

Abstract
Background
Developments in high‐throughput genomic technologies have led to improved understanding of the molecular underpinnings of esophageal squamous cell carcinoma (ESCC). However, there is currently no model that combines the clinical features and gene expression signatures to predict outcomes.
Methods
We obtained data from the GSE53625 database of Chinese ESCC patients who had undergone surgical treatment. The R packages, Limma and WGCNA, were used to identify and construct a co‐expression network of differentially expressed genes, respectively. The Cox regression model was used, and a nomogram prediction model was constructed.
Results
A total of 3654 differentially expressed genes were identified. Bioinformatics enrichment analysis was conducted. Multivariate analysis of the clinical cohort revealed that age and adjuvant therapy were independent factors for survival, and these were entered into the clinical nomogram. After integrating the gene expression profiles, we identified a “2‐gene score” associated with overall survival. The combinational model is composed of clinical data and gene expression profiles. The C‐index of the combined nomogram for predicting survival was statistically higher than the clinical nomogram. The calibration curve revealed that the combined nomogram and actual observation showed better prediction accuracy than the clinical nomogram alone.
Conclusions
The integration of gene expression signatures and clinical variables produced a predictive model for ESCC that performed better than those based exclusively on clinical variables. This approach may provide a novel prediction model for ESCC patients after surgery.
Keywords: ESCC, nomogram, prediction model, prognosis
Introduction
Esophageal cancer is the sixth leading cause of death globally; the two major subtypes are esophageal squamous cell carcinoma (ESCC) and esophageal adenocarcinoma.1, 2 According to epidemiological and biological analysis, ESCC accounts for almost 90% of esophageal cancer cases worldwide, and is prevalent in East Asia, East Africa, and South America. Esophageal adenocarcinoma is more common in the Americas, Europe, and Australia.2, 3 The primary treatment for patients with esophageal cancer includes chemotherapy, chemoradiotherapy, and/or surgical resection.4 Although the incidence of esophageal cancer is declining in most parts of the world, the five‐year survival rate remains < 20%.5, 6, 7 One of the major treatment challenges is the lack of accurate prediction of patient survival, which may lead to the inappropriate treatment prescription.
The gold standard for prognostication in oncology remains the tumor node metastasis (TNM) staging system, which states that solid tumors first spread from the primary site to the lymphatic system and then to distant organs.8 However, the TNM system has some limitations when used in a clinical setting.9 TNM staging can only incorporate tumors, nodes, or metastasis as categorical variables, not continuous variables, which complicates the determination of individual patient prognosis. The TNM system also cannot incorporate other variables that govern prognosis, such as genome or transcriptome differences. Patients classified in the same stage may have variable outcomes. Thus, the development of a more advanced method to predict prognosis based on patient and disease characteristics is necessary.
With the development of high throughput sequencing, it is now possible to screen the genomic, epigenetic, or proteomic characteristics of esophageal cancer, which leads to a better understanding of esophageal cancer biology to improve patient care.10, 11 Jang et al. developed a robust prediction model for recurrence based on an analysis of the expression profile data of small non‐coding RNAs (sncRNAs) from 108 fresh frozen ESCC specimens. They identified that the expression of three different sncRNAs was associated with recurrence‐free survival.12 Qin et al. sequenced 10 whole‐genome and 57 whole‐exome matched tumor‐normal ESCC sample pairs, and found that the amplification of somatic copy number alterations (SCNAs) in several miRNA genes was significantly associated with survival.13 In recent years, long non‐coding RNA (lncRNA), which is a type of RNA molecule > 200 nucleotides with a lack of protein‐coding capacity, has emerged as a new star in the field of oncology.14, 15, 16 Wu et al. identified that lincRNA‐uc002yug.2 may serve as a predictor for esophageal cancer and prognosis.17 However, a prediction model that combines clinical data and gene expression profiles associated with overall or recurrence‐free survival is lacking.
Nowadays, nomograms are widely used as prognostic devices in oncology and medicine, which integrate various prognostic and determinant variables for individual patients.18, 19, 20 In this manuscript, we used clinical and gene expression profiles from the Gene Expression Omnibus (GEO, GSE53625) to analyze the different protein‐coding and long non‐coding genes, respectively. Using the coefficient and regression formula of the multivariate Cox model, we identified several clinical variables and “2‐gene score” (lncRNA) associated with survival duration. Based on the clinical variables and the “2‐gene score,” we constructed a nomogram to predict prognosis. The accuracy of this prediction model was higher than in a model based on clinical variables alone. This model incorporated molecular and clinical/pathological prognostic markers and may refine prognosis assessment.
Methods
Data sources and bioinformatics
The GSE53625gene expression profiles were obtained from GEO (https://www.ncbi.nlm.nih.gov), and included 179 paired tumor‐normal matched samples from ESCC patients treated by resection. The platform of this microarray GPL18109, which incorporates lncRNA and messenger RNA (mRNA) probes, is Agilent‐038314 CBC Homo sapiens lncRNA + mRNA microarray V2.0 (Agilent Technologies, Santa Clara, CA, USA). We re‐annotated this platform mainly focusing on the lncRNA probes according to the database, including ENCODE, CombinedLit, EvoFold, H‐InvDB, imsRNA, hox‐HOX, int‐HOX, nc‐HOX, lncRNAdb, XLOC, NRED, and UCSC.
The Limma package in R software (R Foundation for Statistical Computing, Vienna, Austria) was used to show the different mRNA and lncRNA gene expression between normal and tumor specimens. The list of different transcriptional genes was submitted to the Database for Annotation, Visualization and Integrated Discovery (DAVID) Bioinformatics Resources 6.8 (http://david.abcc.ncifcrf.gov) for Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway and Gene Ontology (GO) biological progress enrichment analysis. The network of the different genes was constructed based on the R package WGCNA (R Foundation) and Cytoscape software (National Institute of General Medical Sciences, Bethesda, MD, USA). The pheatmap package in R software (R Foundation) was used to draw the heatmap, while a receiver operating characteristic (ROC) curve was constructed based on the ROCR package (https://CRAN.R-project.org/package=ROCR). The nomogram was built using the rms package of R statistical software (http://www.R-project.org/).
Statistical analysis
Statistical analysis was performed using SPSS version 20.0 (IBM Corp., Armonk, NY, USA) and P < 0.05 was considered statistically significant. A log‐rank test and the Kaplan–Meier method were used to assess survival. Univariate and multivariate analyses were performed using the Cox model. In the clinical variable Cox model, the following formula was used: variable_1 = 0.363 (Age variable) + 0.564 (TNM stage variable) – 0.582 (adjuvant therapy variable). In the clinical and “2‐gene score” Cox model, the following formula was used: variable_2 = 0.358 (age variable) + 0.605 (TNM stage variable) – 0.605 (adjuvant therapy variable) + 0.723 (RP11‐357H14.20 variable) + 0.295 (RP11‐768G7.2 variable). Based on variable‐1 and variable‐2 scores, patients were assigned into low and high‐risk groups, respectively.
Results
Clinicopathologic characteristics of patients with esophageal squamous cell carcinoma (ESCC)
A total of 179 patients with ESCC were included in the study. The clinical data and gene expression profiles associated with these patients were obtained from the GEO datasets in GSE53625. The baseline characteristics of these patients are listed in Table 1. Approximately half of the patients were aged over 60 years and over 80% were male. More than half of the patients had a history of alcohol consumption and smoking. The tumor was located in the middle esophagus in over half of the patients. The tumor grade was moderate in 54.7% of patients. The percentage of patients in TNM stages I, II, and III were 5.59%, 43.0%, and 51.4%, respectively. A few of the patients suffered from arrhythmia pneumonia and anastomotic leaks. Based on research and the clinical data, patients can benefit from adjuvant therapy.
Table 1.
Clinicopathologic characteristics of ESCC patients
| Characteristics | No. of patients | % |
|---|---|---|
| Age, years | ||
| ≥ 60 | 88 | 49.2 |
| < 60 | 91 | 50.8 |
| Gender | ||
| Male | 146 | 81.6 |
| Female | 33 | 18.4 |
| Tobacco use | ||
| Yes | 114 | 63.7 |
| No | 65 | 36.3 |
| Alcohol use | ||
| Yes | 106 | 59.2 |
| No | 73 | 40.8 |
| Tumor location | ||
| Upper | 20 | 11.2 |
| Middle | 97 | 54.2 |
| Lower | 62 | 34.6 |
| Tumor grade | ||
| Well | 32 | 17.9 |
| Moderately | 98 | 54.7 |
| Poorly | 49 | 27.4 |
| Invasion of adjacent structure | ||
| Yes | 31 | 17.3 |
| No | 148 | 82.7 |
| Lymphatic metastasis | ||
| Yes | 96 | 53.6 |
| No | 83 | 46.4 |
| TNM stage | ||
| I | 10 | 5.59 |
| II | 77 | 43.0 |
| III | 92 | 51.4 |
| Arrhythmia | ||
| Yes | 43 | 24.0 |
| No | 136 | 76.0 |
| Pneumonia | ||
| Yes | 35 | 19.6 |
| No | 164 | 80.4 |
| Anastomotic leak | ||
| Yes | 12 | 6.70 |
| No | 167 | 93.3 |
| Adjuvant therapy | ||
| Yes | 108 | 60.3 |
| No | 45 | 25.1 |
| Unknown | 26 | 14.6 |
ESCC, esophageal squamous cell carcinoma; TNM, tumor node metastasis.
Comprehensive analysis of the differentially expressed protein‐coding genes
To identify potential esophageal cancer‐related genes, we used the Limma package of R software to analyze the different transcriptional genes, based on GSE53625 array data. Fold changes > 2 and adjusted P values of < 0.05 were set to filter different genes. A total of 3654 different protein‐coding and long non‐coding genes were identified (Fig 1a). Among these genes, 3205 coding genes were significantly expressed (Fig 1b), of which 1311 were upregulated in tumors, while 1894 were downregulated (Appendix S1 and S2). We used GO and KEGG pathway analysis (DAVID Bioinformatics Resources 6.8) to explore the main function of differentially expressed protein‐coding genes.21 As shown in Figure 1c, the process related to epidermis development, epithelial cell differentiation, ectoderm development, and epithelium development ranked highest in the enrichment analysis of the GO Biological Process. Extracellular matrix (ECM)‐receptor interaction, focal adhesion, and cell cycle achieved the highest scores in KEGG pathway enrichment analysis (Fig 1d). These results indicated that epithelial cell differentiation, ECM‐receptor interaction, focal adhesion, and cell cycle may play important roles in the progression of ESCC, which is consistent with previous reports.10, 22, 23
Figure 1.
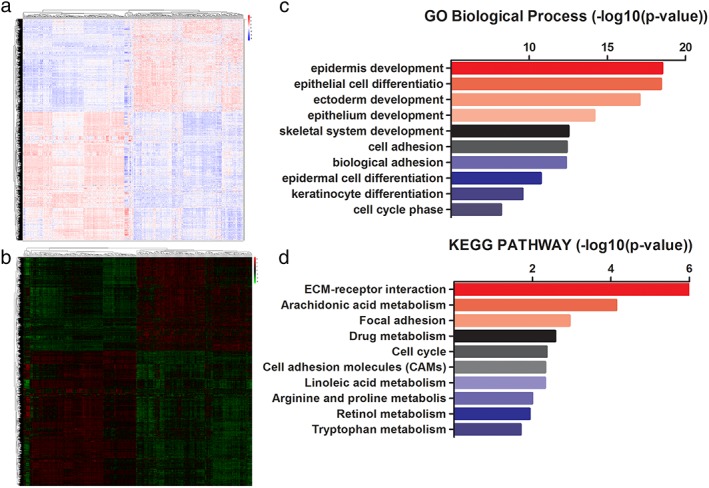
Systematic analysis of differential transcribed genes and bioinformatics analysis of the differentially expressed coding genes. (a) Use of the Limma package (R software) to screen and analyze the differentially expressed genes of paired samples, including coding and non‐coding. (b) The heatmap reveals the significantly differentially expressed coding genes between tumor and normal specimens. (c,d) Bioinformatic analysis of differentially expressed coding genes according to Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway.
Comprehensive analysis of the differential non‐coding genes
Based on the array data, we also identified 449 differentially expressed non‐coding genes (Fig 2a). When comparing the expression profiles of the tumor specimens and matched normal samples, 224 non‐coding genes were upregulated and 225 were downregulated (Appendix S3 and S4). According to the non‐coding RNA database classification,24 we observed that over 60% of differential non‐coding RNAs were long intergenic non‐coding RNAs (lincRNAs) (Fig 2b). The antisense, pseudogene, and processed transcript lincRNAs accounted for 17.1%, 10.2%, and 9.80%, respectively. Increasing evidence shows that lncRNAs play vital roles in cancer processes, which emphasizes the need for investigation of lncRNA function. Methods based on the construction of a coding‐non‐coding co‐expression network have been widely used to predict the probable functions of lncRNAs.25, 26 According to this theoretical coding‐non‐coding co‐expression network, we constructed a network between the differential protein‐coding genes and the differential non‐coding genes to facilitate the prediction of lncRNAs (Fig 2c); 386 coding genes and 79 lncRNAs were implicated in this prediction model (Appendix S5 and S6). Using highly related coding genes based on GO Biological Process enrichment analysis, we observed that lncRNAs may play important roles in the progresses associated with peptide cross‐link, keratinization, and nucleosome assembly (Fig 2d).
Figure 2.
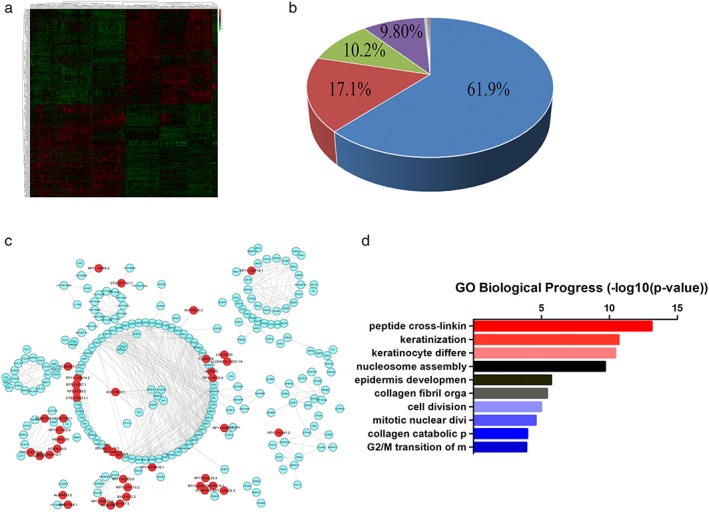
Systematic analysis of differentially expressed non‐coding genes and the prediction of function. (a) The heatmap comprises significantly differentially expressed non‐coding genes. (b) The classification of differentially expressed long non‐coding RNAs (lncRNAs). (c) The network between the different protein‐coding genes and non‐coding genes based on the WGCNA package. (d) The predictive function of lncRNA according to correlation analysis. ( ) long intergenic non‐coding RNA (lincRNA), (
) long intergenic non‐coding RNA (lincRNA), ( ) antisense, (
) antisense, ( ), pseudogene, (
), pseudogene, ( ) Processed transcript, (
) Processed transcript, ( ) misc RNA, (
) misc RNA, ( ) sense_intronic, (
) sense_intronic, ( ) to be experimentally confirmed (TEC), and (
) to be experimentally confirmed (TEC), and ( ) unknown.
) unknown.
Univariate and multivariate analyses of clinical and biological variables based on the Cox model
We first constructed a logistic regression model based on clinicopathologic characteristics. Clinical features including age, gender, tobacco use, alcohol use, tumor location, tumor grade, invasion of adjacent structures, lymphatic metastasis, TNM stage, arrhythmia, pneumonia, anastomotic leak, and adjuvant therapy were entered into univariate analysis (Table 2). We observed that age, tumor grade, invasion of adjacent structures, lymphatic metastasis, TNM stage, and adjuvant therapy were prognostic factors (all P < 0.05). TNM stage had a high correlation with adjacent structures and lymphatic metastasis, thus invasion of adjacent structures and lymphatic metastasis were entered into multivariate analysis. As shown in Figure 3a (left; Table 2), age (hazard ratio [HR] 1.575, 95% CI 1.006–2.467; P = 0.047) and adjuvant therapy (HR 0.520, 95% CI 0.289–0.934; P = 0.029) were independent prognostic factors. Because TNM stage is considered the gold standard for prognostication in clinical practice,8 TNM stage was therefore included in all subsequent analyses. We constructed a Cox model using the new formula: variable_1 = 0.363 (age variable) + 0.564 (TNM stage variable) – 0.582 (adjuvant therapy variable). As shown in Figure 3b (left), patients in the low‐risk group survived longer compared to those in the high‐risk group (P < 0.0001). We also estimated the specificity and sensitivity of variable_1 using an ROC curve. The area under the ROC (AUC) of this new variable was 0.717.
Table 2.
Univariate and multivariable analyses based on the clinical Cox model
| Parameters | Univariate analysis | Multivariable analysis | ||||
|---|---|---|---|---|---|---|
| HR | 95% CI | P | HR | 95% CI | P | |
| Age (≥ 60) | 1.680 | 1.146–2.461 | 0.008 | 1.575 | 1.006–2.467 | 0.047* |
| Gender (female) | 1.277 | 0.789–2.044 | 0.307 | — | — | — |
| Tobacco use | 1.334 | 0.905–1.967 | 0.145 | — | — | — |
| Alcohol use | 1.158 | 0.788–1.700 | 0.456 | — | — | — |
| Tumor location | 0.257 | — | — | — | ||
| Tumor location (middle vs. upper) | 1.669 | 0.905–3.078 | 0.101 | — | — | — |
| Tumor location (lower vs. upper) | 1.135 | 0.740–1.741 | 0.561 | — | — | — |
| Tumor location (middle vs. lower) | 0.680 | 0.385–1.202 | 0.184 | — | — | — |
| Tumor grade | 0.048 | 0.829 | 0.516–1.330 | 0.436 | ||
| Tumor grade (well vs. poorly) | 0.605 | 0.338–1.082 | 0.090 | |||
| Tumor grade (moderately vs. poorly) | 0.613 | 0.401–0.939 | 0.024 | |||
| Tumor grade (moderately vs. well) | 1.014 | 0.587–1.750 | 0.961 | |||
| Invasion of adjacent structure | 1.628 | 1.017–2.605 | 0.042 | 0.852 | 0.610–1.189 | 0.346 |
| Lymphatic metastasis | 2.129 | 1.420–3.192 | 0.000 | 1.528 | 0.931–2.508 | 0.094 |
| TNM stage | 0.001 | — | — | — | ||
| TNM stage (I vs. III) | 0.276 | 0.087–0.879 | 0.029 | — | — | — |
| TNM stage (II vs. III) | 0.492 | 0.327–0.739 | 0.001 | — | — | — |
| TNM stage (II vs. I) | 1.782 | 0.549–5.788 | 0.336 | — | — | — |
| Arrhythmia | 0.893 | 0.580–1.375 | 0.607 | — | — | — |
| Pneumonia | 0.702 | 0.354–1.390 | 0.310 | — | — | — |
| Anastomotic leak | 0.770 | 0.357–1.658 | 0.504 | — | — | — |
| Adjuvant therapy | 0.442 | 0.256–0.762 | 0.003 | 0.520 | 0.289–0.934 | 0.029* |
Indicated P < 0.05. CI, confidence interval; HR, hazard ratio; TNM, tumor node metasta.
Figure 3.
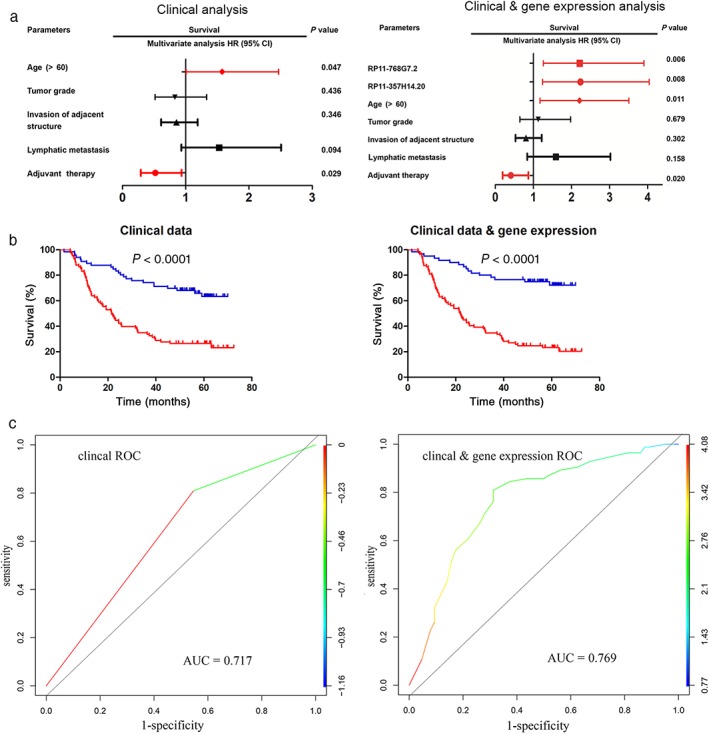
Two logistic regression modeling approaches to predict esophageal squamous cell carcinoma (ESCC) survival after surgery. (a) Multivariate analysis of the overall survival of ESCC patients based on the different Cox models, one based exclusively on the clinical variables, the other based on the integration of clinical variables and a 2‐gene score. (b) Kalpan–Meier survival curves of the two logistic regression models. (c) Receiver operating characteristic (ROC) curves of the two models are presented, and reflect the specificity and sensitivity of the two different comprehensive variables. ( ) Low‐risk and (
) Low‐risk and ( ) High‐risk; (
) High‐risk; ( ) Low‐risk and (
) Low‐risk and ( ) High‐risk. AUC, area under the ROC; CI, confidence interval; HR, hazard ratio.
) High‐risk. AUC, area under the ROC; CI, confidence interval; HR, hazard ratio.
We also constructed a novel Cox model based on the clinical features and gene expression profiles associated with patient survival. An increasing amount of research has indicated that lncRNAs are closely correlated with prognosis. We estimated lncRNAs as the candidate genes in univariable analyses. As shown in Table 3, we distinguished 31 lncRNAs as prognostic factors. Incorporating these candidate lncRNAs into the variables of clinicopathologic characteristics, we observed four independent prognostic factors (Fig 3a, right): age (HR 2.029, 95% CI 1.173–3.508; P = 0.011), adjuvant therapy (HR 0.408, 95% CI 0.192–0.868; P = 0.020), RP11‐357H14.20 (HR 2235, 95% CI 1.237–4.038; P = 0.008), and RP11‐768G7.2 (HR 2.215, 95% CI 1.258–3.903; P = 0.006) (Table 4). Moreover we calculated a new variable (variable_2) according to the novel Cox model: variable_2 = 0.358 (age variable) + 0.605 (TNM stage variable) – 0.605 (adjuvant therapy variable) + 0.723 (RP11‐357H14.20 variable) + 0.295 (RP11‐768G7.2 variable). We also estimated the predictive ability of variable_2. As shown in Figure 3b (right), patients in the high‐risk cohort had poorer long‐term prognosis. The AUC of ROC allowed us to estimate the specificity and sensitivity of variable_2. As shown in Figure 3c, the AUC of variable_2 was 0.769, higher than that of variable_1, indicating that the combination of clinical features and gene expression patterns is a more accurate predictor than clinicopathologic characteristics alone.
Table 3.
Univariate analysis of gene expression profiles correlated with overall survival of ESCC patients
| Number | Ensemble name | logFC | adj.P.Val | ENSG | Type | Univariate analysis | ||
|---|---|---|---|---|---|---|---|---|
| HR | 95% CI | P | ||||||
| 1 | CASC2 | −1.05 | 1.23E‐25 | ENSG00000177640 | Antisense | 1.506 | 1.026–2.209 | 0.036 |
| 2 | FLJ40288 | −2.98 | 6.24E‐82 | ENSG00000183470 | lincRNA | 0.649 | 0.441–0.954 | 0.028 |
| 3 | KB‐1183D5.11 | 1.09 | 3.19E‐11 | ENSG00000215498 | Processed_transcript | 0.676 | 0.461–0.992 | 0.046 |
| 4 | RP11‐357H14.2 | 1.85 | 1.88E‐34 | ENSG00000233283 | Processed_transcript | 1.703 | 1.159–2.503 | 0.007 |
| 5 | RP11‐438N16.1 | 2.19 | 6.6E‐27 | ENSG00000249550 | lincRNA | 0.657 | 0.447–0.965 | 0.032 |
| 6 | RP11‐129M6.1 | 1.38 | 2.22E‐14 | ENSG00000251363 | lincRNA | 0.677 | 0.461–0.996 | 0.047 |
| 7 | AC006296.1 | −1.73 | 9.65E‐21 | ENSG00000251412 | lincRNA | 0.644 | 0.438–0.946 | 0.025 |
| 8 | AC007880.1 | −2.22 | 4.47E‐26 | ENSG00000234572 | lincRNA | 0.652 | 0.444–0.957 | 0.029 |
| 9 | AC092168.4 | −1.21 | 6.69E‐32 | ENSG00000228488 | lincRNA | 0.586 | 0.398–0.865 | 0.007 |
| 10 | AC093850.2 | 4.96 | 6.63E‐96 | ENSG00000230838 | lincRNA | 1.471 | 1.002–2.159 | 0.049 |
| 11 | AF003626.1 | −1.18 | 2.87E‐36 | ENSG00000230153 | lincRNA | 0.629 | 0.428–0.925 | 0.018 |
| 12 | AP000344.3 | −2.11 | 1.18E‐32 | ENSG00000234928 | lincRNA | 0.654 | 0.445–0.961 | 0.031 |
| 13 | AP000473.6 | 1.23 | 3.22E‐16 | ENSG00000237735 | lincRNA | 0.605 | 0.410–0.892 | 0.011 |
| 14 | CTD‐2382E5.1 | 1.13 | 8.53E‐12 | ENSG00000246740 | Antisense | 0.644 | 0.439–0.946 | 0.025 |
| 15 | FRMPD2P1 | −1.93 | 3.84E‐33 | ENSG00000150175 | Pseudogene | 0.614 | 0.418–0.904 | 0.013 |
| 16 | LINC00028 | −1.41 | 6.61E‐33 | ENSG00000233354 | lincRNA | 0.582 | 0.395–0.858 | 0.006 |
| 17 | MAMDC2‐AS1 | −1.71 | 6.45E‐54 | ENSG00000204706 | Antisense | 1.685 | 1.144–2.483 | 0.008 |
| 18 | RP11‐120J1.1 | −1.62 | 8.68E‐33 | ENSG00000225472 | Antisense | 0.635 | 0.431–0.936 | 0.022 |
| 19 | RP11‐225N10.1 | −1.58 | 3.08E‐47 | ENSG00000240063 | Antisense | 0.680 | 0.463–0.999 | 0.049 |
| 20 | RP11‐226F19.5 | 1.10 | 3.34E‐21 | ENSG00000259062 | Antisense | 1.486 | 1.013–2.181 | 0.043 |
| 21 | RP11‐242F24.1 | 1.03 | 1.75E‐46 | ENSG00000228750 | lincRNA | 1.513 | 1.028–2.226 | 0.036 |
| 22 | RP1‐12803.4 | −3.71 | 2.95E‐82 | ENSG00000230248 | lincRNA | 0.638 | 0.432–0.936 | 0.022 |
| 23 | RP11‐411K7.1 | −1.30 | 4.81E‐13 | ENSG00000236740 | Processed_transcript | 0.642 | 0.437–0.943 | 0.024 |
| 24 | RP11‐51M18.1 | 1.59 | 2.44E‐17 | ENSG00000253898 | lincRNA | 0.594 | 0.403–0.876 | 0.009 |
| 25 | RP11‐521B24.3 | 1.09 | 2.49E‐21 | ENSG00000251602 | Antisense | 1.740 | 1.181–2.564 | 0.005 |
| 26 | RP11‐526P5.2 | −1.16 | 3.68E‐12 | ENSG00000235281 | lincRNA | 0.655 | 0.446–0.963 | 0.031 |
| 27 | RP11‐71G12.1 | 1.23 | 1.1E‐10 | ENSG00000229961 | lincRNA | 0.653 | 0.445–0.960 | 0.030 |
| 28 | RP11‐768G7.2 | 1.26 | 3.51E‐31 | ENSG00000241213 | lincRNA | 1.694 | 1.150–2.495 | 0.008 |
| 29 | RP11‐89N17.4 | −1.55 | 4.54E‐41 | ENSG00000236494 | lincRNA | 0.573 | 0.389–0.844 | 0.006 |
| 30 | RP11‐726G1.1 | 1.04 | 2.23E‐19 | ENSG00000214776 | Processed_transcript | 1.497 | 1.019–2.200 | 0.040 |
| 31 | RP11‐69C17.1 | −1.97 | 1.75E‐37 | ENSG00000234962 | lincRNA | 0.674 | 0.459–0.989 | 0.044 |
CI, confidence interval; HR, hazard ratio; lincRNA, long intergenic non‐coding RNA.
Table 4.
Multivariate analysis based on the integration of clinical variables and gene expression signatures in a Cox model
| Parameters | Multivariable analysis | ||
|---|---|---|---|
| HR | 95% CI | P | |
| Age (> 60) | 2.029 | 1.173–3.508 | 0.011* |
| Tumor grade (well vs. poorly) | 1.126 | 0.642–1.976 | 0.679 |
| Invasion of adjacent structure | 0.804 | 0.531–1.217 | 0.302 |
| Lymphatic metastasis | 1.589 | 0.836–3.023 | 0.158 |
| Adjuvant therapy | 0.408 | 0.192–0.868 | 0.020* |
| CASC2 | 0.841 | 0.468‐1.510 | 0.561 |
| FLJ40288 | 0.667 | 0.365–1.219 | 0.188 |
| KB‐1183D5.11 | 0.707 | 0.386–1.292 | 0.259 |
| RP11‐357H14.20 | 2.235 | 1.237–4.038 | 0.008* |
| RP11‐438N16.1 | 0.804 | 0.468–1.384 | 0.432 |
| RP11‐129M6.1 | 0.971 | 0.545–1.730 | 0.920 |
| AC006296.1 | 0.389 | 0.117–1.293 | 0.123 |
| AC007880.1 | 2.347 | 0.749–7.358 | 0.143 |
| AC092168.4 | 1.313 | 0.654–2.636 | 0.444 |
| AC093850.2 | 1.011 | 0.545–1.877 | 0.972 |
| AF003626.1 | 0.742 | 0.386–1.425 | 0.370 |
| AP000344.3 | 1.573 | 0.769–3.218 | 0.215 |
| AP000473.6 | 0.792 | 0.457–1.374 | 0.407 |
| CTD‐2382E5.1 | 1.199 | 0.611–2.353 | 0.597 |
| FRMPD2P1 | 0.614 | 0.158–2.383 | 0.481 |
| LINC00028 | 0.784 | 0.429–1.436 | 0.431 |
| MAMDC2‐AS1 | 1.313 | 0.723–2.385 | 0.372 |
| RP11‐120J1.1 | 0.715 | 0.371–1.377 | 0.316 |
| RP11‐225N10.1 | 0.903 | 0.524–1.557 | 0.714 |
| RP11‐226F19.5 | 1.605 | 0.845–3.051 | 0.149 |
| RP11‐242F24.1 | 0.840 | 0.387–1.823 | 0.659 |
| RP1‐12803.4 | 1.055 | 0.540–2.061 | 0.875 |
| RP11‐411K7.1 | 1.021 | 0.550–1.895 | 0.947 |
| RP11‐51M18.1 | 0.827 | 0.479–1.428 | 0.496 |
| RP11‐521B24.3 | 1.167 | 0.595–2.289 | 0.653 |
| RP11‐526P5.2 | 0.733 | 0.390–1.375 | 0.333 |
| RP11‐71G12.1 | 0.819 | 0.467–1.436 | 0.485 |
| RP11‐768G7.2 | 2.215 | 1.258–3.903 | 0.006* |
| RP11‐89N17.4 | 0.598 | 0.300–1.193 | 0.144 |
| RP11‐726G1.1 | 1.698 | 0.990–2.914 | 0.055 |
| RP11‐69C17.1 | 1.646 | 0.766–3.535 | 0.201 |
Indicated P < 0.05. CI, confidence interval; HR, hazard ratio.
Construction of a novel nomogram to predict survival in ESCC patients
To further assess the predictive ability of the novel Cox model, we built a nomogram using the rms package (R statistical software, R Foundation). Figure 4a shows the prognostic nomogram integrating all of the significant independent factors for overall survival in the clinical cohort. The nomogram illustrated shows the contribution of each variable to predict tumor‐related death at three or five years. The C‐index, reflecting the predictive ability of the nomogram, was 0.639 (95% CI 0.577–0.701). The calibration plot for the probability of survival at three or five years after surgery showed moderate agreement between the predictions made by the nomogram and actual observations (Fig 4b).
Figure 4.
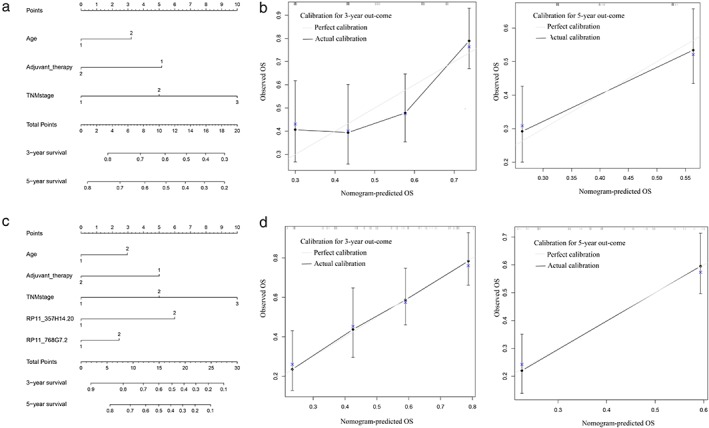
Nomograms of the two Cox models. (a,c) Two models are shown. An individual patient's value is located on each variable axis, and a line is drawn upward to determine the number of points received for each variable value. The sum of these numbers is located on the Total Points axis, and a line is drawn downward to the survival axes to determine the likelihood of three or five‐year survival. (b,d) The calibration curve for predicting patient survival at three or five years in the former and combined nomograms, respectively. Nomogram‐predicted probability of overall survival is plotted on the x‐axis; actual overall survival is plotted on the y‐axis. OS, overall survival; TNM, tumor node metastasis.
To develop a composite prognostic predictor, we assembled the 2‐genes, the independent prognostic factors, and clinical variables in the overall series of ESCC patients, including age, adjuvant therapy, TNM stage, and the 2‐gene score (Fig 4c). The C‐index of this new nomogram was 0.699 (95% CI 0.640–0.758), which was statistically higher than that of the clinical cohort (P < 0.05). The calibration plot for the probability of survival at three or five years showed greater agreement than that of the previous nomogram (Fig 4d). These results indicated that incorporating a 2‐gene score into the clinicopathologic variables improved the prognostic accuracy of survival in ESCC patients after surgery.
Discussion
Population‐based studies have shown that esophageal cancer is predominant in men aged ≥ 60 years, many of whom also have a history of heavy tobacco and alcohol use.27 China has a high‐incidence of ESCC, the most common histological subtype of esophageal cancer.5, 28 The mean ESCC male to female ratio is 3:1.4, 29, 30 Based on these epidemiological characteristics, we chose patients in the GSE53625 dataset, which represents features of ESCC.
In this study, we investigated coding and non‐coding gene expression profiles in ESCC by re‐annotating the microarray probe sets of Agilent‐038314 CBC Homo sapiens. Through differential expression, GO, and KEGG pathway enrichment analysis, we observed that epithelial cell differentiation, ECM‐receptor interaction, and cell cycle may play important roles in the development and progression of ESCC. A previous study showed that cell cycle regulators, such as CCND1, CCNE1, CDK6, or RB1, are frequently altered in ESCC via distinct mechanisms.31 Dysregulated pathways, which are of therapeutic interest in ESCC, include receptor tyrosine kinase signaling, chromatin remodeling, and embryonic pathways.32 Our observations are consistent with these results.
LncRNA, a new star in the field of oncology, also has been widely investigated in ESCC. Li et al. found that linc‐POU3F3, which is highly expressed in ESCC samples, contributes to the development of ESCC by interacting with EZH2 to promote POU3F3 methylation.33 Zhang et al. also reported that lncRNA CCAT1, which shows significantly increased expression in ESCC, could serve as a scaffold for two distinct epigenetic modifications that facilitate cell growth and migration.34 These results indicate that lncRNA may affect the regulation of epigenetics. In this study, we identified 449 non‐coding genes that were closely implicated in the nucleosome assembly bioprocess, which may provide a novel therapeutic target for ESCC.
According to the Cox model of the clinical cohort, age and adjuvant therapy are two independent prognostic factors. Advanced age as a prognostic factor for surgery outcome remains controversial. Some studies have shown that the risk of mortality after esophagectomy is strongly related to patient age and performance status, with poorer long‐term survival among elderly patients.35 However, Alibakhshi et al. reported that esophagectomy outcomes in elderly patients were not significantly different than in young patients.36 The effect of age may be related to comorbidities rather than age itself. The prognosis for ESCC patients with ≥ T2 or N+ after surgery alone is poor and the 10‐year survival rate in stage 1b after surgery is only 50%.37 Thus, adjuvant therapy is recommended, including neoadjuvant or perioperative chemotherapy, radiotherapy, or chemoradiotherapy for ≥ T2 esophageal cancer patients.38
Two logistic regression modeling approaches were used to predict outcomes after surgery, one based exclusively on clinical variables, and the other integrating prognostic gene variables with clinicopathologic characteristics. We identified a “2‐gene score,” of lncRNAs and independent prognostic factors. The combination of 2‐gene score with clinical and pathological features shows better specificity and sensitivity than the clinicopathologic parameters alone for outcome prediction.
Nomograms have been developed to predict prognosis in some cancers, and have proven more accurate than conventional staging systems, such as TNM stage. However, few studies have integrated gene expression profiling and clinical variables to predict outcomes after surgery. The predictive accuracy of the nomogram combination the “2‐gene score” and clinical features was higher than the nomogram based exclusively on clinical variables. The calibration plot of the combined nomogram for the probability of survival at three or five years showed greater agreement than that of the clinical nomogram. The 2‐gene score may more accurately reflect tumor biology than clinicopathologic parameters alone and may enhance the ability to predict outcomes in ESCC patients treated by surgery.
Some limitations of this study should be taken into consideration. The heterogeneity of the tumor samples presents difficulties in detecting the expression of two lncRNAs (RP11‐357H14.20 and RP11‐768G7). The nomogram that was constructed using this one dataset should be validated in another cohort.
In conclusion, the combined nomogram proposed in this study objectively and accurately predicted the prognosis of ESCC patients after surgery. Additional studies are required to determine whether it can be applied in a clinical setting.
Disclosure
No authors report any conflict of interest.
Supporting information
Appendix S1. Differentially expressed coding genes.
Appendix S2. Fold change of differentially expressed coding genes.
Appendix S3. Differentially expressed non‐coding genes.
Appendix S4. Fold change of differentially expressed non‐coding genes.
Appendix S5. Network of differentially expressed coding and non‐coding genes.
Appendix S6. Correlation of node in the network.
Acknowledgments
We wish to thank Han Jing for assistance with data analysis. This research was supported by the National Natural Science Foundation of China (Nos. 81472702, 81501977, 81672294, 81702892), the Foundation of Jiangsu Cancer Hospital (No. ZQ201509, ZK201601), and the Innovation Capability Development Project of Jiangsu Province (No. BM2015004).
Lin Wang, Gaochao Dong and Wenjie Xia contributed equally to this work.
Contributor Information
Lin Xu, Email: xulin_83@hotmail.com.
Feng Jiang, Email: zengnljf@hotmail.com.
References
- 1. Njei B, McCarty TR, Birk JW et al Trends in esophageal cancer survival in United States adults from 1973 to 2009: A SEER database analysis. J Gastroenterol Hepatol 2016; 31: 1141–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Arnold M, Soerjomataram I, Ferlay J, Forman D. Global incidence of oesophageal cancer by histological subtype in 2012. Gut 2015; 64: 381–7. [DOI] [PubMed] [Google Scholar]
- 3. Torre LA, Bray F, Siegel RL, Ferlay J, Lortet‐Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin 2015; 65: 87–108. [DOI] [PubMed] [Google Scholar]
- 4. Smyth EC, Lagergren J, Fitzgerald RC et al Oesophageal cancer. Nat Rev Dis Primers 2017; 3: 17048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chen W, Zheng R, Baade PD et al Cancer statistics in China, 2015. CA Cancer J Clin 2016; 66: 115–32. [DOI] [PubMed] [Google Scholar]
- 6. Gavin AT, Francisci S, Foschi R et al Oesophageal cancer survival in Europe: A EUROCARE‐4 study. Cancer Epidemiol 2012; 36: 505–12. [DOI] [PubMed] [Google Scholar]
- 7. Fitzmaurice C, Dicker D, Pain A et al The global burden of cancer 2013. JAMA Oncol 2015; 1: 505–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gospodarowicz M, Benedet L, Hutter RV et al History and international developments in cancer staging. Cancer Prev Control 1998; 2: 262–8. [PubMed] [Google Scholar]
- 9. Balachandran VP, Gonen M, Smith JJ, DeMatteo RP. Nomograms in oncology: More than meets the eye. Lancet Oncol 2015; 16: e173–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. The Cancer Genome Atlas Research Network . Integrated genomic characterization of oesophageal carcinoma. Nature 2017; 541: 169–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Song Y, Li L, Ou Y et al Identification of genomic alterations in oesophageal squamous cell cancer. Nature 2014; 509: 91–5. [DOI] [PubMed] [Google Scholar]
- 12. Guo JC, Li CQ, Wang QY et al Protein‐coding genes combined with long non‐coding RNAs predict prognosis in esophageal squamous cell carcinoma patients as a novel clinical multi‐dimensional signature. Mol Biosyst 2016; 12: 3467–77. [DOI] [PubMed] [Google Scholar]
- 13. Qin HD, Liao XY, Chen YB et al Genomic characterization of esophageal squamous cell carcinoma reveals critical genes underlying tumorigenesis and poor prognosis. Am J Hum Genet 2016; 98: 709–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Schmitt AM, Chang HY. Long noncoding RNAs in cancer pathways. Cancer Cell 2016; 29: 452–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Huarte M. The emerging role of lncRNAs in cancer. Nat Med 2015; 21: 1253–61. [DOI] [PubMed] [Google Scholar]
- 16. Prensner JR, Chinnaiyan AM. The emergence of lncRNAs in cancer biology. Cancer Discov 2011; 1: 391–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wu H, Zheng J, Deng J et al LincRNA‐uc002yug.2 involves in alternative splicing of RUNX1 and serves as a predictor for esophageal cancer and prognosis. Oncogene 2015; 34: 4723–34. [DOI] [PubMed] [Google Scholar]
- 18. Gandaglia G, Fossati N, Zaffuto E et al Development and internal validation of a novel model to identify the candidates for extended pelvic lymph node dissection in prostate cancer. Eur Urol 2017; 72: 632–40. [DOI] [PubMed] [Google Scholar]
- 19. Necchi A, Sonpavde G, Lo Vullo S et al Nomogram‐based prediction of overall survival in patients with metastatic Urothelial carcinoma receiving first‐line platinum‐based chemotherapy: Retrospective International Study of Invasive/Advanced Cancer of the Urothelium (RISC). Eur Urol 2017; 71: 281–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wang Y, Li J, Xi Y et al Prognostic nomogram for intrahepatic cholangiocarcinoma after partial hepatectomy. J Clin Oncol 2013; 31: 1188–95. [DOI] [PubMed] [Google Scholar]
- 21. Kanehisa M, Goto S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res 2000; 28: 27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Miyazaki T, Kato H, Nakajima M et al FAK overexpression is correlated with tumour invasiveness and lymph node metastasis in oesophageal squamous cell carcinoma. Br J Cancer 2003; 89: 140–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Li X, Jiang C, Wu X et al A systems biology approach to study the biology characteristics of esophageal squamous cell carcinoma by integrating microRNA and messenger RNA expression profiling. Cell Biochem Biophys 2014; 70: 1369–76. [DOI] [PubMed] [Google Scholar]
- 24. Jarroux J, Morillon A, Pinskaya M. History, discovery, and classification of lncRNAs. Adv Exp Med Biol 2017; 1008: 1–46. [DOI] [PubMed] [Google Scholar]
- 25. Guo X, Gao L, Liao Q et al Long non‐coding RNAs function annotation: A global prediction method based on bi‐colored networks. Nucleic Acids Res 2013; 41: e35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Liao Q, Liu C, Yuan X et al Large‐scale prediction of long non‐coding RNA functions in a coding‐non‐coding gene co‐expression network. Nucleic Acids Res 2011; 39: 3864–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Prabhu A, Obi KO, Rubenstein JH. The synergistic effects of alcohol and tobacco consumption on the risk of esophageal squamous cell carcinoma: A meta‐analysis. Am J Gastroenterol 2014; 109: 822–7. [DOI] [PubMed] [Google Scholar]
- 28. Zeng H, Zheng R, Guo Y et al Cancer survival in China, 2003‐2005: A population‐based study. Int J Cancer 2015; 136: 1921–30. [DOI] [PubMed] [Google Scholar]
- 29. Abnet CC, Arnold M, Wei WQ. Epidemiology of esophageal squamous cell carcinoma. (Published erratum appears in Gastroenterology 2018; 155: 1281.) Gastroenterology 2018; 154: 360–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lagergren J, Smyth E, Cunningham D, Lagergren P. Oesophageal cancer. Lancet 2017; 390: 2383–96. [DOI] [PubMed] [Google Scholar]
- 31. Gao YB, Chen ZL, Li JG et al Genetic landscape of esophageal squamous cell carcinoma. Nat Genet 2014; 46: 1097–102. [DOI] [PubMed] [Google Scholar]
- 32. Isohata N, Aoyagi K, Mabuchi T et al Hedgehog and epithelial‐mesenchymal transition signaling in normal and malignant epithelial cells of the esophagus. Int J Cancer 2009; 125: 1212–21. [DOI] [PubMed] [Google Scholar]
- 33. Li W, Zheng J, Deng J et al Increased levels of the long intergenic non‐protein coding RNA POU3F3 promote DNA methylation in esophageal squamous cell carcinoma cells. Gastroenterology 2014; 146: 1714–26.e1715. [DOI] [PubMed] [Google Scholar]
- 34. Zhang E, Han L, Yin D et al H3K27 acetylation activated‐long non‐coding RNA CCAT1 affects cell proliferation and migration by regulating SPRY4 and HOXB13 expression in esophageal squamous cell carcinoma. Nucleic Acids Res 2017; 45: 3086–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Poon RT, Law SY, Chu KM et al Esophagectomy for carcinoma of the esophagus in the elderly: Results of current surgical management. Ann Surg 1998; 227: 357–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Alibakhshi A, Aminian A, Mirsharifi R, Jahangiri Y, Dashti H, Karimian F. The effect of age on the outcome of esophageal cancer surgery. Ann Thorac Med 2009; 4: 71–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Rice TW, Rusch VW, Ishwaran H, Blackstone EH, Worldwide Esophageal Cancer Collaboration . Cancer of the esophagus and esophagogastric junction: Data‐driven staging for the seventh edition of the American Joint Committee on Cancer/International Union Against Cancer Cancer Staging Manuals. Cancer 2010; 116: 3763–73. [DOI] [PubMed] [Google Scholar]
- 38. Lordick F, Mariette C, Haustermans K, Obermannová R, Arnold D, ESMO Guidelines Committee . Oesophageal cancer: ESMO clinical practice guidelines for diagnosis, treatment and follow‐up. Ann Oncol 2016; 27: v50–7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1. Differentially expressed coding genes.
Appendix S2. Fold change of differentially expressed coding genes.
Appendix S3. Differentially expressed non‐coding genes.
Appendix S4. Fold change of differentially expressed non‐coding genes.
Appendix S5. Network of differentially expressed coding and non‐coding genes.
Appendix S6. Correlation of node in the network.