI] [PubMed] [Google Scholar]
- 25. Agarwal K, Brunetto M, Seto WK, Lim YS, Fung S, Marcellin P, et al.; GS‐US‐320‐0110; GS‐US‐320‐0108 Investigators . 96 weeks treatment of tenofovir alafenamide vs. tenofovir disoproxil fumarate for hepatitis B virus infection. J Hepatol 2018;68:672‐681. [DOI] [PubMed] [Google Scholar]
- 26. Wong GL, Tse YK, Wong VW, Yip TC, Tsoi KK, Chan HL. Long‐term safety of oral nucleos(t)ide analogs for patients with chronic hepatitis B: a cohort study of 53,500 subjects. Hepatology 2015;62:684‐693. [DOI] [PubMed] [Google Scholar]
- 27. Antiretroviral Pregnancy Registry Steering Committee . Antiretrovial pregnancy registry interim report for 1 January 1989 through January 2018. www.apregistry.com/forms/interim_report.pdf. Published June 2018. Accessed.
- 28. Tenney DJ, Rose RE, Baldick CJ, Pokornowski KA, Eggers BJ, Fang J, et al. Long‐term monitoring shows hepatitis B virus resistance to entecavir in nucleoside‐naive patients is rare through 5 years of therapy. Hepatology 2009;49:1503‐1514. [DOI] [PubMed] [Google Scholar]
- 29. Liu Y, Corsa AC, Buti M, Cathcart AL, Flaherty JF, Miller MD, et al. No detectable resistance to tenofovir disoproxil fumarate in HBeAg+ and HBeAg‐ patients with chronic hepatitis B after 8 years of treatment. J Viral Hepat 2017;24:68‐74. [DOI] [PubMed] [Google Scholar]
- 30. Buster EH, Hansen BE, Lau GK, Piratvisuth T, Zeuzem S, Steyerberg EW, et al. Factors that predict response of patients with hepatitis B e antigen‐positive chronic hepatitis B to peginterferon‐alfa. Gastroenterology 2009;137:2002‐2009. [DOI] [PubMed] [Google Scholar]
- 31. Konerman MA, Lok AS. Interferon treatment for hepatitis B. Clin Liver Dis 2016;20:645‐665. [DOI] [PubMed] [Google Scholar]
- 32. Lok AS, Trinh H, Carosi G, Akarca US, Gadano A, Habersetzer F, et al. Efficacy of entecavir with or without tenofovir disoproxil fumarate for nucleos(t)ide‐naive patients with chronic hepatitis B. Gastroenterology 2012;143:619‐628.e1. [DOI] [PubMed] [Google Scholar]
- 33. Fung S, Kwan P, Fabri M, Horban A, Pelemis M, Hann HW, et al. Tenofovir disoproxil fumarate (TDF) vs. emtricitabine (FTC)/TDF in lamivudine resistant hepatitis B: a 5‐year randomised study. J Hepatol 2017;66:11‐18. [DOI] [PubMed] [Google Scholar]
- 34. Marcellin P, Ahn SH, Ma X, Caruntu FA, Tak WY, Elkashab M, et al. Study 149 Investigators. Combination of tenofovir disoproxil fumarate and peginterferon alpha‐2a increases loss of hepatitis B surface antigen in patients with chronic hepatitis B. Gastroenterology 2016;150:134‐144.e10. [DOI] [PubMed] [Google Scholar]
- 35. Liaw YF, Jia JD, Chan HL, Han KH, Tanwandee T, Chuang WL, et al. Shorter durations and lower doses of peginterferon alfa‐2a are associated with inferior hepatitis B e antigen seroconversion rates in hepatitis B virus genotypes B or C. Hepatology 2011;54:1591‐1599. [DOI] [PubMed] [Google Scholar]
- 36. Lampertico P, Vigano M, Di Costanzo GG, Sagnelli E, Fasano M, Di Marco V, et al.;PegBeLiver Study Group . Randomised study comparing 48 and 96 weeks peginterferon alpha‐2a therapy in genotype D HBeAg‐negative chronic hepatitis B. Gut 2013;62:290‐298. [DOI] [PubMed] [Google Scholar]
- 37. Chang TT, Lai CL, Kew Yoon S, Lee SS, Coelho HS, Carrilho FJ, et al. Entecavir treatment for up to 5 years in patients with hepatitis B e antigen‐positive chronic hepatitis B. Hepatology 2010;51:422‐430. [DOI] [PubMed] [Google Scholar]
- 38. Marcellin P, Gane E, Buti M, Afdhal N, Sievert W, Jacobson IM, et al. Regression of cirrhosis during treatment with tenofovir disoproxil fumarate for chronic hepatitis B: A 5‐year open‐label follow‐up study. Lancet 2013;381:468‐475. [DOI] [PubMed] [Google Scholar]
- 39. Hadziyannis SJ, Sevastianos V, Rapti I, Vassilopoulos D, Hadziyannis E. Sustained responses and loss of HBsAg in HBeAg‐negative patients with chronic hepatitis B who stop long‐term treatment with adefovir. Gastroenterology 2012;143:629‐636.e1. [DOI] [PubMed] [Google Scholar]
- 40. Jeng WJ, Chen YC, Chien RN, Sheen IS, Liaw YF. Incidence and predictors of hepatitis B surface antigen seroclearance after cessation of nucleos(t)ide analogue therapy in hepatitis B e antigen‐negative chronic hepatitis B. Hepatology 2018;68:425‐434. [DOI] [PubMed] [Google Scholar]
- 41. Berg T, Simon KG, Mauss S, Schott E, Heyne R, Klass DM, et al.; FINITE CHB study investigators . Long‐term response after stopping tenofovir disoproxil fumarate in non‐cirrhotic HBeAg‐negative patients ‐ FINITE study. J Hepatol 2017;67:918‐924. [DOI] [PubMed] [Google Scholar]
- 42. Wang J, Shen T, Huang X, Kumar GR, Chen X, Zeng Z, et al. Serum hepatitis B virus RNA is encapsidated pregenome RNA that may be associated with persistence of viral infection and rebound. J Hepatol 2016;65:700‐710. [DOI] [PubMed] [Google Scholar]
- 43. Buster EH, Flink HJ, Cakaloglu Y, Simon K, Trojan J, Tabak F, et al. Sustained HBeAg and HBsAg loss after long‐term follow‐up of HBeAg‐positive patients treated with peginterferon alpha‐2b. Gastroenterology 2008;135:459‐467. [DOI] [PubMed] [Google Scholar]
- 44. Marcellin P, Bonino F, Lau GK, Farci P, Yurdaydin C, Piratvisuth T, et al.; Peginterferon alfa‐2a in HBeAg‐negative Chronic Hepatitis B Study Group . Sustained response of hepatitis B e antigen‐negative patients 3 years after treatment with peginterferon alpha‐2a. Gastroenterology 2009;136:2169‐2179.e1‐e4.19303414 [Google Scholar]
- 45. Papatheodoridis GV, Sypsa V, Dalekos G, Yurdaydin C, van Boemmel F, Buti M, et al. Eight‐year survival in chronic hepatitis B patients under long‐term entecavir or tenofovir therapy is similar to the general population. J Hepatol 2018;68:1129‐1136. [DOI] [PubMed] [Google Scholar]
- 46. Wong GL, Chan HL, Mak CW, Lee SK, Ip ZM, Lam AT, et al. Entecavir treatment reduces hepatic events and deaths in chronic hepatitis B patients with liver cirrhosis. Hepatology. 2013;58:1537–1547. [DOI] [PubMed] [Google Scholar]
- 47. Papatheodoridis GV, Idilman R, Dalekos GN, Buti M, Chi H, van Boemmel F, et al. The risk of hepatocellular carcinoma decreases after the first 5 years of entecavir or tenofovir in Caucasians with chronic hepatitis B. Hepatology 2017;66:1444‐1453. [DOI] [PubMed] [Google Scholar]