

Abstract
Bile acid (BA) homeostasis is tightly regulated by multiple transcription factors, including farnesoid X receptor (FXR) and small heterodimer partner (SHP). We previously reported that loss of the FXR/SHP axis causes severe intrahepatic cholestasis, similar to human progressive familial intrahepatic cholestasis type 5 (PFIC5). In this study, we found that constitutive androstane receptor (CAR) is endogenously activated in Fxr:Shp double knockout (DKO) mice. To test the hypothesis that CAR activation protects DKO mice from further liver damage, we generated Fxr;Shp;Car triple knockout (TKO) mice. In TKO mice, residual adenosine triphosphate (ATP) binding cassette, subfamily B member 11 (ABCB11; alias bile salt export pump [BSEP]) function and fecal BA excretion are completely impaired, resulting in severe hepatic and biliary damage due to excess BA overload. In addition, we discovered that pharmacologic CAR activation has different effects on intrahepatic cholestasis of different etiologies. In DKO mice, CAR agonist 1,4‐bis[2‐(3,5‐dichloropyridyloxy)]benzene (TCPOBOP; here on TC) treatment attenuated cholestatic liver injury, as expected. However, in the PFIC2 model Bsep knockout (BKO) mice, TC treatment exhibited opposite effects that reflect increased BA accumulation and liver injury. These contrasting results may be linked to differential regulation of systemic cholesterol homeostasis in DKO and BKO livers. TC treatment selectively up‐regulated hepatic cholesterol levels in BKO mice, supporting de novo BA synthesis. Conclusion: CAR activation in DKO mice is generally protective against cholestatic liver injury in these mice, which model PFIC5, but not in the PFIC2 model BKO mice. Our results emphasize the importance of the genetic and physiologic background when implementing targeted therapies to treat intrahepatic cholestasis.
Abbreviations
- ABCB11
adenosine triphosphate binding cassette subfamily B member 11
- ALT
alanine aminotransferase
- ANOVA
analysis of variance
- AST
aspartate aminotransferase
- ATP
adenosine triphosphate
- BA
bile acid
- BKO
bile acid export pump knockout
- BSEP
bile acid export pump
- BW
body weight
- CAR
constitutive androstane receptor
- Ces
carboxylesterase
- CO
corn oil
- CYP7A1
cytochrome P450, family 7, subfamily a, polypeptide 1
- DKO
double knockout
- FXR
farnesoid X receptor
- GGT
gamma‐glutamyltransferase
- GST
glutathione S‐transferase
- HDL
high‐density lipoprotein
- HSD
honestly significant difference
- KEGG
Kyoto Encyclopedia of Genes and Genomes
- KRT19
keratin 19
- LCAT
lecithin cholesterol acyltransferase
- LC/MS‐MS
liquid chromatography–tandem mass spectrometry
- LDL
low‐density lipoprotein
- LPL
lipoprotein lipase
- mRNA
messenger RNA
- MRP
multidrug‐resistance like protein
- NR1H4
nuclear receptor subfamily 1, group H, member 4
- PFIC
progressive familial intrahepatic cholestasis
- PLTP
plasma phospholipid transfer protein
- PXR
pregnane X receptor
- qPCR
quantitative polymerase chain reaction
- RRID
Research Resource Identifier
- SHP
small heterodimer partner
- SREBP/SREBF
sterol regulatory element binding protein/transcription factor
- TBA
total bile acid
- TC
TCPOBOP
- TCPOBOP
1,4‐bis[2‐(3,5‐dichloropyridyloxy)]benzene
- TKO
triple knockout
- WT
wild type
Bile acids (BAs) act as endogenous surfactants required for digestion of fats and fat‐soluble vitamins. BAs are also implicated in multiple biological processes, acting as metabolic hormones to regulate liver functions, including systemic energy homeostasis.1 BAs are primarily synthesized from cholesterol in the liver by a variety of enzymatic reactions in the endoplasmic reticulum, mitochondria, peroxisomes, and cytosol2; they are then excreted, mostly reabsorbed in the intestine, and returned to the liver in a process known as enterohepatic circulation that is crucial for maintaining normal BA levels.3
Hereditary or acquired disruption of enterohepatic BA circulation causes cholestasis. The common hallmark of cholestasis is the accumulation of toxic BAs in the liver and in circulation. This can be due to reduced hepatic BA excretion (intrahepatic) or bile duct obstruction (extrahepatic). Intrahepatic cholestasis reflects dysregulation of membrane BA and phospholipid transport as well as BA synthesis and conjugation processes.4 Classical hereditary defects identify progressive familial intrahepatic cholestasis (PFIC), which is a group of five autosomal recessive disorders caused by gene mutations in adenosine triphosphate 8B1 (ATP8B1) (aliases FIC1, PFIC1), residual adenosine triphosphate binding cassette, subfamily B, member 11 (ABCB11) (aliases bile salt export pump [BSEP], PFIC2), ABCB4 (aliases multidrug resistance 3 [MDR3], PFIC3), tight junction protein 2 (TJP2; alias PFIC4), and nuclear receptor subfamily 1, group H, member 4 (NR1H4; aliases farnesoid X receptor [FXR], PFIC5).5 PFIC typically emerges in infancy and childhood, resulting in end‐stage liver disease and death unless it is properly treated.6
Physiologic levels of BAs are tightly regulated through a multistep feedback loop by FXR (NR1H4) and its downstream target small heterodimer partner (SHP) (NR0B2).7 FXR is directly activated by BAs, like chenodeoxycholic acid, which suppresses de novo BA synthesis and enhances BA excretion, largely through repression of cytochrome P450, family 7, subfamily a, polypeptide 1 (CYP7A1) and induction of BSEP (ABCB11) expression.8 SHP is a direct FXR target gene and acts as a transcriptional repressor of several transcription factors to inhibit Cyp7a1 expression.9 Thus, FXR and SHP activation restrains BA synthesis and facilitates enterohepatic BA circulation to maintain BA homeostasis.
Recently, we reported surprising distinct molecular pathogenesis in two PFIC models, despite common cholestasis phenotypes.10 Combined deletion of FXR and SHP (double knockout [DKO]) causes juvenile onset intrahepatic cholestasis with severe liver damage,11 similar to the human PFIC5 disease.12 DKO mice exhibit substantially higher Cyp7a1 expression and decreased Abcb11 expression compared to Fxr or Shp single knockouts. DKO alters the expression of many genes involved in BA metabolism and BA transport.11 Bile salt export pump knockouts (BSEP KOs), which recapitulate human PFIC2 disease,13 share many aspects of the DKO phenotype, including similarly elevated total hepatic BAs and hepatomegaly. However, DKO and BKO have very different BA composition and gene expression profiles.10 This is primarily due to activation of constitutive androstane receptor (CAR) and pregnane X receptor (PXR) in DKO but not BKO livers.10 However, the detailed molecular mechanisms that account for this striking difference are largely unknown.
Multiple BA‐activated nuclear receptors, such as FXR, CAR, PXR, and vitamin D receptor, play crucial roles in hepatobiliary BA homeostasis.14 Although there is no evidence that BAs directly activate CAR following cholestatic liver injury, CAR activation generally mitigates BA‐induced liver injury.15, 16 Genetic ablation of CAR accelerates liver injury in lithocholic acid‐treated or bile duct‐ligated mice.16, 17 CAR agonist 1,4‐bis[2‐(3,5‐dichloropyridyloxy)]benzene (TC) treatment reverses these phenotypes through enhanced bilirubin and BA clearance.16, 18 However, these studies were mainly based on the extrahepatic cholestasis model induced by surgical biliary obstruction or a hydrophobic BA‐containing diet. CAR function in a genetic model of intrahepatic cholestasis has not been evaluated.
Here, we investigated the selective function of CAR in intrahepatic cholestasis. We found that loss of CAR in DKO mice exacerbated BA‐induced toxicity, implying a protective function of endogenous CAR activation in DKO liver. Interestingly, pharmacologic CAR activation differentially regulated BA homeostasis in DKO and BKO models. Using transcriptomic and proteomic approaches, we analyzed the molecular mechanism underlying the distinct responses of CAR, which may ultimately establish CAR as a selective therapeutic target to treat specific subsets of intrahepatic cholestatic disease.
Materials and Methods
Animal Studies
C57BL/6J and BKO mice were purchased from the Jackson Laboratory (#000664 and #004125, respectively; Bar Harbor, ME). Car KO (Nr1i3–/–) and Fxr:Shp DKO (Nr1h4–/–;Nr0b2–/–) have been described.11, 19 Male mice were used for all experiments. Gallbladder volume was calculated by use of the ellipsoid formula (volume = 0.52 × length × width2). To activate CAR, TC (T1443; Sigma Aldrich, St Louis, MO) was intraperitoneally injected in DKO and BKO mice for 2 weeks (twice per week, 2 mg/kg body weight [BW]). To measure the BA excretion rate, urine, bile, and feces were collected the day after TC treatment. All animal studies and procedures were approved by the Institutional Animal Care and Use Committee of Baylor College of Medicine. The serum liver panel was analyzed by the Center for Comparative Medicine (Baylor College of Medicine).
total BA and Cholesterol Level
To analyze serum BA and cholesterol level, serum was separated from whole blood using a gel barrier collection tube (CAPIJECT 3T‐MGA; Terumo Medical Corporation, Somerset, NJ) and then analyzed with the Total Bile Acid Assay Kit (GWB‐BQK090; GenWay Biotech, San Diego, CA) or the Infinity Total Cholesterol Reagent (TR13421; Thermo Fisher Scientific, Waltham, MA). For hepatic BA analysis, liver homogenate in 70% ethanol was used. For hepatic cholesterol level, liver homogenates were mixed with a 1:2 chloroform:methanol solution and the lipid‐containing chloroform layer was isolated according to the modified Folch method. After evaporation, lipids were resuspended in 100% ethanol containing 1% Tween 20. Uninary, biliary, and fecal BA levels were analyzed in gallbladder bile and dried feces. Serum fractions were collected by high‐performance liquid chromatography to measure lipoprotein lipid profiles (Mouse Metabolic and Phenotyping Core, Baylor College of Medicine).
Real‐Time Quantitative Polymerase Chain Reaction
Tissue RNA was isolated by PureXtract RNAsol reagent (R6101; GenDEPOT, Inc. Katy, TX), and complementary DNA was synthesized by the qScript Reverse Transcriptase Kit (95047; Quantabio, Beverly, MA). KAPA SYBR FAST quantitative polymerase chain reaction (qPCR) reagent (KAPA Biosystems, Wilmington, MA) was used on the LightCycler 480 Real‐Time PCR system (Roche, Basel, Switzerland). Relative expression level to glyceraldehyde‐3‐phosphate dehydrogenase was calculated by the comparative cycle threshold (ΔΔCt) method. Primer sequences are listed in Supporting Table S1.
Proteome Profiling by Liquid Chromatography–Tandem Mass Spectrometry
The detailed procedure has been described.20 Briefly, liver tissue was homogenized in a lysis buffer (50 mM NH5CO3 and 1 mM CaCl2). After denaturation and subsequent trypsinization, peptides were extracted by 50% acetonitrile/0.1% formic acid and 80% acetonitrile/0.1% formic acid solution. The hepatic proteome was profiled by nano‐liquid chromatography–tandem mass spectrometry (LC/MS‐MS) analysis with a nano‐LC1000 coupled to a Thermo Q‐Exactive (Thermo Fisher Scientific). Obtained MS‐MS spectra were searched against the target‐decoy mouse Reference Sequence database in the Proteome Discoverer 1.4 interface (PD1.4; Thermo Fisher Scientific) with the Mascot algorithm (Mascot 2.4; Matrix Science, Boston, MA). The relative amount was calculated by the intensity‐based absolute quantification algorithm and normalized to the intensity‐based fraction of the total.
Immunohistochemistry
Immunohistochemical analysis of keratin 19 (KRT19; alias CK19), ABCB11 (BSEP), ABCC3 (alias multidrug‐resistance like protein 3 [MRP3]), and ABCC4 (alias MRP4) was carried out according to the general guidelines of immunohistochemistry (Abcam, Cambridge, United Kingdom). Briefly, after antigen retrieval in sodium citrate buffer (pH 6.0), sections were incubated with blocking buffer (5% normal goat serum, 30 minutes) followed by diluted anti‐KRT19 (1:200, Ab133496; Abcam; Research Resource Identifier [RRID], AB_11155282), anti‐ABCB11 (1:100, R31844; NSJ Bioreagents, San Diego, CA; RRID, AB_2725751), anti‐ABCC3 (1:20, sc‐59612; Santa Cruz Biotechnology, Dallas, TX; RRID, AB_631970), and anti‐ABCC4 (1:50, sc‐376262; Santa Cruz Biotechnology; RRID, AB_10987858) antibodies at room temperature for 2 hours. Antigen‐antibody complex was then visualized by sequential incubation with biotin‐conjugated secondary antibodies (rabbit, BA‐1000; mouse, BA‐2000; Vector Laboratories, Burlingame, CA; RRID, AB_2313606 and AB_2313581, respectively) for 1.5 hours, Vectastatin Elite ABC Reagent (PK‐7100; Vector Laboratories) for 30 minutes, and Vector NovaRed Peroxidase Substrate Kit (SK‐4800; Vector Laboratories).
Gene Expression Omnibus Data Set
The GSE40120 and GSE20599 data sets were used for TC‐treated and DKO liver gene expression profiles, respectively. To compare the data sets of different platforms (GSE40120, Affymetrix Mouse Expression 430A Array; GSE20599, Illumina mouseRef‐8 v1.1 expression beadchip), data were first sorted by official gene symbols; the most significantly changed gene (lower P value) was then selected for duplicated genes. In total, 11,608 nonduplicated genes were compared. Of these, 513 genes were commonly found in both microarrays (P < 0.01) and further classified into four groups according to the expression pattern. Genes that were commonly increased or decreased in both microarrays (n = 347) were analyzed by DAVID Bioinformatics Resources tools (https://david.ncifcrf.gov; National Institute of Allergy and Infectious Diseases, Rockville, MD). The top four functional categories were displayed (Kyoto Encyclopedia of Genes and Genomes Pathway [KEGG]). Complete lists of KEGG and Gene Ontology‐Biological Pathway analyses are provided in Supporting Table S2.
Statistical Analysis
All results are presented as mean ± SEM, and statistical significance between corn oil (CO‐) or TC‐treated mice in each genotype (wild type [WT], DKO, or BKO) was calculated by the 2‐tailed Student t test. For comparison of three or five groups, 1‐way analysis of variance (ANOVA) followed by the posthoc Tukey honestly significant difference (HSD) test was used.
Results
DKO Cholestasis Activates CAR
Previously, we found that CAR and PXR signaling is activated in DKO liver.10 We first examined the broad DKO gene signature for CAR activation by comparing two microarrays from CAR agonist TC‐treated (GSE40120) and DKO (GSE20599) liver (Fig. 1A). Among 11,608 common genes, we identified 2,185 (19%, DKO) and 1,352 (12%, TC) differentially regulated genes (P < 0.01); 513 genes were detected on both arrays (hypergeometric test, P = 4.47 × 10–70), and 67% (347/513) of these genes shared identical patterns of regulation.
Figure 1.
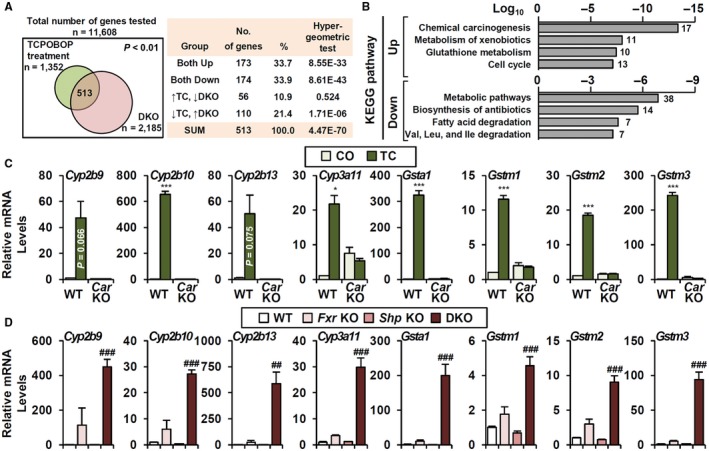
DKO gene signature indicates potential CAR activation. (A) Comparison of DKO and TC‐treated liver microarrays. Among 513 genes that were significantly changed in both microarrays, 347 genes (173 + 174, 67.6%) were regulated in the same manner. (B) KEGG pathway analysis of 347 genes; the top four gene categories in each analysis are shown. (C,D) Representative CYP and GST gene expressions (C) by TC treatment in WT and Car knockout (n = 4) as well as (D) in Fxr knockout, Shp knockout, and DKO (n = 3‐6). Student t test, *P < 0.05, ***P < 0.005 compared to CO‐treated WT. ANOVA followed by Tukey HSD; ##P < 0.01, ###P < 0.005. In some cases, the actual P value is presented.
KEGG pathway analysis showed that the up‐regulated genes are enriched for chemical carcinogenesis and xenobiotic metabolism pathways, including both CYP genes and glutathione S‐transferases (GSTs) as components of their gene signature (Fig. 1B). Many CYPs and GSTs were highly induced by TC treatment and blunted in Car KO (Fig. 1C). Of note, CAR‐sensitive CYP and GST genes were exclusively dependent on the FXR/SHP axis (Fig. 1D).
Loss Of Car Exacerbates Cholestatic Liver Damage And Regulates Ba Homeostasis
CYPs and GSTs are controlled by multiple transcription factors, including the xenobiotic nuclear receptors CAR and PXR.21 To define the CAR‐specific function in DKO liver, we generated Fxr:Shp:Car TKO mice. TKO mice exhibited significantly reduced BW at 3‐5 months of age (Fig. 2A), with severe jaundice compared to DKO mice. Consistent with the ability of CAR activation to promote liver growth,18 the hepatomegaly phenotype of DKO was partially reversed in TKO (Fig. 2B,C) and hepatocyte proliferation was decreased (Supporting Fig. S1). Strikingly, TKO showed greater gallbladder volume and cholecystomegaly (Fig. 2B, arrowhead; Fig. 2D). Immunohistochemical analysis with the biliary marker keratin‐19 (KRT19) indicated enhanced biliary proliferation due to biliary tract damage (Fig. 2E), which was less severe in DKO mice (Fig. 2E and Anakk et al.11).
Figure 2.
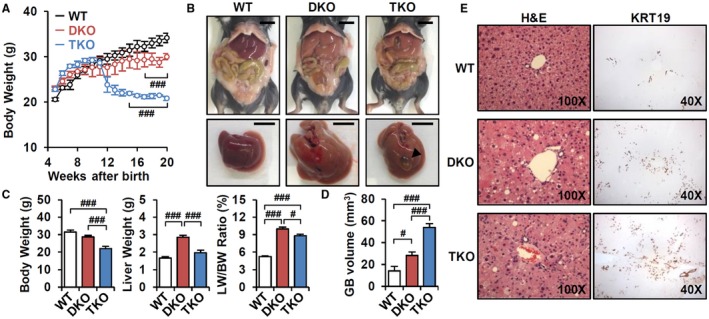
Loss of CAR accelerates cholestatic liver injury. (A) BW changes of WT, DKO, and TKO mice (n = 3‐5). (B) Representative image of whole body and liver at 3 months old. Scale bar, 10 mm; arrowhead, cholecystomegaly. (C) BW, liver weight, liver/BW ratio (%) (n = 8), and (D) gallbladder volume (n = 5) were measured. (E) Hematoxylin and eosin staining (magnification ×100) and KRT19 immunostaining (magnification ×40). ANOVA followed by Tukey HSD; #P < 0.05, ###P < 0.005. Abbreviations: GB, gallbladder; H&E, hematoxylin and eosin; LW, liver weight.
In addition to phenotypic changes, TKO mice exhibited slightly increased serum and hepatic total BA levels (Fig. 3A). Metabolomic analysis of primary BAs revealed elevated levels of most BA species in TKO serum. More specifically, tauro‐cholic acid and tauro‐αβ‐muricholic acid were elevated in TKO liver (Supporting Fig. S2A,B). Increased liver and other tissue injury were further supported by increased serum alanine aminotransferase (ALT), aspartate aminotransferase (AST), lactate dehydrogenase, and bilirubin levels (Fig. 3B). The proportion of direct and indirect bilirubin was not changed (data not shown), indicating that both DKO and TKO mice exhibited conjugated hyperbilirubinemia. Elevated gamma‐glutamyltransferase (GGT) levels indicated biliary tract damage in TKO mice (Fig. 3B), accounting for cholecystomegaly in Fig. 2D. Overall, loss of CAR accelerated cholestatic liver injury in DKO mice, which indicates a protective role of CAR activation in DKO pathogenesis.
Figure 3.
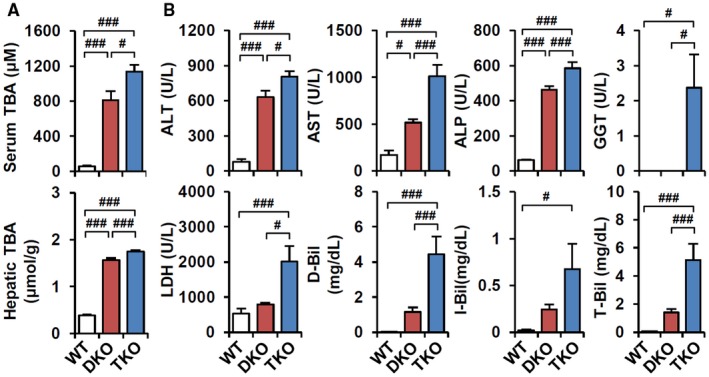
TKO increases BA‐induced liver toxicity. (A) Total BA levels in serum and liver (n = 6‐8). (B) Serum biochemistry analysis (n = 8). ANOVA followed by Tukey HSD; #P < 0.05, ###P < 0.005. Abbreviations: ALP, alkaline phosphatase; D‐Bil, direct bilirubin; I‐Bil, indirect bilirubin; LDH, lactate dehydrogenase; T‐Bil, total bilirubin.
TKO Impairs BA Export Through Blunted BSEP Function
CAR activation induces CYP and GST expression to facilitate clearance of xenobiotics and toxic cellular components, such as BAs.19, 22 We tested whether loss of CAR (TKO) diminishes the induction of CYPs and GSTs (Fig. 1D). In contrast to our expectation, only a few genes induced in DKO livers were strongly decreased in TKO (Cyp2b13, Gstm3, and Cyp3a11; Supporting Figs. S3 and S4), potentially due to the compensatory activation of other transcription factors, including the xenobiotic nuclear receptor PXR.22
To determine the molecular mechanism of accelerated liver injury in TKO mice, we further profiled the messenger RNA (mRNA) levels of genes involved in BA synthesis, hydroxylation, conjugation, and transport (Supporting Fig. S4). Most genes were not altered in TKO livers, except for Abcb11. ABCA11/BSEP is a major canalicular BA transporter that mainly excretes tauro‐conjugated and glyco‐conjugated BAs into bile.23 Decreased mRNA and protein levels of ABCB11 in DKO mice were further suppressed in TKO mice (Fig. 4A,B). Direct immunohistochemical analysis of ABCB11 protein showed that the normal canalicular ABCB11 expression in DKO mice was completely abolished in TKO mice (Fig. 4C). Despite markedly decreased ABCB11 expression relative to WT, DKO mice retained canalicular BA efflux with slightly reduced biliary and fecal BA excretion, but these phenotypes were strongly attenuated in TKO mice (Fig. 4D). Thus, loss of CAR activation in TKO results in loss of residual DKO ABCB11 function, which likely accelerates cholestatic liver injury.
Figure 4.
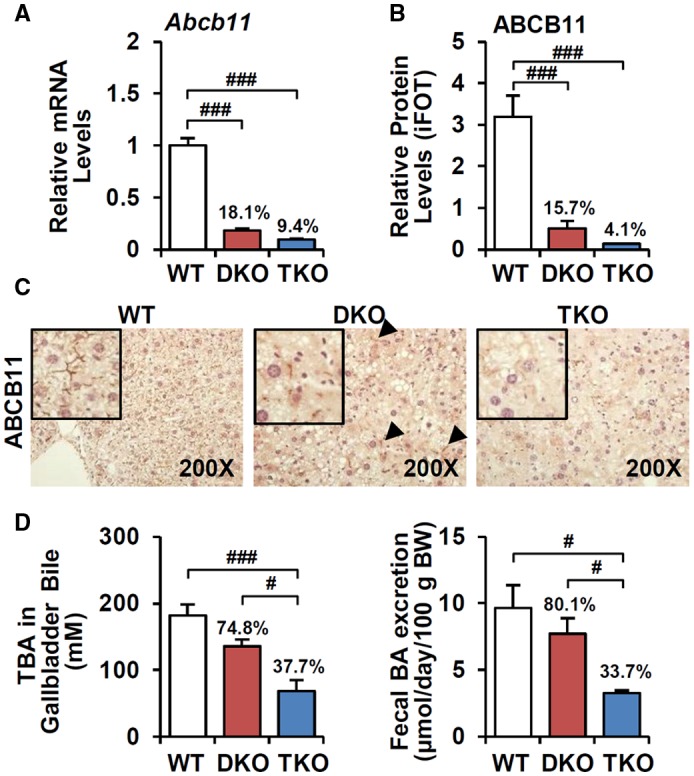
TKO further attenuates ABCB11‐mediated fecal BA excretion. (A) mRNA (n = 8) and (B) protein levels (n = 3) of ABCB11 were analyzed by q‐PCR and proteomics, respectively. Ratio (%) indicates the proportion of ABCB11 expressions compared to WT. (C) Immunostaining of ABCB11 (magnification ×200); arrowhead, residual canalicular ABCB11 expression. (D) BA levels in gallbladder‐collected bile (n = 4) and feces (n = 3). ANOVA followed by Tukey HSD, #P < 0.05, ###P < 0.005. Abbreviation: iFOT, intensity‐based fraction of total.
Pharmacologic CAR Activation Exhibits Distinct Phenotypes in DKO and BKO
This genetic analysis strongly predicts that CAR activation should be protective against DKO cholestasis. We investigated the therapeutic potential of CAR activation in DKO and compared it to another mouse model of intrahepatic cholestasis, BKO (PFIC2 model). BKO mice on a C57BL/6 background progressively develop cholestatic liver damage with elevated ALT and AST levels.13
TC treatment dramatically increased liver size in both DKO and BKO mice (Fig. 5A,B). This effect was expected from the well‐known ability of CAR activation to increase liver weight. In DKO mice, TC reduced serum and hepatic BA levels (Fig. 5C) and partially reversed BA toxicity and liver injury (Fig. 5D). TC treatment did not affect basal BA levels in WT mice. In accord with our previous demonstration of a direct induction of clearance of elevated bilirubin levels on CAR activation,15 TC treatment strongly reduced direct, indirect, and total bilirubin in DKO livers. The very low basal levels of bilirubin in WT mice were not affected. We observed an unexpected increase of GGT levels, likely due to direct TC‐dependent induction of Ggt1 mRNA expression, which was not coupled with biliary tract damage (Supporting Fig. S5).
Figure 5.
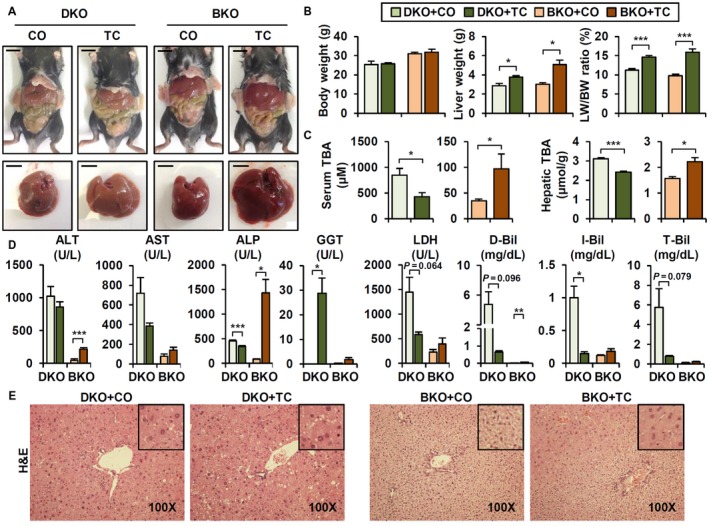
CAR activation has differential effects in DKO and BKO. (A) Representative whole body and liver image. Scale bar, 10 mm. (B) BW, liver weight, and liver/BW ratio (n = 4). (C) Serum and hepatic TBA levels (n = 4). (D) Serum biochemistry (n = 4). (E) Hematoxylin and eosin staining (magnification ×100). Student t test; *P < 0.05, **P < 0.01, ***P < 0.005 compared to CO‐treated group. In some cases, actual P value is presented. Abbreviations: ALP, alkaline phosphatase; D‐Bil, direct bilirubin; I‐Bil, indirect bilirubin; LDH, lactate dehydrogenase; LW, liver weight; T‐Bil, total bilirubin.
TC treatment had different effects in BKO mice. These effects were mixed but generally deleterious, with increased BA levels, accelerated liver and other tissue injury (Fig. 5C,D, brown bar), and hepatocyte hyperplasia (Fig. 5E, right panel; Blanco‐Bose et al.24). For reasons that were not clear, BKO hepatocytes appeared much smaller than those of DKO on hematoxylin and eosin histology (Fig. 5E). TC treatment increased hepatocyte size and apparent ploidy as indicated by nuclear size in pericentral zone 3 hepatocytes (Fig. 5F) where CAR was differentially expressed.
CAR Activation Regulates BA Transporter Expressions
To elucidate the molecular mechanism of distinct TC responses in DKO and BKO mice, we analyzed CYP and GST gene expression. As noted earlier, basal levels were highly up‐regulated in DKO (Fig. 1C) but not BKO mice.10 TC treatment did not further enhance DKO CYP and GST expression (Supporting Fig. S6). As expected, however, TC strongly up‐regulated these genes in BKO mice to levels comparable to those observed in DKO mice (Supporting Fig. S6).
CAR activation induces both canalicular and basolateral BA transporters, including Abcb11/BSEP, Abcc2/MRP2, and Abcc3/MRP3, to enhance BA excretion.21, 25 Both DKO and BKO mice showed a tendency toward increased fecal BA excretion on TC treatment, whereas urinary BAs were increased in DKO mice (Fig. 6A). TC treatment slightly restored Abcb11 mRNA (Fig. 6B) and canalicular ABCB11 expression (Fig. 6C, arrowhead) in DKO, which supports increased BA excretion and reduced BA burden (Fig. 5C). No further induction of other transporters was observed at the mRNA level in DKO livers (Fig. 6B). In BKO, TC treatment enhanced alternative BA transporter Abcc2/MRP2, which further increased fecal BA excretion (Fig. 6A). Also, TC treatment strongly activated BA transport into the circulation, mediated by ABCC3 and ABCC4 (Fig. 6B,C, arrowhead), explaining increased serum BA levels (Fig. 5C).
Figure 6.
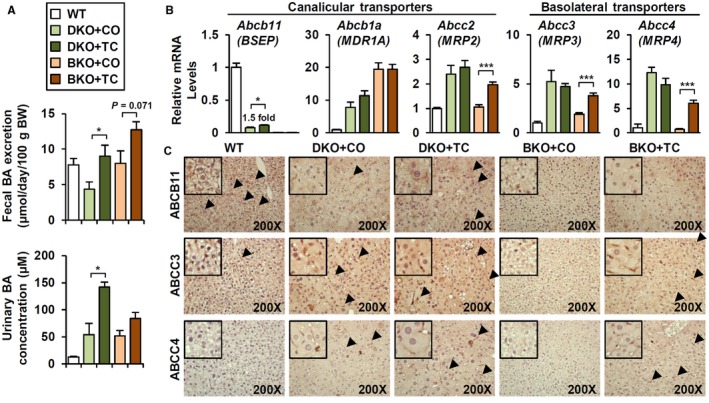
TC treatment enhances canalicular and basolateral BA transporters. (A) Fecal BA excretion (n = 4) and urinary BA concentration (n = 3) in TC‐treated (1 week) DKO and BKO. (B) mRNA levels of BA transporters (n = 4). (C) Immunostaining of ABCB11, ABCC3, and ABCC4 (magnification ×200). Arrowhead, positive area. Student t test; *P < 0.05, ***P < 0.005 compared to CO‐treated group. Abbreviation: MDR, multidrug resistance.
CAR Activation Differentially Modulates Cholesterol Trafficking in DKO and BKO MICE
It remained unclear why TC unexpectedly increased BKO hepatic BA levels (Fig. 5C) despite increased fecal BA excretion and BA transporter expressions (Fig. 6). To address this question, we first investigated whether CAR directly regulates BA absorption in the intestine. No gene markers of intestinal BA homeostasis were affected by TC treatment (Supporting Fig. S7), as previously observed.18, 26 Furthermore, BA synthetic enzymes encoded by Cyp7a1, Cyp7b1, Cyp8b1, and Cyp27a1 were unchanged or even decreased (data not shown), suggesting that neither BA synthesis nor absorption pathways contribute to the BKO phenotype.
Next, we hypothesized that CAR activation could affect cholesterol synthesis and mobilization. Liver BA synthesis is dependent on cholesterol, which is replenished by de novo cholesterol synthesis and lipoprotein uptake.27 CAR activation stimulates reverse cholesterol transport to facilitate elimination of sterols through fecal BA excretion.28 In this scenario, the liver enhances cholesterol uptake and conversion into BAs, with fecal excretion ultimately decreasing systemic cholesterol levels.29, 30 In DKO mice, basal total cholesterol levels were markedly lower than WT and were not induced by TC treatment (Fig. 7A), despite partial recovery of high‐density lipoprotein (HDL) cholesterol levels. In BKO liver and serum, TC increased total cholesterol (Fig. 7A) and HDL cholesterol levels (Fig. 7B). The increase in serum levels argues that the increased hepatic levels are probably not due to defective hepatic secretion; they are also not due to increased activity of cholesterol biosynthesis or uptake pathways in response to TC.
Figure 7.
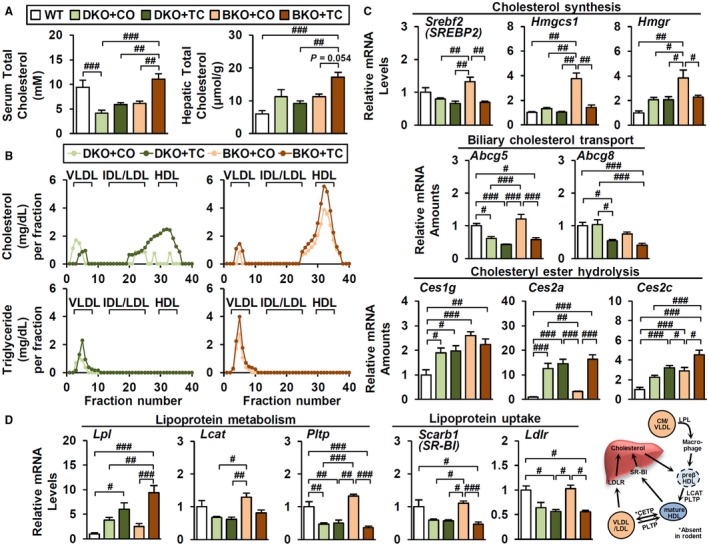
TC differentially controls cholesterol homeostasis in DKO and BKO. (A) Total cholesterol levels in serum and liver (n = 4). (B) Cholesterol (upper) and triglyceride (lower) concentration in lipoprotein fractions. Gene expression profiles involved in (C) cholesterol and (D) lipoprotein metabolism (n = 4). ANOVA followed by Tukey HSD; #P < 0.05, ##P < 0.01, ###P < 0.005. Abbreviations: CETP, cholesteryl ester transfer protein; CM, chylomicrons; Hmgcs1, 3‐hydroxy‐3‐methylglutaryl–coenzyme A synthase 1; Hmgr 3‐hydroxy‐3‐methylglutaryl–coenzyme A reductase; IDL, intermediate density lipoprotein; Ldlr, low‐density lipoprotein receptor; Scarb1, scavenger receptor class B, member 1; VLDL, very low density lipoprotein.
Instead, basal expression of cholesterol synthesis genes, such as sterol regulatory element binding transcription factor 2 (Srebf2; alias sterol regulatory element binding protein 2 [Srebp2]) and its downstream targets 3‐hydroxy‐3‐methylglutaryl–coenzyme A synthase 1 (Hmgcs1) and HMG CoA reductase (Hmgr), was elevated in BKO relative to DKO mice (Fig. 7C). As expected in response to the increased hepatic cholesterol levels (Fig. 7A), this elevated expression was significantly decreased by TC treatment in BKO but not DKO liver. Biliary cholesterol transporter Abcg5 and Abcg8 were reduced by TC treatment, inhibiting cholesterol efflux into bile canaliculi (Fig. 7C). Interestingly, carboxylesterase 2 (Ces2a and Ces2c), which catalyzes hydrolysis of cholesteryl esters into free cholesterol, was markedly induced by TC (Fig. 7C), presumably increasing the free cholesterol pool that serves as the substrate for de novo BA synthesis.
Many genes involved in lipoprotein metabolism were also differentially regulated (Fig. 7D). Basal expression of HDL metabolism (lecithin cholesterol acyltransferase [Lcat] and plasma phospholipid transfer protein [Pltp]) and HDL/low‐density lipoprotein (LDL) receptor genes (Scarb1, Ldlr) was higher in BKO relative to DKO and was decreased by TC (Fig. 7D). Because these genes are also SREBP targets, this is an expected consequence of increased hepatic cholesterol accumulation after TC treatment. In contrast to these expected responses, TC robustly increased expression of hepatic lipoprotein lipase (Lpl) in TC‐treated BKO livers but not in DKO or control livers (Fig. 7D and data not shown). LPL is a key enzyme that hydrolyzes triglycerides in chylomicrons and very low‐density lipoproteins31 and has been linked with pre‐β HDL formation, HDL maturation, and uptake.32, 33, 34, 35 Although LPL is not normally expressed at significant levels in mouse liver, it can be induced in response to liver X receptor activation to promote lipid accumulation.36 A recent report suggests that induced LPL expression also contributes to liver fat accumulation in a novel tree shrew model in response to a diet high in fat and cholesterol.37 It is possible that increased liver LPL expression contributes to elevated BKO liver cholesterol and that the elevated substrate levels contribute to increased hepatic BA levels in response to TC treatment.
Discussion
In DKO livers, pathologic BA overload directly and indirectly controls many adaptive programs that relieve cholestatic liver injury; this enhances BA and bilirubin metabolism/clearance.11 A transcriptomic analysis indicated that the xenobiotic nuclear receptor CAR is endogenously activated to induce CYP and GST expression (Fig. 1). However, in TKO mice lacking CAR in addition to FXR and SHP, we did not observe a significant decrease in induction of a range of known CAR target genes (Supporting Figs. S3 and S4C). This negative result is consistent with both the known functional redundancy of CAR and PXR in the induction of a range of drug metabolism genes22 and the direct activation of PXR by elevated BA levels.38 In accord with this, we previously found that combined CAR/PXR deletion in the DKO background (Fxr;Shp;Car;Pxr quadruple knockout) completely abolished CYP gene induction.10 This impairs bilirubin‐conjugation processes and results in a dramatic accumulation of indirect bilirubin. However, up‐regulation of GST expression was not lost in the quadruple knockouts and may be due to activation of other transcription factors, including aryl hydrocarbon receptor and nuclear transcription factor E2‐related factor 2.39, 40 Further studies are required to test the functional roles of these additional factors in the DKO livers.
Despite the subtle impact on CYP and GST expressions, TKO mice exhibited even more dramatic pathologies than DKO mice. Due to the limited capacity of hepatocyte proliferation (Supporting Fig. S1), TKO liver did not properly respond to BA overload and displayed more aggressive cholestasis (Figs. 2 and 3). In addition to intrahepatic damage, TKO mice showed cholecystomegaly, accounting for biliary damage as a result of high BA levels (Fig. 2D,E). Although this phenotype is rare in PFICs, a case of giant gallbladder associated with PFIC2 has been reported.41 No direct evidence has established that CAR controls gallbladder pathophysiology, but our study aligns with other data that demonstrate CAR activation protects against cholesterol gallstone disease.42 However, the molecular mechanism underlying the cholecystomegaly phenotype needs to be investigated in the context of liver–gallbladder–gut crosstalk as well as BA–cholesterol metabolism.
The broad gene expression profile of BA homeostasis consistently indicates loss of ABCB11 function in TKO mice (Fig. 4). In addition to mRNA and protein expression (Fig. 4A,B), immunohistochemical analysis confirmed complete loss of ABCB11 expression (Fig. 4C), resulting in the reduction of fecal BA excretion in TKO mice (Fig. 4D). Because ABCB11 mainly facilitates the export of monovalent BAs,43 total ablation of ABCB11 function in TKO also corresponded with the selective increase of tauro‐conjugated BAs in liver (Supporting Fig. S2B). Because Car single knockout did not reduce Abcb11 expression under normal conditions (data not shown), this effect is disease‐context dependent and only observed in the DKO cholestasis background. Overall, these changes conferred by loss of CAR function may explain the aggressive phenotypes of TKO mice (Supporting Fig. S8A).
Several studies demonstrate the protective effect of CAR in extrahepatic cholestasis.17, 18 Thus, we initially expected that the CAR agonist TC would ameliorate BA‐induced liver injury in different contexts of intrahepatic cholestasis. However, TC treatment exerted very different effects in the DKO and BKO models (Fig. 5). In DKO, TC treatment slightly restored canalicular BA transport and further enhanced basolateral BA transport (Fig. 7B), which contributed to the elimination of hepatic BAs through fecal and urinary excretion (Supporting Fig. S8B). These effects collectively resolved BA overload and liver damage.
BKO mice have a higher basal expression of genes involved in de novo cholesterol synthesis as well as cholesterol transport into the liver (Fig. 7C,D). The increased basal expression of HDL maturation/remodeling genes (LCAT, PLTP) may contribute to hepatic cholesterol accumulation by TC. The suppression of these genes in response to TC (Fig. 7C,D) is expected as a secondary consequence of increased hepatic cholesterol levels. Many cholesterol metabolism genes are primarily regulated by SREBPs, which are in turn suppressed by cellular cholesterol levels (Fig. 7A) in a negative feedback loop.44, 45, 46 The basis for the marked increase in hepatic cholesterol BKO livers in response to TC is not clear but could be linked to increased expression of hepatic LPL.
Why does TC have a marginal effect on the DKO cholesterol level? Young DKO mice show higher cholesterol levels,11 but as cholestasis progresses, circulating cholesterol levels become lower than the WT after 3 months (Fig. 7A), reflecting a systemic cholesterol depletion possibly by prolonged pathologic activation of de novo BA synthesis. This may be related to chronic CAR activation somehow decreasing basal cholesterol levels.30, 47 Under this cholesterol‐depleted condition, TC does not sufficiently increase total cholesterol levels and subsequent BA synthesis, despite elevated HDL cholesterol level (Fig. 7A).
In conclusion, loss of CAR activation in DKO accelerates cholestatic liver injury due to BA accumulation, possibly through blunted ABCB11 expression and canalicular BA transport. Pharmacologic CAR activation ameliorates DKO cholestasis, relieving BA overload partially by restored fecal BA excretion. Surprisingly, BKO mice unexpectedly show increased hepatic cholesterol levels that potentially promote de novo BA synthesis, which ultimately exacerbates cholestasis phenotypes. These contrasting results predict the therapeutic potential of CAR agonists for treating heterogeneous intrahepatic cholestasis.
Potential conflict of interest
Nothing to report.
Supporting information
Acknowledgment
We thank Dr. Pradip K. Saha (Baylor College of Medicine) for analyzing the lipoproteins, Dr. Nunzia Pastore for the KRT19 antibody (Baylor College of Medicine), and Dr. Jae Man Lee (Kyungpook National University, Korea) for helpful discussions.
Supported by the R.P. Doherty Jr.–Welch Chair in Science (Q‐0022 to D.D.M.), the Assistant Secretary of Defense for Health Affairs endorsed by the Department of Defense through the Peer Reviewed Medical Research Program/Discovery Award (No. W81XWH‐18‐1‐0126 to K.H.K.), the National Institutes of Health (R01DK114356 to S.M.H.), and the American Diabetes Association (1‐18‐IBS‐105 to S.M.H.).
References
Author names in bold designate shared co‐first authorship.
- 1. Trauner M, Claudel T, Fickert P, Moustafa T, Wagner M. Bile acids as regulators of hepatic lipid and glucose metabolism. Dig Dis 2010;28:220‐224. [DOI] [PubMed] [Google Scholar]
- 2. Chiang JY. Bile acid regulation of gene expression: Roles of nuclear hormone receptors. Endocr Rev 2002;23:443‐463. [DOI] [PubMed] [Google Scholar]
- 3. Molinaro A, Wahlstrom A, Marschall HU. Role of bile acids in metabolic control. Trends Endocrinol Metab 2018;29:31‐41. [DOI] [PubMed] [Google Scholar]
- 4. Balistreri WF, Bezerra JA, Jansen P, Karpen SJ, Shneider BL, Suchy FJ. Intrahepatic cholestasis: summary of an American Association for the Study of Liver Diseases single‐topic conference. Hepatology 2005;42:222‐235. [DOI] [PubMed] [Google Scholar]
- 5. Schady DA, Finegold MJ. Contemporary evaluation of the pediatric liver biopsy. Gastroenterol Clin North Am 2017;46:233‐252. [DOI] [PubMed] [Google Scholar]
- 6. Srivastava A. Progressive familial intrahepatic cholestasis. J Clin Exp Hepatol 2014;4:25‐36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wagner M, Zollner G, Trauner M. Nuclear receptors in liver disease. Hepatology 2011;53:1023‐1034. [DOI] [PubMed] [Google Scholar]
- 8. Wang H, Chen J, Hollister K, Sowers LC, Forman BM. Endogenous bile acids are ligands for the nuclear receptor FXR/BAR. Mol Cell 1999;3:543‐553. [DOI] [PubMed] [Google Scholar]
- 9. Kir S, Zhang Y, Gerard RD, Kliewer SA, Mangelsdorf DJ. Nuclear receptors HNF4alpha and LRH‐1 cooperate in regulating Cyp7a1 in vivo. J Biol Chem 2012;287:41334‐41341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kim KH, Choi JM, Li F, Arizpe A, Wooton‐Kee CR, Anakk S, et al. Xenobiotic nuclear receptor signaling determines molecular pathogenesis of progressive familial intrahepatic cholestasis. Endocrinology 2018;159:2435‐2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Anakk S, Watanabe M, Ochsner SA, McKenna NJ, Finegold MJ, Moore DD. Combined deletion of Fxr and Shp in mice induces Cyp17a1 and results in juvenile onset cholestasis. J Clin Invest 2011;121:86‐95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gomez‐Ospina N, Potter CJ, Xiao R, Manickam K, Kim MS, Kim KH, et al. Mutations in the nuclear bile acid receptor FXR cause progressive familial intrahepatic cholestasis. Nat Commun 2016;7:10713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhang Y, Li F, Patterson AD, Wang Y, Krausz KW, Neale G, et al. Abcb11 deficiency induces cholestasis coupled to impaired beta‐fatty acid oxidation in mice. J Biol Chem 2012;287:24784‐24794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Halilbasic E, Baghdasaryan A, Trauner M. Nuclear receptors as drug targets in cholestatic liver diseases. Clin Liver Dis 2013;17:161‐189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Huang W, Zhang J, Chua SS, Qatanani M, Han Y, Granata R, et al. Induction of bilirubin clearance by the constitutive androstane receptor (CAR). Proc Natl Acad Sci U S A 2003;100:4156‐4161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhang J, Huang W, Qatanani M, Evans RM, Moore DD. The constitutive androstane receptor and pregnane X receptor function coordinately to prevent bile acid‐induced hepatotoxicity. J Biol Chem 2004;279:49517‐49522. [DOI] [PubMed] [Google Scholar]
- 17. Stedman CA, Liddle C, Coulter SA, Sonoda J, Alvarez JG, Moore DD, et al. Nuclear receptors constitutive androstane receptor and pregnane X receptor ameliorate cholestatic liver injury. Proc Natl Acad Sci U S A 2005;102:2063‐2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wagner M, Halilbasic E, Marschall HU, Zollner G, Fickert P, Langner C, et al. CAR and PXR agonists stimulate hepatic bile acid and bilirubin detoxification and elimination pathways in mice. Hepatology 2005;42:420‐430. [DOI] [PubMed] [Google Scholar]
- 19. Wei P, Zhang J, Egan‐Hafley M, Liang S, Moore DD. The nuclear receptor CAR mediates specific xenobiotic induction of drug metabolism. Nature 2000;407:920‐923. [DOI] [PubMed] [Google Scholar]
- 20. Jung SY, Choi JM, Rousseaux MW, Malovannaya A, Kim JJ, Kutzera J, et al. An anatomically resolved mouse brain proteome reveals Parkinson disease‐relevant pathways. Mol Cell Proteomics 2017;16:581‐593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Aleksunes LM, Klaassen CD. Coordinated regulation of hepatic phase I and II drug‐metabolizing genes and transporters using AhR‐, CAR‐, PXR‐, PPARalpha‐, and Nrf2‐null mice. Drug Metab Dispos 2012;40:1366‐1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wei P, Zhang J, Dowhan DH, Han Y, Moore DD. Specific and overlapping functions of the nuclear hormone receptors CAR and PXR in xenobiotic response. Pharmacogenomics J 2002;2:117‐126. [DOI] [PubMed] [Google Scholar]
- 23. Thomas C, Pellicciari R, Pruzanski M, Auwerx J, Schoonjans K. Targeting bile‐acid signalling for metabolic diseases. Nat Rev Drug Discov 2008;7:678‐693. [DOI] [PubMed] [Google Scholar]
- 24. Blanco‐Bose WE, Murphy MJ, Ehninger A, Offner S, Dubey C, Huang W, et al. C‐Myc and its target FoxM1 are critical downstream effectors of constitutive androstane receptor (CAR) mediated direct liver hyperplasia. Hepatology 2008;48:1302‐1311. [DOI] [PubMed] [Google Scholar]
- 25. Lickteig AJ, Csanaky IL, Pratt‐Hyatt M, Klaassen CD. Activation of constitutive androstane receptor (CAR) in mice results in maintained biliary excretion of bile acids despite a marked decrease of bile acids in liver. Toxicol Sci 2016;151:403‐418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Park S, Cheng SL, Cui JY. Characterizing drug‐metabolizing enzymes and transporters that are bona fide CAR‐target genes in mouse intestine. Acta Pharm Sin B 2016;6:475‐491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jolley CD, Dietschy JM, Turley SD. Induction of bile acid synthesis by cholesterol and cholestyramine feeding is unimpaired in mice deficient in apolipoprotein AI. Hepatology 2000;32:1309‐1316. [DOI] [PubMed] [Google Scholar]
- 28. Sberna AL, Assem M, Gautier T, Grober J, Guiu B, Jeannin A, et al. Constitutive androstane receptor activation stimulates faecal bile acid excretion and reverse cholesterol transport in mice. J Hepatol 2011;55:154‐161. [DOI] [PubMed] [Google Scholar]
- 29. Masson D, Qatanani M, Sberna AL, Xiao R, Pais de Barros JP, Grober J, et al. Activation of the constitutive androstane receptor decreases HDL in wild‐type and human apoA‐I transgenic mice. J Lipid Res 2008;49:1682‐1691. [DOI] [PubMed] [Google Scholar]
- 30. Sberna AL, Assem M, Xiao R, Ayers S, Gautier T, Guiu B, et al. Constitutive androstane receptor activation decreases plasma apolipoprotein B‐containing lipoproteins and atherosclerosis in low‐density lipoprotein receptor‐deficient mice. Arterioscler Thromb Vasc Biol 2011;31:2232‐2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hayden MR, Ma Y. Molecular genetics of human lipoprotein lipase deficiency. Mol Cell Biochem 1992;113:171‐176. [DOI] [PubMed] [Google Scholar]
- 32. Clee SM, Zhang H, Bissada N, Miao L, Ehrenborg E, Benlian P, et al. Relationship between lipoprotein lipase and high density lipoprotein cholesterol in mice: modulation by cholesteryl ester transfer protein and dietary status. J Lipid Res 1997;38:2079‐2089. [PubMed] [Google Scholar]
- 33. Tsutsumi K, Inoue Y, Shima A, Iwasaki K, Kawamura M, Murase T. The novel compound NO‐1886 increases lipoprotein lipase activity with resulting elevation of high density lipoprotein cholesterol, and long‐term administration inhibits atherogenesis in the coronary arteries of rats with experimental atherosclerosis. J Clin Invest 1993;92:411‐417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Rinninger F, Brundert M, Brosch I, Donarski N, Budzinski RM, Greten H. Lipoprotein lipase mediates an increase in selective uptake of HDL‐associated cholesteryl esters by cells in culture independent of scavenger receptor BI. J Lipid Res 2001;42:1740‐1751. [PubMed] [Google Scholar]
- 35. Camont L, Chapman MJ, Kontush A. Biological activities of HDL subpopulations and their relevance to cardiovascular disease. Trends Mol Med 2011;17:594‐603. [DOI] [PubMed] [Google Scholar]
- 36. Zhang Y, Repa JJ, Gauthier K, Mangelsdorf DJ. Regulation of lipoprotein lipase by the oxysterol receptors. LXRalpha and LXRbeta. J Biol Chem 2001;276:43018‐43024. [DOI] [PubMed] [Google Scholar]
- 37. Zhang L, Zhang Z, Li Y, Liao S, Wu X, Chang Q, et al. Cholesterol induces lipoprotein lipase expression in a tree shrew (Tupaia belangeri chinensis) model of non‐alcoholic fatty liver disease. Sci Rep 2015;5:15970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Staudinger JL, Goodwin B, Jones SA, Hawkins‐Brown D, MacKenzie KI, LaTour A, et al. The nuclear receptor PXR is a lithocholic acid sensor that protects against liver toxicity. Proc Natl Acad Sci U S A 2001;98:3369‐3374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yeager RL, Reisman SA, Aleksunes LM, Klaassen CD. Introducing the “TCDD‐inducible AhR‐Nrf2 gene battery”. Toxicol Sci 2009;111:238‐246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Anwar‐Mohamed A, Degenhardt OS, El Gendy MA, Seubert JM, Kleeberger SR, El‐Kadi AO. The effect of Nrf2 knockout on the constitutive expression of drug metabolizing enzymes and transporters in C57Bl/6 mice livers. Toxicol In Vitro 2011;25:785‐795. [DOI] [PubMed] [Google Scholar]
- 41. Panaro F, Chastaing L, Navarro F. Education and imaging. Hepatobiliary and pancreatic: giant gallbladder associated with Byler's disease. J Gastroenterol Hepatol 2012;27:620. [DOI] [PubMed] [Google Scholar]
- 42. Cheng S, Zou M, Liu Q, Kuang J, Shen J, Pu S, et al. Activation of constitutive androstane receptor prevents cholesterol gallstone formation. Am J Pathol 2017;187:808‐818. [DOI] [PubMed] [Google Scholar]
- 43. Trauner M, Boyer JL. Bile salt transporters: molecular characterization, function, and regulation. Physiol Rev 2003;83:633‐671. [DOI] [PubMed] [Google Scholar]
- 44. Madison BB. Srebp2: a master regulator of sterol and fatty acid synthesis. J Lipid Res 2016;57:333‐335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Okazaki H, Goldstein JL, Brown MS, Liang G. LXR‐SREBP‐1c‐phospholipid transfer protein axis controls very low density lipoprotein (VLDL) particle size. J Biol Chem 2010;285:6801‐6810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Treguier M, Doucet C, Moreau M, Dachet C, Thillet J, Chapman MJ, et al. Transcription factor sterol regulatory element binding protein 2 regulates scavenger receptor Cla‐1 gene expression. Arterioscler Thromb Vasc Biol 2004;24:2358‐2364. [DOI] [PubMed] [Google Scholar]
- 47. Rezen T, Tamasi V, Lovgren‐Sandblom A, Bjorkhem I, Meyer UA, Rozman D. Effect of CAR activation on selected metabolic pathways in normal and hyperlipidemic mouse livers. BMC Genom 2009;10:384. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials