

Abstract
Background
A high lymphatic metastasis rate and strong local invasive ability are the key characteristics of esophageal squamous cell carcinoma (ESCC) that affect patient survival, and long non‐coding RNAs (lncRNAs) may play a crucial role. We performed genome‐wide analysis of lncRNAs to identify novel biomarkers associated with local invasion and lymphatic metastasis in ESCC.
Methods
Six pairs of ESCC tumor and para‐tumor tissues were subjected to microarray analysis to identify differentially expressed lncRNAs, and 25 pairs of tissues samples were used to verify the effectiveness of screened lncRNAs using quantitative reverse transcription PCR. The correlations between verified lncRNAs and clinicopathological characteristics were analyzed to confirm specific lncRNAs associated with the local invasion and lymphatic metastasis of ESCC, and gene co‐expression analysis was used to predict potential mechanisms.
Results
Microarray analysis identified 1850 lncRNAs with significant differential expression in ESCC. Of 22 lncRNAs selected for quantitative reverse transcription PCR verification, four were significantly upregulated and one was significantly downregulated in ESCC cancer compared to para‐cancer tissues. ENST00000508406.1 was significantly associated with T, N, and tumor node metastasis stages, and NR_037652.1 was significantly associated with N stage. Moreover, 49 lncRNA‐messenger RNA pairs were significantly associated with the two dysregulated lncRNAs and possibly involved in the regulation of local invasion and lymphatic metastasis of ESCC.
Conclusion
The present genome‐wide analysis identified two novel and tumor‐specific lncRNAs for predicting ESCC local invasion and lymphatic metastasis, providing insight into the potential underlying mechanism, which warrants further investigation.
Keywords: Esophageal squamous cell carcinoma, long non‐coding RNA, microarray analysis, quantitative reverse transcription PCR
Introduction
Esophageal carcinoma (EC) is one of the most aggressive cancer types, ranking sixth in cancer‐related mortality and eighth in cancer incidence worldwide.1, 2 Esophageal squamous cell carcinoma (ESCC) is the predominant histological type, accounting for 80–90% of cases, especially in China.3, 4 Despite recent advances in surgical and irradiation techniques and pharmaceutical treatment for EC, the five‐year overall survival rate of patients with advanced ESCC remains 20%.5, 6 This is mainly because of the high lymphatic metastasis rate and strong local invasive ability of ESCC, even in the early stages.5 Molecular basis determines esophageal carcinoma development and progression,7 and a better understanding of the potential molecular mechanisms underlying ESCC, especially concerning tumorigenesis, progression, and lymphatic metastasis, is urgently needed to improve the survival rate.
Long non‐coding RNAs (lncRNAs) are a novel class of RNA molecules longer than 200 nucleotides that lack protein‐coding ability.8, 9 LncRNAs regulate transcription by mediating target gene activation or silencing through chromatin modification.10 Accumulating evidence suggests that lncRNAs play critical roles in many biological processes, including embryonic stem cell development,11 Th‐cell differentiation,12 cancer cell apoptosis, and metastasis.13 LncRNAs can act as oncogenes or tumor suppressors,14 and play important roles in carcinogenesis and cancer proliferation, invasion, and metastasis, as well as cancer prognosis.15, 16 LncRNA expression is tissue‐specific, and lncRNA dysregulation is present in various types of cancer.17, 18, 19 Therefore, lncRNAs are potential biomarkers for cancer diagnosis and prediction of prognosis.
Dysregulation of the expression of several lncRNAs is associated with ESCC tumorigenesis, progression, and regional lymphatic metastasis.20, 21, 22, 23, 24, 25, 26, 27, 28, 29 However, these lncRNAs were mostly identified in in vitro studies and lack application value in clinical practice. Specific lncRNAs that can be used as biomarkers for the early diagnosis of ESCC and to predict tumor invasive ability and regional lymphatic metastasis, which are key factors for making treatment decisions and for improving the survival rate, have not been identified to date.
Based on the tissue specific expression of lncRNAs and their critical roles in biological processes associated with ESCC, we performed microarray analysis to explore the expression profiles of dysregulated lncRNAs in ESCC cancer tissues and para‐cancer tissues. The aim of the study was to identify tumor‐specific lncRNAs that could be used for the early diagnosis of ESCC and to predict tumor invasive ability and lymphatic metastasis to guide the clinical management of patients with ESCC.
Methods
Samples
Primary cancer tissues and para‐cancer tissues were collected from patients with EC. Tissues were frozen in liquid nitrogen immediately after surgical resection until used in the experiments. Patient samples were selected according to the following criteria: radical EC resection had been performed and ESCC was pathologically confirmed postoperatively; chemotherapy or radiotherapy had not been administered; and patients did not have any other serious diseases and were expected to survive > 6 months. The para‐cancer tissues were adjacent esophageal tissues confirmed as being from a microscopically negative tumor margin. Eligible patients were recruited from the Shandong Provincial Hospital, the Shandong Cancer Hospital Affiliated to Shandong University, and the Second Hospital of Shandong University between August 2016 and February 2017. Written informed consent was obtained from all patients, and the Ethics Committees of the three hospitals approved the study.
RNA extraction, quality control, amplification, and labeling
Total RNA was extracted from cancer and para‐cancer tissues using the TRIzol reagent (Invitrogen, Carlsbad, CA, USA) and purified using the mirVana miRNA Isolation Kit (Ambion, Austin, TX, USA) according to the manufacturer's protocol. To ensure that total RNAs met the requirements for the experiment, RNA purity and concentration were quantified using the Nano Drop ND‐1000 Spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA), and RNA integrity was assessed with the Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA). High yields of labeled complementary DNA for further array hybridization were produced with the CapitalBio cRNA Amplification and Labeling Kit (CapitalBio Technology, Beijing, China) according to the manufacturer's protocol.
Microarray analysis
Human lncRNA/mRNA Array version 4.0 (CapitalBio Technology) was designed with four identical arrays per slide (4 × 180K format) for the global profiling of approximately 41 000 human lncRNAs and 34 000 human messenger RNAs (mRNAs) in each array. LncRNA and mRNA target sequences were merged from almost all relevant databases, including GENCODE/Ensembl, Human lincRNA Catalog,30 RefSeq, UCSC, NRED (ncRNA Expression Database), LNCipedia, H‐InvDB, and LncRNAs‐a (Enhancer‐like). Each RNA was detected by probes, and was performed three times. The array also contained 4974 Agilent control probes.
After array hybridization, Agilent Feature Extraction version 10.7 (Agilent Technologies) was used to analyze the array images and obtain the raw data. Raw data summarization, normalization, and quality control were performed using Agilent GeneSpring version 13.0 (Agilent Technologies). A threshold value of ≥ 2 and ≤ ‐2‐fold change and a Benjamini–Hochberg corrected P value of 0.05 was used to select differentially expressed genes. The adjust data function of CLUSTER 3.0 (Stanford University School of Medicine, Stanford, CA, USA) was used for Log2 transformation and to determine the median of data. Further hierarchical clustering analysis was performed to detect gene expression patterns. Tree visualization was performed using Java Treeview (Stanford University School of Medicine). CapitalBio Technology performed all microarray works.
Gene ontology enrichment and pathway analysis
The Gene Ontology (GO) project provides a controlled vocabulary to describe the genes and gene products of any organism (http://www.geneontology.org), and categorizes the roles of mRNAs into three domains: biological process, cellular component, and molecular function. A Fisher's exact test was used to determine whether the overlap between the differential expression list and the GO annotation list was greater than that expected by chance. Pathway analysis is functional analysis that maps genes to Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways. A Fisher's exact test was used to select a significant pathway, and the threshold of significance was defined by the P value; the lower the P value, the more significant the pathway.
Gene co‐expression analysis
The correlation between differentially expressed lncRNAs and mRNAs was determined with the normalized signature data using Pearson's correlation analysis, and significant lncRNA‐mRNA pairs were identified. The top 1000 gene pairs with Pearson correlation coefficients > 0.95 were selected to construct the coding‐non‐coding gene co‐expression (CNC) network using open source bioinformatics Cytoscape software (http://www.cytoscape.org/download.php). In the network, a gene “degree” was defined as the number of times one specific gene was linked to other genes. This is the simplest and most important measure of gene centrality within a network and determines the relative importance of a gene.31
Long non‐coding RNA (lncRNA) screening and quantitative reverse transcription PCR verification
Based on the results of microarray and gene co‐expression analyses, lncRNAs were screened for further verification according to the following criteria: (i) fold change > 2.5; (ii) Benjamini–Hochberg corrected P < 0.05; (iii) the raw signal intensity of each lncRNA > 50 and with a “detected” Feature Extraction Flag; and (iv) lncRNA‐mRNA pairs with correlation coefficients > 0.99 according to the gene co‐expression analysis. After the lncRNA screen, 25 pairs of tissue samples that met the selection criteria were selected for verification by quantitative reverse transcription (qRT) PCR.
Correlation analysis between verified lncRNAs and clinicopathological characteristics
To identify clinically significant lncRNAs capable of predicting tumor invasive ability and regional lymphatic metastasis in ESCC, the correlations between verified lncRNAs and clinicopathological characteristics were also analyzed. The clinicopathological characteristics analyzed included gender, age, T stage, N stage, tumor node metastasis (TNM) stage, tumor size, tumor location, tumor differentiation degree, and tumor clinical subtype. The potential mechanisms of the significant lncRNAs screened were analyzed according to the results of gene co‐expression analysis. Postoperative stage was defined according to the 2017 American Joint Committee on Cancer (AJCC) TNM staging classification system.
Statistical analysis
Statistical analysis was performed using SPSS version 20.0 (IBM Corp., Armonk, NY, USA). LncRNA expression levels in verified samples were presented as the mean ± standard deviation (SD). Differences in lncRNA expression between the groups were compared using the paired samples t‐test. Correlations between verified lncRNAs and clinicopathological characteristics were determined using Spearman's correlation analysis. All P values were two‐sided, and P < 0.05 was considered statistically significant.
Results
Sample selection
According to the selection criteria, six pairs of tissues samples (including ESCC and para‐cancer tissues) with similar clinicopathological characteristics were selected for microarray analysis, and 25 pairs of tissue samples were used to verify the effectiveness of the screened lncRNAs. The clinicopathological characteristics of the samples used for microarray analysis are detailed in Table 1.
Table 1.
Clinical information of samples for microarray analysis
| Sample | Age (years) | Gender | Primary location | Pathology grade | Clinical subtype | Tumor size (cm3) | Stage† |
|---|---|---|---|---|---|---|---|
| 1 | 58 | Male | Lower thoracic esophagus | Moderate differentiation | Ulcerative type | 5 × 3 × 1.2 | pT3N3M0 |
| 2 | 73 | Male | Mid‐thoracic esophagus | Moderate differentiation | Ulcerative type | 4 × 3.5 × 1.2 | pT2N1M0 |
| 3 | 75 | Male | Lower thoracic esophagus | Poor differentiation | Ulcerative type | 3 × 2.5 × 1 | pT3N2M0 |
| 4 | 56 | Female | Lower thoracic esophagus | Moderate differentiation | Ulcerative type | 5 × 4 × 1.3 | pT3N1M0 |
| 5 | 62 | Male | Mid‐thoracic esophagus | Poor differentiation | Ulcerative type | 6 × 3 × 0.5 | pT3N0M0 |
| 6 | 67 | Female | Lower thoracic esophagus | Poor differentiation | Ulcerative type | 3 × 1.5 × 0.8 | pT3N0M0 |
Postoperative pathological stage.
Overview of the microarray analysis
To evaluate the reliability of samples, the results of microarray analysis were expressed in cluster, principal component analysis, and box plots (Fig 1). These plots confirmed that the samples were of high quality with good representativeness and could meet the requirements of the experiment. Collectively, these findings indicated that the results of the experiment were reliable.
Figure 1.
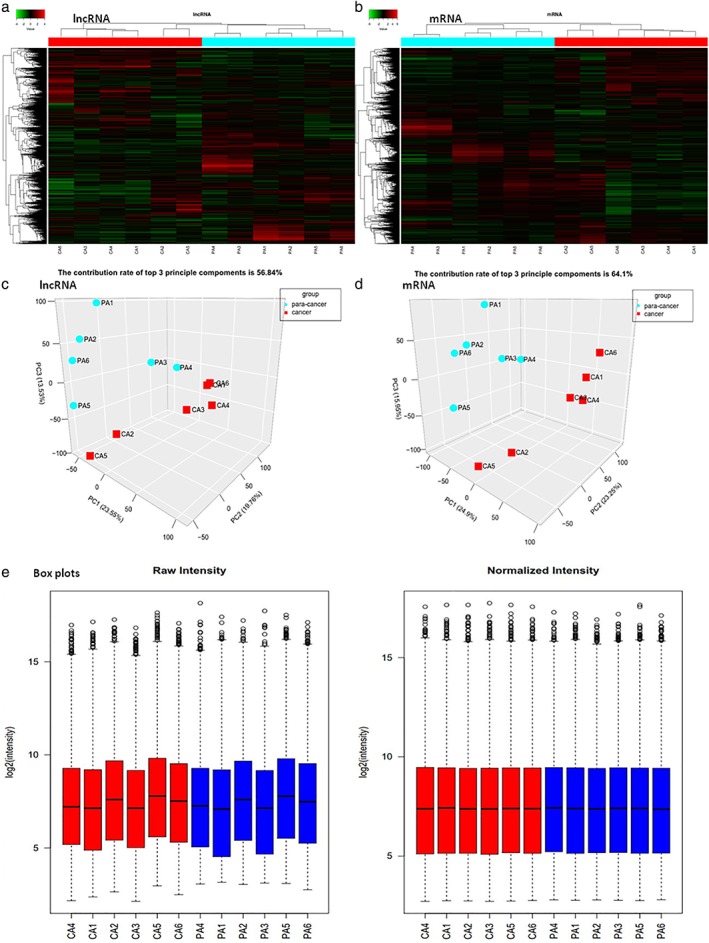
Plots of the microarray analysis data of the samples. Hierarchical cluster analysis plots of the expression of (a) long non‐coding RNAs (lncRNAs) and (b) messenger RNAs (mRNAs) in esophageal squamous cell carcinoma (ESCC) and para‐cancer tissues show good similarities among cancer tissues and among para‐cancer tissues, respectively. Red denotes high relative expression and green denotes low relative expression. Each RNA is represented by a single row of colored boxes and each sample by a single column. The three‐dimensional principal component analysis plots for normalized data of (c) lncRNAs and (d) mRNAs of the samples, respectively, shows good similarities among cancer tissues and among para‐cancer tissues. Groups ( ) para‐cancer and (
) para‐cancer and ( ) cancer. (e) The box plots for overall gene expression level in each sample before and after normalization show overall gene expression levels among all samples tended to be the same after normalization.
) cancer. (e) The box plots for overall gene expression level in each sample before and after normalization show overall gene expression levels among all samples tended to be the same after normalization.
The comparative microarray analysis identified 1850 lncRNAs and 2261 mRNAs showing significant differential expression (fold change ≥ 2.0, P < 0.05). Among them, 962 lncRNAs and 1394 mRNAs were upregulated and 888 lncRNAs and 867 mRNAs were downregulated in ESCC cancer tissues compared to para‐cancer tissues. The results were expressed in cluster, scatter, and volcano plots (Fig 2). To further evaluate the expression patterns of the lncRNAs, the classification of dysregulated lncRNAs was analyzed. The results showed that 950 intergenic lncRNAs were differentially expressed, indicating that most lncRNAs were intergenic lncRNAs.
Figure 2.
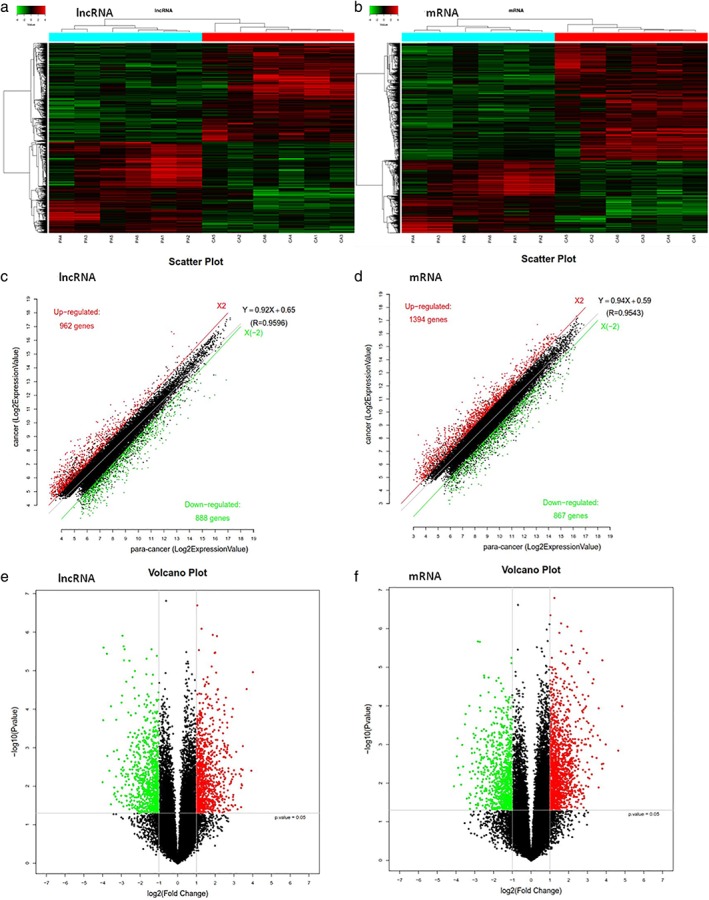
Hierarchical cluster analysis plots of the samples show the differentially expressed (a) long non‐coding RNAs (lncRNAs) and (b) messenger RNAs (mRNAs) in esophageal squamous cell carcinoma (ESCC) tissues compared to para‐cancer tissues. Red denotes high relative expression and green low relative expression. Each RNA is represented by a single row of colored boxes and each sample by a single column. The scatter and volcano plots illustrate the distributions of the data in the (c,e) lncRNA and (d,f) mRNA profiles. The values of the x and y axes in the scatter plot are the averaged normalized signal values of the group (log2 scaled). Red dots represent significantly upregulated genes, green dots represent significantly downregulated genes, and black dots represent genes without significant differences in the scatter and volcano plots. All of these plots directly reflect the quantity and reliability of the genes with significant differences.
Gene ontology and pathway analyses
The results of GO analysis showed that the dysregulated transcripts were mainly involved in biological processes; one fraction was defined as cellular components and a small number was involved in molecular function (Fig 3). Among the involved biological processes, cellular, single‐organism, and metabolic processes, biological regulation, and regulation of biological processes were the five most significant processes associated with the dysregulated mRNAs. Among the cellular components, cell parts, cell, and organelle were the three most significant component processes associated with the dysregulated mRNAs. Among molecular functions, binding and catalytic activity were the two most significant molecular functions of the dysregulated mRNAs.
Figure 3.
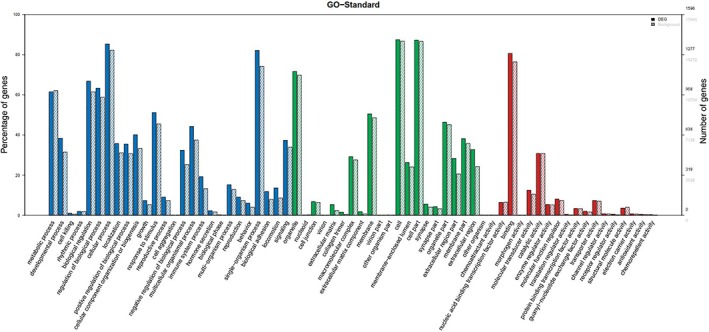
Gene ontology (GO) enrichment analysis of long non‐coding RNA (lncRNAs) target genes (messenger RNAs). The x‐axis represents GO terms including biological process (blue), cell component (green), and molecular function (red), while the y‐axis includes three parts, the percentage of genes denoted in the left, numbers of differentially expressed genes (DEGs, black number), and background genes (gray number, representing all genes in chips) in the right. The solid histogram represents DEGs and the virtual histogram represents background genes. This plot shows the enrichment expression of DEGs.
In KEGG pathway analysis, among the top 20 significantly enriched KEGG pathways, cytokine‐cytokine receptor interaction, transcriptional misregulation in cancer, DNA replication, cell cycle, Toll‐like receptor, and viral carcinogenesis were the significant pathways involved in the pathogenesis of cancer and associated with the dysregulated mRNAs (Fig 4).
Figure 4.
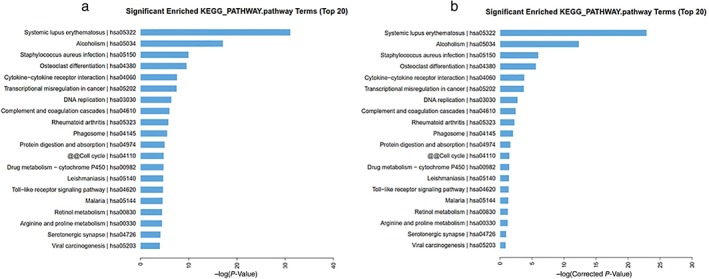
The top 20 significantly enriched Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways of messenger RNAs according to (a) P and (b) q (corrected P) values. ( ) KEGG pathway.
) KEGG pathway.
Gene co‐expression analysis
According to the results of gene co‐expression analysis, 1759 differentially expressed lncRNAs and 1929 mRNAs showed a significant correlation, and there were 74 420 significant lncRNA‐mRNA pairs with Pearson's correlation coefficients > 0.90. Among these, 110 lncRNA‐mRNA pairs showed Pearson's correlation coefficients > 0.99. Among these significantly related pairs, we selected the top 1000 gene pairs with Pearson correlation coefficients > 0.95 to construct the CNC network, as shown in Figure 5. Further target gene and transcription factor prediction analyses identified 1626 lncRNAs among all differentially expressed lncRNAs that had potential regulatory value in the pathogenesis of ESCC.
Figure 5.
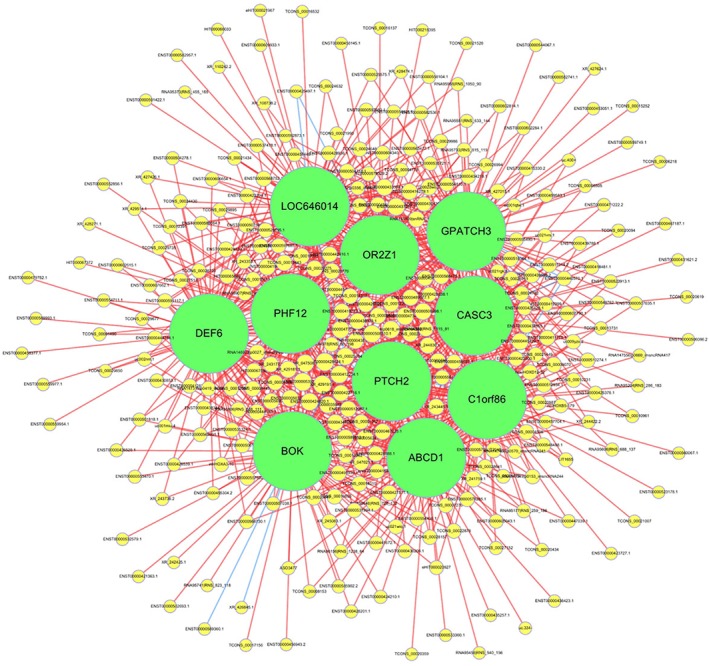
The coding‐non‐coding gene co‐expression network. This network was constructed with the top 1000 significant long non‐coding RNA (lncRNA)‐messenger RNA (mRNAs) pairs with Pearson correlation coefficients > 0.95. Yellow circles represent lncRNAs, green circles represent mRNAs, red lines represent positive correlation, and blue lines represent negative correlation. The size of the circle in the network represents a gene “degree,” defined as the number of times one specific gene is linked to other genes.
LncRNA screening and qRT‐PCR verification
Eleven lncRNAs among the differentially expressed lncRNAs were selected according to the screening criteria for further qRT‐PCR verification. In order to verify a greater number of lncRNAs, an additional 11 lncRNAs with a “detected” Feature Extraction Flag in the raw signal intensity were also randomly selected for further qRT‐PCR verification. Thus, a total of 22 lncRNAs, including 10 upregulated and 12 downregulated lncRNAs, were verified. Twenty‐five pairs of tissues samples were used to verify the effectiveness of the selected lncRNAs (the clinicopathological characteristics of these samples are detailed in Table 2). The results of qRT‐PCR verification showed that 15 lncRNAs showed an expression tendency of 68.18%, which was consistent with the microarray data. Among the seven inconsistently expressed lncRNAs, three were selected according to the screening criteria and four were selected randomly. Among the 22 lncRNAs verified, four were significantly upregulated, whereas two were significantly downregulated in ESCC tissues compared to para‐cancer tissues. Among the six significantly dysregulated lncRNAs, two were selected according to the screening criteria and four were selected randomly. However, among the six significantly dysregulated lncRNAs, the expression tendency of ENST00000501715.2 was inconsistent between the qRT‐PCR and microarray data, and was therefore excluded. Finally, four significantly upregulated lncRNAs and one significantly downregulated lncRNA were identified as potentially involved in the pathogenesis of ESCC, and the correlations between these five‐lncRNAs and clinicopathological characteristics were further analyzed. The expression data of the 22 lncRNAs verified are detailed in Table 3.
Table 2.
Clinicopathological characteristics of samples for verification
| Characteristics | N | Characteristics | N | Characteristics | N |
|---|---|---|---|---|---|
| Median age (range) (year) | 63 (46–76) | Gender | N stage | ||
| Tumor size (mean ± SD) (cm3) | 9.329 ± 7.081 | Male | 19 | N0 | 14 |
| Tumor location | Female | 6 | N1 | 6 | |
| Upper thoracic esophagus | 2 | Tumor clinical subtype | N2–3 | 5 | |
| Mid‐thoracic esophagus | 12 | Ulcerative type | 9 | pTNM stage† | |
| Lower thoracic esophagus | 11 | Medullary type | 12 | IIA | 8 |
| Tumor differentiation degree | Fungating type | 4 | IIB | 7 | |
| Well differentiation | 1 | T stage | IIIA | 1 | |
| Moderate differentiation | 14 | T2 | 6 | IIIB | 8 |
| Poor differentiation | 10 | T3 | 19 | IVA | 1 |
Postoperative pathological stage.
pTNM, pathological tumor node metastasis; SD, standard deviation.
Table 3.
Expression data of verified lncRNAs in verified samples
| Group | LncRNA ID | Expression level in cancer tissues (mean ± SD) | Expression level in para‐cancer tissues (mean ± SD) | Expression tendency in cancer tissues | P | Consistency with microarray data |
|---|---|---|---|---|---|---|
| Group A† | ENST00000416890.1 | 1.155 ± 2.874 | 0.654 ± 0.725 | Up | 0.402 | Consistent |
| ENST00000501715.2 | 3.376 ± 3.113 | 6.957 ± 6.027 | Down | 0.002 | Inconsistent | |
| XR_243745.1 | 3.830 ± 6.141 | 2.413 ± 4.154 | Up | 0.359 | Inconsistent | |
| ENST00000479752.1 | 1.892 ± 1.988 | 2.063 ± 4.119 | Down | 0.847 | Consistent | |
| ENST00000508406.1 | 13.641 ± 10.523 | 6.785 ± 8.970 | Up | 0.002 | Consistent | |
| ENST00000609933.1 | 5.641 ± 8.994 | 2.671 ± 3.850 | Up | 0.138 | Inconsistent | |
| NR_108085.1 | 2.511 ± 3.377 | 2.740 ± 4.043 | Down | 0.831 | Consistent | |
| ENST00000424322.1 | 7.306 ± 12.128 | 1.564 ± 1.920 | Up | 0.022 | Consistent | |
| ENST00000508572.1 | 5.909 ± 9.968 | 1.562 ± 1.445 | Up | 0.040 | Consistent | |
| XR_243353.1 | 0.478 ± 0.760 | 0.367 ± 0.597 | Up | 0.578 | Inconsistent | |
| ENST00000593452.1 | 6.448 ± 11.957 | 3.812 ± 6.757 | Up | 0.231 | Consistent | |
| Group B‡ | ENST00000497988.1 | 1.402 ± 2.531 | 1.270 ± 1.739 | Up | 0.815 | Inconsistent |
| TCONS_00018449 | 0.802 ± 0.990 | 0.924 ± 1.534 | Down | 0.738 | Consistent | |
| NR_037652.1 | 0.008 ± 0.036 | 0.161 ± 0.337 | Down | 0.024 | Consistent | |
| ENST00000589257.1 | 0.761 ± 1.385 | 0.926 ± 2.575 | Down | 0.779 | Consistent | |
| ENST00000440357.1 | 0.982 ± 1.431 | 2.282 ± 3.594 | Down | 0.100 | Consistent | |
| RNA95373|RNS_455_165 | 2.689 ± 2.570 | 1.522 ± 2.390 | Up | 0.114 | Inconsistent | |
| ENST00000578897.1 | 2.087 ± 2.939 | 2.434 ± 3.399 | Down | 0.680 | Consistent | |
| RNA95551|RNS_633_144 | 0.157 ± 0.275 | 0.287 ± 0.364 | Down | 0.182 | Inconsistent | |
| uc031qqx.1 | 4.529 ± 5.219 | 2.293 ± 4.086 | Up | 0.123 | Consistent | |
| ENST00000603948.1 | 5.690 ± 8.703 | 1.678 ± 1.651 | Up | 0.037 | Consistent | |
| ENST00000607933.1 | 3.730 ± 4.047 | 1.824 ± 2.886 | Up | 0.083 | Consistent |
Long non‐coding RNAs (lncRNAs) were randomly selected with a “detected” Feature Extraction Flag in the raw signal intensity.
lncRNAs were selected according to the screening criteria for lncRNAs.
SD, standard deviation.
Correlations between verified lncRNAs and clinicopathological characteristics
Analysis of the correlations of the five significantly dysregulated lncRNAs with clinicopathological characteristics showed that the upregulated ENST00000508406.1 was significantly associated with T, N, and TNM stages, and the downregulated NR_037652.1 was significantly associated with N stage, whereas the other three lncRNAs were not (Table 4). Furthermore, the gene co‐expression analysis confirmed 49 lncRNA‐mRNA pairs and 45 mRNAs that were significantly associated with ENST00000508406.1 and NR_037652.1, and were possibly involved in the mechanism underlying the regulation of local invasion and lymphatic metastasis of ESCC.
Table 4.
Correlations between significantly dysregulated lncRNAs and clinicopathological characteristics
| Clinicopathological characteristics | ENST00000508406.1 | ENST00000424322.1 | ENST00000508572.1 | ENST00000603948.1 | NR_037652.1 | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| R† | P | R† | P | R† | P | R† | P | R† | P | |
| Gender | 0.286 | 0.166 | −0.026 | 0.902 | −0.052 | 0.805 | −0.026 | 0.902 | 0.169 | 0.420 |
| Age | 0.056‡ | 0.791 | 0.217‡ | 0.299 | 0.269‡ | 0.194 | 0.207‡ | 0.322 | −0.300‡ | 0.145 |
| T stage | −0.455 | 0.022 | 0.091 | 0.666 | −0.026 | 0.902 | −0.052 | 0.805 | 0.286 | 0.166 |
| N stage | −0.472 | 0.017 | 0.050 | 0.812 | −0.003 | 0.989 | −0.148 | 0.479 | 0.454 | 0.023 |
| TNM stage | −0.447 | 0.025 | 0.002 | 0.991 | −0.035 | 0.870 | −0.162 | 0.439 | 0.308 | 0.134 |
| Tumor size | −0.341‡ | 0.095 | −0.141‡ | 0.500 | −0.164‡ | 0.432 | 0.284‡ | 0.169 | −0.054‡ | 0.798 |
| Tumor location | −0.226 | 0.277 | −0.094 | 0.655 | −0.143 | 0.495 | −0.145 | 0.488 | 0.185 | 0.377 |
| Pathology grade | −0.048 | 0.821 | −0.053 | 0.802 | −0.085 | 0.688 | −0.040 | 0.851 | 0.193 | 0.355 |
| Clinical subtype | 0.308 | 0.134 | −0.108 | 0.606 | −0.191 | 0.362 | −0.063 | 0.763 | −0.025 | 0.906 |
R = correlation coefficient.
Pearson's correlation coefficient. lncRNA, long non‐coding RNA; TNM, tumor node metastasis.
Discussion
Esophageal squamous cell carcinoma is an aggressive malignancy characterized by high degrees of local invasion and regional lymphatic metastasis, leading to poor overall survival.5, 6 Intrinsic molecular distinction determines the biological behavior of cancer.7 Among numerous molecules that play key roles in ESCC, lncRNAs have attracted increasing attention because their aberrant expression is associated with ESCC carcinogenesis, invasion, and metastasis.20, 21, 22, 23, 24, 25, 26, 27, 28, 29 Several lncRNAs involved in ESCC have been reported, including HOTAIR,20 MALAT1,21 NMR,22 CASC9,23 PEG10,24 HOTTIP,25 SPRY4‐IT1,26 PCAT‐1,27 AK001796,28 and CCAT2.29 HOTAIR directly downregulates the expression of WIF‐1 and activates the Wnt/β‐catenin signaling pathway to promote the migration and invasion of ESCC cells. MALAT1 promotes the malignant development of ESCC by targeting β‐catenin via Ezh2. NMR promotes tumor progression via NSUN2 and BPTF, and CASC9 promotes tumor metastasis by upregulating LAMC2 through interaction with the CREB‐binding protein. HOTTIP promotes tumor metastasis by inducing epithelial‐mesenchymal transition, and AK001796 contributes to tumor growth by regulating the expression of p53. Although these dysregulated lncRNAs in ESCC have been extensively studied, most are also involved in other types of cancer. For example, HOTAIR is involved in many cancers32 including lung, breast, hepatocellular, and cervical cancer. PEG10 is involved in hypopharyngeal squamous cell carcinoma,33 pancreatic cancer,34 and diffuse large B cell lymphoma.35 Therefore, these lncRNAs lack tumor specificity. Although the function of these dysregulated lncRNAs in ESCC has previously been reported, most reports were based on in vitro studies, and most of these lncRNAs failed to perform their functions in vivo. Therefore, these lncRNAs are of limited clinical value. Genome‐wide analysis of lncRNAs allows the identification of dysregulated lncRNAs in ESCC to obtain novel and tumor‐specific lncRNAs for the prediction of ESCC invasion and lymphatic metastasis. In the present study, we performed genome‐wide analysis of lncRNAs to identify tumor‐specific biomarkers of clinical value for the diagnosis and treatment of ESCC.
We used strict quality control for sample selection, pathological sampling, storage, RNA extraction, and the process of microarray analysis to exclude any possible interference with the experimental results. The use of cluster, principal component analysis, and box plots show the high quality and good representativeness of the samples, reflecting the reliability of the experimental results.
The comparative microarray analysis identified 1850 lncRNAs and 2261 mRNAs that were differentially expressed, and target gene and transcription factor prediction analysis showed that 1626 lncRNAs had potential regulatory functions in ESCC. GO analysis showed that dysregulated transcripts were mainly involved in biological processes. Further KEGG pathway analysis identified at least six main signaling pathways associated with the carcinogenesis, invasion, and metastasis of cancer, especially transcriptional misregulation in the cancer (hsa05202) pathway, which is associated with many solid tumors and contributes to the tumorigenic process. Finally, gene co‐expression analysis identified 110 lncRNA‐mRNA pairs showing a significant correlation, with Pearson's correlation coefficients > 0.99. Taken together, these results provide valuable research clues for further studies.
Although we identified 1626 lncRNAs that had potential regulatory value in ESCC, only 11 lncRNAs were selected according to the screening criteria, indicating that some significantly dysregulated lncRNAs may not have been included. Thus, we randomly selected an additional 11 lncRNAs for qRT‐PCR verification for a total of 22 lncRNAs, including 10 upregulated and 12 downregulated lncRNAs. An additional 25 samples were selected during the verification process, therefore, the verification results not only tested the consistency between the qRT‐PCR results and microarray data as previously reported,36, 37 but also verified the expression levels of these selected lncRNAs in ESCC. The qRT‐PCR verification results showed good consistency with the microarray data, and were similar to previously reported data.36, 37
In addition, five lncRNAs that have not been reported in previous studies were identified via qRT‐PCR as significantly dysregulated in ESCC. Further correlation analysis between these five lncRNAs and clinicopathological characteristics showed that upregulated ENST00000508406.1 was significantly associated with T, N, and TNM stages, and downregulated NR_037652.1 was significantly associated with N stage, indicating that they might be involved in the regulation of local invasion and lymphatic metastasis of ESCC. Further gene co‐expression analysis confirmed 49 lncRNA‐mRNA pairs and 45 mRNAs that were significantly associated with ENST00000508406.1 and NR_037652.1, and were possibly involved in the mechanism underlying the regulation of local invasion and lymphatic metastasis of ESCC. Finally, in the two identified lncRNAs associated with local invasion and lymphatic metastasis of ESCC, NR_037652.1 was selected according to the screening criteria, whereas ENST00000508406.1 was selected randomly, indicating that some lncRNAs with potential regulatory value among 1626 lncRNAs might have been missed and are worthy of further investigation.
Local relapse and regional lymphatic metastasis are important causes of failure after radical resection and irradiation of ESCC,38, 39 leading to poor survival rates.40 Regional lymphatic metastasis of ESCC involves a wide region from the neck to the upper abdomen.41 Because of this pattern of lymphatic metastasis, it is difficult to remove and irradiate all regional lymph nodes via radical resection and irradiation, which can also have severe implications. Therefore, selective lymph node dissection and regional lymphatic irradiation is of value in selected patients; however, this procedure requires a method to effectively predict which patients will develop regional lymphatic metastasis. LncRNAs are involved in the regulation of tumor invasion and lymphatic metastasis and could therefore be useful to predict local invasion and regional lymphatic metastasis in ESCC.9, 13, 15, 16
In summary, using genome‐wide analysis we identified two novel and tumor‐specific lncRNAs associated with local invasion and regional lymphatic metastasis that could be of predictive value as biomarkers in ESCC. These two lncRNAs are worthy of further investigation in a large cohort of ESCC samples. In addition, the lncRNA‐mRNA pairs and mRNAs identified in the gene co‐expression analysis provide significant clues for further study of the potential underlying mechanisms.
Disclosure
No authors report any conflict of interest.
Acknowledgments
This study was supported by the Key Research and Development Plan of Shandong Province (grant no.2016GSF201140 and 2016GSF201155) and the Clinical Medical Science and Technology Innovation Plan of Jinan City (grant no. 201506005 and 201602145).
References
- 1. Wheeler JB, Reed CE. Epidemiology of esophageal cancer. Surg Clin North Am 2012; 92: 1077–87. [DOI] [PubMed] [Google Scholar]
- 2. Ferlay J, Soerjomataram I, Dikshit R et al Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer 2015; 136: E359–86. [DOI] [PubMed] [Google Scholar]
- 3. Arnold M, Soerjomataram I, Ferlay J, Forman D. Global incidence of oesophageal cancer by histological subtype in 2012. Gut 2015; 64: 381–7. [DOI] [PubMed] [Google Scholar]
- 4. Chen W, Zheng R, Baade PD et al Cancer statistics in China, 2015. CA Cancer J Clin 2016; 66: 115–32. [DOI] [PubMed] [Google Scholar]
- 5. Pennathur A, Gibson MK, Jobe BA, Luketich JD. Oesophageal carcinoma. Lancet 2013; 381: 400–12. [DOI] [PubMed] [Google Scholar]
- 6. Rustgi A, El‐Serag HB. Esophageal carcinoma. N Engl J Med 2015; 372: 1472–3. [DOI] [PubMed] [Google Scholar]
- 7. Denlinger CE, Thompson RK. Molecular basis of esophageal cancer development and progression. Surg Clin North Am 2012; 92: 1089–103. [DOI] [PubMed] [Google Scholar]
- 8. Djebali S, Davis CA, Merkel A et al Landscape of transcription in human cells. Nature 2012; 489: 101–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ponting CP, Oliver PL, Reik W. Evolution and functions of long noncoding RNAs. Cell 2009; 136: 629–41. [DOI] [PubMed] [Google Scholar]
- 10. Paul J, Duerksen JD. Chromatin‐associated RNA content of heterochromatin and euchromatin. Mol Cell Biochem 1975; 9: 9–16. [DOI] [PubMed] [Google Scholar]
- 11. Yin Y, Yan P, Lu J et al Opposing roles for the lncRNA haunt and its genomic locus in regulating HOXA gene activation during embryonic stem cell differentiation. Cell Stem Cell 2015; 16: 504–16. [DOI] [PubMed] [Google Scholar]
- 12. Spurlock CF 3rd, Tossberg JT, Guo Y, Collier SP, Crooke PS 3rd, Aune TM. Expression and functions of long noncoding RNAs during human T helper cell differentiation. Nat Commun 2015; 6: 6932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yarmishyn AA, Kurochkin IV. Long noncoding RNAs: A potential novel class of cancer biomarkers. Front Genet 2015; 6: 145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wapinski O, Chang HY. Long noncoding RNAs and human disease. Trends Cell Biol 2011; 21: 354–61. [DOI] [PubMed] [Google Scholar]
- 15. Prensner JR, Chinnaiyan AM. The emergence of lncRNAs in cancer biology. Cancer Discov 2011; 1: 391–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gutschner T, Diederichs S. The hallmarks of cancer: A long non‐coding RNA point of view. RNA Biol 2012; 9: 703–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kim HS, Minna JD, White MA. GWAS meets TCGA to illuminate mechanisms of cancer predisposition. Cell 2013; 152: 387–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tang Y, Cheung BB, Atmadibrata B et al The regulatory role of long noncoding RNAs in cancer. Cancer Lett 2017; 391: 12–9. [DOI] [PubMed] [Google Scholar]
- 19. Bach DH, Lee SK. Long noncoding RNAs in cancer cells. Cancer Lett 2018; 419: 152–66. [DOI] [PubMed] [Google Scholar]
- 20. Song W, Zou SB. Prognostic role of lncRNA HOTAIR in esophageal squamous cell carcinoma. Clin Chim Acta 2016; 463: 169–73. [DOI] [PubMed] [Google Scholar]
- 21. Wang W, Zhu Y, Li S et al Long noncoding RNA MALAT1 promotes malignant development of esophageal squamous cell carcinoma by targeting beta‐catenin via Ezh2. Oncotarget 2016; 7: 25668–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Li Y, Li J, Luo M et al Novel long noncoding RNA NMR promotes tumor progression via NSUN2 and BPTF in esophageal squamous cell carcinoma. Cancer Lett 2018; 430: 57–66. [DOI] [PubMed] [Google Scholar]
- 23. Liang Y, Chen X, Wu Y et al LncRNA CASC9 promotes esophageal squamous cell carcinoma metastasis through upregulating LAMC2 expression by interacting with the CREB‐binding protein. Cell Death Differ 2018. 10.1038/s41418-018-0084-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zang W, Wang T, Huang J et al Long noncoding RNA PEG10 regulates proliferation and invasion of esophageal cancer cells. Cancer Gene Ther 2015; 22: 138–44. [DOI] [PubMed] [Google Scholar]
- 25. Chen X, Han H, Li Y, Zhang Q, Mo K, Chen S. Upregulation of long noncoding RNA HOTTIP promotes metastasis of esophageal squamous cell carcinoma via induction of EMT. Oncotarget 2016; 7: 84480–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cui F, Wu D, He X, Wang W, Xi J, Wang M. Long noncoding RNA SPRY4‐IT1 promotes esophageal squamous cell carcinoma cell proliferation, invasion, and epithelial‐mesenchymal transition. Tumour Biol 2016; 37: 10871–6. [DOI] [PubMed] [Google Scholar]
- 27. Shi WH, Wu QQ, Li SQ et al Upregulation of the long noncoding RNA PCAT‐1 correlates with advanced clinical stage and poor prognosis in esophageal squamous carcinoma. Tumour Biol 2015; 36: 2501–7. [DOI] [PubMed] [Google Scholar]
- 28. Liu B, Pan CF, Yao GL, Wei K, Xia Y, Chen YJ. The long non‐coding RNA AK001796 contributes to tumor growth via regulating expression of p53 in esophageal squamous cell carcinoma. Cancer Cell Int 2018; 18: 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wang J, Qiu M, Xu Y et al Long noncoding RNA CCAT2 correlates with smoking in esophageal squamous cell carcinoma. Tumour Biol 2015; 36: 5523–8. [DOI] [PubMed] [Google Scholar]
- 30. Ørom UA, Derrien T, Beringer M et al Long noncoding RNAs with enhancer‐like function in human cells. Cell 2010; 143: 46–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Barabási AL, Oltvai ZN. Network biology: Understanding the cell's functional organization. Nat Rev Genet 2004; 5: 101–13. [DOI] [PubMed] [Google Scholar]
- 32. Tang Q, Hann SS. HOTAIR: An oncogenic long non‐coding RNA in human cancer. Cell Physiol Biochem 2018; 47: 893–913. [DOI] [PubMed] [Google Scholar]
- 33. Zhao M, Sun D, Li X et al Overexpression of long noncoding RNA PEG10 promotes proliferation, invasion and metastasis of hypopharyngeal squamous cell carcinoma. Oncol Lett 2017; 14: 2919–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Peng YP, Zhu Y, Yin LD et al PEG10 overexpression induced by E2F‐1 promotes cell proliferation, migration, and invasion in pancreatic cancer. J Exp Clin Cancer Res 2017; 36: 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Peng W, Fan H, Wu G, Wu J, Feng J. Upregulation of long noncoding RNA PEG10 associates with poor prognosis in diffuse large B cell lymphoma with facilitating tumorigenicity. Clin Exp Med 2016; 16: 177–82. [DOI] [PubMed] [Google Scholar]
- 36. Chen D, Sun Q, Cheng X et al Genome‐wide analysis of long noncoding RNA (lncRNA) expression in colorectal cancer tissues from patients with liver metastasis. Cancer Med 2016; 5: 1629–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Shen Z, Du C, Zang R et al Microarray expression profiling of dysregulated long non‐coding RNAs in Hirschsprung's disease reveals their potential role in molecular diagnosis. Neurogastroenterol Motil 2016; 28: 266–73. [DOI] [PubMed] [Google Scholar]
- 38. Mariette C, Balon JM, Piessen G, Fabre S, Van Seuningen I, Triboulet JP. Pattern of recurrence following complete resection of esophageal carcinoma and factors predictive of recurrent disease. Cancer 2003; 97: 1616–23. [DOI] [PubMed] [Google Scholar]
- 39. Nakagawa S, Kanda T, Kosugi S, Ohashi M, Suzuki T, Hatakeyama K. Recurrence pattern of squamous cell carcinoma of the thoracic esophagus after extended radical esophagectomy with three‐field lymphadenectomy. J Am Coll Surg 2004; 198: 205–11. [DOI] [PubMed] [Google Scholar]
- 40. Wang H, Deng F, Liu Q, Ma Y. Prognostic significance of lymph node metastasis in esophageal squamous cell carcinoma. Pathol Res Pract 2017; 213: 842–7. [DOI] [PubMed] [Google Scholar]
- 41. Tachimori Y. Pattern of lymph node metastases of squamous cell esophageal cancer based on the anatomical lymphatic drainage system: Efficacy of lymph node dissection according to tumor location. J Thorac Dis 2017; 9 (Suppl. 8): S724–S30. [DOI] [PMC free article] [PubMed] [Google Scholar]