

Abstract
Objective
Although there is growing awareness of the long-term cognitive effects of repetitive mild traumatic brain injury (rmTBI; eg, sports concussions), whether repeated concussions cause long-term cognitive deficits remains controversial. Moreover, whether cognitive deficits depend on increased amyloid β deposition and tau phosphorylation or are worsened by the apolipoprotein E4 allele remains unknown. Here, we use an experimental model of rmTBI to address these clinical controversies.
Methods
A weight drop rmTBI model was used that results in cognitive deficits without loss of consciousness, seizures, or gross or microscopic evidence of brain damage. Cognitive function was assessed using a Morris water maze (MWM) paradigm. Immunostaining and enzyme-linked immunosorbent assay (ELISA) were used to assess amyloid β deposition and tau hyperphosphorylation. Brain volume and white matter integrity were assessed by magnetic resonance imaging (MRI).
Results
Mice subjected to rmTBI daily or weekly but not biweekly or monthly had persistent cognitive deficits as long as 1 year after injuries. Long-term cognitive deficits were associated with increased astrocytosis but not tau phosphorylation or amyloid β (by ELISA); plaques or tangles (by immunohistochemistry); or brain volume loss or changes in white matter integrity (by MRI). APOE4 was not associated with worse MWM performance after rmTBI.
Interpretation
Within the vulnerable time period between injuries, rmTBI produces long-term cognitive deficits independent of increased amyloid β or tau phosphorylation. In this model, cognitive outcome is not influenced by APOE4 status. The data have implications for the long-term mental health of athletes who suffer multiple concussions.
Repetitive mild traumatic brain injury (rmTBI), such as those suffered by athletes sustaining concussions, has been associated with long-term neurological impairment, including memory disturbances, parkinsonism, behavioral abnormalities, personality changes, speech irregularities, and gait abnormalities.1–5 In some cases, rmTBI may be associated with long-term brain volume loss, as well as histological changes, including tau-immunoreactive neurofibrillary tangles, the hallmark of chronic traumatic encephalopathy (CTE),6 and amyloid β (Aβ) deposition, the hallmark of Alzheimer disease (AD).4,7 Whether tau tangles and Aβ deposits are causative or merely associated with poor cognitive outcome after rmTBI, however, is controversial. Several studies suggest that it is soluble rather than insoluble tau and Aβ species that cause cognitive decline in other neurodegenerative disorders.8,9
The effect of the cholesterol and lipid transport protein apolipoprotein E epsilon 4 (APOE4) on long-term cognitive outcome after repetitive brain injuries is also controversial.10,11 It is unclear whether APOE4 and TBI operate in a synergistic manner to increase the risk of poor outcome or, alternatively, act as independent (but additive) risk factors, or whether APOE4 influences outcome after mild TBI at all.
We have previously reported a model of rmTBI that produces long-term cognitive dysfunction without mortality, intracranial hemorrhage, or evidence of cell death.12 In that model, injury was associated with loss of consciousness (LOC) and brief convulsions in approximately ¼ to ⅕ of mice.12 The majority of patients with sport-related concussions, however, do not experience LOC13 or convulsions.14 In the current study, we used a milder injury level that is not associated with LOC or convulsions, to test the following hypotheses: (1) rmTBI, even in the absence of LOC and convulsions, is associated with acute and long-term deficits in cognitive function; (2) increased intervals between rmTBIs protect against long-term cognitive deficits; (3) worse Morris water maze (MWM) performance after rmTBI is associated with white matter injury and brain volume loss detectable by magnetic resonance imaging (MRI); (4) the APOE4 allele is associated with worse acute and long-term MWM performance after rmTBI; and (5) that worse MWM performance after rmTBI is associated with increased Aβ deposition and tau accumulation in injured brains.
Materials and Methods
All experiments were approved by the Massachusetts General Hospital Institutional Animal Care and Use Committee and complied with the NIH Guide for the Care and Use of Laboratory Animals. Transgenic mice that express targeted replacement of the mouse ApoE allele with human APOE415 were obtained from Jackson Laboratories (Bar Harbor, ME). A total of 45 adult APOE4 and 168 wild-type (WT) C57BL/6 mice were used for the experiments.
rmTBI
The mouse rmTBI model was used as previously described with important modifications.12 Briefly, mice (3-month-old males) were anesthetized for 45 seconds using 4.5% isoflurane in a 70:30 mixture of nitrous oxide and oxygen. Anesthetized mice were placed on a delicate task wiper (Kimwipe; Kimberly-Clark, Irving, TX) and grasped by the tail. The head was placed directly under a hollow guide tube 28 inches in length. A 54g metal bolt was used to deliver the impact to the dorsal aspect of the skull. At impact, the mouse head readily penetrated the Kimwipe, resulting in a rotational acceleration of the head. Sham-injured age-matched control mice underwent anesthesia but not concussive injury. All mice recovered in room air. LOC was defined as the time from removal of anesthesia to spontaneous righting. Anesthesia exposure for each mouse was strictly controlled to 45 seconds exposure. LOC times reflected the effects of anesthesia as well as the effects of rmTBI.
Mice were randomized to receive injury or sham injury. Mice randomized to injury underwent 1 of the following injury regimens: (1) 5 daily concussive injuries (n = 7–10/group), (2) 7 concussive injuries in 9 days (n = 8–10/group), (3) 1 concussive injury weekly for 5 weeks (n = 16), (4) 1 concussive injury biweekly for 10 weeks (n = 14), (5) 1 concussive injury monthly for 5 months (n = 16), and (6) 1 concussive injury (n = 15). Sham-injured controls underwent anesthesia exposures (n = 4–12/group) at the same time intervals as injured mice. For all behavioral testing, experimenters were blinded to injury and genotype status, using color coding stored in a password-protected computer. Details of the study flow are found in Figure 1.
FIGURE 1.
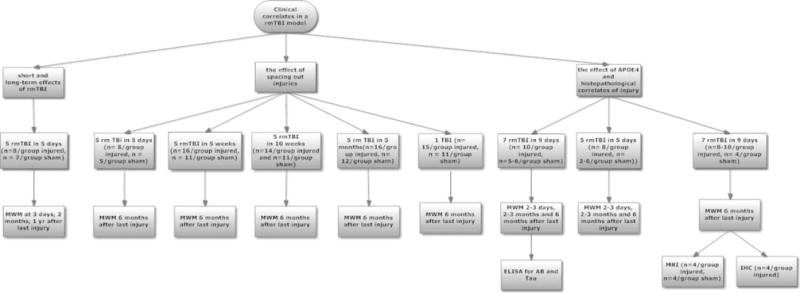
Study flow diagram. AB = amyloid β; ELISA = enzyme-linked immunosorbent assay; IHC = immunohistochemistry; MWM = Morris water maze; rmTBI = repetitive mild traumatic brain injury.
Assessment of Spatial Learning and Memory
Spatial learning and memory were assessed using a MWM paradigm as previously described.16 Each mouse was subjected to a maximum of 2 series of 4 trials per day. For probe trials, mice were placed in the pool with the platform removed. The time that the animal swam in the target quadrant was recorded (maximum 60 seconds). For visible trials, the platform was marked by red tape and placed 0.5cm above the water level. Swim speeds were measured by Any Maze software (Stoeling, Wood Dale, IL). When mice underwent repeat MWM testing, 2 to 3 months or 6 months after their final injury, the platform was moved to a different quadrant than that used previously.
Determination of Brain Soluble Aβ40 and Soluble Tau
Soluble Aβ40 and phosphorylated tau were assessed 6 months after the last of 7 injuries (n = 6/group) or sham injury (n = 2– 6/group). We assessed Aβ40 levels rather than Aβ42 because Aβ40 levels are normally higher in brain tissue.17 Soluble Aβ40 and soluble phosphorylated tau were determined in whole cortices and hippocampi using a sandwich enzyme-linked immunosorbent assay (ELISA; WAKO, Richmond, VA and Invitrogen, Grand Island, NY) per the manufacturer’s directions. Data were expressed as picomoles per gram of protein (Aβ40) and picograms per gram of protein (tau and phosphorylated tau).
Immunohistochemistry
To assess for Aβ plaques, brains were frozen and 12lm coronal sections were cut. Sections of cortex and hippocampus were fixed in ethanol, washed in phosphate-buffered saline (PBS; pH 7.4), and incubated overnight at room temperature with pan-amyloid antibody in 1.5% goat serum (Invitrogen, Grand Island, NY). After incubation with biotinylated antirabbit immunoglobulin G in 1.5% goat serum (Vector Laboratories, Burlingame, CA), slides were washed, and NovaRED Peroxidase Substrate (Vector Laboratories) was applied. Brain sections were photographed using the Mirax Midi Slide Scanner, and analysis was performed using Mirax Viewer (Zeiss, Oberkochen, Germany).
For the remainder of the immunohistochemical analyses, mice were anesthetized and transcardially perfused with 4% paraformaldehyde 6 months after injury. The brain was post-fixed for 24 hours in 4% paraformaldehyde, and cryoprotected in 30% sucrose for 24 hours. Coronal sections were cut (20mm) and mounted on poly-L-lysine–coated slides. Sections were blocked with 2% fetal bovine serum in 50mM Tris, pH 7.4, 150mM NaCl.
For tau tangles, slides were incubated with goat antitau (Chemicon, Rosemont, IL), Tau-1 (kindly provided by L. Binder, University of Chicago), AT8 and AT270 (Thermo Scientific, Waltham, MA), or PHF1 (a generous gift of P. Davies, Albert Einstein College of Medicine). For axonal pathology, slides were incubated with SMI-31 (Sternberger Monoclonal, Baltimore, MD). Subsequently, sections were incubated with biotin-conjugated secondary antibodies (Vector Laboratories) followed by amplification with the avidin–biotin method and visualization with 3,3′-diaminobenzidine as chromogen (Vectastain ABC kit; Vector Laboratories). The same sections were additionally stained with hematoxylin and eosin (H&E).
To assess for microglia and astrocytes, brain sections were rehydrated in PBS for 10 minutes and antigen retrieval was performed by immersing slides in ~90°C citric acid buffer (10mM citric acid with 0.05% Tween 20, adjusted to pH 6.0 with NaOH) for 30 minutes. Sections were then permeabilized and blocked for 1 hour in permeabilization buffer (PBS containing 4% normal goat serum and 0.1% Triton-X). Brain sections were incubated overnight at 4°C with rabbit primary antibodies specific for glial fibrillary acidic protein (GFAP; G9269; Sigma-Aldrich, St Louis, MO) or IBA-1 (019–19741; Wako Chemicals USA, Richmond, VA), diluted 1:200 in permeabilization buffer. The following day, slides were rinsed 3× in PBS. For fluorescent staining, slides were incubated for 2 hours at room temperature with Alexa Fluor 568 goat anti-rabbit secondary antibody (A-11011; Invitrogen, Carlsbad, CA) diluted 1:200 in permeabilization buffer. Brain sections were photographed on a Eclipse T300 fluorescence microscope (Nikon, Tokyo, Japan), using 568/585nm excitation/emission filters.
Microglia and astrocytes were quantitated in mice at 6 months after rmTBI (5 rmTBI in 5 days) or sham injury (n = 5/group) in ×400 fields randomly chosen from cortex, CA3, or central corpus callosum in 3 brain sections spanning anterior to posterior hippocampus. Left and right hemispheric regions were equally represented in cortical and hippocampal counts. A total of 6 fields per mouse were counted for cortex and hippocampus and 3 fields for corpus callosum. Neurons were assessed qualitatively by Hoechst staining.
MRI
To assess changes in brain volume and white matter integrity, MRI was undertaken 6 months after final injury or final sham injury (n = 4 per group). Imaging employed a 9.4T magnet and a 4-channel phased array receiver coil inside a volume radiofrequency transmitter (Bruker BioSpin Corporation, Billerica, MA). All scans were acquired at an isotropic in-plane resolution of 150μm with 400μm coronal slices that covered the brain from olfactory bulb to cerebellum.
To search for focal lesions of the type that should appear bright on T2-weighted images at late postinjury time points,18 we employed fast spin-echo imaging. Additionally, to calculate quantitative T2, we acquired multiple images with stepped echo time values (10, 30, 50 milliseconds) using a conventional spin-echo sequence. White matter evaluation included diffusion tensor imaging and the magnetization transfer ratio (MTR) method. The total imaging time per animal was about 1 hour.
Analysis resampled all MRI data onto the Allen Mouse Brain Atlas19 at 250μm coronal levels and a resolution of 125μm in coronal planes. Data were converted to map-based values of T2, fractional anisotropy (FA), and MTR, respectively. Data were assessed using statistical comparisons across groups for each voxel and for regions of interest (ROIs). Gray matter ROIs derived from the brain atlas included (1) whole brain, (2) whole striatum, (3) whole hippocampus, and (4) whole cortex posterior to the coronal slice that is 1.5mm anterior to bregma. White matter ROIs for assessing FA and MTR included (1) all white matter as segmented from the atlas, (2) all voxels with a group-averaged FA >0.25 in either cohort, (3) midline corpus callosum on slices from 1.0 to 2.5mm posterior to bregma, and (4) bilateral internal capsule on the same slices.
Statistical Analyses
Data are mean ± standard error of the mean. ELISA data were analyzed by analysis of variance (ANOVA) in a model that included injury status (injury vs sham injury) and APOE status (APOE4 vs WT). MWM data were analyzed by repeated measures ANOVA (group × time). MRI data were analyzed by ANOVA, with a Bonferroni correction for multiple comparisons. LOC time was analyzed by ANOVA. Cell count data were analyzed by t test or rank sum as appropriate. Statistical significance was considered p < 0.05.
Results
There were no convulsions after injury. There was no statistically significant difference in mean LOC times (times from recovery from anesthesia plus time from recovery from sham injury or rmTBI) between injured (7 hits in 9 days) and sham-injured mice (51.8 ± 1.8 seconds vs 62.2 ± 6.7 seconds). There were no differences in swim speed between injured (5 hits 5 days) and sham-injured mice (0.26 ± 0.01m/s for injured mice vs 0.28 ± 0.01m/s for sham-injured mice, p = 0.2). APOE4 was associated with longer LOC times in both injured (61.5 ± 3.2 seconds for injured APOE4 vs 42.1 ± 1.1 seconds for injured WT, p = 0.04) and sham-injured mice (88.2 ± 14.3 seconds for sham-injured APOE4 vs 61.5 ± 3.2 seconds for sham-injured WT, p < 0.001).
rmTBI Produces a Long-Term Cognitive Deficit
Injured WT mice (5 hit, 5 days) assessed 2 to 3 days, 2 months, and 1 year after the last injury performed worse than sham-injured WT mice on hidden trials of the MWM (Fig 2). There was no difference in probe trial performance between injured and sham-injured mice at 2 to 3 days after injury (21 ± 3 seconds vs 16 ± 2 seconds, p = 0.2) or 1 year after injury (13 ± 3 seconds vs 16 ± 3 seconds, p = 0.4), but at 2 months after injury, injured mice performed worse than sham-injured mice (14 ± 4 seconds vs 19 ± 4 seconds, p = 0.03).
FIGURE 2.
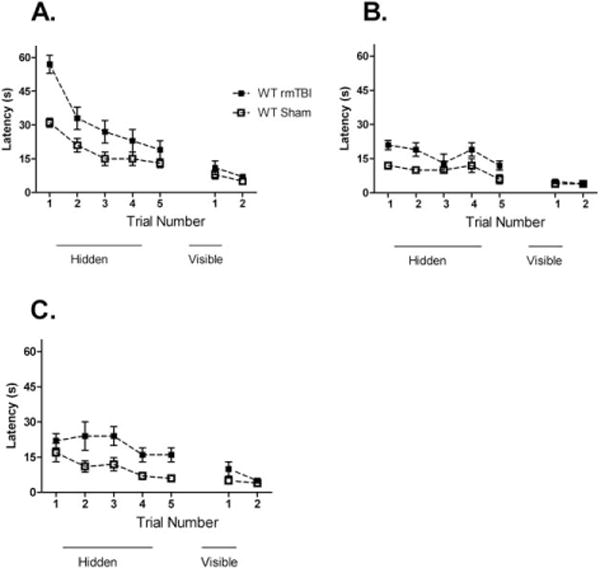
Morris water maze performance in wild-type (WT) mice that underwent 5 repetitive mild traumatic brain injuries (rmTBIs) in 5 days. (A) Three days after the last injury, injured mice had worse performance on hidden platform testing than sham-injured mice (n = 8/group, p = 0.01), but there was no difference in injured versus sham-injured mice on visible platform testing. (B) Two months after the last injury, injured mice persisted with deficits in hidden platform performance compared to sham-injured mice (n = 8/group, p = 0.01), with no differences in visible platform testing. (C) One year after injury, injured mice demonstrated worse hidden platform performance compared to sham-injured mice (n = 7–8/group, p = 0.02), with no differences in visible platform performance.
Rest Intervals between rmTBI Injuries Protect against Long-Term Cognitive Deficits
After sustaining 5 daily or 5 weekly injuries, injured mice performed worse than sham-injured mice at 6 months on hidden trials of the MWM. There were no differences between injured and sham-injured mice in probe trial performance after 5 daily (21 ± 1 seconds vs 20 ± 1 seconds, p = 0.8) or 5 weekly injuries (19 ± 2 seconds vs 21 ± 2 seconds, p = 0.8). After sustaining 5 biweekly injuries, 5 monthly injuries, or 1 injury, however, there was no difference in performance in hidden platform trials between injured and sham-injured mice (Fig 3). There were no differences in probe trial performance in injured versus sham-injured mice after 5 biweekly injuries (18 ± 2 seconds vs 15 ± 2 seconds, p = 0.4), 5 monthly injuries (18 ± 2 seconds vs 18 ± 3 seconds, p = 1), or 1 injury (18 ± 2 seconds vs 21 ± 2 seconds, p = 0.2).
FIGURE 3.
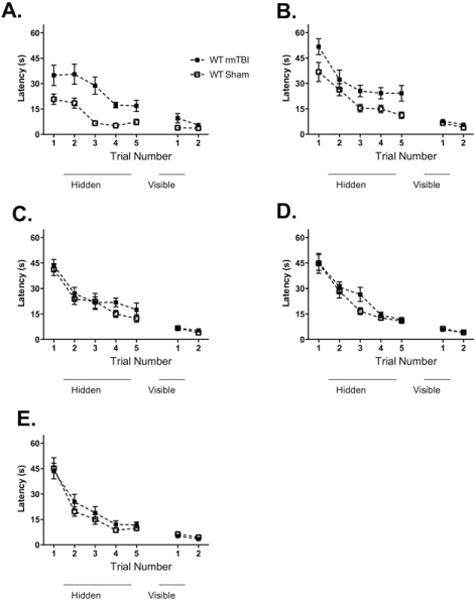
The effect of time interval between repetitive mild traumatic brain injuries (rmTBIs) on Morris water maze performance 6 months after the last injury. All groups demonstrated time-dependent improvement in hidden platform performance. There were no group differences in visual platform performance. Mice that underwent 5 concussive injuries in 5 days (A) and weekly concussive injuries for 5 weeks (B) had deficits in hidden platform performance when compared to sham-injured mice (n = 5–8/group, p = 0.001 and n = 11–16/group, p = 0.002, respectively). There were no differences in hidden platform performance between injured and sham-injured mice that underwent biweekly injuries for 10 weeks (n = 11–14, p = 0.1; C), 5 monthly injuries (n = 12–16/group, p = 0.2; D), or 1 injury (n = 11–15/group, p = 0.4; E). WT = wild type.
rmTBI Is Not Associated with White Matter Changes or Volume Loss on MRI
In mice subjected to 7 rmTBI over 9 days, all imaging characteristics at 6 months after final injury were strikingly similar between injured and sham-injured mice. There were no differences in group-average brain volumes, FA, or MTR (Fig 4). No focal lesions were observed on T2-weighted MRI scans for any animal. Voxel-wise statistics revealed no differences between groups. No differences between injured and sham-injured groups were seen in T2, FA, or MTR for any regions of interest.
FIGURE 4.
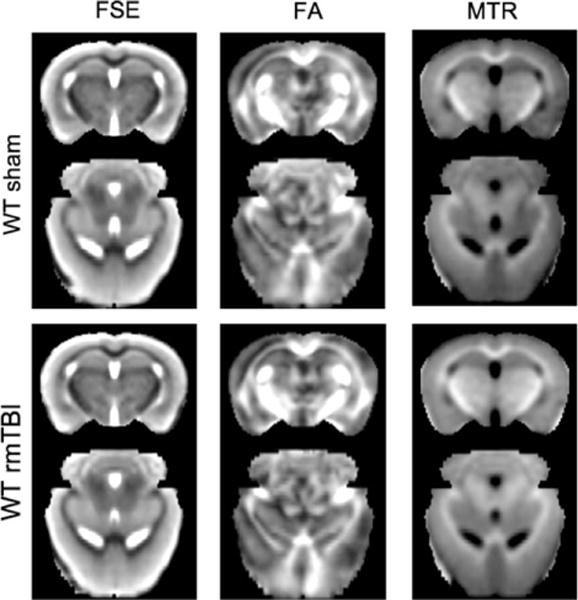
Comparisons of group-averaged brain data between sham-injured (top) and repetitive mild traumatic brain injury (rmTBI)-injured (bottom) mice (n = 4/group) using fast spin-echo (FSE) images, fractional anisotropy (FA) computed from diffusion tensor imaging, and magnetization transfer ratio (MTR). Group averages were obtained at a coronal slice 2mm posterior to bregma and an axial level 1.75mm ventral to bregma. WT = wild type.
APOE4 Is Not Associated with Worse Outcome after rmTBI
In all experiments, injured WT and APOE4 mice performed worse than their respective sham-injured controls on hidden trials of the MWM. Following 5 concussions in 5 days, there was no difference between injured APOE4 and injured WT animals on hidden trials of the MWM (Fig 5A). There was no difference in probe trial performance in injured APOE4 versus injured WT mice (20 ± 4 seconds vs 17 ± 3 seconds, p = 0.4). To test whether increasing the number of injuries might result in differences in cognitive outcome between APOE4 and WT mice, a separate group of mice were subjected to 7 rmTBIs over 9 days. There was no difference in hidden platform trials between injured APOE4 and WT mice tested 2 to 3 days after the last injury (see Fig 5B). There was no difference in probe trial performance in injured APOE4 versus injured WT mice (18 ± 2 seconds vs 18 ± 2 seconds, p = 1).
FIGURE 5.
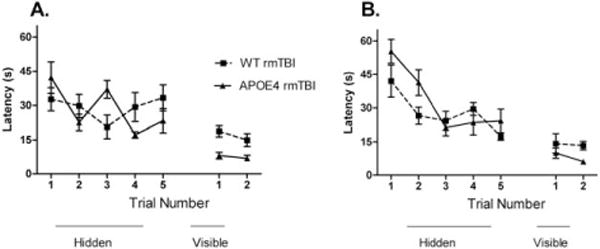
There were no genotype-specific differences in Morris water maze (MWM) performance after repetitive mild traumatic brain injury (rmTBI). All sham-injured and injured groups demonstrated time-dependent improvements in latency to the hidden platform, indicating the ability to learn the MWM paradigm before and after rmTBI (p < 0.05 for time for all groups). Injured mice performed worse than their sham-injured controls (p < 0.05 for all groups, data not shown). (A) MWM performance in APOE4 versus wild-type (WT) mice immediately after last injury in mice that underwent 5 rmTBIs in 5 days. Hidden platform trial performance did not differ between injured APOE4 and injured WT animals (p = 0.9) or between sham-injured APOE4 and sham-injured WT animals (p = 0.3, data not shown). Injured WT animals had worse performance on visual platform testing compared to injured APOE4 mice (p = 0.005). (B) MWM performance immediately after last injury in mice that underwent 7 rmTBIs in 9 days. Hidden platform trial performance did not differ between injured APOE4 and injured WT animals (p = 0.1), but was better in sham-injured APOE4 compared to sham-injured WT animals (p = 0.003, data not shown).
To assess for possible time-dependent differences in cognitive function, mice were retested in the MWM with the platform location changed at 2 to 3 months postinjury and again at 6 months postinjury. Surprisingly, WT mice subjected to 5 injuries had worse performance than injured APOE4 mice on hidden platform trials 2 months after injury (Fig 6A), but there was no difference in probe performance in injured APOE4 versus WT mice (11 ± 3 seconds vs 12 ± 2 seconds, p = 0.8). In contrast, hidden trial performance was similar between WT and APOE4 mice subjected to 7 hits over 9 days, 2 months and 6 months after the last injury (see Fig 6B, C), and there was also no difference in probe trial performance in injured APOE4 versus WT mice at 2 months (24 ± 2 seconds vs 22 ± 2 seconds, p = 0.8) or at 6 months (9 ± 1 seconds vs 11 ± 1 seconds, p = 0.2). A second cohort of APOE4 and WT mice also subjected to 7 rmTBIs in 9 days were tested only once at 6 months after their last injury; no difference was seen between injured APOE4 and injured WT mice (see Fig 6D).
FIGURE 6.
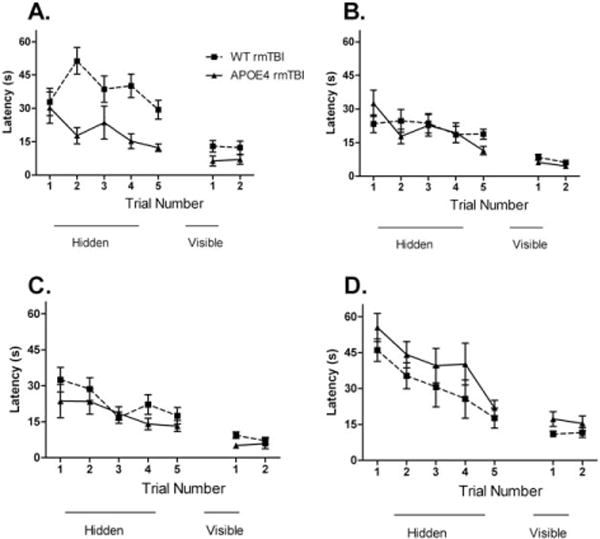
APOE4 does not worsen long-term Morris water maze (MWM) performance after repetitive mild traumatic brain injury (rmTBI). There were no baseline differences in MWM performance in sham-injured APOE4 versus wild-type (WT) mice (data not shown). (A) MWM performance 2 months after last injury in mice that underwent 5 rmTBIs in 5 days. Injured WT mice performed worse than injured APOE4 mice (p = 0.003) and WT sham-injured controls (p = 0.001, data not shown). Injured WT animals had worse performance on visible platform testing compared to injured APOE4 animals (p = 0.04). (B) MWM performance 2 months after last injury in mice that underwent 7 rmTBIs in 9 days. There were no differences in hidden platform performance between injured APOE4 mice and injured WT mice (p = 0.8). There were no differences in visual platform performance in injured APOE4 versus injured WT mice (p = 0.06). (C) MWM performance 6 months after last injury in mice that underwent daily 7 concussions in 9 days. There were no differences between injured APOE4 animals and injured WT animals (p = 0.2) on hidden platform performance. On visual platform testing, there were no differences in performance in injured APOE4 versus injured WT mice (p = 0.1). (D) Mice that were naive to the MWM when tested 6 months after their final injury showed no differences in hidden platform performance between WT and APOE4 mice (p = 0.1).
Assessment of Aβ
Six months after the final injury (7 rmTBs in 9 days), there was no significant difference in cortical soluble Aβ40 between injured and sham-injured mice (p = 0.1) or APOE4 mice versus WT mice (p = 0.2; Fig 7A). In hippocampi, soluble Aβ40 did not differ between injured and sham-injured animals (p = 0.3) or between APOE4 and WT (p = 0.1) at 6 months after final injury (see Fig 7B). Neither APOE4 nor WT mice had Aβ deposits 6 months after injury (Fig 8).
FIGURE 7.
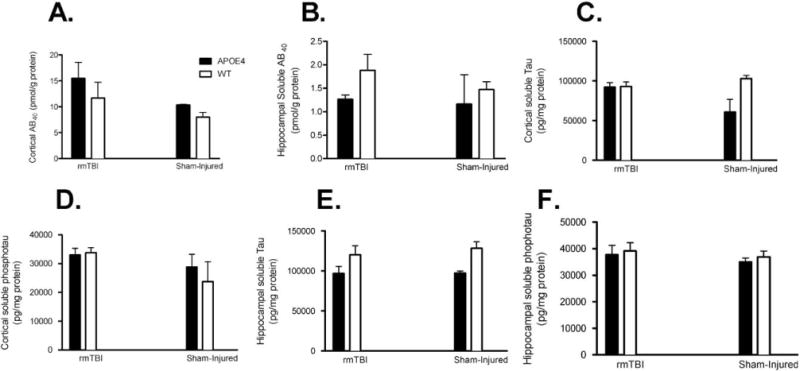
Enzyme-linked immunosorbent assay testing for soluble amyloid β (Aβ) 40, total tau, and phosphorylated tau in cortical and hippocampal samples 6 months after final injury. (A) There was no difference in cortical soluble Aβ40 in injured APOE4 versus injured wild-type (WT) animals (16 ± 3.0pmol/g vs 12 ± 3.0pmol/g, p = 0.4), injured APOE4 versus sham-injured APOE4 mice (16 ± 3.0pmol/g vs 10 ± 0.1pmol/g, p = 0.4), or injured WT versus sham-injured WT animals (12 ± 3.0pmol/g vs 8.0 ± 0.9pmol/g, p = 0.3). (B) There was no difference in hippocampal soluble Aβ40 in injured APOE4 versus injured WT animals (1.3 ± 0.1pmol/g vs 1.9 ± 0.3pmol/g, p = 0.1), injured APOE4 versus sham-injured APOE4 mice (1.3 ± 0.1pmol/g vs 1.2 ± 0.6pmol/g, p = 0.8), or injured WT versus sham-injured WT animals (12 ± 3.0pmol/g vs 8.0 ± 0.9pmol/g, p = 0.3). (C) Total tau was not elevated in injured APOE4 versus sham-injured APOE4 mice (92,245 ± 5,468pg/mg vs 60,911 ± 15,606pg/mg, p = 0.05), injured APOE4 versus injured WT animals (92,445 ± 5,468pg/mg vs 93,042 ± 5,587pg/mg, p = 0.9), or injured WT versus sham-injured WT mice (93,402 ± 5,587pg/mg vs 102,904 ± 4,056pg/mg, p = 0.2). (D) No differences were seen in cortical soluble phosphorylated tau injured APOE4 versus sham-injured APOE4 mice (33,019 ± 2,267pg/mg vs 28,785 ± 4,398pg/mg, p = 0.4), injured APOE4 versus injured WT animals (33,019 ± 2,267pg/mg vs 33,767 ± 1,760pg/mg, p = 0.8), or injured WT versus sham-injured WT mice (33,767 ± 1,760pg/mg vs 28,785 ± 4,398pg/mg, p = 0.2). (E) Total tau was not elevated in hippocampal samples from injured APOE4 versus sham-injured APOE4 mice (96,792 ± 8,683pg/mg vs 97,125 ± 2,510pg/mg, p = 1), injured APOE4 versus injured WT animals (96,792 ± 8,683pg/mg vs 120,074 ± 11,200pg/mg, p = 0.4), or injured WT versus sham-injured WT mice (120,074 ± 11,200pg/mg vs 128,117 ± 8,031pg/mg, p = 0.6). (F) No differences were seen in hippocampal soluble phosphorylated tau in injured APOE4 versus sham-injured APOE4 mice (37,784 ± 3,543pg/mg vs 35,032 ± 1426pg/mg, p = 0.7), injured APOE4 versus injured WT animals (37,784 ± 3,543pg/mg vs 39,189 ± 3,086pg/mg, p = 0.5), or injured WT versus sham-injured WT mice (39,189 ± 3,086pg/mg vs 36,903 ± 2,190pg/mg, p = 0.6).
FIGURE 8.

No focal amyloid β (Aβ) deposition is seen 6 months after repetitive mild traumatic brain injury. Representative images show no focal deposition of Aβ in injured wild-type (A) or APOE4 (B) mice compared to an Alzheimer disease transgenic naive positive control (C). [Color figure can be viewed in the online issue, which is available at www.annalsofneurology.org.]
Assessment of Tau
There was no significant difference in total tau protein (p = 0.7) or cortical soluble phosphorylated tau (p = 0.1) between injured (7 rmTBIs in 9 days) and sham-injured mice 6 months after final injury. Similarly, there was no significant difference in cortical soluble total tau protein between APOE4 and WT mice (p = 0.1, see Fig 7C) or cortical soluble phosphorylated tau (p = 0.8, see Fig 7D) between APOE4 and WT mice. Neither hippocampal soluble total tau (p = 0.6) nor phosphorylated tau (p = 0.4) was different in injured and sham-injured mice. Hippocampal total tau was elevated in WT mice compared to APOE4 mice (p = 0.02), but there were no differences between APOE4 and WT mice in phosphorylated tau (p = 0.6, see Fig 7E, F). Immunohistochemical analysis using phosphorylation-independent and phosphospecific anti-tau antibodies revealed no evidence of neurofibrillary tangle formation in injured WT or APOE4 mice.
Assessment of Microglia, Astrocytes, and Neurons
Quantitative analysis showed no difference in IBA-1–immunoreactive cells in cortex (p = 0.3), corpus callosum (p = 0.1), or hippocampus (p = 0.5) between sham-injured and injured mice (5 rmTBI in 5 days) at 6 months. Compared to sham-injured mice, injured mice had increased GFAP-positive cells in cortex (p = 0.009), corpus callosum (p = 0.02), and hippocampus (p = 0.04; Fig 9A, B, D). Compared to sham-injured mice, Hoechst staining did not reveal gross neuron loss in CA1, CA3, or dentate gyrus brain regions in injured mice (see Fig 9C and data not shown).
FIGURE 9.
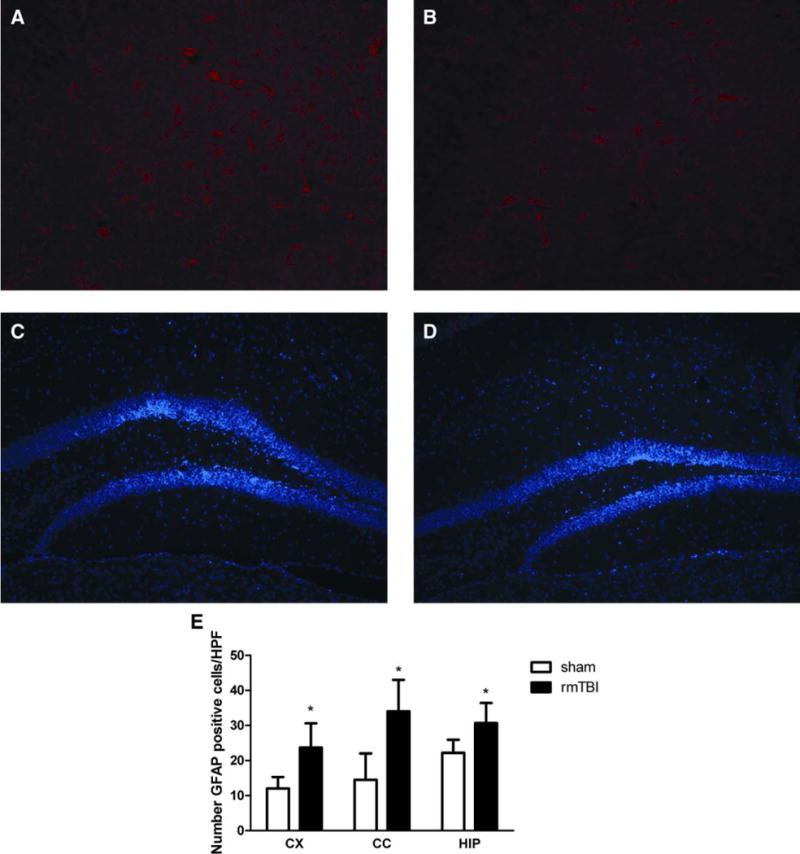
Increased astrocytosis after repetitive mild traumatic brain injury (rmTBI; 5 rmTBIs in 5 days) versus sham-injured mice. Representative photomicrographs show immunoreactivity of glial fibrillary acidic protein (GFAP; red) and Hoechst staining (blue) in injured versus sham-injured mice. Increased GFAP immunoreactivity is demonstrated in hippocampus of injured (A) versus sham-injured mice (B). No qualitative differences in neuronal density by Hoechst staining in hippocampus (dentate gyrus is shown) was observed between injured (C) and sham-injured (D) mice. (E) Quantitative analysis of GFAP-positive cells counted in cortex (CX), corpus callosum (CC), and hippocampus (HIP) of injured (5 rmTBIs in 5 days, n = 5) and sham-injured mice (n = 5) 6 months after injury (mean ± standard deviation); *p = 0.009 for CX, *p = 0.02 for CC, and *p = 0.04 for HIP. HPF = high-power magnification field. [Color figure can be viewed in the online issue, which is available at www.annalsofneurology.org.]
Assessment of Axonal Pathology
Consistent with the chronic nature of our injury model, examination of H&E- and SMI31-immunostained sections failed to reveal significant numbers of spheroids in major white matter tracts in injured mice (7 rmTBI in 9 days) 6 months after injury.
Discussion
Using a clinically relevant mouse model of rmTBI that does not produce LOC or seizures, we demonstrate that permanent cognitive deficits occur after 5 or 7 injuries depending on interinjury interval. Notably, long-term cognitive deficits occur in the absence of significant Aβ or tau accumulation, in the absence of structural correlates on MRI, and independent of the human APOE4 allele. While prior studies have implicated both tau and Aβ in the pathogenesis of cognitive decline after rmTBI, we demonstrate that cognitive deficits after rmTBI can occur in the absence of the histopathologic findings of CTE or AD, suggesting that severe histopathological manifestations of CTE/AD are not required for lifelong cognitive deficits. Furthermore, we demonstrate that the APOE4 allele does not exacerbate the cognitive deficits in this model, calling into question whether APOE4 alone is sufficient to cause worse outcome after rmTBI. These results have implications for athletes who sustain multiple concussions over the course of their careers.
APOE4 transgenic mice had prolonged LOC after anesthesia, irrespective of rmTBI. This suggests a role of APOE in the clearance of or sensitivity to inhaled anesthetics in the mouse brain. A prior clinical study has suggested that APOE4 may influence sensitivity to inhalational anesthetics in humans, with APOE4 carriers demonstrating prolonged cognitive deficits after inhaled anesthesia exposure.20 In our study, we found only LOC but not cognitive dysfunction associated with APOE4 status.
We previously demonstrated persistent cognitive deficits after multiple, mild concussive injuries in mice associated with a brief LOC and occasional convulsions.12 As both LOC and seizures are rarely associated with sport-related concussions, we reduced the level of injury in the current study, yet there remained a cumulative effect of rmTBIs on cognitive function, both acutely after injury and chronically. Increasing the time interval between rmTBIs was protective against long-term sequelae, suggesting that there is a vulnerable time period during which repeated injuries produce lasting effects even at the milder level of injury employed here. It is important to note that probe trial performance was not consistently associated with injury, suggesting that mechanisms other than hippocampus-dependent spatial memory are affected by rmTBI, a finding similar to those we have previously reported.21 Moreover, both injured and sham-injured mice demonstrated impaired probe trial performance that worsened over time, possibly due to the long-term effects of repeated anesthesia exposure. A recent study has demonstrated a similar effect of isoflurane exposure on probe trial performance.22 Nonetheless, injured mice showed worse MWM performance than sham-injured mice, demonstrating an effect of injury in addition to those of anesthesia.
Our data add to the growing body of literature suggesting that the effects of multiple concussions are both cumulative and long term.23–28 Prior experimental models have demonstrated acute23–27 and chronic28 functional and histopathological changes after rmTBI. The protective effect of spacing between injuries likely has significant clinical relevance to athletes at risk for rmTBI; however, further studies are needed to assess the influence of injury severity on the length of time defining the vulnerable period between repeated injuries.
Despite robust cognitive deficits, mice undergoing daily rmTBI had no histopathological evidence of disruption of major white matter tracts and no structural correlates on MRI 6 months after injury. Whereas prior human studies have shown chronic white matter changes associated with rmTBI,29,30 we found no differences in FA or brain volume between injured and sham-injured mice, despite reports of volume loss in human studies of mTBI.31 Differences in the complexity of the human brain, as well as differences in injury level reported in human studies versus that of the current study, may account for the lack of brain volume loss in our rmTBI model. However, additional detailed analysis of axonal numbers and morphology would be required to address more fully the contribution of axonal pathology to the behavioral abnormalities we observe.
We did not find an association between cognitive outcome and the APOE4 allele in this model. Using a controlled cortical impact model, we previously demonstrated worse long-term cognitive function in mice homozygous for APOE4.32 Our current study did not find an association between cognitive outcome and the APOE4 allele after rmTBI. These findings are in contrast to prior epidemiological studies in boxers and football players11,33 demonstrating worse outcome for APOE4 carriers after rmTBI. Our findings are consistent, however, with recent studies that have challenged whether APOE4 is a risk factor for worse outcomes across the spectrum of injury, including mild to moderate TBI.34–36 Further investigation into the potential effect of APOE4 on outcome after rmTBI, particularly at higher injury severity levels, is warranted before consideration of APOE4 screening of athletes in high-risk sports.37
Cognitive deficits after rmTBI occurred independently of Aβ or tau accumulation. Studies of retired athletes with decreased cognitive function who played sports associated with frequent, repetitive blows to the head have shown increased Aβ and tau accumulation.6,38 Although tau accumulation is undoubtedly associated with CTE, whether the pathological findings (Aβ and tau accumulation) are causative of or merely associated with cognitive dysfunction in CTE is unknown.39 Our data show that CTE pathology is not required to produce long-term cognitive deficits after rmTBI. These data are in contrast to 1 recent study using a rmTBI model similar to ours that reported increased phosphotau in injured brain at 30 days after the last injury.25 We did not find significant increases in phosphotau or Aβ in brain 6 months after rmTBI despite persistent cognitive deficits. These data suggest that athletes with multiple concussions may be at risk for cognitive decline through mechanisms independent of tau and Aβ, but do not rule out the association between rmTBI and CTE and AD.
Although we found no MRI changes, evidence of axonal pathology, or histopathological evidence of CTE, our injury model did result in robust increase in GFAP-positive cells in several brain regions in mice subjected to 5 rmTBI in 5 days. Although we cannot rule out differences in GFAP antigenicity as well, the increased number of GFAP-positive cells appears to be a biomarker for rmTBI. Our findings are corroborated by prior studies that have demonstrated astrocyte proliferation after brain injury by various mechanisms.40–42 It is not certain in our model whether astrocytes have a protective role after injury43 or could offer a potential therapeutic target for the fixed cognitive deficits in this model.
This study has several limitations. The 6-month outcome data provide proof of principle but cannot be used to predict the interinjury interval related to long-term sequelae of multiple concussions in humans. We also did not follow the mice long enough to fully assess development of AD or CTE pathology after rmTBI. APOE4 transgenic mice that express human APOE4 in astrocytes do so in the absence of APOE3. Other transgenic mice have been utilized to assess the effect of APOE4 on outcome after TBI, and the behavioral phenotypes may differ between APOE4 models.44–46 Another limitation is that we used purchased WT mice rather than WTs generated from heterozygous to heterozygous breeding or APOE4 controls. However, homozygous APOE4 transgenic mice are congenic with C57Bl/6 mice undergoing at least 6 backcrosses (Jackson Laboratories, September 21, 2012).
In conclusion, we have shown that rmTBI associated with astrocyte proliferation but not LOC or convulsions can produce long-term deficits in cognitive function independent of APOE4 status and significant Aβ or tau accumulation. These data suggest that athletes may be at greater risk of cognitive decline than previously thought after rmTBI.
Acknowledgments
This study was supported by the NFL Charities Medical Grant (R.M., W.P.M., M.W.), NICHD T32HD040128-06 (W.P.M.), Charles Hood Foundation (R.M.), and NIH National Institute of Neurological Disorders and Stroke R21NS075226 (M.W.).
Footnotes
Potential Conflicts of Interest
Nothing to report.
References
- 1.Plassman BL, Havlik RJ, Steffens DC, et al. Documented head injury in early adulthood and risk of Alzheimer’s disease and other dementias. Neurology. 2000;55:1158–1166. doi: 10.1212/wnl.55.8.1158. [DOI] [PubMed] [Google Scholar]
- 2.Bower JH, Maraganore DM, Peterson BJ, et al. Head trauma preceding PD: a case-control study. Neurology. 2003;60:1610–1615. doi: 10.1212/01.wnl.0000068008.78394.2c. [DOI] [PubMed] [Google Scholar]
- 3.Critchley M. Medical aspects of boxing, particularly from a neurological standpoint. Br Med J. 1957;1:357–362. doi: 10.1136/bmj.1.5015.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Grahmann H, Ule G. Diagnosis of chronic cerebral symptoms in boxers (dementia pugilistica & traumatic encephalopathy of boxers) [in German] Psychiatr Neurol (Basel) 1957;134:261–283. [PubMed] [Google Scholar]
- 5.Martland H. Punch drunk. JAMA. 1928;91:1103–1107. [Google Scholar]
- 6.McKee AC, Cantu RC, Nowinski CJ, et al. Chronic traumatic encephalopathy in athletes: progressive tauopathy after repetitive head injury. J Neuropathol Exp Neurol. 2009;68:709–735. doi: 10.1097/NEN.0b013e3181a9d503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Uryu K, Laurer H, McIntosh T, et al. Repetitive mild brain trauma accelerates Abeta deposition, lipid peroxidation, and cognitive impairment in a transgenic mouse model of Alzheimer amyloidosis. J Neurosci. 2002;22:446–454. doi: 10.1523/JNEUROSCI.22-02-00446.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Oddo S, Vasilevko V, Caccamo A, et al. Reduction of soluble Abeta and tau, but not soluble Abeta alone, ameliorates cognitive decline in transgenic mice with plaques and tangles. J Biol Chem. 2006;281:39413–39423. doi: 10.1074/jbc.M608485200. [DOI] [PubMed] [Google Scholar]
- 9.Shankar GM, Li S, Mehta TH, et al. Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat Med. 2008;14:837–842. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mayeux R, Ottman R, Maestre G, et al. Synergistic effects of traumatic head injury and apolipoprotein-epsilon 4 in patients with Alzheimer’s disease. Neurology. 1995;45(3 pt 1):555–557. doi: 10.1212/wnl.45.3.555. [DOI] [PubMed] [Google Scholar]
- 11.Jordan BD, Relkin NR, Ravdin LD, et al. Apolipoprotein E epsilon4 associated with chronic traumatic brain injury in boxing. JAMA. 1997;278:136–140. [PubMed] [Google Scholar]
- 12.Meehan WP, III, Zhang J, Mannix R, Whalen MJ. Increasing recovery time between injuries improves cognitive outcome after repetitive mild concussive brain injuries in mice. Neurosurgery. 2012;71:885–891. doi: 10.1227/NEU.0b013e318265a439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhao L, Han W, Steiner C. Sports related concussions, 2008 Statistical brief #114. Rockville MD: Healthcare Cost and Utilization Project; 2006. [PubMed] [Google Scholar]
- 14.McCrory PR, Berkovic SF. Video analysis of acute motor and convulsive manifestations in sport-related concussion. Neurology. 2000;54:1488–1491. doi: 10.1212/wnl.54.7.1488. [DOI] [PubMed] [Google Scholar]
- 15.Holtzman DM, Bales KR, Tenkova T, et al. Apolipoprotein E isoform-dependent amyloid deposition and neuritic degeneration in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci U S A. 2000;97:2892–2897. doi: 10.1073/pnas.050004797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Morris R. Developments of a water-maze procedure for studying spatial learning in the rat. J Neurosci Methods. 1984;11:47–60. doi: 10.1016/0165-0270(84)90007-4. [DOI] [PubMed] [Google Scholar]
- 17.Wang J, Dickson DW, Trojanowski JQ, Lee VM. The levels of soluble versus insoluble brain Abeta distinguish Alzheimer’s disease from normal and pathologic aging. Exp Neurol. 1999;158:328–337. doi: 10.1006/exnr.1999.7085. [DOI] [PubMed] [Google Scholar]
- 18.Kida K, Minamishima S, Wang H, et al. Sodium sulfide prevents water diffusion abnormality in the brain and improves long term outcome after cardiac arrest in mice. Resuscitation. 2012;83:1292–1297. doi: 10.1016/j.resuscitation.2012.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lein ES, Hawrylycz MJ, Ao N, et al. Genome-wide atlas of gene expression in the adult mouse brain. Nature. 2007;445:168–176. doi: 10.1038/nature05453. [DOI] [PubMed] [Google Scholar]
- 20.Cai Y, Hu H, Liu P, et al. Association between the apolipoprotein E4 and postoperative cognitive dysfunction in elderly patients undergoing intravenous anesthesia and inhalation anesthesia. Anesthesiology. 2012;116:84–93. doi: 10.1097/ALN.0b013e31823da7a2. [DOI] [PubMed] [Google Scholar]
- 21.Khuman J MWI, Whalen M. TNF-α and Fas receptors mediate cognitive deficits independent of cell death after closed head injury in mice; Paper presented at: Second Joint Symposium of International and National Neurotrauma Societies; September 7–11, 2009; Santa Barbara, CA. [Google Scholar]
- 22.Callaway JK, Jones NC, Royse CF. Isoflurane induces cognitive deficits in the Morris water maze task in rats. Eur J Anaesthesiol. 2012;29:239–245. doi: 10.1097/EJA.0b013e32835103c1. [DOI] [PubMed] [Google Scholar]
- 23.Creeley CE, Wozniak DF, Bayly PV, et al. Multiple episodes of mild traumatic brain injury result in impaired cognitive performance in mice. Acad Emerg Med. 2004;11:809–819. doi: 10.1111/j.1553-2712.2004.tb00761.x. [DOI] [PubMed] [Google Scholar]
- 24.DeFord SM, Wilson MS, Rice AC, et al. Repeated mild brain injuries result in cognitive impairment in B6C3F1 mice. J Neurotrauma. 2002;19:427–438. doi: 10.1089/08977150252932389. [DOI] [PubMed] [Google Scholar]
- 25.Kane MJ, Angoa-Perez M, Briggs DI, et al. A mouse model of human repetitive mild traumatic brain injury. J Neurosci Methods. 2012;203:41–49. doi: 10.1016/j.jneumeth.2011.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mouzon BC, Chaytow H, Crynen G, et al. Repetitive mild traumatic brain injury in a mouse model produces learning and memory deficits accompanied by histological changes. J Neurotrauma. 2012;29:2761–2773. doi: 10.1089/neu.2012.2498. [DOI] [PubMed] [Google Scholar]
- 27.Shitaka Y, Tran HT, Bennett RE, et al. Repetitive closed-skull traumatic brain injury in mice causes persistent multifocal axonal injury and microglial reactivity. J Neuropathol Exp Neurol. 2011;70:551–567. doi: 10.1097/NEN.0b013e31821f891f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yoshiyama Y, Uryu K, Higuchi M, et al. Enhanced neurofibrillary tangle formation, cerebral atrophy, and cognitive deficits induced by repetitive mild brain injury in a transgenic tauopathy mouse model. J Neurotrauma. 2005;22:1134–1141. doi: 10.1089/neu.2005.22.1134. [DOI] [PubMed] [Google Scholar]
- 29.Chappell MH, Ulug AM, Zhang L, et al. Distribution of microstructural damage in the brains of professional boxers: a diffusion MRI study. J Magn Reson Imaging. 2006;24:537–542. doi: 10.1002/jmri.20656. [DOI] [PubMed] [Google Scholar]
- 30.Lipton ML, Gellella E, Lo C, et al. Multifocal white matter ultrastructural abnormalities in mild traumatic brain injury with cognitive disability: a voxel-wise analysis of diffusion tensor imaging. J Neurotrauma. 2008;25:1335–1342. doi: 10.1089/neu.2008.0547. [DOI] [PubMed] [Google Scholar]
- 31.Levine B, Kovacevic N, Nica EI, et al. The Toronto traumatic brain injury study: injury severity and quantified MRI. Neurology. 2008;70:771–778. doi: 10.1212/01.wnl.0000304108.32283.aa. [DOI] [PubMed] [Google Scholar]
- 32.Mannix RC, Zhang J, Park J, et al. Age-dependent effect of apolipoprotein E4 on functional outcome after controlled cortical impact in mice. J Cereb Blood Flow Metab. 2011;31:351–361. doi: 10.1038/jcbfm.2010.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kutner KC, Erlanger DM, Tsai J, et al. Lower cognitive performance of older football players possessing apolipoprotein E epsilon4. Neurosurgery. 2000;47:651–657. doi: 10.1097/00006123-200009000-00026. discussion 657–658. [DOI] [PubMed] [Google Scholar]
- 34.Chamelian L, Reis M, Feinstein A. Six-month recovery from mild to moderate traumatic brain Injury: the role of APOE-epsilon4 allele. Brain. 2004;127(pt 12):2621–2628. doi: 10.1093/brain/awh296. [DOI] [PubMed] [Google Scholar]
- 35.Moran LM, Taylor HG, Ganesaligam K, et al. Apolipoprotein E4 as a predictor of outcomes in pediatric mild traumatic brain injury. J Neurotrauma. 2009;26:1489–1495. doi: 10.1089/neu.2008.0767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Collie A, Maruff P, Falleti M. APOE influences on neuropsychological function after mild head injury: within-person comparisons. Neurology. 2004;63:2460. doi: 10.1212/wnl.63.12.2460. author reply 2460. [DOI] [PubMed] [Google Scholar]
- 37.Jordan BD. Genetic susceptibility to brain injury in sports: a role for genetic testing in athletes? Phys Sportsmed. 1998;26:25–26. doi: 10.3810/psm.1998.02.928. [DOI] [PubMed] [Google Scholar]
- 38.De Beaumont L, Theoret H, Mongeon D, et al. Brain function decline in healthy retired athletes who sustained their last sports concussion in early adulthood. Brain. 2009;132(pt 3):695–708. doi: 10.1093/brain/awn347. [DOI] [PubMed] [Google Scholar]
- 39.Johnson VE, Stewart W, Smith DH. Widespread tau and amyloid-Beta pathology many years after a single traumatic brain injury in humans. Brain Pathol. 2012;22:142–149. doi: 10.1111/j.1750-3639.2011.00513.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kernie SG, Erwin TM, Parada LF. Brain remodeling due to neuronal and astrocytic proliferation after controlled cortical injury in mice. J Neurosci Res. 2001;66:317–326. doi: 10.1002/jnr.10013. [DOI] [PubMed] [Google Scholar]
- 41.Barreto GE, Sun X, Xu L, Giffard RG. Astrocyte proliferation following stroke in the mouse depends on distance from the infarct. PLoS One. 2011;6:e27881. doi: 10.1371/journal.pone.0027881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hinkle DA, Baldwin SA, Scheff SW, Wise PM. GFAP and S100beta expression in the cortex and hippocampus in response to mild cortical contusion. J Neurotrauma. 1997;14:729–738. doi: 10.1089/neu.1997.14.729. [DOI] [PubMed] [Google Scholar]
- 43.Myer DJ, Gurkoff GG, Lee SM, et al. Essential protective roles of reactive astrocytes in traumatic brain injury. Brain. 2006;129(pt 10):2761–2772. doi: 10.1093/brain/awl165. [DOI] [PubMed] [Google Scholar]
- 44.Hartman RE, Wozniak DF, Nardi A, et al. Behavioral phenotyping of GFAP-apoE3 and -apoE4 transgenic mice: apoE4 mice show profound working memory impairments in the absence of Alzheimer’s-like neuropathology. Exp Neurol. 2001;170:326–344. doi: 10.1006/exnr.2001.7715. [DOI] [PubMed] [Google Scholar]
- 45.Raber J, Wong D, Buttini M, et al. Isoform-specific effects of human apolipoprotein E on brain function revealed in ApoE knockout mice: increased susceptibility of females. Proc Natl Acad Sci U S A. 1998;95:10914–10919. doi: 10.1073/pnas.95.18.10914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Raber J, Wong D, Yu GQ, et al. Apolipoprotein E and cognitive performance. Nature. 2000;404:352–354. doi: 10.1038/35006165. [DOI] [PubMed] [Google Scholar]