

Abstract
Like other unsaturated lipids in cell membranes and lipoproteins, cholesterol (Ch) is susceptible to oxidative modification, including photodynamic oxidation. There is a sustained interest in the pathogenic properties of Ch oxides such as those generated by photooxidation. Singlet oxygen (1O2)-mediated Ch photooxidation (Type II mechanism) gives rise to three hydroperoxide (ChOOH) isomers: 5α-OOH, 6α-OOH, and 6β-OOH, the 5α-OOH yield far exceeding that of the others. 5α-OOH detection is relatively straightforward and serves as a definitive indicator of 1O2 involvement in a reaction, photochemical or otherwise. Like all lipid hydroperoxides (LOOHs), ChOOHs can disrupt membrane or lipoprotein structure/function on their own, but subsequent light-independent reactions may either intensify or attenuate such effects. Such reactions include (a) one-electron reduction to redox-active free radical intermediates, (b) two-electron reduction to redox-silent alcohols, and (c) translocation to other lipid compartments, where (a) or (b) may take place. In addition to these effects, ChOOHs may act as signaling molecules in reactions that affect cell fates. Although processes a-c have been well studied for ChOOHs, signaling activity is still poorly understood compared with that of hydrogen peroxide. This review focuses on these various aspects Ch photo-peroxidation and its biological consequences.
Keywords: cholesterol, cholesterol oxidation, lipid peroxidation, singlet oxygen, free radicals, translocation, glutathione peroxidase
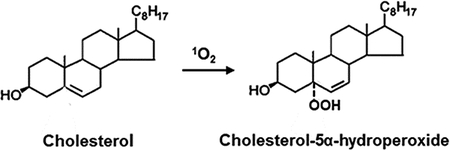
Cholesterol-5α-hydroperoxide (5α-OOH) is the major product of singlet oxygen (1O2)-mediated photosensitized peroxidation of cholesterol, a monoenoic lipid in cell membranes and lipoproteins. In the presence of redox-active metal ions and reductants, 5α-OOH undergoes one-electron reduction to free radical species that can induce light-independent chain peroxidation reactions which exacerbate oxidative damage/dysfunction. Selenoperoxidase GPx4-catalyzed two-electron reduction of 5α-OOH opposes these effects, yet 5α-OOH remains one of the most noxious of the known natural lipid hydroperoxides. These and related aspects of photodynamic cholesterol peroxidation are discussed in this review.
Introduction
Cholesterol (cholest-5-en-3β-ol; abbreviated as Ch) is a sterol lipid found naturally in all animal cells and lipoproteins. Most of the Ch (>80%) in mammalian cells is located in the plasma membrane, where it comprises 45–50 mol % of the total lipid (1,2). In low density lipoprotein (LDL), free Ch and esterified Ch (i.e. Ch with a fatty acid at the C3 position) account for ~19 mol % and ~55 mol %, respectively, of the total lipid. As a monounsaturated lipid with a double bond at the 5–6 position, Ch is susceptible to non-enzymatic oxidation (autoxidation), but less readily than a polyunsaturated phospholipid such as 1-palmitoyl-2-linoleoyl-sn-glycero-3-phosphocholine. Nevertheless, the relatively abundant Ch in plasma membranes compared with other cellular membranes makes it an important oxidative target in the former. Unlike natural phospholipids, Ch exists as a single molecular species, and its relatively few oxidation products are easier to separate and characterize than those of phospholipids (3,4). Cholesterol oxides (ChOX) generated by chemical oxidants, ionizing radiation, or photodynamic action have been well studied in a variety of reaction systems, ranging from liposomal membranes and lipoproteins to mammalian cells (3–7). The authors review their extensive work in this area, with a special emphasis on photodynamically generated ChOX species.
Cholesterol Hydroperoxides Generated by Photodynamic Action
Lipid hydroperoxides (LOOHs) are important intermediates in the non-enzymatic oxidation of all unsaturated lipids, including phospholipids and Ch (4,8,9). In free radical-mediated reactions, including Type I photooxidation reactions, where O2 levels are relatively low, Ch gives rise to two epimeric ring hydroperoxides (ChOOHs): 3β-hydroxycholest-5-ene-7α-hydroperoxide (7α-OOH) and 3β-hydroxycholest-5-ene-7β-hydroperoxide (7β-OOH) (3,9–11) (Fig. 1). Both of these species arise via abstraction of a C7 allylic hydrogen from the Ch molecule. Abstraction could be accomplished by a strong oxidant such as the radical anion of a proximal photosensitizer (Fig. 3) or a reactive oxygen species (ROS) that it generates, e.g. hydroxyl radical (HO∙) (12). Rapid addition of O2 to the resulting C7 radical, followed by hydrogen abstraction from another Ch or proximal other lipid gives 7α-OOH. The latter gradually isomerizes to 7β-OOH, which is thermodynamically more stable (11). Redox-active 7α-OOH and 7β-OOH are typically accompanied by their downstream diol reduction products: cholest-5-ene-3β,7α-diol (7α-OH) and cholest-5-ene-3β,7β-diol (7β-OH), along with 3β-hydroxycholest-5-ene-7-one (7-ketone) and cholestan-5α/5β,6α/6β-epoxy-3β-ol (epimeric 5,6-epoxides) (Fig. 1). All of these species can potentially arise during Type I photoperoxidation of Ch, 7α-OOH and 7β-OOH as unstable intermediates.
Fig. 1.
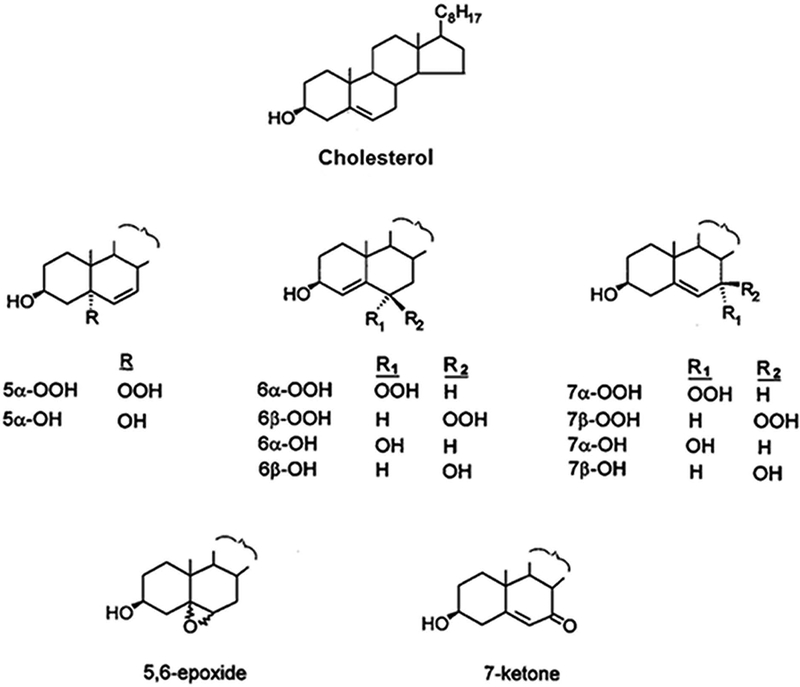
Structures of cholesterol, its relevant redox-active hydroperoxides (ChOOHs), and corresponding redox-inactive alcohols (ChOHs).
Fig. 3.
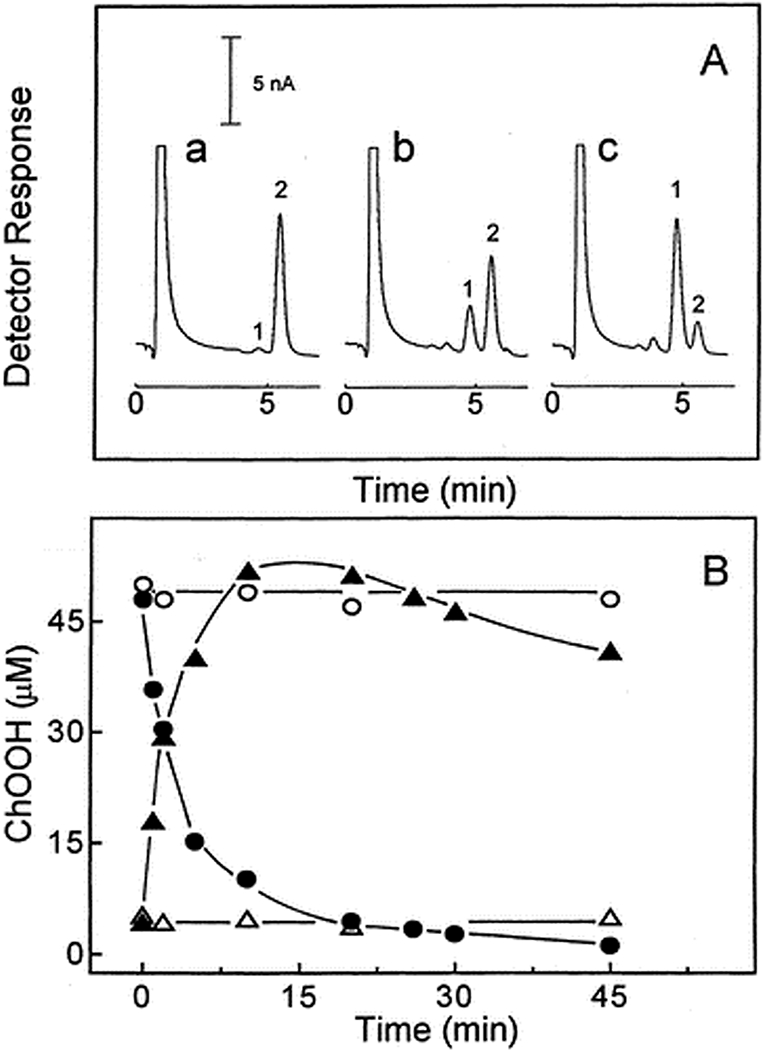
One-electron degradation of 5α-OOH with concurrent formation of 7α/7β-OOH in unilamellar POPC/Ch/5α-OOH (100/75/5 by mol) liposomes during incubation with ascorbate (1 mM) and Fe(HQ)3 (1 µM) in Chelexed PBS at 37°C. (A) HPLC-EC(Hg) profiles of lipid extracts after 0 min (a), 5 min (b), and 30 min (c). (B) 5α-OOH (⬤,○) and 7α/7β-OOH (π,ρ) levels over time with Fe(HQ)3 present (⬤, π) or absent (○,ρ). Reproduced from Korytowski et al. (2000) Chem. Res. Toxicol. 13:1265 (with permission).
In 1∆gO2 singlet oxygen (1O2)-mediated Type II photooxidation reactions, where O2 levels are relatively high, three primary ChOOH species are generated: 3β-hydroxy-5α-cholest-6-ene-5-hydroperoxide (5α-OOH), 3β-hydroxycholest-4-ene-6α-hydroperoxide (6α-OOH), and 3β-hydroxycholest-4-ene-6β-hydroperoxide (6β-OOH) (Fig. 1). Each arises via the ene addition of 1O2 at the Ch double bond, with all atoms in the target molecule being retained (13–15). The generation rate of 5α-OOH is usually much greater than that of 6α- or 6β-OOH. This is probably due to steric hindrance which results in less favorable 1O2 attack at the C6 position (16). While 7α- and 7β-OOH do not arise directly from 1O2 attack, they may appear indirectly via allylic rearrangement of 5α-OOH, which becomes more favorable as the extent of lipid peroxidation increases in organized systems. Cholesteryl esters bearing an unsaturated fatty acyl group would be susceptible to oxidation in this group as well as in the Ch A/B ring portion. If an esterified polyunsaturated fatty acid such as linoleic or linolenic were present, its oxidation rate would be expected to exceed that of the monoenoic Ch moiety; however, it appears that no confirmation of this has been reported, at least for photodynamic oxidation.
One-Electron Reduction of ChOOHs: Exacerbation of Oxidative Damage/Dysfunction
If catalytic metal ions and/or reductants are absent or inaccessible, Type II 1O2-derived ChOOHs, like phospholipid counterparts (PLOOHs), will accumulate linearly with increasing sensitizer concentration or light fluence (4,6,17). The same may apply for Type I ChOOH formation, although a light-generated sensitizer radical anion might reduce the ChOOH to an oxyradical which begets new LOOHs/ChOOHs via chain peroxidation (see below), although this has not been rigorously tested for Type I chemistry. If a properly chelated metal ion (e.g. iron) and a reductant such as ascorbate or cysteine are available, the primary ChOOH (like LOOHs in general) can undergo one-electron reduction to an oxyl radical (ChO∙), which may directly abstract an allylic hydrogen from a neighboring lipid, thereby triggering a wave of free radical-mediated (chain) peroxidation (Fig. 2). Alternatively, the ChO∙ might undergo rapid one-electron rearrangement with O2 addition to give an epoxyallylic peroxyl radical (OChOO∙) which initiates chain peroxidation. Although the latter mechanism has been demonstrated for relatively simple fatty acid model systems (18), there appears to be no similar existing evidence for Ch or fatty acids in phospholipid form. Chain peroxidation induced by one-electron reduction of primary or “priming” ChOOHs in a membrane bilayer would produce new ChOOHs/LOOHs that feed into the overall process (Fig. 2). These secondary reactions would exacerbate the damaging effects of primary peroxidation alone, e.g. 1O2-mediated 5α-OOH formation in a photodynamic system. Fig. 3 illustrates this by showing that liposomes containing an unsaturated phospholipid (POPC), Ch, and priming 5α-OOH underwent a rapid loss of the latter during incubation with ascorbate and a catalytic amount of lipophilic iron chelate, ferric-8-hydroxyquinoline [Fe(HQ)3]. 7α/7β-OOH accumulated concurrently, reached a steady state level after ~15 min, and declined thereafter, evidently because it also began to be consumed by one-electron reduction and free Ch became limiting. Thus, 7α/7β-OOH served as a reporter of free radical-mediated lipid peroxidation triggered by 5α-OOH one-electron turnover. Consistent results were obtained when cultured cells were monitored after a photodynamic challenge. For example, exposure of merocyanine 540-sensitized leukemia L1210 cells to increasing fluences of broad-band visible light, followed by dark incubation, resulted in a >3-log loss of clonogenicity (6). When introduced immediately after irradiation, the chain-breaking antioxidant butylated hydroxytoluene (BHT) inhibited the cell killing in substantial, dose-dependent fashion. However, di-tert-butyltoluene (DBT), a non-phenolic analogue used as a control, had no effect. Importantly, BHT also suppressed post-irradiation chain peroxidation in these cells, as indicated by increasing 7-OOH/5α-OOH ratio (5). Of related interest, treating cells with an iron chelator/redox inhibitor (desferrioxamine) before a photodynamic challenge strongly attenuated the extent of photokilling and increase in 7-OOH/5α-OOH ratio, whereas addition of a lipophilic iron chelate (ferric-8-hydroxyquinoline) in trace amounts did the opposite (5). Collectively, these findings support the notion that while primary LOOHs/ChOOHs can be deleterious on their own (depending on levels attained), they may induce secondary, light independent reactions that are potentially more damaging. In other words, there could be an after-light continuum in the cytotoxic activity initiated by photodynamic events, primary peroxide formation being the main driving force. This possibility needs to be better recognized in photo-cytotoxicity studies, e.g. those involving anti-cancer photodynamic therapy (PDT).
Fig. 2.
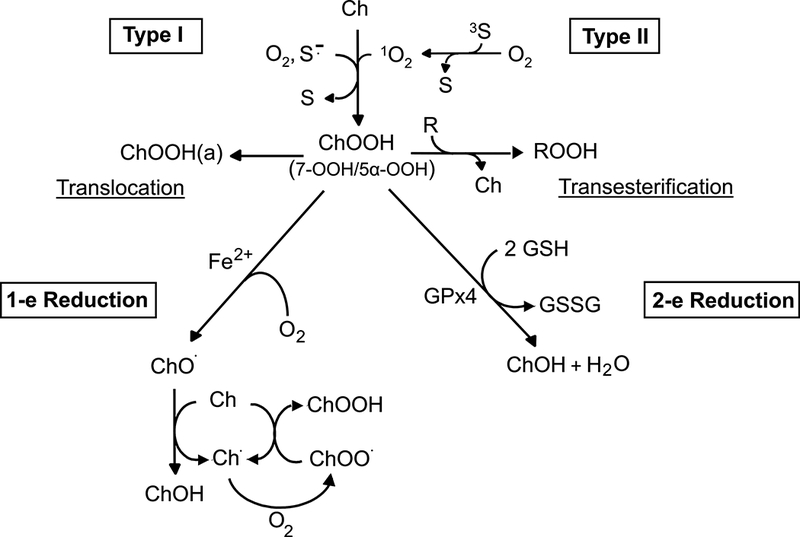
Type I (free radical) versus Type II (1O2) photoperoxidation of cholesterol to hydroperoxide (ChOOH) intermediates, which can have different fates in membrane and lipoprotein environments, including transesterification, translocation, iron-catalyzed one-electron reduction to chain-initiating/propagating free radicals, and Gpx4-catalyzed two-electron reduction to redox-inert alcohols.
The preceding comments about damaging peroxide turnover require some qualification, based on a more recent comparison of the chain-initiating potency of different ChOOH isomers. When unilamellar liposomes containing POPC, Ch, and 5α-OOH, 6β-OOH, or 7α-OOH (each at the same starting level) were incubated with Fe(HQ)3 and ascorbate, all three hydroperoxides decayed at the same rate (16,19). This implied equal ability to undergo one-electron reduction. However, when chain-initiating potency was assessed, using [14C]Ch as an endogenous sensor (see below), 5α-OOH and 7α-OOH exhibited nearly equal potency, whereas 6β-OOH showed very little activity (19). Because of its scarcity, 6α-OOH was not tested, but is expected to mimic 6β-OOH as a relatively poor initiator. The contrasting initiation ability of 5α-OOH and 6β-OOH was also observed at the cellular level. Thus, when L1210 cells were challenged with 5α-OOH- vs. 6β-OOH-containing liposomes, chain peroxidation and cytotoxicity caused by the latter were substantially less than those by the former (19). Since 5α-OOH and 6β-OOH decay equally rapidly in the presence iron and ascorbate, their oxyl radicals should appear at the same rate. Why, then, is the chain-initiating potency of 6β-O∙ so low compared with that of 5α-O∙? Two possible explanations have been offered (19): (i) The oxidizing potential of 6β-O∙, i.e. its ability to abstract an allylic hydrogen from another lipid, might be much lower than that of 5α-O∙; (ii) Improper orientation of 6β-O∙ in the bilayer may substantially slow its access to neighboring chain-propagating lipids. The latter kinetic argument is more plausible because 6β-OOH can induce at least some chain peroxidation after prolonged membrane incubation with iron and a reductant (19). One should note that even if 6β-OOH were as efficient as 5α-OOH in triggering chain reactions, 6β-OOH would still have a relatively small effect, given its low yield in Type II photooxidation reactions.
Two-Electron Reduction of ChOOHs: Alleviation of Oxidative Damage/Dysfunction
Unlike superoxide (O2-) and hydrogen peroxide (H2O2), which are inactivated by superoxide dismutases and catalase/peroxidases, respectively, 1O2 has no known primary enzymatic scavengers. In the case of cytotoxicity due to photogenerated 1O2, therefore, secondary or back-up defenses need to be engaged. This would typically involve enzymatic reduction or removal of oxidized groups at damage sites, followed by repair or replacement processes (4,6,8). Four intracellular enzymes with glutathione (GSH)-dependent peroxidatic activity have been implicated in LOOH reduction/detoxification: glutathione peroxidase-1 (GPx1), glutathione peroxidase-4 (GPx4), glutathione-S-transferase type-α (GSTα), and peroxiredoxin-6 (Prx6) (4). GPx1 and GPx4 (formerly known as phospholipid hydroperoxide glutathione peroxidase, PHGPx) have been studied far more extensively than the others. Each GPx isotype has an active site selenocysteine residue (Sec), which participates in the two-electron reduction of peroxides to redox-inactive alcohols. Monomeric GPx4 (~20 kDa) is functionally quite distinct from homotetrameric GPx1 (~82 kDa), the most abundant seleno-peroxidase in mammalian cells. Whereas GPx4 with GSH can act directly on LOOHs in membrane and lipoprotein environments, GPx1 lacks this ability (4,20–22). Instead, GPx1 acts on H2O2 and relatively polar LOOHs such as fatty acid hydroperoxides, typically after they are released from peroxidized membrane phospholipids by phospholipase-A2 (PLA2) hydrolysis (23). Consistent with this, two general enzymatic mechanisms have been proposed for elimination of PLOOH peroxide groups in membranes or lipoproteins: (i) excision/reduction/repair, where sequential action of PLA2 and GPx1 is followed by acyltransferase-catalyzed re-acylation of the lysolipid; and (ii) reduction/excision/repair, where sequential action of GPx4 and PLA2 is followed by re-acylation (4,23). Existing evidence favors the latter mechanism over the former, although studies in this important area are ongoing.
As discovered by the authors and collaborators (24,25), GPx4, but not GPx1, can readily catalyze not only the two-electron reduction of PLOOHs, but ChOOHs as well. To date, GPx4 appears to be the only enzyme capable of catalyzing ChOOH reductive detoxification, and regardless of whether these peroxides are membrane-bound or in Triton-X100 micellar form. Studies in the authors’ laboratory have revealed a broad range of reactivity with GSH/GPx4 for various ChOOH isomers; collectively, however, they reacted much more slowly than PLOOHs. The first-order rate constants for hydroperoxide decay were found to increase in the following order: 5α-OOH<<7α/β-OOH<6β-OOH<PLOOH (26). This pattern was observed for purified GPx4 from rat testis and also for GPx4 in homogenates of selenium-replete L1210 cells, whereas selenium-starved cells showed little activity (Fig. 4A). Importantly, the order of cytotoxicity for selenium-replete cells under similar incubation conditions (Fig. 4B) was diametrically opposite to that of GPx4-catalyzed detoxification. Thus, 5α-OOH with the slowest two-electron reduction rate was the most cytotoxic, whereas POPC-OOH with the fastest rate was the least cytotoxic. Note that 6β-OOH was the least toxic among the ChOOHs; although it was reduced relatively rapidly by GSH/GPx4, this probably mattered little compared with its sluggish one-electron reduction (see preceding section). It should be emphasized that very slow 5α-OOH detoxification, combined with rapid generation under 1O2 stress conditions (e.g. UVA radiation, PDT, etc.), would make 5α-OOH the most physiologically deleterious of the LOOHs described. A plausible explanation for slow inactivation is that 5α-OOH is a tertiary hydroperoxide, and as such might not interact with GPx4 as readily as PLOOHs, 6α/6β-OOH, or 7α/β-OOH, which are all secondary hydroperoxides (16).
Fig. 4.
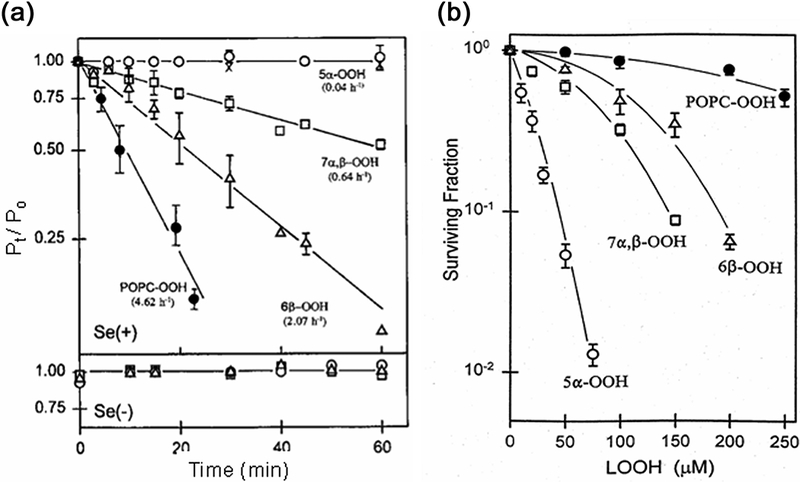
Ability of GSH/GPx4 in lysates of selenium-replete [Se(+)] L1210 cells to (A) reduce and (B) detoxify different ChOOH species versus a PLOOH species. (A) Reaction mixtures contained the same concentration of hydroperoxide (5α-OOH, 6β-OOH, 7α/7β-OOH, or POPC-OOH), GSH, desferrioxamine, EDTA, and cell lysate in Triton-X100/PBS at 37°C. At the indicated times, lipid were extracted and analyzed by HPLC-EC(Hg). Se(−): lysates of selenium-starved cells; (x): controls lacking GSH or lysate. Plotted data are means ± SEM (n=3). (B) Se(+) cells treated with ChOOH- or POPC-OOH-bearing liposomes. Lipid extracts were analyzed 20 h after incubation. Data are means ± deviation (n=2). Reproduced in part from Korytowski et al. (1996) Biochemistry 35:8670 (with permission).
Cholesterol as a Mechanistic Probe and Chain Peroxidation Reporter
Mechanistic information based on detection of 5α-OOH as a 1O2 reporter and 7α/β-OOH as a free radical reporter has been acquired for a variety of isolated membrane and cellular systems (5,27). High sensitivity/specificity analytical techniques are essential for monitoring these ChOOHs, the yields of which are typically quite low. An key example is reverse-phase high performance liquid chromatography with mercury cathode electrochemical detection [HPLC-EC(Hg)], which was developed in the authors’ laboratory (28,29). Using an appropriate column and deoxygenated mobile phase, one can separate and detect all of the ChOOHs described at very high sensitivity; for example, 7α/β-OOH has a detection limit of ~0.2 pmol (29). With suitable care in sample preparation, HPLC-EC(Hg) is amenable to analysis of complex systems, e.g. lipid extracts from photodynamically-stressed cells. Mechanistic assignment based on 5α-OOH for 1O2 and 7α/β-OOH for free radicals is usually very dependable, although some uncertainties may exist. For example, 5α-OOH can isomerize to 7α-OOH, but this is usually very slow in moderately peroxidized membranes (28,29). Also, 5α-OOH might possibly derive from 1O2 arising via downstream Russell-type reactions, i.e. peroxyl radical disproportionation reactions (30,31). Detecting both reporters in such cases could lead to confusion, e.g. co-existence of Type I and Type II photooxidation mechanisms. Up to now, however, no de novo 5α-OOH has been detected in membranes undergoing chain peroxidation (16), so we believe that it strictly reports on primary 1O2 formation in such systems.
In addition to using specific ChOOHs as diagnostics, the authors have used cholesterol oxide intermediates/products called “ChOX” to obtain additional mechanistic information. A novel approach was devised whereby radiolabeled Ch is exploited as a natural probe for assessing overall chain peroxidation or the chain-initiating potency of pre-existing primary PLOOHs/ChOOHs. In this approach, [14C]Ch is first incorporated into a lipid matrix such as liposomal membrane, cell membrane, or lipoprotein. During chain reactions induced by an oxidative challenge in the presence of reducible metal ions or reductive turnover of pre-existing LOOHs, the probe gives rise to [14C]ChOX species. The latter are conveniently detected and quantified by high performance thin layer chromatography with phosphorimaging detection (HPTLC-PI) (32,33). Individual ChOX species include redox-active 7α/β-OOH as well as redox-inactive 7α/β-OH, 7-ketone, and 5,6-epoxide. The overall levels attained can reflect the chain-initiating potency of a primary peroxide such as PDT-generated 5α-OOH as well as the oxidizability of surrounding lipids and availability of metal ions and reductants. There are many advantages of using [14C]Ch as a natural sensor. First of all, oxidative degradation of Ch has been studied extensively for many years and most of its oxidation products (oxides/oxysterols) have been well characterized (3,7,13,14,34). This was facilitated by the fact that Ch, unlike natural phospholipids, exists as a single molecular species, making separation and identification of its oxidation products much more straightforward than for phospholipids, each of which can exist in multiple molecular forms. A second advantage of [14C]Ch is that it translocate spontaneously and relatively rapidly from a liposome or serum albumin carrier to an acceptor, e.g. cell membrane. Transfer rate can be increased using a protein transporter such as recombinant sterol carrier protein-2 (SCP-2), which is easily removed sufficient transfer uptake is achieved. For mammalian cells, most of the [14C]Ch transferred in this way will reside in the plasma membrane, allowing chain peroxidation activity to be probed specifically in this compartment (32,33). Another plus about using [14C]Ch is that it avoids the potential artifacts associated with exogenous probes such as spin traps and fluorophores (34,35), which might perturb lipid organization or act as antioxidants in systems being studied. On the other hand, such probes have the advantage of being non-invasive, i.e. signals are obtained directly without having to solubilize and extract lipids. Nevertheless, the [14C]Ch approach is highly sensitive and straightforward in application and has been successfully applied to a number of reaction systems, ranging from liposomes and lipoproteins to membranes of living cells (33).
Spontaneous and Protein-Mediated ChOOH Translocation
As first demonstrated by the authors and associates about 20 years ago, one-electron or two-electron reduction of a primary ChOOH such as 5α-OOH is not necessarily limited to its membrane of origin, but can extend to other membranes via a translocation process (37–39). Transfer from lipoproteins to membranes and vice-versa has also been shown to occur (39). All LOOHs are capable of spontaneous translocation, but ChOOHs move from a donor to acceptor membrane/lipoprotein far more rapidly than PLOOHs with the same peroxyl group content, i.e. one per molecule (39). The parent lipids can also translocate, Ch much more rapidly than any given PL; however, peroxidation increases each rate substantially due to increased hydrophilicity of the peroxide products (40). An experiment with photoperoxidized erythrocyte membrane (ghost) donors and small unilamellar liposome acceptors in ~10-fold lipid molar excess showed that the rate constant for overall ChOOH transfer exceeded that of parent Ch by >60-fold (38). Transfer occurred via an aqueous pool, departure from the donor membrane being the rate-limiting step (40). For HPLC-EC(Hg)-assessed individual ChOOHs, the first-order transfer rate constants were found to decrease in the following order: 7α/7β-OOH > 5α-OOH > 6α-OOH > 6β-OOH, which correlated with their elution rate (or hydrophilicity) order on reverse-phase HPLC (38,39). ChOOH or PLOOH translocation to an acceptor membrane could put the latter at greater risk of chain peroxidative damage if ferric iron and reductants are more available there. On the other hand, if the GSH/GPx4 system is more available in the acceptor compartment, translocation might become beneficial by promoting LOOH detoxification.
Although pro-oxidant ChOOH translocation may play a significant role in various types of oxidative challenge, it could be particularly important for 1O2-mediated photodynamic reactions. Special attention should be directed to 5α-OOH translocation in this regard. In a cell under 1O2 attack, e.g. a skin fibroblast exposed to UVA (320–400 nm) radiation, 5α-OOH would be generated much more rapidly than other ChOOHs, but detoxified relatively slowly by GSH/GPx4 (see Fig. 4A). PLOOHs would also be generated rapidly, e.g. in a plasma membrane exposed to 1O2, but detoxified rapidly by GSH/GPx4 (24,25). Moreover, PLOOHs translocate far more slowly than all ChOOHs, including 5α-OOH (39). This being the case, intermembrane transfer of 5α-OOH, whether spontaneous or protein-mediated (see below), would clearly be favored over that of other LOOHs, and would thus greatly expand its range as a cytotoxic oxidant. Up to now, such possibilities for 5α-OOH have not been well recognized in the photobiology field.
Lipid transfer proteins are known to play key roles in lipid metabolism, membrane biogenesis, and membrane function (41). A well-known example is sterol-carrier protein-2 (SCP-2), a 13.2 kDa protein that can facilitate intracellular translocation of relatively low polarity lipids such as Ch and other sterols, along with various phospholipids (42,43). Because of its broad ligand recognition, SCP-2 is often referred to as a non-specific lipid transfer protein. Seminal studies in the authors’ laboratory revealed that the rate of intermembrane ChOOH or PLOOH transfer could be substantially increased over the spontaneous rate when SCP-2 was present. As shown in Fig. 5, recombinant SCP-2 significantly accelerated the movement of Ch and various ChOOHs from photoperoxidized red cell ghosts to unilamellar liposomes (44). The first-order rate constants (corrected for spontaneous transfer) decreased in the following rank order: 7α/7β-OOH > 5α-OOH > 6α-OOH > 6β-OOH > Ch, which was the same as that for spontaneous transfer (see above). This was the first known evidence for enhanced ChOOH transfer by a lipid transfer protein. Subsequent work revealed that a transfectant clone of hepatoma cells expressing ~10-fold more SCP-2 than vector controls, imported liposomal 7α-OOH more rapidly than the latter and delivered it preferentially to mitochondria over other subcellular compartments (45). Such delivery resulted in relatively rapid peroxidation of mitochondrial lipids, membrane depolarization, and apoptotic cell death (45). These effects were primarily attributed to damaging free radical (chain) reactions triggered by one-electron reduction of SCP-2-delivered 7α-OOH. Mitochondria are rich in redox iron, which would have favored these reactions. No significant damage or cytotoxicity was observed when redox-inactive 7α-OH (a SCP-2 ligand) or redox-active tert-butyl hydroperoxide (a non-SCP-2 ligand) were used, demonstrating the necessity for both protein recognition and redox activation. When attached to the sterol binding pocket of SCP-2, 7α-OOH was probably protected from redox turnover in transit, allowing it to survive delivery through the cytosol to/into mitochondria. The molecular basis for mitochondrial targeting of SCP-2-borne ChOOH is unknown, but presumably reflects the existence of some outer membrane recognition site for the protein.
Fig. 5.
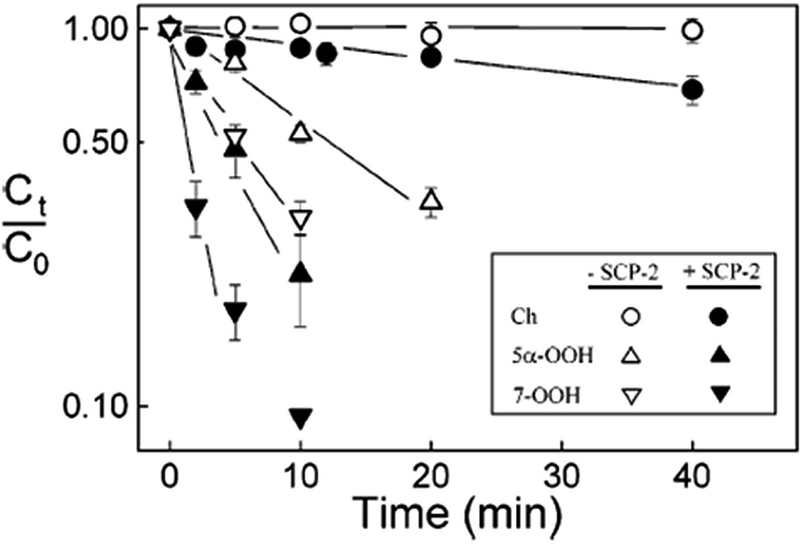
SCP-2-accelerated intrmembrane transfer of Ch and ChOOHs. Reaction mixtures contained photoperoxidized erythrocyte ghosts, and DMPC/DCP (100:1 by mol) liposome acceptors in redox metal ion-depleted PBS containing or lacking SCP-2. Plots: HPLC-EC(Hg)-assessed Ch and ChOOH departure from ghosts during 37°C incubation. Plotted values are means ± SEM (n=3). Reproduced from Vila et al. (2004) Biochemistry 43:12592 (with permission).
Proteins of the steroidogenic acute regulatory (StAR) family have also been implicated in potentially deleterious ChOOH transfer (46–49). These proteins contain a unique sterol binding (START) domain and play a crucial role in steroid hormone synthesis by steroidogenic cells (50) as well as reverse cholesterol transport (RCT) by vascular macrophages for maintenance of Ch homeostasis (51). Most StAR proteins bind a single Ch or similar sterol molecule with high specificity, and thus are distinct from SCP-2 or other non-specific lipid transporters (52). StarD4 is a cytosolic member of the StAR family whose proposed function is to deliver Ch to the mitochondrial outer membrane, where it is recognized by StarD1 and transferred to the inner membrane for metabolism to pregnenolone in steroidogenic cells or 27-hydroxycholesterol (27-OH) in macrophages (53,54). We (the authors) postulated that under pathological conditions associated with oxidative stress, including stress from certain therapeutic regimens, oxidized LDL (oxLDL)-derived ChOOHs might be caught up in StAR-mediated trafficking to/into mitochondria, thereby triggering peroxidative damage that impairs Ch metabolism. Korytowski et al. (55) provided the first supporting evidence by using testicular MA-10 cells, which exhibited a strong upregulation of StarD4 and StarD1 after cAMP stimulation. When incubated with 7α-OOH-containing liposomes, stimulated cells trafficked more of the peroxide to mitochondria than non-stimulated controls, resulting in a greater loss of membrane potential and progesterone output, along with more extensive apoptotic cell death (55). StarD1 knockdown using siRNA strongly blunted 7α-OOH uptake and membrane depolarization, confirming StarD1’s role in the mitochondrial damage/dysfunction. Similarly to SCP-2, StarD4/D1 would be expected to protect ligated ChOOH against premature reductive turnover in transit, allowing the bulk of this turnover to occur after release in mitochondria.
In more recent work, mitochondrial impairment due to inauspicious StAR-mediated ChOOH trafficking was studied in the context of cardiovascular disease, specifically atherogenesis. Using human THP-1 monocyte-derived macrophages, Korytowski et al. (49) showed that cAMP stimulation resulted in a robust upregulation of StarD1 and also the ABCA1/G1 transporters, which localize to the plasma membrane and mediate Ch efflux to extracellular acceptors such as apoA1 and HDL (49). The major functional consequences of exposing stimulated cells to liposome-borne 7α-OOH were (i) enhanced mitochondrial uptake of this ChOX; (ii) greater chain lipid peroxidation localized to mitochondria; (iii) significant activity loss of 27-hydroxylase (CYP27A1), which generates 27-OH, a key agonist for ABCA1/G1 expression and RCT operation; (iv) diminished 27-OH yield from Ch; (v) diminished ABCA1/G1 expression; and (vi) increasing cell loss to intrinsic (mitochondrial-initiated) apoptosis during exposure to 7α-OOH (49). Collectively, these findings with MA-10 cells and THP-1 macrophages were the first to show that a ChOOH like 7α-OOH can impair crucial mitochondrial functions by integrating into a natural trafficking pathway (46–49). Although most of this work was done with free-radical-derived 7α-OOH, similar damage/dysfunction would be expected if 5α-OOH were used, since this ChOX can also be trafficked by the StAR system. A long lifetime because of low susceptibility to enzymatic detoxification and protection against reductive turnover when StarD1/D4-bound (see above), would favor “stealthy” 5α-OOH delivery to mitochondrial targets. Chronic skin exposure to 1O2-generating UVA might conceivably generate sufficient 5α-OOH in vivo to act in this fashion, but this remains to be documented.
ChOOHs as Stress Signaling Mediators
It is now clear that many LOOHs, including ChOOHs, may not only induce/propagate oxidative damage in biological systems, but at relatively low levels may act in a more subtle manner characterized by reversible signaling processes. As oxidative pressure intensifies, the outcome may change accordingly, e.g. (i) no net damage if antioxidant capacity is sufficient; (ii) relatively minor damage that induces cytoprotective or pro-survival responses; (iii) more extensive damage that exceeds cytoprotective antioxidant capacity and triggers programmed (apoptotic or autophagic) cell death; or (iv) extensive membrane damage that results in necrotic cell death because programmed responses are no longer possible (8). The tipping point between cell survival and death would occur at some point between responses (ii) and (iii). This often involves a signaling cascade induced by oxidation of a low pKa cysteine thiol group to a sulfenic acid (-CySOH) form, followed by reaction (resolution) with another cysteine residue to give a disulfide link. The modified protein would either gain or lose activity, and this could be reversed by a reductant such as glutathione (GSH) or thioredoxin, qualifying this event as part of a signaling pathway (55,56). Hydrogen peroxide (H2O2) is a well-established initiator of redox signaling cascades, and much more is known about cysteine-mediated H2O2 signaling than that of most other natural peroxides, including PLOOHs and ChOOHs. The discovery that these LOOHs can translocate from sites of origin (40) has suggested new possibilities for disseminated redox signaling which could enhance our understanding of this phenomenon beyond what is known about H2O2 signaling via simple diffusion. One important distinguishing factor is that amphiphilic LOOHs should move mainly between low polarity compartments in cells, i.e. membranes, whereas hydrophilic H2O2, which has no known protein transporters, would be confined to aqueous compartments. Accordingly, one can postulate that sensor proteins on or near membranes would be the preferred targets of mobilized LOOHs. By contrast, H2O2 would preferentially target relatively hydrophilic, non-membrane-associated sensors. While there are several well-known examples of the latter, including thioredoxins, peroxiredoxins, and protein tyrosine phosphatases (55–57), membrane-associated analogues specifically activated by LOOHs are yet to be identified. Because PLOOH and ChOOH molecules are larger than H2O2, they would diffuse more slowly to sensor targets. LOOH transit would be even slower if mediated by transfer proteins. However, there would be distinct advantages for signaling in the latter case, e.g. (i) longer peroxide lifetime due to sequestration against reductive turnover in transit, and (ii) site-specific LOOH delivery dictated by precise transfer protein-sensor protein interactions. PLOOHs and ChOOHs may also have a longer lifetime in transit because only one enzyme, GPx4, could potentially inactivate them, whereas H2O2 can be rapidly decomposed by much more abundant GPx1 as well as catalase and peroxiredoxins. Largely due to difficulties in establishing it experimentally, genuine LOOH-mediated signaling remains largely unexplored except for a few well qualified examples (58,59).
As indicated in the preceding section, 5α-OOH is the most rapidly generated ChOOH in photodynamic systems and the slowest to be redox-inactivated by GPx4. Thus, 5α-OOH’s relatively long lifetime, combined with its ability to be dispersed by protein transporters, would make it one of the most active and far-reaching LOOHs in terms of cytotoxicity as well as redox signaling. One intriguing possibility regarding 5α-OOH signaling activity relates to peroxide modification/inactivation of PTEN, the tumor-suppressor enzyme that down-regulates PI3K/Akt-promoted survival/expansion of many tumor cells (60,61). Most studies have associated PTEN inactivation with H2O2 produced, for example, by NADPH oxidase (NOX) enzymes (61,62). However, in cells irradiated in the presence of a membrane-associated 1O2-generating sensitizer (e.g. 5-aminolevulinate-induced protoporphyrin IX), high yield 5α-OOH might reach a sensor like PTEN via translocation, resulting in its inactivation. This is one of many signaling possibilities for this ChOOH. Our understanding of 5α-OOH’s unique characteristics along these lines is still far from complete, and much more remains to be learned in an effort to better understand its role in pathologic as well as therapeutic photobiology.
Summary and Perspectives
We have discussed the special features of Ch peroxidation and the ChOOH species produced, with an emphasis on photoperoxidation reactions. The hydroperoxide most associated with oxidative stress damage/dysfunction or oxidative signaling is H2O2. This ROS is generated by NADPH-dependent oxidase (NOX) enzymes and during mitochondrial electron transport (54,55), but is usually not a prominent intermediate in photootxidation reactions, with the possible exception of free radical Type I reactions (12). When O2 is sufficient, most photooxidations proceed via 1O2-generating Type II reactions. Unlike H2O2, 1O2 has no enzymatic scavengers, so protection against this ROS must occur at the secondary level, e.g. GPx4-catalyzed reduction of 1O2 adducts such as 5α-OOH and 6α/6β-OOH (25,26). Like these ChOOHs, H2O2 may undergo toxic one-electron reduction at/near its site of origin or diffuse to other sites where this may occur. Such movement would be a random process, since H2O2 has no known protein transporters. In contrast, CHOOHs can be trafficked intracellularly by protein carriers such as SCP-2 and StARs, as discussed. In mammalian cells under oxidative pressure, ChOOHs arising in the Ch-rich plasma membrane might be transported to endoplasmic reticulum or mitochondria, thus permitting dissemination of one-electron oxidative damage. Unlike H2O2, which could encounter potent scavengers (catalase, various peroxidases), protein-bound ChOOHs would be effectively shielded from degradation until released at target sites. Similar arguments would hold for more subtle ChOOH vs. H2O2 signaling activity, where ChOOH “sensor” protein analogous to those involved in H2O2 signaling would be engaged. Based on peroxide polarity, one can predict that ChOOH sensors would be localized at relatively hydrophobic sites compared with H2O2 sensors (56). These arguments about ChOOH signaling are still largely hypothetical, given that specific receptors/sensors for such species have yet to be identified. An import goal for future investigation is to achieve a better mechanistic understanding of the diverse ChOOH activities described in this review.
Acknowledgements
Studies in the authors’ laboratories were supported by USPHS Grants CA72630, TW001386, and CA70823 (to A.W.G.) and Polish National Center for Science Grant NCN-2014/13/B/NZ3/00833 (to W.K.). We thank Peter Geiger, Andrew Vila, Tamas Kriska, Vlad Levchenko, Jared Schmitt, Magda Niziolek, Kasia Wawak, and Anna Pilat for their many valuable contributions to the studies supported by these grants.
Footnotes
Conflict of Interest
The authors declare that they have no conflicts of interest with any of the work described.
This article is part of a Special Issue celebrating Photochemistry and Photobiology’s 55th Anniversary.
References
- 1.Bloch K (1983) Sterol structure and membrane function. Crit. Rev. Biochem 14, 47–92. [DOI] [PubMed] [Google Scholar]
- 2.Fielding CJ and Fielding PE (1985) Biochemistry of Lipids and Membranes, Edited by Vance DE and Vance JE, Benjamin/Cummings, Menlo Park. [Google Scholar]
- 3.Smith LL (1981) Cholesterol Autoxidation. Plenum Press, New York. [Google Scholar]
- 4.Girotti AW (1998) Lipid hydroperoxide generation, turnover, and effector action in biological systems. J. Lipid Res 39, 1529–1542. [PubMed] [Google Scholar]
- 5.Geiger PG, Korytowski W, Lin F and Girotti AW (1997) Lipid peroxidation in photodynamically stressed mammalian cells: use of cholesterol hydroperoxides as mechanistic reporters. Free Radic. Biol. Med 23, 57–68. [DOI] [PubMed] [Google Scholar]
- 6.Girotti AW (2001) Photosensitized oxidation of membrane lipids: reaction pathways, cytotoxic effects, and cytoprotective mechanism. J. Photochem. Photobiol. B 63, 103–113. [DOI] [PubMed] [Google Scholar]
- 7.Kulig W, Olżyńska A, Jurkiewicz P, Kantola AM, Komulainen S, Manna M, Pourmousa M, Vazdar M, Cwiklik L, Rog T, Khelashvili G, Harries D, Telkki VV, Hof M, Vattulainen I.and Jungwirth P (2015) Cholesterol under oxidative stress-How lipid membranes sense oxidation as cholesterol is being replaced by oxysterols. Free Radic. Biol. Med 84, 30–41 [DOI] [PubMed] [Google Scholar]
- 8.Girotti AW and Korytowski W (2014) Generation and reactivity of lipid hydroperoxides in biological systems. In: The Chemistry of Peroxides, Vol. 3, Edited by Liebman JF and Greer A, Ch. 18, pp. 747–767. [Google Scholar]
- 9.Girotti AW and Korytowski W (2017) Cholesterol hydroperoxide generation, translocation, and reductive turnover in biological systems. Cell Biochem. Biophys 75, 413–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Smith LL, Teng JL, Kulig MJ and Hill FL (1973) Sterol metabolism XXIII. Cholesterol oxidation by radiation-induced processes. J. Org. Chem 38, 1763–1765. [DOI] [PubMed] [Google Scholar]
- 11.Teng JL, Kulig MJ, Smith LL, Kan G and van Lier JE (1973) Sterol metabolism XX. Cholesterol 7-hydroperoxide. J. Org. Chem 38, 119–123. [DOI] [PubMed] [Google Scholar]
- 12.Foote CS (1976) Photosensitized oxidation and singlet oxygen: consequences in biological systems. In: Free Radicals in Biology, Vol. 2, Edited by Pryor WA, Ch. 3, pp. 85–133. [Google Scholar]
- 13.Schenck GO, Gollnick K and Neumuller OA (1957) Zur photosensibilisieren autoxydation der steroide. Darstellung von steroid-hydroperoxyden mittels phototoxischer photosensibilisatoren. Justus Liebigs. Ann. Chem 603, 46–59. [Google Scholar]
- 14.Kulig MJ and Smith LL (1973) Sterol metabolism XXV. Cholesterol oxidation by singlet molecular oxygen. J. Org. Chem 38, 3639–3642. [DOI] [PubMed] [Google Scholar]
- 15.Suwa K, Kimura T and Schaap AP (1977) Reactivity of singlet molecular oxygen with cholesterol in a phospholipid membrane matrix. A model for oxidative damage on membranes. Biophys. Res. Commun 75, 785–792. [DOI] [PubMed] [Google Scholar]
- 16.Korytowski W and Girotti AW (1999) Singlet oxygen adducts of cholesterol: photogeneration and reductive turnover in membrane systems. Photochem. Photobiol 70, 484–489. [PubMed] [Google Scholar]
- 17.Geiger PG, Korytowski W and Girotti AW (1995) Photodynamically generated 3-β-hydroxy-5α-cholest-6-ene-5-hydroperoxide: toxic reactivity in membranes and susceptibility to enzymatic detoxification. Photochem Photobiol. 62, 580–587. [DOI] [PubMed] [Google Scholar]
- 18.Wilcox AL and Marnett LJ (1993). Polyunsaturated fatty acid alkoxyl radicals exist as carbon-centered epoxyallylic radicals: a key step in hydroperoxide-amplified lipid peroxidation. Chem. Res. Toxicol 6, 413–416. [DOI] [PubMed] [Google Scholar]
- 19.Korytowski W, Schmitt JC and Girotti AW (2010) Surprising inability of singlet oxygen-generated 6-hydroperoxycholesterol to induce damaging free radical lipid peroxidation in cell membranes. Photochem. Photobiol 86, 747–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ursini F and Bindoli A (1984) The role of selenium peroxidases in the protection against oxidative damage of membranes. Chem. Phys. Lipids 44, 255–276. [DOI] [PubMed] [Google Scholar]
- 21.Ursini F, Maiorino M and Sevanian A (1991) Membrane hydroperoxides. In: Oxidative Stress: Oxidants and Antioxidants, Edited by Sies H, Academic Press, New York, pp. 319–336. [Google Scholar]
- 22.Grossman A and Wendel A (1983) Non-reactivity of the selenoenzyme glutathione peroxidase with enzymatically hydroperoxidized phospholipids. Eur. J. Biochem 135, 549–552. [DOI] [PubMed] [Google Scholar]
- 23.Van Kuijk EJGM, Sevanian A, Hendelman GJ and Dratz EA (1987) A new role for phospholipase A2: protection of membranes from lipid peroxidation damage. Trends Biochem. Sci 12, 31–34. [Google Scholar]
- 24.Thomas JP, Maiorino M, Ursini F and Girotti AW (1990) Protective action of phospholipid hydroperoxide glutathione peroxidase against membrane-damaging lipid peroxidation. J. Biol. Chem 265, 454–461. [PubMed] [Google Scholar]
- 25.Thomas JP, Geiger PG, Maiorino M, Ursini F and Girotti AW (1990) Enzymatic reduction of phospholipid and cholesterol hydroperoxides in artificial bilayers and lipoproteins. Biochim. Biophys. Acta 1045, 252–260. [DOI] [PubMed] [Google Scholar]
- 26.Korytowski W, Geiger PG and Girotti AW (1996) Enzymatic reducibility in relation to cytotoxicity for various cholesterol hydroperoxides. Biochemistry 35, 8670–8679. [DOI] [PubMed] [Google Scholar]
- 27.Girotti AW and Korytowski W (2000) Cholesterol as a singlet oxygen detector in biological systems. Methods Enzymol. 319, 85–100. [DOI] [PubMed] [Google Scholar]
- 28.Korytowski W, Geiger PG and Girotti AW (1995) High-performance liquid chromatography with mercury cathode electrochemical detection: application to lipid hydroperoxide analysis. J. Chromatogr. B 670, 189–197. [DOI] [PubMed] [Google Scholar]
- 29.Korytowski W, Geiger PG and Girotti AW (1999) Lipid hydroperoxide analysis by high-performance liquid chromatography with mercury cathode electrochemical detection. Methods Enzymol. 300, 23–33. [DOI] [PubMed] [Google Scholar]
- 30.Howard JA and Ingold KV (1968) The self-reaction of sec-butylhydroperoxide radicals: confirmation of the Russell mechanism. J. Am. Chem. Soc 90, 1056–1058. [Google Scholar]
- 31.Uemi M, Ronsein GE, Prado FM, Motta FD, Miyamoto S, Medeiros MH and Di Mascio P (2011) Cholesterol hydroperoxides generate singlet molecular oxygen [O(2) ((1)Δ(g))]: near-IR emission, (18)O-labeled hydroperoxides, and mass spectrometry. Chem. Res. Toxicol 24, 887–895. [DOI] [PubMed] [Google Scholar]
- 32.Korytowski W, Wrona M and Girotti AW (1999) Radiolabeled cholesterol as a reporter for assessing one-electron turnover of lipid hydroperoxides. Anal. Biochem 270, 123–132. [DOI] [PubMed] [Google Scholar]
- 33.Girotti AW and Korytowski W (2016) Cholesterol as a natural probe for free radical-mediated lipid peroxidation in biological membranes and lipoproteins. J. Chromatogr. B 1019, 202–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Iuliano L (2011) Pathways of cholesterol oxidation via non-enzymatic mechanisms. Chem. Phys. Lipids 164, 457–468. [DOI] [PubMed] [Google Scholar]
- 35.Wagner BA, Buettner GR and Burns CP (1994) Free radical-mediated lipid peroxidation in cells: oxidizability is a function of cell lipid bis-allylic hydrogen content. Biochemistry 33, 4449–4453. [DOI] [PubMed] [Google Scholar]
- 36. Ritov VB, Banni S, Yalowich JC, Day BW, Claycamp H, Corongiu FP and Kagan VE (1996) Non-random peroxidation of different classes of membrane phospholipids in live cells detected by metabolically integrated cis-parinaric acid. Biochim. Biophys. Acta 1238, 127–140. [DOI] [PubMed] [Google Scholar]
- 37.Vila A, Korytowski W and Girotti AW (2000) Dissemination of peroxidative stress via intermembrane transfer of lipid hydroperoxides: model studies with cholesterol hydroperoxides. Arch. Biochem. Biophys 380, 208–218. [DOI] [PubMed] [Google Scholar]
- 38.Vila A, Korytowski W and Girotti AW (2001) Spontaneous intermembrane transfer of various cholesterol-derived hydroperoxide species: kinetic studies with model membranes and cells. Biochemistry 40, 14715–14726. [DOI] [PubMed] [Google Scholar]
- 39.Vila A, Korytowski W and Girotti AW (2002) Spontaneous transfer of phospholipid and cholesterol hydroperoxides between cell membranes and low density lipoprotein: assessment of reaction kinetics and prooxidant effects. Biochemistry 41, 13705–13716. [DOI] [PubMed] [Google Scholar]
- 40.Girotti AW (2008) Translocation as a means of disseminating lipid hydroperoxide-induced oxidative damage and effector action. Free Radic. Biol. Med 44, 956–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wirtz KWA (1991. Photpholipid transfer proteins. Ann. Rev. Biochem 60, 73–99. [DOI] [PubMed] [Google Scholar]
- 42.Noland BJ, Arebalo RE, Hansbury E and Scallen TJ (1980) Purification and properties of sterol carrier protein-2. J Biol Chem. 255, 4282–4289. [PubMed] [Google Scholar]
- 43.Gallegos AM, Atshaves BP, Storey SM, Starodub O, Petrescu AD, Huang H, McIntosh AL, Martin GG, Chao H, Kier AB and Schroeder F (2001) Gene structure, intracellular localization, and functional roles of sterol carrier protein-2. Prog Lipid Res. 40, 498–563. [DOI] [PubMed] [Google Scholar]
- 44.Vila A, Levchenko VV, Korytowski W and Girotti AW (2004) Sterol carrier protein-2-facilitated intermembrane transfer of cholesterol- and phospholipid-derived hydroperoxides. Biochemistry 43, 12592–12605. [DOI] [PubMed] [Google Scholar]
- 45.Kriska T, Levchenko VV, Korytowski W, Atshaves BP, Schroeder F and Girotti AW (2006) Intracellular dissemination of peroxidative stress: internalization, transport, and lethal targeting of a cholesterol hydroperoxide species by sterol carrier protein-2-overexpressing hepatoma cells. J. Biol. Chem 281, 23643–23651. [DOI] [PubMed] [Google Scholar]
- 46.Korytowski W, Rodriguez-Agudo D, Pilat A and Girotti AW (2010) StarD4-mediated translocation of 7-hydroperoxycholesterol to isolated mitochondria: deleterious effects and implications for steroidogenesis under oxidative stress conditions. Biochem. Biophys. Res. Commun 392, 58–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Korytowski W, Pilat A, Schmitt JC and Girotti AW (2013) Deleterious cholesterol hydroperoxide trafficking in steroidogenic acute regulatory (StAR) protein-expressing MA-10 Leydig cells: implications for oxidative stress-impaired steroidogenesis. J. Biol. Chem 288, 11509–11519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Korytowski W, Wawak K, Pabisz P, Schmitt JC and Girotti AW. (2014) Macrophage mitochondrial damage from StAR transport of 7-hydroperoxycholesterol: implications for oxidative stress-impaired reverse cholesterol transport. FEBS Lett. 588, 65–70. [DOI] [PubMed] [Google Scholar]
- 49.Korytowski W, Wawak K, Pabisz P, Schmitt JC, Chadwick AC, Sahoo D, Girotti AW. (2015) Impairment of macrophage cholesterol efflux by cholesterol hydroperoxide trafficking: implications for atherogenesis under oxidative stress. Arterioscler Thromb Vasc Biol. 35, 2104–2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rone MB, Fan J and Papadopoulos V (2009) Cholesterol transport in steroid biosynthesis: role of protein-protein interactions and implications in disease states. Biochim. Biophys. Acta 1791, 646–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cuchel M and Rader DJ (2006) Macrophage reverse cholesterol transport: key to the regression of atherosclerosis? Circulation. 113, 2548–2555. [DOI] [PubMed] [Google Scholar]
- 52.Tsujishita Y, Hurley JH. (2000) Structure and lipid transport mechanism of a StAR-related domain. Nat Struct Biol 7, 408–414. [DOI] [PubMed] [Google Scholar]
- 53.Soffientini U and Graham A (2016) Intracellular cholesterol transport proteins: roles in health and disease. Clin. Sci 130, 1843–1859. [DOI] [PubMed] [Google Scholar]
- 54.Graham A (2015) Mitochondrial regulation of macrophage cholesterol homeostasis. Free Radic. Biol. Med 89, 982–992. [DOI] [PubMed] [Google Scholar]
- 55.Stone JR and Yang S (2006) Hydrogen peroxide: a signaling messenger. Antioxid Redox Signal. 8, 243–270. [DOI] [PubMed] [Google Scholar]
- 56.Veal EA, Day AM and Morgan BA (2007) Hydrogen peroxide sensing and signaling. Mol. Cell 26, 1–14. [DOI] [PubMed] [Google Scholar]
- 57.Paulsen CE and Carroll KS (2010) Orchestrating redox signaling networks through regulatory cysteine switches. ACS Chem. Biol 5, 47–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tyurina YY, Tyurin VA, Zhao Q, Djukic M, Quinn PJ, Pitt BR and Kagan VE (2004) Oxidation of phosphatidylserine: a mechanism for plasma membrane phospholipid scrambling during apoptosis? Biochem. Biophys. Res. Commun 324, 1059–1064. [DOI] [PubMed] [Google Scholar]
- 59.Korytowski W, Basova LV, Pilat A, Kernstock RM and Girotti AW. (2011) Permeabilization of the mitochondrial outer membrane by Bax/truncated Bid (tBid) proteins as sensitized by cardiolipin hydroperoxide translocation: mechanistic implications for the intrinsic pathway of oxidative apoptosis. J Biol Chem. 286, 26334–26343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Myers MP, Stolarov JP, Eng C, Li J, Wang SI, Wigler MH, Parsons R and Tonks NK (1997) P-TEN, the tumor suppressor from human chromosome 10q23, is a dual-specificity phosphatase. Proc Natl Acad Sci U S A. 94, 9052–9057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Verrastro I, Tween-Jensen K, Woscholski R, Spickett CM and Pitt AR (2016) Reversible oxidation of phosphatase and tensin homolog (PTEN) alters its interactions with signaling and regulatory proteins. Free Radic. Biol. Med 90, 24–34. [DOI] [PubMed] [Google Scholar]
- 62.Lee SR, Yang KS, Kwon J, Lee C, Jeong W and Rhee SG (2002) Reversible inactivation of the tumor suppressor PTEN by H2O2. J Biol Chem. 277, 20336–20342. [DOI] [PubMed] [Google Scholar]