

Abstract
This Article describes the molecular recognition of peptides containing an N-terminal methionine (Met) by the synthetic receptor cucurbit[8]-uril (Q8) in aqueous solution and with submicromolar affinity. Prior work established that Q8 binds with high affinity to peptides containing aromatic amino acids, either by simultaneous binding of two aromatic residues, one from each of two different peptides, or by simultaneous binding of an aromatic residue and its immediate neighbor on the same peptide. The additional binding interface of two neighboring residues suggested the possibility of targeting nonaromatic peptides, which have thus far bound only weakly to synthetic receptors. A peptide library designed to test this hypothesis was synthesized and screened qualitatively for Q8 binding using a fluorescent indicator displacement assay. The large fluorescence response observed for several Met-terminated peptides suggested strong binding, which was confirmed quantitatively by the determination of submicromolar equilibrium dissociation constant values for Q8 binding to MLA, MYA, and MFA using isothermal titration calorimetry (ITC). This discovery of high affinity binding to Met-terminated peptides and, more generally, to nonaromatic peptides prompted a detailed investigation of the determinants of binding in this system using ITC, electrospray ionization mass spectrometry, and 1H NMR spectroscopy for 25 purified peptides. The studies establish the sequence determinants required for high-affinity binding of Met-terminated peptides and demonstrate that cucurbit[n]uril-mediated peptide recognition does not require an aromatic residue for high affinity. These results, combined with the known ability of cucurbit[n]urils to target N-termini and disordered loops in folded proteins, suggest that Q8 could be used to target unmodified, Met-terminated proteins.
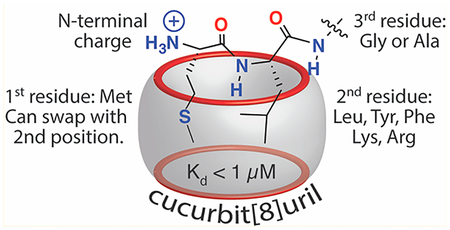
Graphical Abstract
INTRODUCTION
The development of synthetic compounds that bind target proteins with high affinity and selectivity drives many areas of basic and applied chemical biology.1–7 Examples of successful approaches to protein recognition include small molecules that bind inside protein cavities,4 aptamers and mimics of protein secondary structure that bind to large areas on protein surfaces,1,6,8 and compounds that bind to relatively small areas(i.e., “hotspots”) on protein surfaces.1,6,9 These approaches typically require the selection of high-affinity ligands from combinatorial libraries or the optimization of ligand design on the basis of detailed knowledge of the tertiary structure of the target protein. Protein structure remains difficult to predict from the sequence of amino acids, and thus we assert that design principles for protein recognition based primarily on the sequence of amino acids would be especially useful for their predictive power.
A strong and selective protein interaction requires a large binding interface,10 which typically comprises several amino acid residues that are neighbors in the folded protein but not typically in the amino acid sequence. Therefore, to predict recognition based on sequence, the binding site should be small enough (2–3 amino acid residues) so that the residues involved are adjacent in the sequence. In this situation, each amino acid residue in the complex would need to contribute significantly to the complexation energy, that is, to be efficient ligands. Aromatic ligands are particularly efficient due to their large, flat surfaces, which can provide substantial hydrophobic, van der Waals, and/or electrostatic interactions (e.g., cation–π).11–14 It is not surprising, therefore, that aromatic residues are commonly found to be important for stabilizing protein interactions.9,15–17
Synthetic receptors are well suited to binding small sites on proteins due to their ability to encapsulate their binding partners within a concave cavity and thus create an extensive binding interface. Among the synthetic receptors reported to bind peptides noncovalently in aqueous solution,18 submicromolar equilibrium dissociation constants have been observed only with aromatic peptides, and only for two classes of host: Fujita’s coordination cages19 and cucurbit[n]urils (Qn’s, Figure 1). Qn’s are particularly effective at targeting aromatic residues in peptides and proteins with submicromolar affinities.20,21 The hydrophobic cavity and two constricted, C=O-lined portals drive the binding of guests with nonpolar and cationic groups. Cucurbit[7]uril (Q7) and cucurbit[8]uril (Q8) typically bind to a single aromatic residue at the N-terminal position of the polypeptide by including the aromatic side chain within the hydrophobic cavity of the Qn and interacting with the N-terminal ammonium group via ion–dipole interactions with the C=O groups at the Qn portal. Although this motif targets only a single residue, it affords an extraordinary degree of selectivity because the N-terminal residue is a unique binding epitope in the polypeptide chain comprising the N-terminal ammonium group proximal to the side chain of the first residue. Remarkably, this selectivity is sufficient to enable protein recognition in simple and complex mixtures.22,23
Figure 1.

(a) Chemical formula of cucurbit[8]uril. (b) Schematic of the complex of Q8 with the peptide YLA. (c) Semiempirical model of the Q8 YLA complex based on 2D-NMR data. (d) Peptide library screened in this study.
Despite these successes, the utility of Qn-based peptide and protein recognition is inherently limited by the single-residue binding site. Recently, we reported the discovery that Q8 can bind with high affinity and selectivity to certain peptides that fold to include the side chains of two neighboring residues within the Q8 cavity (Figure 1).24 This “pair inclusion” motif enabled the submicromolar binding of the Tyr-containing peptides YLA, YYA, YFA, and YKA. The additional binding interface generated by the side chains of the N-terminal Tyr and its immediate neighbor suggests that the ligand efficiency of each residue can be reduced somewhat. Therefore, we hypothesized that an N-terminal aromatic residue may not be necessary for high-affinity binding if two sufficiently large residues could be included simultaneously. To test this hypothesis, we describe here a further exploration of the pair inclusion motif for peptide binding by Q8.
RESULTS AND DISCUSSION
Design and Screen of a Peptide Library.
A peptide library was designed to explore the scope and limitations of the pair inclusion motif. Past studies showed that only aromatic residues at the first position in the chain yielded high affinity binding to Q7 and Q8,20,21 presumably due to their ligand efficiency. The discovery of the pair inclusion motif, however, inspired us to ask whether nonaromatic N-terminal residues may bind to Q8 with high affinity when paired with a second residue. The wealth of Qn literature teaches us that nonpolar and cationic functional groups are preferred for guest binding,25–27 and we know that the displacement of high-energy water molecules from the Qn cavity is the dominant driving force for guest binding.28 Therefore, the library of 144 tripeptides (Figure 1) of sequence X1-X2-Ala was designed to vary the first position (i.e., X1) among amino acids with large side chains that are hydrophobic or cationic, including Tyr, Phe, Ile, Leu, Met, Pro, Arg, and Lys. The second position in the chain was varied (i.e., X2) among 18 genetically encoded amino acids. Trp was omitted due to its incompatibility with the fluorescence assay. Cys was omitted due to its propensity to form disulfide bonds. All peptides contained Ala at the third position and a C-terminal primary amide group.
The peptide library was synthesized by Fmoc-scheme solid-phase synthesis on Rink amide resin using Synphase Lanterns to enable parallel synthesis (see the Supporting Information for experimental details), as described previously.24,29 The library was screened for binding to Q8 using a fluorescent indicator displacement assay, as described previously.30 The competitive displacement of MBBI from Q8 by a peptide analyte causes an increase in the observed fluorescence.24 In all cases, we observed an increase in fluorescence intensity upon the addition of peptide to a mixture of Q8 and MBBI (Figure 2 and Table S1). Relative increases in fluorescence correlate to binding affinity and ranged from 11.6% to 44.3% at sample concentrations of 200 μM peptide, 40 μM Q8, and 40 μM MBBI in 10 mM sodium phosphate, pH 7.0. In general, the observed changes in fluorescence intensity were greater for peptides containing the Tyr, Phe, Ile, and Leu at the N-terminus as well as peptides containing Tyr, Phe, Leu, and Lys at the second position. It is important to note that peptides containing N-terminal Phe are known to form stable 2:1 peptide:Q8 complexes,31 which can also displace MBBI and lead to a large increase in fluorescence.24 The series containing N-terminal Pro showed a weak response overall. By contrast, the series containing N-terminal Met, Lys, and Arg displayed a large fluorescence increase for only a few sequences, which suggested significant sequence selectivity.
Figure 2.

Fluorescence assay for the binding of Q8 to the library of 144 peptides of sequence Var1-Var2-Ala. The density of each circle correlates to the average degree of fluorescence enhancement observed upon adding 200 μM peptide to 40 μM Q8 MBBI complex at room temperature in 10 mM sodium phosphate, pH 7.0.
In Depth Study of Met-Terminated Tripeptides.
We were particularly excited about the potential to recognize Met-terminated peptides because eukaryotic proteins are expressed with N-terminal Met. Therefore, we conducted a detailed investigation of the thermodynamic and structural determinants of binding for a representative series of purified Met-terminated tripeptides. MFA, MYA, MLA, and MKA were selected as leads from the fluorescence screen that represent aromatic, aliphatic, and basic side chains at the second position. The peptide MAA was included as a control containing an N-terminal Met and a minimal side chain at the second position. The binding thermodynamics of Q8 to these peptides were determined by isothermal titration calorimetry (ITC) at 300 K in 10 mM sodium phosphate, pH 7.0 (Table 1 and Figures S1, S4, S7, S13, and S14).
Table 1.
Thermodynamic Data for the Binding of Q8 to MXA Peptides
| peptide | Kd (μM)a | ΔHa (kcal mol−1) | −TΔSb (kcal mol−1) |
|---|---|---|---|
| MFA | 0.14 (±0.01) | −20.1 (±0.1) | 10.6 (±0.1) |
| MYA | 0.25 (±0.01) | −18.2 (±0.2) | 9.3 (±0.1) |
| MLA | 0.72 (±0.09) | −15.8 (±2.6) | 7.4 (±2.5) |
| MKA | 2.6 (±0.3) | −13.7 (±0.5) | 6.0 (±0.6) |
| MAA | >100 |
Mean values measured from at least three ITC experiments at 300 K in 10 mM sodium phosphate, pH 7.0. Standard deviations are in parentheses.
Entropic contributions to the free energy of binding were calculated from the Kd and ΔH values, with error propagated from those of Kd and ΔH.
The ITC results show that Q8 binds to MFA, MYA, MLA, and MKA, with equilibrium dissociation constant (Kd) values ranging from 0.14 to 2.6 μM. MAA showed weak binding (Kd > 100 μM). The peptide:Q8 stoichiometry for complexes containing MFA, MYA, MLA, and MKA was determined to be 1:1 (Q8:pepide). All 1:1 complexes in this study were confirmed by electrospray ionization time-of-flight mass spectrometry (ESI-TOF-MS, Figures S26–S50). Binding was enthalpically driven and entropically unfavorable. These results confirm the qualitative trends observed in the fluorescence screen, and they suggest a mode of binding similar to the Q8· YLA complex. On the basis of the significantly higher affinity of Q8 for MFA, MYA, MLA, and MKA versus MAA, we conclude that a second large residue is necessary for binding.
We were surprised to observe such high affinities for these complexes due the lack of an N-terminal aromatic residues. To the best of our knowledge, this is the first report of submicromolar binding of a synthetic receptor to peptides without an aromatic residue or to peptides with an N-terminal Met residue. These results significantly expand the scope of peptide sequences that can be targeted predictively by synthetic agents. It also opens the door to targeting natural and recombinant eukaryotic proteins, which are all translated with an N-terminal Met. Given the potential utility of this discovery, the remainder of this Article is concerned with determining the sequence–activity relationships for the binding of Q8 to Met-terminated peptides.
Sequence–Activity Relationships for Pair Inclusion in Tripeptides.
Prior work on the Q8 YLA system demonstrated >100-fold reduction in binding affinity to Tyr-Leu when separating the Tyr and Leu residues by an Ala (e.g., YAL), or when moving the Tyr-Leu binding site away from the N-terminus (e.g., AYL).24 Given the similar results observed thus far between the Q8 YLA and Q8·MXA complexes, we wanted to confirm that these sequence–activity relationships also apply to the Met-terminated peptides. We were also curious about the effects of reversing the sequence order of the inclusion pair (e.g., YLA vs LYA), which had not been addressed previously. To answer these questions, we designed three series of peptides of sequence MAX, AMX, and XMA, as sequence variants of the parent MXA, with X chosen to be Tyr, Leu, and Lys to represent aromatic, aliphatic, and basic residues, respectively. Thermodynamic constants for the binding of these nine peptides to Q8 were determined by ITC at 300 K in 10 mM sodium phosphate, pH 7.0 (Table 2 and Figures S2, S3, S5, S6, and S8–S12).
Table 2.
Thermodynamic Data for the Binding of Q8 to MXA versus MAX, AMX, and XMA Peptides
| peptide | Kd (μM)b | ΔHb (kcal mol−1) | −TΔSc (kcal mol−1) |
|---|---|---|---|
| MYAa | 0.25 (±0.01) | −18.2 (±0.2) | 9.3 (±0.1) |
| MAY | >100 | ||
| AMY | 6.1 (±0.5) | −13.8 (±0.2) | 6.6 (±0.3) |
| YMA | 1.3 (±0.2) | −16.6 (±0.8) | 8.5 (±0.8) |
| MLAa | 0.72 (±0.1) | −15.8 (±2.6) | 7.4 (±2.5) |
| MAL | >100 | ||
| AML | >100 | ||
| LMA | 0.60 (±0.11) | −12.1 (±0.2) | 3.5 (±0.2) |
| MKAa | 2.6 (±0.4) | −13.7 (±0.5) | 6.0 (±0.6) |
| MAK | >100 | ||
| AMK | >100 | ||
| KMA | 0.89 (±0.01) | −10.9 (±0.2) | 2.6 (±0.2) |
Shown again for reference.
Mean values measured from at least three ITC experiments at 300 K in 10 mM sodium phosphate, pH 7.0. Standard deviations are in parentheses.
Entropic contributions to the free energy of binding were calculated from the Kd and ΔH values, with error propagated from those of Kd and ΔH.
As compared to the MXA peptides, the MAX peptides retain the N-terminal Met but separate the target pair of residues by an Ala. The ITC data show that this change reduces binding affinity considerably, with Kd values >100 μM for all MAX peptides. Therefore, we conclude that the target residues need to be adjacent in sequence to achieve high affinity binding.
As compared to the MXA peptides, the AMX peptides keep the target pair of residues together but move the pair one residue away from the N-terminus. The ITC data show that this change reduces binding affinity considerably, with Kd values >100 μM for AML and AMK. The Kd value for AMY is 6.1 μM, which is a 24-fold weaker affinity than MYA but still considerably stronger than AML or AMK. In prior work, we reported that Q8 binds AYL with a Kd value of 3.1 μM under conditions identical to those reported here. These results show that Tyr can increase binding affinity for Q8 in several contexts.
As compared to the MXA peptides, the XMA peptides keep the target pair adjacent in sequence and at the N-terminus but reverse the sequence order of the target pair. The ITC data show that this change does not significantly impact the binding affinity. Q8 binding is 5-fold weaker for YMA than for MYA, 3-fold stronger for KMA than for MKA, and approximately the same for LMA and MLA. This result supports the conclusion that binding affinity is governed to a greater extent by the identity of the residues in the target pair than by their sequence order.
Effects of the Third Residue.
Given the preference of Q8 for binding a pair of neighboring residues at the N-terminus, we wanted to study the effects of the third residue on binding. We knew from the Q8 YLA study that the alanine methyl group experiences a downfield NMR chemical shift perturbation upon Q8 binding, suggesting that it could be interfering sterically with complex formation. Therefore, we designed a series of peptides that substitute Ala with Gly at the third position. In a tripeptide, however, the third position is also the C-terminus, and thus we extended the chain to five residues. Finally, we installed Tyr at the C-terminal position to facilitate quantifying the peptides by UV spectroscopy. As predicted from prior work20 and confirmed by the ITC and NMR results described below, we know that this C-terminal Gly-Tyr sequence binds weakly to Q8 and does not compete effectively with pair inclusion.
The ITC data (Table 3 and Figures S16–S21) show that the change from MXA to MXGGY increases binding affinity 2–4- fold, which we attribute to a reduction in steric hindrance by removing the methyl side chain. To further explore this effect, we designed two additional peptides, MKAGY and MKVGY, which systematically increase steric bulk at the third position within a pentapeptide. As expected, the binding affinity decreases as a function of the size of the side chain at the third position, with a 15-fold difference in affinity between MKGGY and MKVGY.
Table 3.
Thermodynamic Data for the Binding of Q8 to MXA versus MXGGY and MXZGY Peptides
| peptide | Kd (μM)b | ΔHb (kcal mol−1) | −TΔSc (kcal mol−1) |
|---|---|---|---|
| MYAa | 0.25 (±0.01) | −18.2 (±0.2) | 9.3 (±0.1) |
| MYGGY | 0.16 (±0.01) | −19.4 (±0.4) | 10.1 (± 0.4) |
| MLAa | 0.72 (±0.09) | −15.8 (±2.5) | 7.4 (±2.5) |
| MLGGY | 0.30 (±0.01) | −16.9 (±0.1) | 7.9 (±0.1) |
| LMAa | 0.60 (±0.11) | −12.1 (±0.2) | 3.5 (±0.2) |
| LMGGY | 0.16 (±0.02) | −22.9 (±0.2) | 13.5 (±0.3) |
| MKAa | 2.6 (±0.4) | −13.7 (±0.5) | 6.0 (±0.6) |
| MKGGY | 0.42 (±0.02) | −16.9 (±0.1) | 8.2 (±0.1) |
| MKAGY | 0.89 (±0.07) | −16.7 (±0.3) | 8.3 (±0.4) |
| MKVGY | 6.1 (±0.4) | −15.9 (±0.5) | 8.8 (±0.5) |
Shown again for reference.
Mean values measured from at least three ITC experiments at 300 K in 10 mM sodium phosphate, pH 7.0. Standard deviations are in parentheses.
Entropic contributions to the free energy of binding were calculated from the Kd and ΔH values, with error propagated from those of Kd and ΔH.
Additional Sequence Determinants at the Second Position.
To further elucidate the sequence–activity relationships in this binding motif, we asked whether other hydrophobic and basic residues could be tolerated at the second position. As analogues of MLGGY, the peptides MIGGY and MVGGY were tested for binding to Q8, and surprisingly showed low affinity by ITC (Table 4 and Figures S15 and S22–S25). We believe this is due to branching at the β carbon that occurs in Val and Ile residues but not in Leu. On the basis of the published semiempirical model of the Q8 YLA complex,24 that branching would force an alkyl group directly into the portal of Q8, thereby causing an unfavorable steric interaction. As an analogue to MKGGY, the peptide MRGGY was tested for binding to Q8 by ITC and found to bind 5-fold less tightly than MKGGY. We obtained MHGGY commercially but were unable to solubilize it. We speculate that His should bind poorly because its cationic side chain would be destabilized upon encapsulation within the hydrophobic Q8 cavity, whereas side chains of Lys and Arg are long enough to thread the cationic groups all of the way through Q8. Ser and Gly residues were also installed at the second position, and the resulting MSGGY and MGGGY peptides showed low affinity for Q8 by ITC. These results support the need for a large side chain at the second position that is either hydrophobic or basic but not branched at the β carbon.
Table 4.
Thermodynamic Data for the Binding of Q8 to MXGGY Peptides
| peptide | Kd (μM)b | ΔHb (kcal mol−1) | −TΔSc (kcal mol−1) |
|---|---|---|---|
| MLGGYa | 0.30 (±0.01) | −16.9 (±0.4) | 7.9 (±0.4) |
| MIGGY | >100 | ||
| MVGGY | >100 | ||
| MKGGYa | 0.42 (±0.02) | −16.9 (±0.1) | 8.2 (±0.1) |
| MRGGY | 2.1 (±0.2) | −12.4 (±0.4) | 4.6 (±0.5) |
| MSGGY | >100 | ||
| MGGGY | >100 |
Shown again for reference.
Mean values measured from at least three ITC experiments at 300 K in 10 mM sodium phosphate, pH 7.0. Standard deviations are in parentheses.
Entropic contributions to the free energy of binding were calculated from the Kd and ΔH values, with error propagated from those of Kd and ΔH.
Structural Characterization.
1H NMR spectroscopy was used to characterize the 25 purified peptides described above (Figures 3a and S51–S111). For each peptide, we include an overlay of one-dimensional spectra of the peptide in the absence of Q8, at a mole ratio of 0.5:1 Q8:peptide (which, importantly, is also 2:1 peptide:Q8 ratio), and at a mole ratio of 1:1 Q8:peptide. The mole ratios were determined by comparing peak integrations in the NMR spectra. When necessary, two-dimensional 1H–1H correlation spectroscopy (COSY) was used to assign the signals of bound and unbound peptides. Through-space interactions were characterized using two-dimensional 1H–1H nuclear Overhauser effect spectroscopy (NOESY) for some of the pure peptide samples and two-dimensional 1H–1H rotating frame nuclear Overhauser effect spectroscopy (ROESY) for some of the Q8 peptide complexes.
Figure 3.

(a) 500 MHz 1H NMR titrations of samples containing (a) a 1:1 mixture of Q8:peptide, (b) a 0.5:1 mixture of Q8:peptide, and (c) peptide. Spectra were acquired at 25 °C in D2O. (b) 500 MHz 2D ROESY spectra of samples containing (a) a 1:1 mixture of Q8:MYA and (b) a 1:1 mixture of Q8:MLA. The regions of the spectra displayed here reveal NOE crosspeaks between protons on neighboring side chains. Spectra were acquired at 25 °C in D2O solution.
The NMR data were analyzed for changes to the peptide spectra induced by the addition of Q8, including (1) perturbation in chemical shift, in which upfield perturbation indicates the positioning of that group within the cavity of Q8, and downfield perturbation indicates the positioning of that group proximal to carbonyl groups whether at the portal or in the peptide backbone;32 (2) change in the resolution of the signals corresponding to pairs of geminal protons upon binding; and (3) the kinetics of chemical exchange on the NMR time scale. These spectral characteristics are discussed here for each of the following categories of peptide structure:(1) the Met residue, which is present in all peptides; (2) the residue adjacent to Met for peptides that bind with high affinity (e.g., Leu in MLA or Lys in MKVGY); (3) the residue adjacent to the target pair (e.g., Ala in MLA or Val in MKVGY); and (4) C-terminal Tyr residues in the pentapeptides.
For the Met residues in all peptides, we observe an upfield perturbation in chemical shifts of the signals corresponding to the side chain protons. This indicates that the Met side chain of all peptides is included within the Q8 cavity. For tripeptides that bind with measurable affinity to Q8, vide supra, we observe increased resolution of signals corresponding to the β and γ geminal protons in the Met side chain upon Q8 binding. When the neighboring residue is Tyr or Phe, then the separation between geminal proton pairs on Met is greater than when the neighboring residue is Lys or Leu. We believe this difference is due to chemical shift anisotropy from the proximal aromatic ring.
There are differences in the kinetics of chemical exchange on the NMR time scale for the various mixtures of Q8 and peptide. Exchange is slow for all peptides containing the MY pair, but fast for MFA. Exchange is slow for MKGGY, MKAGY, and KMA but fast for MKVGY and MKA. Exchange is slow for AML but fast for MLA. In addition, all weak binders (Kd > 100 μM) as well as the peptides MRGGY, LMA, and LMGGY are fast exchange. The compounds in this study have fairly similar binding constants. For high-affinity binding, the relative kinetics of chemical exchange on the NMR time scale are dictated by the relationship between the unimolecular dissociation rate constant and the difference in chemical shift between the bound and unbound states (Δδ). Therefore, for functional groups with similar Δδ values, as observed for many complexes here, the observed differences in chemical exchange kinetics reflect differences in the association rate constants. Unfortunately, the NMR data alone are insufficient to draw deeper conclusions about the effects of peptide structure on the binding kinetics.
For peptides that bind tightly to Q8, we characterized the structural effects of the residue neighboring Met upon addition of Q8. We observe upfield perturbation in the chemical shifts for signals corresponding to protons on the side chains of Tyr, Phe, Leu, and Lys upon Q8 addition. In general, this result demonstrates the inclusion of these residues along with the neighboring Met within the Q8 cavity, i.e., pair inclusion. For peptides containing MY and MF sequences, it is interesting to note that the signals corresponding to the β protons on Tyr and Phe (i.e., proton c in Figure 3a) perturb downfield, suggesting their positioning near the portal carbonyl oxygens or near a peptide carbonyl in the complex. For peptides containing ML, the signals corresponding to the two β protons of Leu (i.e., proton q in Figure 3b) perturb in opposite directions upon binding Q8, leading to unusually large separation and suggesting that these protons project in different directions in the complex. 2D ROESY data show NOE crosspeaks between protons on the neighboring side chains of Met and Leu in the Q8 MLA complex, and of the side chains of Met and Tyr in the Q8 MYA and Q8 YMA complexes (Figures 3b and S60, S68, and S74). These NOEs are not present in the absence of Q8, as observed in the NOESY spectra (Figures S58, S66, and S72).
The thermodynamic data presented in Table 3 led us to conclude that the residue adjacent to the binding pair can impact the binding affinity, likely due to steric effects. When that residue is Ala, the NMR signal corresponding to the Ala methyl protons perturbs downfield upon the addition of Q8. This result was also observed previously for the Q8 YLA complex and indicates the methyl group is positioned near the Q8 portal. In MKVGY, the signal corresponding to one of the two Val methyl groups perturbs downfield upon addition of Q8. These data support our hypothesis that there may be steric interactions between the Q8 portal and the side chain of the third residue.
In the pentapeptide series, we included a Tyr residue at the C-terminus to aid in quantifying the peptides by UV spectroscopy. Prior work led us to predict that we would observe negligible competition for Q8 binding by this residue. To support this prediction, we studied the effects of Q8 addition on the NMR signals corresponding to the side chain of the C-terminal Tyr residues. For the pentapeptides containing an N-terminal inclusion pair (e.g., MLGGY), we observe minimal changes to the spectra of the C-terminal Tyr upon Q8 addition. In MYGGY, there are two Tyr residues, and chemical exchange is slow on the NMR time scale. We observe upfield perturbation of one of these sets of signals at a 1:1 Q8:peptide ratio, and we believe these signals correspond to the Tyr in the MY inclusion pair. For pentapeptides that do not contain a strong-binding inclusion pair, we observe significant broadening and upfield chemical shift perturbation for the C-terminal Tyr residue. This result is consistent with the absence of a competitive binding site.
CONCLUSIONS
This study establishes a novel approach to the molecular recognition of Met-terminated peptides and nonaromatic peptides with submicromolar affinity in aqueous solution. It also shows that cucurbit[n]uril-mediated peptide recognition does not require an aromatic residue for high affinity. In the absence of an aromatic residue, additional binding energy can be generated by the inclusion of the side chains of two immediately neighboring residues within the cavity of Q8. The detailed sequence–activity relationships presented here suggest several rules for targeting Met-containing peptides with Q8 (Figure 4). The residue paired with Met should be large and either hydrophobic or cationic (i.e., Leu, Lys, Arg, Tyr, or Phe), but it cannot have branching at the β carbon (i.e., Val, Ile). The sequence order of the target pair is not as important as its amino acid composition. Targeting the pair at the N-terminus enhances binding affinity in all cases and is necessary for nonaromatic target pairs. The residue adjacent to the target pair on the C-terminal side should be as small as possible to minimize steric interactions.
Figure 4.

Summary schematic of the sequence determinants that lead to high-affinity binding to Q8.
Methionyl aminopeptidase (MetAP) is known to remove N-terminal Met from newly translated proteins when the second residue is small and neutral.33 In the pair inclusion motif, Q8 binds tightly to N-terminal Met only when the second residue is large and hydrophobic or cationic, which should withstand degradation by MetAP. This characteristic combined with the ability of cucurbit[n]urils to target N-termini22 and disordered loops34 in folded proteins suggests that Q8 could be useful for targeting newly translated and unmodified proteins. More generally, the high affinity binding of recombinant proteins via a two-residue target site would constitute a truly minimal protein affinity tag that should have a negligible impact on protein structure and function.
Supplementary Material
ACKNOWLEDGMENTS
We gratefully acknowledge Elena Boms and Prof. Jeffery W. Kelly for valuable discussions. This work was supported in part by grants from the Welch Foundation (W-1640, W-0031), the National Institutes of Health (R15-GM126511–01), the National Science Foundation (CHE-1309978, CHE-1726441), and Trinity University. Z.H. was a Beckman Scholar, a Semmes Distinguished Scholar, and a Goldwater Scholar. C.W.B. acknowledges the Institute for Basic Science (IBS-R019-D1) as well as the BK21 Plus Program as funded by the Ministry of Education and the National Research Foundation of Korea for their support.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.8b07865.
Experimental details, thermodynamic binding data, mass spectrometry data, and NMR and fluorescence spectroscopy (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Smith B Synthetic Receptors for Biomolecules: Design Principles and Applications; Royal Society of Chemistry: London, UK, 2015. [Google Scholar]
- (2).Kubota R; Hamachi I Chem. Soc. Rev 2015, 44, 4454–4471. [DOI] [PubMed] [Google Scholar]
- (3).Hugget J; O’Grady J Molecular Diagnostics: Current Research and Applications; Horizon Press: Norfolk, UK, 2014. [Google Scholar]
- (4).Patrick GL An Introduction to Medininal Chemistry, 5th ed.; Oxford University Press: New York, 2005. [Google Scholar]
- (5).Bertozzi CR Acc. Chem. Res 2011, 44, 651–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Yin H; Hamilton AD Angew. Chem., Int. Ed 2005, 44, 4130–4163. [DOI] [PubMed] [Google Scholar]
- (7).Peczuh MW; Hamilton AD Chem. Rev 2000, 100, 2479–2494. [DOI] [PubMed] [Google Scholar]
- (8).Bunka DHJ; Stockley PG Nat. Rev. Microbiol 2006, 4, 588–596. [DOI] [PubMed] [Google Scholar]
- (9).Moriera IS; Fernandes PA; Ramos MJ Proteins: Struct., Funct., Genet 2007, 68, 803–812. [DOI] [PubMed] [Google Scholar]
- (10).Day ES; Cote SM; Whitty A Biochemistry 2012, 51, 9124–9136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Meyer EA; Castellano RK; Diederich F Angew. Chem., Int. Ed 2003, 42, 1210–1250. [DOI] [PubMed] [Google Scholar]
- (12).Salonen LM; Ellermann M; Diederich F Angew. Chem., Int. Ed 2011, 50, 4808–4842. [DOI] [PubMed] [Google Scholar]
- (13).Martinez CR; Iverson BL, Chem BL. Sci 2012, 3, 2191–2201. [Google Scholar]
- (14).Mahadevi AS; Sastry GN Chem. Rev 2013, 113, 2100–2138. [DOI] [PubMed] [Google Scholar]
- (15).Ma B; Elkayam T; Wolfson H; Nussinov R Proc. Natl. Acad. Sci. U. S. A 2003, 100, 5772–5777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Espinoza-Fonseca LM Mol. BioSyst 2012, 8, 237–246. [DOI] [PubMed] [Google Scholar]
- (17).Rahman MM; Muhseen ZT; Junaid M; Zhang H Curr. Protein Pept. Sci 2015, 16, 502–512. [DOI] [PubMed] [Google Scholar]
- (18).By contrast, submicromolar binding of His-containing peptides has been observed using coordinate-covalent interactions. See: Hortalá MA; Fabbrizzi L; Marcotte N; Stomeo F; Taglietti A J. Am. Chem. Soc 2003, 125, 20–21. [DOI] [PubMed] [Google Scholar]
- (19).Tashiro S; Tominaga M; Kawano M; Therrien B; Ozeki T; Fujita MJ Am. Chem. Soc 2005, 127, 4546–4547. [DOI] [PubMed] [Google Scholar]
- (20).Urbach AR; Ramalingam V Isr. J. Chem 2011, 51, 664. [Google Scholar]
- (21).Bockus AT; Urbach AR In Aromatic Interactions: Frontiers in Knowledge and Application; Johnson DW, Hof F, Eds.; Royal Society of Chemistry: Cambridge, UK, 2017. [Google Scholar]
- (22).Chinai JM; Taylor AB; Ryno LM; Hargreaves ND; Morris CA; Hart PJ; Urbach AR J. Am. Chem. Soc 2011, 133, 8810–8813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Li W; Bockus AT; Vinciguerra B; Isaacs L; Urbach AR Chem. Commun 2016, 52, 8537–8540. [DOI] [PubMed] [Google Scholar]
- (24).Smith LC Smith; Leach DG; Blaylock BE; Ali OA; Urbach AR J. Am. Chem. Soc 2015, 137, 3663–3669. [DOI] [PubMed] [Google Scholar]
- (25).Mock WL Top. Curr. Chem 1995, 175, 1–24. [Google Scholar]
- (26).Lagona K; Mukhopadhyay P; Chakrabarti S; Isaacs L Angew. Chem., Int. Ed 2005, 44, 4844–4870. [DOI] [PubMed] [Google Scholar]
- (27).Barrow SJ; Kasera S; Rowland MJ; del Barrio J; Scherman OA Chem. Rev 2015, 115, 12320–12406. [DOI] [PubMed] [Google Scholar]
- (28).Biedermann F; Nau WM; Schneider H-J Angew. Chem., Int. Ed 2014, 53, 11158–11171. [DOI] [PubMed] [Google Scholar]
- (29).Ali OA; Olson EM; Urbach AR Supramol. Chem 2013, 25, 863–368. [Google Scholar]
- (30).Biedermann F; Rauwald U; Cziferszky M; Williams KA; Gann LD; Guo BY; Urbach AR; Bielawski CW; Scherman OA Chem. - Eur. J 2010, 16, 13716–13722. [DOI] [PubMed] [Google Scholar]
- (31).Heitmann LM; Taylor AD; Hart PJ; Urbach AR J. Am. Chem. Soc 2006, 128, 12574–12581. [DOI] [PubMed] [Google Scholar]
- (32).Moon K; Kaifer AE Org. Lett 2004, 6, 185–188. [DOI] [PubMed] [Google Scholar]
- (33).Hirel P-H; Schmitter J-M; Dessen P; Fayat G; Blanquet S Proc. Natl. Acad. Sci. U. S. A 1989, 86, 8247–8251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Sonzini S; Marcozzi A; Gubeli RJ; van der Walle CF; Ravn P; Herrmann A; Scherman OA Angew. Chem., Int. Ed 2016, 55, 14000–14004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.