

Abstract
The gut microbiome, the multispecies community of microbes that exists in the gastrointestinal tract, encodes several orders of magnitude more functional genes than the human genome. It also plays a pivotal role in human health, in part due to metabolism of environmental, dietary, and host‐derived substrates, which produce bioactive metabolites. Perturbations to the composition and associated metabolic output of the gut microbiome have been associated with a number of chronic liver diseases, including nonalcoholic fatty liver disease (NAFLD). Here, we review the rapidly evolving suite of next‐generation techniques used for studying gut microbiome composition, functional gene content, and bioactive products and discuss relationships with the pathogenesis of NAFLD.
Abbreviations
- AAA
aromatic amino acid
- BCAA
branched‐chain amino acid
- FMT
fecal microbial transplantation
- FXR
farnesoid X receptor
- LC
liquid chromatography
- mRNA
messenger RNA
- MS
mass spectrometry
- NAFL
nonalcoholic fatty liver
- NAFLD
nonalcoholic fatty liver disease
- NASH
nonalcoholic steatohepatitis
- rRNA
ribosomal RNA
- SCFA
short‐chain fatty acid
- TMA
trimethylamine
- TMAO
trimethylamine‐N‐oxide
The human gastrointestinal tract harbors trillions of bacterial, fungal, and archaeal cells in addition to viral particles. Collectively termed the “gut microbiome,” members of these microbial communities engage each other and the human host by exchanging signaling molecules and substrates, interactions that are now emerging as critically important in defining host health.1 (Table 1) The healthy gut microbiome comprises a large diversity of phylogenetically distinct microbial species, with even greater interindividual diversity observed at the subspecies or strain level.2 Although culture‐independent studies of the gut microbiome have more traditionally focused on bacteria, investigation of fungal and viral species has increased, leading to their recognition as integral members of the gut microbiome and implicating trans‐kingdom interactions in microbiome composition and function.3, 4
Table 1.
Definitions Of Key Terms
| Term | Definition |
|---|---|
| Microbiota | Microbes (e.g., bacteria, fungi, viruses) that collectively inhabit an ecosystem. |
| Microbiome | Collection of all genomes of microbes in an ecosystem. |
| Biomarker‐based sequencing | Process of cataloguing microbes through analysis of sequence variation in a single ubiquitous gene. |
| Whole metagenome sequencing | Process of cataloguing all the genetic material present in a biologic sample. |
| Metatranscriptomics | Process of profiling all expressed genes (RNA) in a biologic sample. |
| Metaproteomics | Study of all proteins found in a biologic sample. |
| Metabolomics | Study of all small‐molecule chemicals found in a biologic sample. |
The healthy adult gut microbiome encodes several orders of magnitude more functional genes than the human genome, considerably expanding the functional capacity of the human superorganism.5 Whereas average genomic differences between individual humans are small, the unique collection of microbial species and strains within an individual’s gut microbiome confers a high degree of interpersonal functional capacity.6 As the field of gut microbiome research has developed, it has become clear that the gut microbiome plays a pivotal role in defining host health status. This is achieved through more traditional host–microbial interactions involving expression of virulence factors and production of cell‐associated ligands (e.g., lipopolysaccharides) that influence microbial sensing and response systems (e.g., toll‐like receptors), which function as immune gatekeepers.7 However, it is increasingly recognized that the products of microbial primary and secondary metabolism also modulate host cellular physiology and immune responses, thus offering an additional avenue by which the gut microbiome and its products may influence host health. Among the known gut microbiome products that influence host cell responses are short‐chain fatty acids (SCFAs) that serve as energy sources for host cells, interspecies communication or quorum‐sensing molecules, complex macrolide and non‐ribosomal peptides with antimicrobial and immunomodulatory capacity, as well as vitamins and neuro‐signaling molecules.1, 8
That the gut microbiome is central to human health is underscored by an ever‐increasing body of literature indicating that perturbations to the composition, function, or metabolic output of the gut microbiome are associated with a wide array of chronic disease states.1 Given that the liver receives the majority of its blood supply from the intestine through the portal circulation, it is particularly exposed to gut microbiome‐derived products. Unsurprisingly, the gut microbiome has been implicated in the development of chronic liver diseases, including nonalcoholic fatty liver disease (NAFLD).9 Here, we review the following:
Current methods for studying gut microbiome composition, function, and metabolic productivity.
Factors that shape the gut microbiome.
Gut microbial metabolism and its effect on human health.
The influence of gut microbial metabolism in the pathogenesis of NAFLD.
Potential therapeutic implications of the gut microbiome in the management of NAFLD.
Current Methods in Microbiome Research
A variety of methods exist for characterizing the gut microbiome. Each of these tools, including advantages and current limitations, is reviewed in Table 2. Sequencing methods include biomarker gene‐based sequencing that permit an assessment of microbial community composition as well as untargeted “shotgun” approaches that enable determination of the genes encoded (metagenomics) or expressed (metatranscriptomics) by the microbiome. We also review mass spectrometry (MS)‐based platforms that permit identification of proteins (metaproteomics) or metabolites (metabolomics) produced by the microbiome. Each of these methods has been comprehensively reviewed elsewhere,10, 11, 12, 13 and therefore this review provides an overview of each approach (Fig. 1).
Table 2.
Advantages And Limitations Of Methods For Studying The Microbiome
| Method | Advantages | Limitations |
|---|---|---|
| Sequencing methods | ||
| Biomarker‐based sequencing |
|
|
| Whole (“shotgun”) metagenome sequencing |
|
|
| Metatranscriptomics |
|
|
| Mass spectrometry‐based methods | ||
| Metaproteomics |
|
|
| Metabolomics |
|
|
Figure 1.
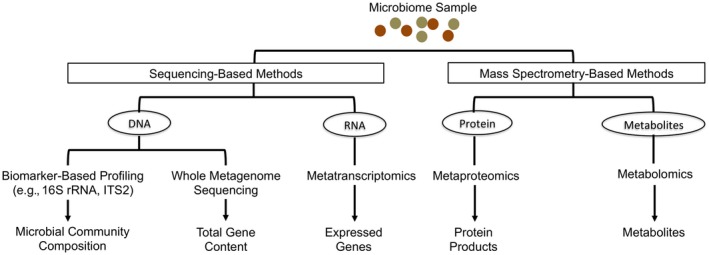
Methods for studying gut microbiome composition, function, and metabolic productivity.
Biomarker‐Based Microbiota Profiling
Many studies have catalogued the identity of distinct microbes present in a specimen through determination of sequence variants in a specific polymerase chain reaction–amplified microbial biomarker gene. Commonly used biomarker genes include the 16S ribosomal RNA (16S rRNA) gene and the internal transcribed spacer 2 region of bacteria and fungi, respectively. Due to the lack of a conserved biomarker gene across known viral species, no comparable approach exists for viruses; their detection currently relies on species‐specific genes or the reassembly of viral genomes from shotgun metagenomic sequencing efforts. Biomarker‐based sequencing offers the opportunity to economically characterize microbial community membership, structure, and taxonomic relative abundance and is particularly useful for large sample sizes and when the microbe:host ratio is low (e.g., mucosal biopsies). Inferential algorithms based on reference bacterial genomes enable predictions of evolutionarily conserved functional traits from 16S rRNA sequences.14 Although a useful tool, poor species‐level resolution in 16S rRNA gene sequencing, limited availability of closed bacterial genomes, and horizontal gene transfer across microorganisms may limit such in silico metagenomic predictions.15
Whole Metagenome Sequencing
In contrast with biomarker‐based sequencing, shotgun metagenomic sequencing does not focus on a single biomarker gene but rather DNA sequences arising from genomes within a microbial community. Sequenced fragments are either mapped to a reference genomic database or undergo de novo assembly to produce contiguous regions of microbial genomes, which subsequently undergo functional gene annotation using specialized platforms.16 Thus, metagenomic sequencing permits parallel identification of microbial species and their encoded functional genes within a microbiome. More recent human metagenomic studies have demonstrated the capacity to follow gut microbiomes at strain‐level resolution, permitting assessment of microbial population dynamics in the neonatal gut17 and tracking of microbial species (and their functional genes) that engraft following fecal microbial transplantation (FMT).18 Although the cost of sequencing has declined dramatically over the past decade, the need for very large numbers of sequence reads per sample to permit sufficient community coverage and metagenomic reconstruction coupled with the computational needs to facilitate analysis of such large data sets have resulted in shotgun metagenomics being more commonly applied to smaller representative sample sets.
Metatranscriptomics
Although DNA‐based metagenomic sequencing permits identification of microbes and encoded genes within a microbiome, it does not provide information on microbial gene expression. Indeed, even though microbiome perturbation is commonly associated with established disease, progression and severity may be mechanistically linked to changes in the transcriptional program of an otherwise compositionally stable pathogenic microbiome. Metatranscriptomic shotgun sequencing (also commonly referred to as RNA sequencing [RNA‐seq]) determines the gene expression profile of a microbiome, which, like other microbiota or microbiome profiling approaches, offers greatest utility when examined longitudinally and when related to specific host conditions (e.g., disease remission or flare) or, for example, in response to dietary inputs.19 Due to the need for high‐quality and high‐quantity RNA for metatranscriptomics, samples must be appropriately preserved at the time of sample collection, i.e., treated with a nucleic acid preservative. Following RNA isolation, rRNA, which can represent upwards of 75% of total RNA, is selectively depleted to enrich for messenger RNA (mRNA), long noncoding RNA, and microRNA. The mRNA pool is then fragmented and reverse transcribed to produce complementary DNA for sequencing. Bioinformatic tools similar to those used for shotgun metagenomics are then used to map, annotate, and quantify gene expression profiles derived from these data sets.16, 20
Metatranscriptomic analysis permits identification of the specific organisms responsible for expression, even if the specific gene or pathway of interest is more broadly encoded within the metagenome. In addition, it also permits directionality to be determined, i.e., whether host or microbial genes are expressed. The fundamental difference between turnover rates of RNA and DNA (minutes or less versus hours or more) underpins how metatranscriptome profiles may better capture contemporaneous microbiome responses to host exposures. This is particularly true for the gut microbiome, which is frequently exposed to nutritional and xenobiotic substrates, many of which have been shown to exert substantial effects on gut microbiome composition and functional gene capacity.21, 22 However, it should be noted that mRNA stability, which is inherently low in prokaryotes,23 may differ across distinct microbial species, leading to differential mRNA degradation across microbes that could skew metatranscriptome profiles.24
Metaproteomics
Facilitated by high‐throughput MS, metaproteomics profiles the complement of proteins produced by a microbiome, offering a complementary view of microbiome function. Given that proteins are inherently more stable than mRNA and represent the end products of posttranscriptional and posttranslational regulatory mechanisms, this approach may also provide a more accurate view of microbial productivity.
Metaproteomics currently uses both gel‐based and gel‐independent liquid chromatography (LC) separations prior to tandem MS (MS/MS)‐based peptide identification. Whole proteomes obtained following cellular lysis can be fractionated by centrifugation based on cellular localization or chemical properties (e.g., phosphorylation) prior to peptide and protein separation and identification. To identify peptides and proteins, high‐throughput proteomics software pipelines25 use search algorithms26 to compare MS/MS spectra to protein databases or to predicted intact masses and fragmentation patterns generated in silico from the corresponding metagenome.
Nonuniform peptide ionization and/or selective peptide loss during MS detection makes direct quantification of shotgun proteomic data challenging.27 To address this technical challenge, much effort has been directed toward stable isotope labeling,28 which enables quantification of the abundance of labeled protein under different states, and label‐free MS methods,29 which exploit intrinsic MS measurement metrics to quantify proteins. Apart from permitting quantitative proteomics, stable isotope labeling and label‐free MS methods have grown in popularity due to their simplicity, relatively low cost, and universal applicability.
Metabolomics
Metabolomics comprises complementary targeted and untargeted analytical chemistry techniques, including nuclear magnetic resonance spectrometry, gas chromatography‐MS (GC‐MS), and LC‐MS, that yield vast amounts of information regarding the composition of both inorganic and organic compounds associated with a microbiome. These include but are not limited to amino acids, lipids, sugars, organic acids, secondary bile acids, and more complex structures, such as macrolides and polyketides. Similar to metaproteomics, spectra are compared with existing databases to facilitate metabolite identification and, in some cases, source tracking.30, 31
Traditionally, throughput and range of protein and metabolite analyses lag far behind that of nucleic acid sequencing platforms (current platforms are predominantly based on MS analyses coupled to slow front‐end separations, such as GC and LC) and are hobbled by relatively underdeveloped databases. Fortunately, recent advances in rapid gas‐phase ion mobility spectrometry separations, which distinguish molecular conformations, have helped to decrease the long front‐end separation times and increase throughput of metabolite detection.32 In parallel, efforts in the field are focused on expanding current databases.
Integrating “Omics” Data
No single “omics” approach can fully decipher a mechanistic pathway in disease pathogenesis, and combining results from different platforms strengthens insight into microbiome–host interactions.33 The fundamental challenge in “multi‐omic” approaches is that these processes generate immense amounts of multidimensional data. As such, novel statistical methods for integrated data analysis that combine disparate data sets and standardized quality control metrics are constantly evolving. In addition to challenges with analytical approaches, data storage associated with such data sets requires robust computational resources and infrastructure. However, as the field advances and data sets increase in complexity and complementarity, the application of systems biology approaches promises to expand our mechanistic understanding of the gut microbiome across temporal and health gradients.
Factors That Shape the Gut Microbiome
Microbiome studies, especially those performed in human subjects, must account for potential confounding factors; therefore, an understanding of the factors that shape the gut microbiome is critical to experimental design.34 (Fig. 2) The healthy gut microbiome rapidly diversifies over the first several years of life,35, 36 typically remains compositionally stable in adulthood,37, 38 and exhibits diversity loss in the elderly.39 The stability of functional gene content encoded by the gut microbiome is less well studied. Mehta et al.19 recently performed metagenomic and metatranscriptomic sequencing of stool samples collected 6 months apart from 308 adults to examine temporal stability of gut microbial function. Although strain composition and the core microbial metagenome were relatively stable over time, unsurprisingly, transcriptomic variance exceeded metagenomic variance. These findings suggest that a single DNA‐based measurement of the adult gut microbiome can likely yield reliable information regarding composition and gene content but that the metatranscriptome (and thus the associated metabolic products) is more dynamic and related to contemporary exposures.
Figure 2.
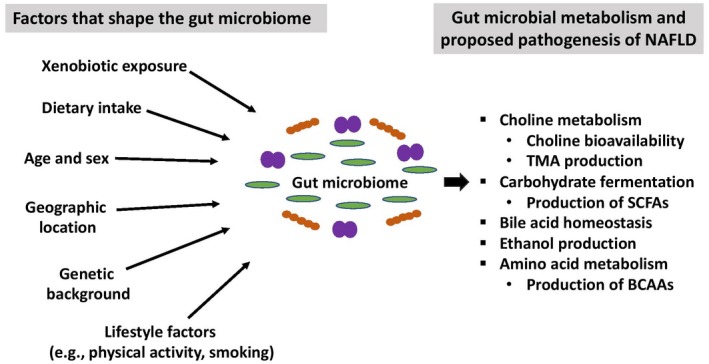
Factors that shape the gut microbial community and postulated mechanistic pathways linking gut microbial metabolism to the development of NAFLD.
Although the host genome has been shown to influence the composition of the gut microbiome,40, 41 environmental influences play a more significant role in shaping the human gut microbiome.42 In studies of the gut microbiota of monozygotic twins, fewer than half of detected bacterial taxa are shared, indicating that a minority of microbial taxa are inherited.35 Apart from host genetics, a number of endogenous and exogenous factors shape microbial communities in the gut. These include dietary and alcohol intake,43, 44 smoking,45, 46 sex and hormonal cycles,47 diurnal variation,48 physical activity,49 and xenobiotic exposure (including antibiotics).22, 50
A combined analysis of large patient cohorts from three continents (Europe, Asia, and North America) revealed country‐specific microbial signatures, suggesting that culture and geographic location significantly impact the gut microbiome composition and functional genes it encodes.51 These geographic microbial signatures are also known to be related to differences in diet, with functional gene enrichments in both babies and adults across geographically distinct sites closely linked with the proportion of plant polysaccharides and simple sugars consumed in the diet.35, 52, 53, 54 Indeed, short‐term dietary interventions in healthy adults modulate the gut microbiota, in part due to providing a competitive advantage to gut microbes capable of extracting energy from the specific dietary components consumed.1 Although there is currently no standardized approach to account for dietary data in human microbiome studies, this is an area of ongoing investigation.55 Nevertheless, there is sufficient compelling evidence to suggest that dietary intake along with other environmental factors that may shape the gut microbiome must be accounted for in human microbiome studies.
The Gut Microbiome in NAFLD
NAFLD, the liver manifestation of obesity and the metabolic syndrome, encompasses a spectrum of histopathologic abnormalities ranging from simple steatosis to nonalcoholic steatohepatitis (NASH). NASH, a more severe phenotype of disease, is associated with increased risk for development of cirrhosis, hepatocellular carcinoma, and liver‐related mortality.56 Among the 100 million adults with NAFLD in the United States, 15% to 20% have NASH.13 Despite the overwhelming burden of NAFLD, there are no approved pharmacologic therapies for NAFLD in the United States.57 Although the mechanisms that lead to the development of NAFLD and progression to NASH are not fully elucidated, an ever‐growing body of literature suggests that the gut microbiome is implicated in the pathogenesis of NAFLD.
Studies in animal models have provided the most compelling evidence for a causal role of the gut microbiome in the pathogenesis of NAFLD. Germ‐free mice lacking a gut microbiome are resistant to the development of hepatic steatosis,58 and FMT can transmit hepatic steatosis in murine models.59 Moreover, administration of probiotics or antibiotics to mice, which can dramatically alter gut microbiome composition and has also been shown to influence metabolic output in humans,60 inhibits development of high‐fat diet‐induced hepatic steatosis.61, 62
A number of cross‐sectional human studies have demonstrated an association between gut microbiota perturbation and the full clinical spectrum of NAFLD, including nonalcoholic fatty liver without steatohepatitis (NAFL),63, 64, 65, 66, 67, 68, 69, 70, 71 NASH,64, 65, 68, 69, 72, 73, 74, 75, 76 NAFLD‐related advanced fibrosis (fibrosis stage >2),68, 76, 77, 78, 79 and NAFLD‐related cirrhosis and hepatocellular carcinoma.77 Several of these studies were performed in pediatric cohorts,63, 64, 75 and an overwhelming majority of these studies used biomarker‐based sequencing to profile the gut microbiota. The majority of these studies revealed a decrease in overall bacterial diversity in NAFLD, similar to that reported in obesity.80 However, as for other chronic diseases,81, 82, 83 human microbiome studies in NAFLD have not identified a unique microbial signature. The most consistent finding among studies is that higher disease severity, and in particular advanced fibrosis, is associated with enrichment of members of the Bacteroides and Escherichia genera. Moreover, the gut microbiome in adults with NAFLD‐related cirrhosis as compared to adults with cirrhosis secondary to other etiologies comprises a higher abundance of Porphyromonadaceae and Bacterioidaceae and lower abundance of Veillonellaceae. 84
Discordant results between studies could be attributable to a variety of factors, such as differences in patient age (adults versus children/adolescents) and comorbidities (e.g., body mass index, diabetes), geographic location, definition of NAFLD clinical phenotypes, and microbial functional redundancy as well as technical differences in stool sample handling, sequencing, or statistical analysis methods between studies. This is particularly pertinent because many of the published studies did not measure or adjust for factors known to affect the gut microbiome. The inability to find a distinct disease‐associated microbiota signature is not unique to NAFLD82 and may also be attributable to a failure to detect overrepresented and underrepresented microbial taxa when statistical analyses are limited to diversity indices and/or comparisons at the phylum or family level (the predominant analytical approaches employed in NAFLD studies). Secondly, microbial biomarker sequencing studies fail to provide information on the functional gene content or products of the microbiome, thus potentially missing the crucial microbial signature associated with disease. This was demonstrated in a study of lean and obese individuals who were better differentiated by their gut metagenome as opposed to their microbiota profile.85 Finally, recent biomarker sequencing studies of large patient cohorts have demonstrated that several distinct microbiota conformations are evident in patients with a given clinical indication and are related to differences in immunologic profiles and clinical features of disease.81, 86 This implies that within a given diseased patient population several pathogenic microbiomes may exist; however, such possibilities remain unexplored in the field of NAFLD microbiome research.
Among the NAFLD human microbiome studies performed to date, a few have used one or more omic techniques and thus have added new perspectives on functional attributes of the gut microbiome in NAFLD pathogenesis.63, 64, 69, 70, 75, 78, 79 Da Silva et al.69 performed targeted profiling of eight fecal metabolites of interest and found that a higher concentration of two fecal SCFAs, propionate and isobutyric acid, differentiated adults with NAFL from healthy controls. Del Chierico et al.64 performed an integrated analysis of the gut microbiota (by 16S rRNA‐seq) and gut metabolome (by targeted metabolomics) in a pediatric cohort with the following clinical phenotypes: NAFL, NASH, and obese and healthy controls. In multivariate analysis, children with NASH had lower levels of the genus Oscillospira, increased levels of the genera Dorea and Ruminococcus, and higher levels of two gut metabolites, 2‐butanone and 4‐methyl‐2‐pentanone, when compared to controls.
Loomba et al.79 were the first to examine the whole gut metagenome, which was integrated with serum metabolomics, in adults with NAFLD. The prospective cohort study included 86 patients with biopsy‐proven NAFLD, of which 72 had no or minimal fibrosis (stages 0‐2) and 14 had advanced fibrosis (stages 3‐4). Whereas differential abundances in microbial gene pathways and serum metabolites did not achieve statistical significance after correction for multiple testing, 37 bacterial species, including Escherichia coli and Bacteroides vulgatas, were differentially represented between minimal and advanced fibrosis phenotypes. When these bacterial species were incorporated into a model with patient age, body mass index, and a microbial diversity index, the model possessed an area under the receiver operating characteristic curve of 0.936 for detecting advanced fibrosis. This finding is concordant with a prior observation that B. vulgatus is a contributor to insulin resistance.87 A more recent study by Caussy et al.78 identified a shared gene effect of serum 3‐(4‐hydroxyphenyl)lactate, a microbial‐derived metabolite, with NAFLD‐related advanced fibrosis. Although the functional significance of this metabolite is unknown, 3‐(4‐hydroxyphenyl)lactate was strongly correlated with the abundance of seven bacterial species that were previously associated with advanced fibrosis, including B. caccae, E. coli, and Clostridium sporogenes. 79
Gut Microbial Metabolism and Interactions With the Human Host
Gut microbial metabolites have now been linked with specific disease states: autism spectrum disorder (4‐ethylphenylsulfate)88 and obesity‐associated hepatocellular carcinoma (deoxycholic acid)89 in murine models as well as childhood atopic asthma (12,13‐dihydroxy‐9Z‐octadecenoic acid [12,13 Di‐HOME])90 and cardiovascular disease (trimethylamine‐N‐oxide [TMAO])91 in human subjects. A review of the current evidence of the causal link between the gut microbiota and cardiovascular disease provides an example of how mechanistic approaches to studying the gut microbiome not only yield insight into disease pathogenesis but may also provide the foundation by which microbiome‐targeted therapies can be developed.
Gut microbial metabolism of dietary methylamines contributes to the formation of the following three metabolites: choline, betaine, and trimethylamine (TMA).92 TMA is metabolized by the liver to generate TMAO, and manipulation of the gut microbiota, either through FMT or administration of specific bacterial strains, alters TMAO levels in murine models.93, 94 In recent years, many human studies have examined the association between serum levels of the microbial‐derived metabolite TMAO and cardiovascular disease. Elevated serum TMAO is associated with the degree of atherosclerotic burden and risk of incident cardiovascular events among adults presenting with chest pain and those undergoing coronary angiography.95, 96 More recent evidence suggests that elevated serum TMAO levels are also linked to adipose tissue dysfunction97 and chronic kidney disease.98 Subsequent mechanistic studies identified the biologic function of TMAO to include endothelial dysfunction and enhancement of platelet hyperreactivity.99, 100 Oral application of a structural analogue of choline, 3,3‐dimethyl‐1‐butanol, inhibited microbial TMA production, lowered serum TMAO levels, and prevented development of atherosclerotic lesions, despite a proatherosclerosis diet, in an animal model.101 Together, these data support that a microbial‐derived metabolite is an important contributor to a variety of interrelated cardiometabolic diseases and that targeting the gut microbial capacity to produce this metabolite offers a potential novel strategy for disease prevention or treatment.
Gut Microbial Metabolism in the Pathogenesis of NAFLD
Unlike in cardiovascular disease, precise microbiome–host mechanisms in NAFLD are not well delineated. However, based on animal studies, many of which were performed in models of obesity and insulin resistance, several potential mechanistic links between the gut microbiome and NAFLD have emerged. Postulated mechanisms include dysregulation of methylamine metabolism, carbohydrate fermentation and generation of SCFAs, bile acid metabolism, endogenous ethanol production, and amino acid metabolism. The gut microbiota may also contribute to NAFLD through intestinal barrier dysfunction and lipopolysaccharide‐induced activation of toll‐like receptor pathways of innate immune signaling,102 but for the purpose of this review, discussion is limited to disease mechanisms potentially mediated by microbial metabolism (Fig. 2).
Methylamine Metabolism
In addition to the role in cardiovascular disease, gut microbial metabolism of dietary methylamines, specifically choline, may also contribute to the development of NAFLD. Choline deficiency contributes to the development of fatty liver disease through multiple mechanisms, including abnormal phospholipid synthesis, defective very low‐density lipoprotein secretion, and alterations in the enterohepatic circulation of bile acids.103 Susceptibility to choline deficiency was previously attributed to factors such as low dietary choline intake, estrogen status, and single‐nucleotide polymorphisms in specific host genes for choline metabolism.103 However, the discovery of gut microbial metabolism of choline to produce TMA provides an additional mechanism for low choline bioavailability.
A high‐fat diet leads to increased gut microbial metabolism of choline and the development of hepatic steatosis in mice.104 Manipulation of dietary choline intake in 15 female human subjects resulted in variations in the abundance of choline‐metabolizing gut microbial species; abundance of these specific microbes along with a single‐nucleotide polymorphism of a host choline metabolism gene accurately predicted the degree to which subjects developed fatty liver while on a choline‐deficient diet.105 Subsequent human studies have revealed varying associations between by‐products of methylamine metabolism and NAFLD severity,106, 107 but additional studies are necessary to decipher the exact role of gut microbial methylamine metabolism in NAFLD pathogenesis.
Fermentation of Indigestible Carbohydrates and Production of SCFAs
Complex carbohydrates undergo microbial fermentation predominantly in the colon, resulting in the production of SCFAs, including acetate, propionate, and butyrate. SCFAs provide the majority of energy needs for intestinal epithelial cells, but they also cross the intestinal epithelial barrier and mediate a variety of biologic activities, including modulation of immune function and exerting antiproliferative effects, in part by signaling through G protein‐coupled receptors in multiple tissue sites.108, 109
Fecal SCFA concentrations are increased in genetically obese mice,80 and germ‐free mice with low levels of gut SCFAs are protected from diet‐induced obesity.110 These data suggest that significantly elevated fecal SCFAs, indicative of increased energy extraction from dietary input, are a hallmark of obesity. However, in contrast to these findings, an increase in butyrate‐producing bacteria has been shown to prevent diet‐induced hepatic steatosis and bile acid dysregulation and its associated hepatitis in murine models.111, 112 Moreover, in humans, increasing colonic propionate prevented weight gain in overweight adults,113 and the beneficial metabolic effects of FMT from lean to obese donors were primarily associated with an increased abundance of butyrate‐producing bacterial strains in the gut.114 These contradictory findings are likely attributable to differing end‐organ effects of individual or combinations of SCFAs, including varying effects on energy expenditure, production of satiety hormones, and central appetite regulation.108
Bile Acid Transformations by the Gut Microbiota
Bile acids are steroid‐derived molecules synthesized in the liver and secreted into the intestinal lumen that facilitate lipid absorption and additionally regulate metabolism by activation of a number of host receptors, including farnesoid X receptor (FXR) and the membrane‐associated G protein‐coupled receptor (TGR5).115 Distinct bile acids have varying affinities for host receptors and may act either as receptor agonists or antagonists, with subsequent effects on the regulation of basal metabolism and energy expenditure, lipid metabolism, nitric oxide synthesis, intestinal motility and permeability, and immune response.116, 117
In addition to their effects on the host, bile acids prevent intestinal bacterial overgrowth, both directly through membrane‐damaging effects and indirectly through induction of antimicrobial protein expression; thus, they also play an important role in shaping gut microbiome membership.118 Indeed, microbial bile acid resistance offers a fitness advantage in the gut and is commonly complemented by the capacity to deconjugate bile acids and convert primary bile acids to secondary bile acids. Germ‐free mice or antibiotic‐treated mice exhibit low concentrations of conjugated bile acids, pointing to a central role of gut microbiome in regulating bile acid composition.
Animal models have revealed that lean and obese mice have a differential abundance of various bile acids.119 Human studies have noted elevated total serum bile acids, with an altered ratio of secondary to primary bile acids, in adults with NAFLD.73, 120, 121 Of interest, administration of the FXR agonist obeticholic acid led to improvement in NASH and insulin sensitivity in randomized controlled trials in adults, indicating an important role for bile acid signaling in NAFLD and the potential for therapeutic intervention targeting bile acid receptors.122, 123 Thus, dysregulation of the reciprocal relationship between the human gut microbiome and bile acids has the capacity to significantly modulate FXR and TGR5 signaling, modulating host metabolism and immunity, and offers an attractive mechanistic hypothesis linking the gut microbiome and metabolic diseases, such as NAFLD.124 For a comprehensive overview of gut microbiome–bile acid crosstalk in the pathogenesis of metabolic disease, we direct the reader to two recent reviews.115, 125
Endogenous Ethanol Production
Hepatic genes involved in alcohol metabolism, including alcohol dehydrogenase, are up‐regulated in NAFLD.126, 127 A number of studies have demonstrated that children and adolescents with NAFLD have increased serum ethanol levels,63, 64, 75 although this finding is not consistent across all studies.69 The gut microbiome can generate ethanol from dietary precursors, and manipulation of the gut microbiome is known to alter endogenous ethanol production.128 A common finding in NAFL and NASH is enrichment in Escherichia and/or Lactobacillus genera, 65, 68, 69, 70, 71, 74, 75, 77, 78, 79 which have the capability to produce ethanol. These findings suggest that increased endogenous ethanol production by the gut microbiome could contribute to the pathogenesis of NAFLD. However, a recent study demonstrated that elevated serum ethanol may also be attributed to insulin‐dependent impairments of alcohol dehydrogenase activity in the liver, independent of endogenous alcohol production by the gut microbiome,129 pointing to the existence of distinct disease mechanisms that may converge on a common clinical manifestation and are plausibly linked to gut microbiome functional differentials within this patient population.
Amino Acid Biosynthesis and Metabolism
The gut microbiome influences amino acid homeostasis, in part due to biosynthesis and metabolism of aromatic amino acids (AAAs) and branched‐chain amino acids (BCAAs). Elevated serum BCAA, postulated to be secondary to perturbations in gut microbial metabolism, has been identified as a potential biomarker for insulin resistance in several cohort studies.130, 131 Specifically, two bacterial species, Prevotella copri and B. vulgatus, are associated with enriched biosynthetic potential for BCAAs and a reduced potential for BCAA transport into bacterial cells in adults with insulin resistance.87 Intriguingly, a more recent study found that B. vulgatus was one of the most abundant gut microbes in adults with NAFLD‐related advanced fibrosis.79
Perturbations in microbial metabolism of AAAs and BCAAs and ensuing alterations in the serum metabolite profile have more recently been identified in NAFLD.70, 132 Hoyles et al.70 performed an integrated analysis of the gut metagenome, hepatic transcriptome, and serum and urine metabolomes in a cohort of women with obesity but without diabetes. Compared to those without NAFL, women with NAFL had significant alterations in the gut metagenome, including differences in BCAA and AAA pathways, as well as in the serum metabolome. Specifically, serum phenylacetic acid, an AAA‐derived microbial metabolite, was strongly correlated with hepatic steatosis in this cohort. This metabolite was subsequently found to induce hepatic steatosis both in vitro (primary culture of human hepatocytes) and in vivo (murine model), suggesting a potential causal role in NAFL pathogenesis.70 This study provides a proof‐of‐concept prototype of how integrated multi‐omic analyses in human subjects combined with in vitro and in vivo mechanistic studies can facilitate identification of possible microbial‐driven mechanistic pathways in NAFLD.
Gut Microbiome‐Targeted Therapy in the Management of NAFLD
The prospect of targeting the gut microbiome in the management of NAFLD offers a novel strategy for disease prevention or management. A diverse array of strategies to manipulate the gut microbiome exist and include dietary interventions, probiotics (culture of living microorganisms), prebiotics (fermentable dietary fibers that stimulate the growth and survival of probiotics), synbiotics (combination of probiotics and prebiotics), antibiotics, and FMT.1 Moreover, bariatric surgery results in rapid and sustained shifts in the gut microbiome.133 Damms‐Machado et al.134 found that the gut microbiome in adults with obesity who underwent bariatric surgery shifted within 6 months after surgery toward a microbial profile found in adults with normal body weight. Recent studies have revealed the existence of distinct and niche‐specific microbial communities along the length of the gastrointestinal tract and that the composition of these communities influence successful engraftment of exogenous microbes,135, 136 suggesting that future microbial interventions may be personalized based on an individual’s gut microbiome. However, large‐scale interventional trials with therapies targeting the gut microbiome in NAFLD are lacking, and a better understanding of the complexities of microbial metabolism and microbe–host interactions is necessary to inform the development of gut microbiome‐targeted therapies for this patient population.
Conclusions
An ever‐growing body of preclinical and clinical literature supports that changes to the composition, function, and/or metabolic output of the gut microbiome are associated with NAFLD; however, the exact mechanistic links remain elusive. Integrated multi‐omic analyses, based on high‐throughput sequencing and MS platforms, are now yielding more comprehensive views of gut microbiome functional capacity and biochemical output in NAFLD. Additional longitudinal prospective cohort studies, which are designed to account for various environmental factors that can shape the gut microbiome and are complemented by in vitro and in vivo mechanistic studies, are necessary to delineate microbiome–host interactions that contribute to NAFLD pathogenesis. Improved knowledge of the contribution of gut microbial metabolism to liver health could yield the foundation for the development of novel microbiota‐targeted precision interventions for the treatment or management of NAFLD.
Potential conflict of interest
Dr. Terrault has institutional grant support from Allergan and Gilead Sciences. Dr. Lynch is a cofounder, is a board member, owns stock in, is employed by, and is a consultant for Siolta Therapeutics and serves on the scientific advisory board of Bloom Science, Inc.
Supported by an Institutional National Research Service Award Hepatology Training Grant from the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) (2T32DK060414‐16 to S.R.S.), the NIDDK (U01 to N.A.T.), and the National Institutes of Health (awards DH082147, P0515267, AI113916, AI133765, and DA040532 to S.V.L.).
References
- 1. Lynch SV, Pedersen O. The human intestinal microbiome in health and disease. New Engl J Med 2016;375:2369‐2379. [DOI] [PubMed] [Google Scholar]
- 2. Human Microbiome Project Consortium . Structure, function and diversity of the healthy human microbiome. Nature 2012;486:207‐214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Carding SR, Davis N, Hoyles L. Review article: The human intestinal virome in health and disease. Aliment Pharmacol Ther 2017;46:800‐815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Nash AK, Auchtung TA, Wong MC, Smith DP, Gesell JR, Ross MC, et al. The gut mycobiome of the Human Microbiome Project healthy cohort. Microbiome 2017;5:153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, et al.; MetaHIT Consortium . A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010;464:59‐65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Franzosa EA, Huang K, Meadow JF, Gevers D, Lemon KP, Bohannan BJ, et al. Identifying personal microbiomes using metagenomic codes. Proc Natl Acad Sci U S A 2015;112:E2930‐E2938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cani PD, Amar J, Iglesias MA, Poggi M, Knauf C, Bastelica D, et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 2007;56:1761‐1772. [DOI] [PubMed] [Google Scholar]
- 8. Shibata N, Kunisawa J, Kiyono H. Dietary and microbial metabolites in the regulation of host immunity. Front Microbiol 2017;8:2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tripathi A, Debelius J, Brenner DA, Karin M, Loomba R, Schnabl B, et al. The gut‐liver axis and the intersection with the microbiome. Nat Rev Gastroenterol Hepatol 2018;15:397‐411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bashiardes S, Zilberman‐Schapira G, Elinav E. Use of metatranscriptomics in microbiome research. Bioinform Biol Insights 2016;10:19‐25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Quince C, Walker AW, Simpson JT, Loman NJ, Segata N. Shotgun metagenomics, from sampling to analysis. Nat Biotechnol 2017;35:833‐844. [DOI] [PubMed] [Google Scholar]
- 12. Petriz BA, Franco OL. Metaproteomics as a complementary approach to gut microbiota in health and disease. Front Chem 2017;5:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Martinez KB, Leone V, Chang EB. Microbial metabolites in health and disease: navigating the unknown in search of function. J Biol Chem 2017;292:8553‐8559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Langille MG, Zaneveld J, Caporaso JG, McDonald D, Knights D, Reyes JA, et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol 2013;31:814‐821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Polz MF, Alm EJ, Hanage WP. Horizontal gene transfer and the evolution of bacterial and archaeal population structure. Trends Genet 2013;29:170‐175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Abubucker S, Segata N, Goll J, Schubert AM, Izard J, Cantarel BL, et al. Metabolic reconstruction for metagenomic data and its application to the human microbiome. PLoS Comput Biol 2012;8:e1002358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Brown CT, Olm MR, Thomas BC, Banfield JF. Measurement of bacterial replication rates in microbial communities. Nat Biotechnol 2016;34:1256‐1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Moss EL, Falconer SB, Tkachenko E, Wang M, Systrom H, Mahabamunuge J, et al. Long‐term taxonomic and functional divergence from donor bacterial strains following fecal microbiota transplantation in immunocompromised patients. PLoS One 2017;12:e0182585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mehta RS, Abu‐Ali GS, Drew DA, Lloyd‐Price J, Subramanian A, Lochhead P, et al. Stability of the human faecal microbiome in a cohort of adult men. Nat Microbiol 2018;3:347‐355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Westreich ST, Korf I, Mills DA, Lemay DG. SAMSA: a comprehensive metatranscriptome analysis pipeline. BMC Bioinformatics 2016;17:399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Turnbaugh PJ, Ridaura VK, Faith JJ, Rey FE, Knight R, Gordon JI. The effect of diet on the human gut microbiome: a metagenomic analysis in humanized gnotobiotic mice. Sci Transl Med 2009;1:6ra14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Maurice CF, Haiser HJ, Turnbaugh PJ. Xenobiotics shape the physiology and gene expression of the active human gut microbiome. Cell 2013;152:39‐50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rauhut R, Klug G. mRNA degradation in bacteria. FEMS Microbiol Rev 1999;23:353‐370. [DOI] [PubMed] [Google Scholar]
- 24. Stark L, Giersch T, Wunschiers R. Efficiency of RNA extraction from selected bacteria in the context of biogas production and metatranscriptomics. Anaerobe 2014;29:85‐90. [DOI] [PubMed] [Google Scholar]
- 25. Deutsch EW. The PeptideAtlas project. Methods Mol Biol 2010;604:285‐296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fenyo D, Beavis RC. A method for assessing the statistical significance of mass spectrometry‐based protein identifications using general scoring schemes. Anal Chem 2003;75:768‐774. [DOI] [PubMed] [Google Scholar]
- 27. Ong SE, Mann M. Mass spectrometry‐based proteomics turns quantitative. Nat Chem Biol 2005;1:252‐262. [DOI] [PubMed] [Google Scholar]
- 28. Chahrour O, Cobice D, Malone J. Stable isotope labelling methods in mass spectrometry‐based quantitative proteomics. J Pharm Biomed Anal 2015;113:2‐20. [DOI] [PubMed] [Google Scholar]
- 29. Lu CY, Wu CY, Lin CH. Protein identification by syringe pump‐driven reversed‐phase LC‐MS/MS. Anal Biochem 2007;368:123‐129. [DOI] [PubMed] [Google Scholar]
- 30. Larive CK, Barding GA Jr, Dinges MM. NMR spectroscopy for metabolomics and metabolic profiling. Anal Chem 2015;87:133‐146. [DOI] [PubMed] [Google Scholar]
- 31. Wishart DS, Jewison T, Guo AC, Wilson M, Knox C, Liu Y, et al. HMDB 3.0—The Human Metabolome Database in 2013. Nucleic Acids Res 2013;41:D801‐D807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Baker ES, Burnum‐Johnson KE, Ibrahim YM, Orton DJ, Monroe ME, Kelly RT, et al. Enhancing bottom‐up and top‐down proteomic measurements with ion mobility separations. Proteomics 2015;15:2766‐2776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Franzosa EA, Hsu T, Sirota‐Madi A, Shafquat A, Abu‐Ali G, Morgan XC, et al. Sequencing and beyond: integrating molecular ‘omics’ for microbial community profiling. Nat Rev Microbiol 2015;13:360‐372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kim D, Hofstaedter CE, Zhao C, Mattei L, Tanes C, Clarke E, et al. Optimizing methods and dodging pitfalls in microbiome research. Microbiome 2017;5:52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yatsunenko T, Rey FE, Manary MJ, Trehan I, Dominguez‐Bello MG, Contreras M, et al. Human gut microbiome viewed across age and geography. Nature 2012;486:222‐227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Backhed F, Roswall J, Peng Y, Feng Q, Jia H, Kovatcheva‐Datchary P, et al. Dynamics and stabilization of the human gut microbiome during the first year of life. Cell Host Microbe 2015;17:690‐703. Erratum. In: Cell Host Microbe 2015;17:852. [DOI] [PubMed] [Google Scholar]
- 37. Faith JJ, Guruge JL, Charbonneau M, Subramanian S, Seedorf H, Goodman AL, et al. The long‐term stability of the human gut microbiota. Science 2013;341:1237439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lozupone CA, Stombaugh JI, Gordon JI, Jansson JK, Knight R. Diversity, stability and resilience of the human gut microbiota. Nature 2012;489:220‐230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Odamaki T, Kato K, Sugahara H, Hashikura N, Takahashi S, Xiao JZ, et al. Age‐related changes in gut microbiota composition from newborn to centenarian: a cross‐sectional study. BMC Microbiol 2016;16:90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Goodrich JK, Waters JL, Poole AC, Sutter JL, Koren O, Blekhman R, et al. Human genetics shape the gut microbiome. Cell 2014;159:789‐799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kolde R, Franzosa EA, Rahnavard G, Hall AB, Vlamakis H, Stevens C, et al. Host genetic variation and its microbiome interactions within the Human Microbiome Project. Genome Med 2018;10:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Rothschild D, Weissbrod O, Barkan E, Kurilshikov A, Korem T, Zeevi D, et al. Environment dominates over host genetics in shaping human gut microbiota. Nature 2018;555:210‐215. [DOI] [PubMed] [Google Scholar]
- 43. Starkel P, Leclercq S, de Timary P, Schnabl B. Intestinal dysbiosis and permeability: the yin and yang in alcohol dependence and alcoholic liver disease. Clin Sci (Lond) 2018;132:199‐212. [DOI] [PubMed] [Google Scholar]
- 44. Dong TS, Gupta A. Influence of early life, diet, and the environment on the microbiome. Clin Gastroenterol Hepatol 2018; doi: 10.1016/j.cgh.2018.08.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Shanahan ER, Shah A, Koloski N, Walker MM, Talley NJ, Morrison M, et al. Influence of cigarette smoking on the human duodenal mucosa‐associated microbiota. Microbiome 2018;6:150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lee SH, Yun Y, Kim SJ, Lee EJ, Chang Y, Ryu S, et al. Association between cigarette smoking status and composition of gut microbiota: population‐based cross‐sectional study. J Clin Med 2018;7:E282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Koren O, Goodrich JK, Cullender TC, Spor A, Laitinen K, Bäckhed HK, et al. Host remodeling of the gut microbiome and metabolic changes during pregnancy. Cell 2012;150:470‐480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Liang X, Bushman FD, FitzGerald GA. Rhythmicity of the intestinal microbiota is regulated by gender and the host circadian clock. Proc Natl Acad Sci U S A 2015;112:10479‐10484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Bressa C, Bailén‐Andrino M, Pérez‐Santiago J, González‐Soltero R, Pérez M, Montalvo‐Lominchar MG, et al. Differences in gut microbiota profile between women with active lifestyle and sedentary women. PLoS One 2017;12:e0171352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Maier L, Pruteanu M, Kuhn M, Zeller G, Telzerow A, Anderson EE, et al. Extensive impact of non‐antibiotic drugs on human gut bacteria. Nature 2018;555:623‐628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Li J, Jia H, Cai X, Zhong H, Feng Q, Sunagawa S, et al; MetaHIT Consortium . An integrated catalog of reference genes in the human gut microbiome. Nature Biotechnol 2014;32:834‐841. [DOI] [PubMed] [Google Scholar]
- 52. Mardinoglu A, Wu H, Bjornson E, Zhang C, Hakkarainen A, Rasanen SM, et al. An integrated understanding of the rapid metabolic benefits of a carbohydrate‐restricted diet on hepatic steatosis in humans. Cell Metab 2018;27:559‐571.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Christensen L, Roager HM, Astrup A, Hjorth MF. Microbial enterotypes in personalized nutrition and obesity management. Am J Clin Nutr 2018;108:645‐651. [DOI] [PubMed] [Google Scholar]
- 54. Wu GD, Chen J, Hoffmann C, Bittinger K, Chen YY, Keilbaugh SA, et al. Linking long‐term dietary patterns with gut microbial enterotypes. Science 2011;334:105‐108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Bowyer RCE, Jackson MA, Pallister T, Skinner J, Spector TD, Welch AA, et al. Use of dietary indices to control for diet in human gut microbiota studies. Microbiome 2018;6:77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Diehl AM, Day C. Cause, pathogenesis, and treatment of nonalcoholic steatohepatitis. N Engl J Med 2017;377:2063‐2072. [DOI] [PubMed] [Google Scholar]
- 57. Younossi ZM, Loomba R, Rinella ME, Bugianesi E, Marchesini G, Neuschwander‐Tetri BA, et al. Current and future therapeutic regimens for non‐alcoholic fatty liver disease and non‐alcoholic steatohepatitis. Hepatology 2018;68:361‐371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Bäckhed F, Manchester JK, Semenkovich CF, Gordon JI. Mechanisms underlying the resistance to diet‐induced obesity in germ‐free mice. Proc Natl Acad Sci U S A 2007;104:979‐984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Le Roy T, Llopis M, Lepage P, Bruneau A, Rabot S, Bevilacqua C, et al. Intestinal microbiota determines development of non‐alcoholic fatty liver disease in mice. Gut 2013;62:1787‐1794. [DOI] [PubMed] [Google Scholar]
- 60. Durack J, Kimes NE, Lin DL, Rauch M, McKean M, McCauley K, et al. Delayed gut microbiota development in high‐risk for asthma infants is temporarily modifiable by Lactobacillus supplementation. Nat Commun 2018;9:707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Okubo H, Sakoda H, Kushiyama A, Fujishiro M, Nakatsu Y, Fukushima T, et al. Lactobacillus casei strain Shirota protects against nonalcoholic steatohepatitis development in a rodent model. Am J Physiol Gastrointest Liver Physiol 2013;305:G911‐G918. [DOI] [PubMed] [Google Scholar]
- 62. Mencarelli A, Cipriani S, Renga B, Bruno A, D'Amore C, Distrutti E, et al. VSL#3 resets insulin signaling and protects against NASH and atherosclerosis in a model of genetic dyslipidemia and intestinal inflammation. PLoS One 2012;7:e45425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Michail S, Lin M, Frey MR, Fanter R, Paliy O, Hilbush B, et al. Altered gut microbial energy and metabolism in children with non‐alcoholic fatty liver disease. FEMS Microbiol Ecol 2015;91:1‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Del Chierico F, Nobili V, Vernocchi P, Russo A, Stefanis C, Gnani D, et al. Gut microbiota profiling of pediatric nonalcoholic fatty liver disease and obese patients unveiled by an integrated meta‐omics‐based approach. Hepatology 2017;65:451‐464. [DOI] [PubMed] [Google Scholar]
- 65. Nobili V, Putignani L, Mosca A, Chierico FD, Vernocchi P, Alisi A, et al. Bifidobacteria and lactobacilli in the gut microbiome of children with non‐alcoholic fatty liver disease: Which strains act as health players? Arch Med Sci 2018;14:81‐87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Raman M, Ahmed I, Gillevet PM, Probert CS, Ratcliffe NM, Smith S, et al. Fecal microbiome and volatile organic compound metabolome in obese humans with nonalcoholic fatty liver disease. Clin Gastroenterol Hepatol 2013;11:868‐875.e3. [DOI] [PubMed] [Google Scholar]
- 67. Wang B, Jiang X, Cao M, Ge J, Bao Q, Tang L, et al. Altered fecal microbiota correlates with liver biochemistry in nonobese patients with non‐alcoholic fatty liver disease. Sci Rep 2016;6:32002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Shen F, Zheng RD, Sun XQ, Ding WJ, Wang XY, Fan JG. Gut microbiota dysbiosis in patients with non‐alcoholic fatty liver disease. Hepatobiliary Pancreat Dis Int 2017;16:375‐381. [DOI] [PubMed] [Google Scholar]
- 69. Da Silva HE, Teterina A, Comelli EM, Taibi A, Arendt BM, Fischer SE, et al. Nonalcoholic fatty liver disease is associated with dysbiosis independent of body mass index and insulin resistance. Sci Rep 2018;8:1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Hoyles L, Fernandez‐Real JM, Federici M, Serino M, Abbott J, Charpentier J, et al. Molecular phenomics and metagenomics of hepatic steatosis in non‐diabetic obese women. Nat Med 2018;24:1070‐1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Jiang W, Wu N, Wang X, Chi Y, Zhang Y, Qiu X, et al. Dysbiosis gut microbiota associated with inflammation and impaired mucosal immune function in intestine of humans with non‐alcoholic fatty liver disease. Sci Rep 2015;5:8096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Wong VW, Tse CH, Lam TT, Wong GL, Chim AM, Chu WC, et al. Molecular characterization of the fecal microbiota in patients with nonalcoholic steatohepatitis: a longitudinal study. PLoS One 2013;8:e62885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Mouzaki M, Wang AY, Bandsma R, Comelli EM, Arendt BM, Zhang L, et al. Bile acids and dysbiosis in non‐alcoholic fatty liver disease. PLoS One 2016;11:e0151829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Duarte SMB, Stefano JT, Miele L, Ponziani FR, Souza‐Basqueira M, Okada LSRR, et al. Gut microbiome composition in lean patients with NASH is associated with liver damage independent of caloric intake: a prospective pilot study. Nutr Metab Cardiovasc Dis 2018;28:369‐384. [DOI] [PubMed] [Google Scholar]
- 75. Zhu L, Baker SS, Gill C, Liu W, Alkhouri R, Baker RD, et al. Characterization of gut microbiomes in nonalcoholic steatohepatitis (NASH) patients: a connection between endogenous alcohol and NASH. Hepatology 2013;57:601‐609. [DOI] [PubMed] [Google Scholar]
- 76. Boursier J, Mueller O, Barret M, Machado M, Fizanne L, Araujo‐Perez F, et al. The severity of nonalcoholic fatty liver disease is associated with gut dysbiosis and shift in the metabolic function of the gut microbiota. Hepatology 2016;63:764‐775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Ponziani FR, Bhoori S, Castelli C, Putignani L, Rivoltini L, Del Chierico F, et al. Hepatocellular carcinoma is associated with gut microbiota profile and inflammation in non‐alcoholic fatty liver disease. Hepatology 2018; doi: 10.1002/hep.30036. [DOI] [PubMed] [Google Scholar]
- 78. Caussy C, Hsu C, Lo M‐T, Liu A, Bettencourt R, Ajmera VH, et al.; Genetics of NAFLD in Twins Consortium . Link between gut‐microbiome derived metabolite and shared gene‐effects with hepatic steatosis and fibrosis in NAFLD. Hepatology 2018; doi: 10.1002/hep.29892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Loomba R, Seguritan V, Li W, Long T, Klitgord N, Bhatt A, et al. Gut microbiome‐based metagenomic signature for non‐invasive detection of advanced fibrosis in human nonalcoholic fatty liver disease. Cell Metab 2017;25:1054‐1062.e1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. An obesity‐associated gut microbiome with increased capacity for energy harvest. Nature 2006;444:1027‐1031. [DOI] [PubMed] [Google Scholar]
- 81. Cope EK, Goldberg AN, Pletcher SD, Lynch SV. Compositionally and functionally distinct sinus microbiota in chronic rhinosinusitis patients have immunological and clinically divergent consequences. Microbiome 2017;5:53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Finucane MM, Sharpton TJ, Laurent TJ, Pollard KS. A taxonomic signature of obesity in the microbiome? Getting to the guts of the matter. PLoS One 2014;9:e84689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Mar JS, LaMere BJ, Lin DL, Levan S, Nazareth M, Mahadevan U, et al. Disease severity and immune activity relate to distinct interkingdom gut microbiome states in ethnically distinct ulcerative colitis patients. MBio 2016;7:pii:e01072‐16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Bajaj JS, Heuman DM, Hylemon PB, Sanyal AJ, White MB, Monteith P, et al. Altered profile of human gut microbiome is associated with cirrhosis and its complications. J Hepatol 2014;60:940‐947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Le Chatelier E, Nielsen T, Qin J, Prifti E, Hildebrand F, Falony G, et al. Richness of human gut microbiome correlates with metabolic markers. Nature 2013;500:541‐546. [DOI] [PubMed] [Google Scholar]
- 86. Shenoy MK, Iwai S, Lin DL, Worodria W, Ayakaka I, Byanyima P, et al. Immune response and mortality risk relate to distinct lung microbiomes in patients with HIV and pneumonia. Am J Respir Crit Care Med 2017;195:104‐114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Pedersen HK, Gudmundsdottir V, Nielsen HB, Hyotylainen T, Nielsen T, Jensen BA, et al. Human gut microbes impact host serum metabolome and insulin sensitivity. Nature 2016;535:376‐381. [DOI] [PubMed] [Google Scholar]
- 88. Hsiao EY, McBride SW, Hsien S, Sharon G, Hyde ER, McCue T, et al. Microbiota modulate behavioral and physiological abnormalities associated with neurodevelopmental disorders. Cell 2013;155:1451‐1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Yoshimoto S, Loo TM, Atarashi K, Kanda H, Sato S, Oyadomari S, et al. Obesity‐induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature 2013;499:97‐101. [DOI] [PubMed] [Google Scholar]
- 90. Fujimura KE, Sitarik AR, Havstad S, Lin DL, Levan S, Fadrosh D, et al. Neonatal gut microbiota associates with childhood multisensitized atopy and T cell differentiation. Nat Med 2016;22:1187‐1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Tang WH, Wang Z, Levison BS, Koeth RA, Britt EB, Fu X, et al. Intestinal microbial metabolism of phosphatidylcholine and cardiovascular risk. N Engl J Med 2013;368:1575‐1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Romano KA, Vivas EI, Amador‐Noguez D, Rey FE. Intestinal microbiota composition modulates choline bioavailability from diet and accumulation of the proatherogenic metabolite trimethylamine‐N‐oxide. MBio 2015;6:e02481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Qiu L, Yang D, Tao X, Yu J, Xiong H, Wei H. Enterobacter aerogenes ZDY01 attenuates choline‐induced trimethylamine N‐oxide levels by remodeling gut microbiota in mice. J Microbiol Biotechnol 2017;27:1491‐1499. [DOI] [PubMed] [Google Scholar]
- 94. Gregory JC, Buffa JA, Org E, Wang Z, Levison BS, Zhu W, et al. Transmission of atherosclerosis susceptibility with gut microbial transplantation. J Biol Chem 2015;290:5647‐5660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Senthong V, Li XS, Hudec T, Coughlin J, Wu Y, Levison B, et al. Plasma trimethylamine N‐oxide, a gut microbe‐generated phosphatidylcholine metabolite, is associated with atherosclerotic burden. J Am Coll Cardiol 2016;67:2620‐2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Senthong V, Wang Z, Li XS, Fan Y, Wu Y, Tang WH, et al. Intestinal microbiota‐generated metabolite trimethylamine‐N‐oxide and 5‐year mortality risk in stable coronary artery disease: the contributory role of intestinal microbiota in a COURAGE‐like patient cohort. J Am Heart Assoc 2016;5:pii: e002816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Schugar RC, Shih DM, Warrier M, Helsley RN, Burrows A, Ferguson D, et al. The TMAO‐producing enzyme flavin‐containing monooxygenase 3 regulates obesity and the beiging of white adipose tissue. Cell Rep 2017;19:2451‐2461. Erratum. In: Cell Rep 2017;20:279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Xu KY, Xia GH, Lu JQ, Chen MX, Zhen X, Wang S, et al. Impaired renal function and dysbiosis of gut microbiota contribute to increased trimethylamine‐N‐oxide in chronic kidney disease patients. Sci Rep 2017;7:1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Zhu W, Gregory JC, Org E, Buffa JA, Gupta N, Wang Z, et al. Gut microbial metabolite TMAO enhances platelet hyperreactivity and thrombosis risk. Cell 2016;165:111‐124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Boini KM, Hussain T, Li PL, Koka S. Trimethylamine‐N‐oxide instigates NLRP3 inflammasome activation and endothelial dysfunction. Cell Physiol Biochem 2017;44:152‐162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Wang Z, Roberts AB, Buffa JA, Levison BS, Zhu W, Org E, et al. Non‐lethal inhibition of gut microbial trimethylamine production for the treatment of atherosclerosis. Cell 2015;163:1585‐1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Schnabl B, Brenner DA. Interactions between the intestinal microbiome and liver diseases. Gastroenterology 2014;146:1513‐1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Sherriff JL, O'Sullivan TA, Properzi C, Oddo J‐L, Adams LA. Choline, its potential role in nonalcoholic fatty liver disease, and the case for human and bacterial genes. Adv Nutr 2016;7:5‐13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Dumas M‐E, Barton RH, Toye A, Cloarec O, Blancher C, Rothwell A, et al. Metabolic profiling reveals a contribution of gut microbiota to fatty liver phenotype in insulin‐resistant mice. Proc Natl Acad Sci U S A 2006;103:12511‐12516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Spencer MD, Hamp TJ, Reid RW, Fischer LM, Zeisel SH, Fodor AA. Association between composition of the human gastrointestinal microbiome and development of fatty liver with choline deficiency. Gastroenterology 2011;140:976‐986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Chen YM, Liu Y, Zhou RF, Chen XL, Wang C, Tan XY, et al. Associations of gut‐flora‐dependent metabolite trimethylamine‐N‐oxide, betaine and choline with non‐alcoholic fatty liver disease in adults. Sci 2016;6:19076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Imajo K, Fujita K, Yoneda M, Shinohara Y, Suzuki K, Mawatari H, et al. Plasma free choline is a novel non‐invasive biomarker for early‐stage non‐alcoholic steatohepatitis: a multi‐center validation study. Hepatol Res 2012;42:757‐766. [DOI] [PubMed] [Google Scholar]
- 108. Canfora EE, Jocken JW, Blaak EE. Short‐chain fatty acids in control of body weight and insulin sensitivity. Nat Rev Endocrinol 2015;11:577‐591. [DOI] [PubMed] [Google Scholar]
- 109. Trompette A, Gollwitzer ES, Yadava K, Sichelstiel AK, Sprenger N, Ngom‐Bru C, et al. Gut microbiota metabolism of dietary fiber influences allergic airway disease and hematopoiesis. Nat Med 2014;20:159‐166. [DOI] [PubMed] [Google Scholar]
- 110. Bäckhed F, Ding H, Wang T, Hooper LV, Koh GY, Nagy A, et al. The gut microbiota as an environmental factor that regulates fat storage. Proc Natl Acad Sci U S A 2004;101:15718‐15723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Endo H, Niioka M, Kobayashi N, Tanaka M, Watanabe T. Butyrate‐producing probiotics reduce nonalcoholic fatty liver disease progression in rats: new insight into the probiotics for the gut‐liver axis. PLoS One 2013;8:e63388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Sheng L, Jena PK, Hu Y, Liu H‐X, Nagar N, Kalanetra KM, et al. Hepatic inflammation caused by dysregulated bile acid synthesis is reversible by butyrate supplementation. J Pathol 2017;243:431‐441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Chambers ES, Viardot A, Psichas A, Morrison DJ, Murphy KG, Zac‐Varghese SE, et al. Effects of targeted delivery of propionate to the human colon on appetite regulation, body weight maintenance and adiposity in overweight adults. Gut 2015;64:1744‐1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Vrieze A, Van Nood E, Holleman F, Salojärvi J, Kootte RS, Bartelsman JF, et al. Transfer of intestinal microbiota from lean donors increases insulin sensitivity in individuals with metabolic syndrome. Gastroenterology 2012;143:913‐916.e917. [DOI] [PubMed] [Google Scholar]
- 115. Molinaro A, Wahlström A, Marschall HU. Role of bile acids in metabolic control. Trends Endocrinol Metab 2018;29:31‐41. [DOI] [PubMed] [Google Scholar]
- 116. Duboc H, Tache Y, Hofmann AF. The bile acid TGR5 membrane receptor: from basic research to clinical application. Dig Liver Dis 2014;46:302‐312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Han CY. Update on FXR biology: promising therapeutic target? Int J Mol Sci 2018;19: pii: E2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Slijepcevic D, van de Graaf SF. Bile acid uptake transporters as targets for therapy. Dig Dis 2017;35:251‐258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Ridaura VK, Faith JJ, Rey FE, Cheng J, Duncan AE, Kau AL, et al. Gut microbiota from twins discordant for obesity modulate metabolism in mice. Science 2013;341:1241214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Jiao N, Baker SS, Chapa‐Rodriguez A, Liu W, Nugent CA, Tsompana M, et al. Suppressed hepatic bile acid signalling despite elevated production of primary and secondary bile acids in NAFLD. Gut 2018;67:1881‐1891. [DOI] [PubMed] [Google Scholar]
- 121. Ferslew BC, Xie G, Johnston CK, Su M, Stewart PW, Jia W, et al. Altered bile acid metabolome in patients with nonalcoholic steatohepatitis. Dig Dis Sci 2015;60:3318‐3328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Neuschwander‐Tetri BA, Loomba R, Sanyal AJ, Lavine JE, Van Natta ML, Abdelmalek MF, et al.; NASH Clinical Research Network . Farnesoid X nuclear receptor ligand obeticholic acid for non‐cirrhotic, non‐alcoholic steatohepatitis (FLINT): a multicentre, randomised, placebo‐controlled trial. Lancet 2015;385:956‐965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Mudaliar S, Henry RR, Sanyal AJ, Morrow L, Marschall HU, Kipnes M, et al. Efficacy and safety of the farnesoid X receptor agonist obeticholic acid in patients with type 2 diabetes and nonalcoholic fatty liver disease. Gastroenterology 2013;145:574‐582.e571. [DOI] [PubMed] [Google Scholar]
- 124. Ridlon JM, Harris SC, Bhowmik S, Kang DJ, Hylemon PB. Consequences of bile salt biotransformations by intestinal bacteria. Gut Microbes 2016;7:22‐39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Chavez‐Talavera O, Tailleux A, Lefebvre P, Staels B. Bile acid control of metabolism and inflammation in obesity, type 2 diabetes, dyslipidemia, and nonalcoholic fatty liver disease. Gastroenterology 2017;152:1679‐1694.e1673. [DOI] [PubMed] [Google Scholar]
- 126. Baker SS, Baker RD, Liu W, Nowak NJ, Zhu L. Role of alcohol metabolism in non‐alcoholic steatohepatitis. PLoS One 2010;5:e9570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Zhu R, Baker SS, Moylan CA, Abdelmalek MF, Guy CD, Zamboni F, et al. Systematic transcriptome analysis reveals elevated expression of alcohol‐metabolizing genes in NAFLD livers. J Pathol 2016;238:531‐542. [DOI] [PubMed] [Google Scholar]
- 128. Elshaghabee FM, Bockelmann W, Meske D, Vrese M, Walte HG, Schrezenmeir J, et al. Ethanol production by selected intestinal microorganisms and lactic acid bacteria growing under different nutritional conditions. Front Microbiol 2016;7:47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Engstler AJ, Aumiller T, Degen C, Dürr M, Weiss E, Maier IB, et al. Insulin resistance alters hepatic ethanol metabolism: studies in mice and children with non‐alcoholic fatty liver disease. Gut 2016;65:1564‐1571. [DOI] [PubMed] [Google Scholar]
- 130. Menni C, Fauman E, Erte I, Perry JR, Kastenmuller G, Shin SY, et al. Biomarkers for type 2 diabetes and impaired fasting glucose using a nontargeted metabolomics approach. Diabetes 2013;62:4270‐4276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Wang TJ, Larson MG, Vasan RS, Cheng S, Rhee EP, McCabe E, et al. Metabolite profiles and the risk of developing diabetes. Nat Med 2011;17:448‐453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Gaggini M, Carli F, Rosso C, Buzzigoli E, Marietti M, Della Latta V, et al. Altered amino acid concentrations in NAFLD: impact of obesity and insulin resistance. Hepatology 2018;67:145‐158. [DOI] [PubMed] [Google Scholar]
- 133. Guo Y, Huang ZP, Liu CQ, Qi L, Sheng Y, Zou DJ. Modulation of the gut microbiome: a systematic review of the effect of bariatric surgery. Eur J Endocrinol 2018;178:43‐56. [DOI] [PubMed] [Google Scholar]
- 134. Damms‐Machado A, Mitra S, Schollenberger AE, Kramer KM, Meile T, Konigsrainer A, et al. Effects of surgical and dietary weight loss therapy for obesity on gut microbiota composition and nutrient absorption. Biomed Res Int 2015;2015:806248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Suez J, Zmora N, Zilberman‐Schapira G, Mor U, Dori‐Bachash M, Bashiardes S, et al. Post‐antibiotic gut mucosal microbiome reconstitution is impaired by probiotics and improved by autologous FMT. Cell 2018;174:1406‐1423.e16. [DOI] [PubMed] [Google Scholar]
- 136. Zmora N, Zilberman‐Schapira G, Suez J, Mor U, Dori‐Bachash M, Bashiardes S, et al. Personalized gut mucosal colonization resistance to empiric probiotics is associated with unique host and microbiome features. Cell 2018;174:1388‐1405.e21. [DOI] [PubMed] [Google Scholar]