

Abstract
Background
This randomized, placebo-controlled, crossover trial examined the antidepressant efficacy of the muscarinic antagonist scopolamine in major depressive disorder subjects with more severe and refractory forms of major depressive disorder relative to previous reports.
Methods
Participants included 23 medication-free major depressive disorder subjects (12 F/11 M, 20–55 years) currently experiencing a major depressive episode. Subjects had scored ≥20 on the Montgomery-Asberg Depression Rating Scale. Following a single-blind, placebo lead-in, participants were randomized to receive 2 counterbalanced blocks of 3 i.v. infusions of scopolamine (4 μg/kg) and placebo in a double-blind manner. The primary and secondary outcomes were the Montgomery-Asberg Depression Rating Scale and the Hamilton Anxiety Rating Scale, respectively. Magnetoencephalography and plasma brain-derived neurotrophic factor concentrations were obtained prior to and after each treatment phase.
Results
As assessed by both the Montgomery-Asberg Depression Rating Scale and Hamilton Anxiety Rating Scale, scopolamine had no significant antidepressant or anxiolytic effects relative to placebo. No significant drug vs placebo effects were seen in magnetoencephalography gamma power or brain-derived neurotrophic factor plasma concentrations, and brain-derived neurotrophic factor changes did not correlate with change in Montgomery-Asberg Depression Rating Scale score in response to scopolamine.
Conclusions
These results do not support the efficacy of scopolamine for more severe or refractory forms of depression. No pre- to post-infusion changes in plasma brain-derived neurotrophic factor were detected, and magnetoencephalography gamma power changed only in the placebo lead-in, suggesting that these biomarker measures were not affected by scopolamine in this cohort. While difficult to interpret given the lack of antidepressant response, the findings suggest that the neurobiological effects of ketamine and scopolamine are at least partly distinct.
Keywords: scopolamine, major depressive disorder, ketamine, biomarkers
Significance Statement
In this crossover trial of subjects with major depressive disorder (MDD), scopolamine administration was not associated with significant improvement in depressive or anxious symptoms compared to placebo. Subjects in this trial may have been more treatment-resistant than in previous scopolamine studies. Neurophysiological markers such as magnetoencephalography (MEG) gamma power and plasma brain derived neurotrophic factor (BDNF) levels—which have been shown to change in response to ketamine administration—demonstrated no significant association with scopolamine administration. These findings do not provide evidence that scopolamine and ketamine exert similar neurobiological effects.
Introduction
Major depressive disorder (MDD) is a major public health concern. Almost one-third of individuals in the US will experience a major depressive episode at some point in their lives (Kessler et al., 2012). Of those who screen positive for depression, more than 70% will not receive appropriate treatment (Olfson et al., 2016), perhaps discouraged by the significant lag time between starting medications and symptomatic relief or by the relative lack of efficacy of current treatment options. For instance, up to one-third of MDD subjects will not achieve symptom remission even after 4 antidepressant trials (Trivedi et al., 2006a, 2006b). Psychopharmacological research over the past few decades has not significantly advanced the number of approved drug treatments for depression beyond the monoaminergic interventions in use for more than 50 years. Thus, the need to develop new and rapid antidepressant treatments is great and will likely require targeting novel neurobiological substrates.
The cholinergic neurotransmitter system, both muscarinic and nicotinic receptors, has been implicated as a potential substrate of rapid antidepressant response. The muscarinic cholinergic system in particular has considerable preclinical and clinical evidence to support its role in regulating mood symptoms (Drevets et al., 2013). Several decades ago, Janowsky and colleagues hypothesized that an adrenergic/cholinergic balance underlies mood disorders (Janowsky et al., 1972). In preclinical studies, rats bred selectively for supersensitivity of the muscarinic receptors (the Flinders Sensitive Line) displayed behavioral depressive-like symptoms in the presence of agents that increase cholinergic function, including increased behavioral despair in the forced swim test (Overstreet et al., 1992). Anticholinergic agents such as scopolamine were found to reduce behavioral despair in animals (Betin et al., 1982). In humans, challenge studies found that the anticholinesterase inhibitor physostigmine increased cholinergic activity and exacerbated depressive symptoms in currently depressed MDD subjects (Janowsky et al., 1972, 1974; Risch et al., 1981; Nurnberger et al., 1983). Physiologic studies found that neuroendocrine and pupillary responses to physostigmine were abnormally increased in depressed individuals (Janowsky et al., 1985; Dilsaver, 1986; Rubin et al., 1999), and genetic studies found that a variation in the type 2 muscarinic (M2) cholinergic receptor gene was associated with an elevated incidence or severity of MDD (Comings et al., 2002; Wang et al., 2004; Cannon et al., 2011).
In treatment trials, open-label administration of the muscarinic receptor antagonist scopolamine (0.4 mg i.m.) to 10 depressed patients and 10 healthy controls before bedtime for 3 consecutive nights was found to have a small but statistically significant antidepressant effect on the second morning of treatment (Gillin et al., 1991). Furey and Drevets, investigating the role of the cholinergic system in the cognitive symptoms of depression, unexpectedly found that scopolamine had rapid antidepressant effects in depressed subjects. In the first of the 2 studies (Furey and Drevets, 2006), 19 medication-free subjects meeting DSM-IV criteria for recurrent MDD or bipolar disorder (subjects were not required to be treatment resistant) were included in a double-blind, placebo-controlled pilot outpatient study; 6 subjects had been chronically ill (more than 2 years) and one was unresponsive to previous treatment. Scopolamine hydrobromide (4 µg/kg) administered i.v. over 15 minutes rapidly reduced the severity of depressive symptoms within 3 to 5 days after the first treatment (effect size 2.2–3.4), although subjects reported marked improvements in clinical symptoms by the evening or the morning after scopolamine administration. A subsequent study (Drevets and Furey, 2010) replicated this initial finding; scopolamine’s antidepressant efficacy was observed within 3 to 5 days in 23 outpatients with MDD (effect size 1.2–1.7). Across both groups (i.e., placebo/active and active/placebo) of this second double-blind, placebo-controlled, crossover study, 13 of 22 subjects were chronically ill (more than 2 years duration), and 6 of 22 had not responded to treatment in a previous depressive episode (one subject dropped out prior to receiving any intervention).
Ketamine, a glutamatergic modulator with antagonistic properties at the N-methyl-D-aspartate receptor, has similarly demonstrated rapid, though short-lived, antidepressant effects (Kishimoto et al., 2016). In one study, effect sizes were 1.46 at day 1 and 0.68 at 1 week post-ketamine infusion (Zarate et al., 2006). Preclinical studies have found that ketamine and scopolamine produce comparable antidepressant effects, suggesting that the 2 agents may share common cellular and molecular mechanisms that rapidly increase extracellular glutamate. For instance, both drugs are thought to exert their effects on gamma-aminobutyric acid (GABA)-ergic interneurons that synapse on presynaptic glutamatergic neurons in the prefrontal cortex; ketamine blocks interneuron N-methyl-D-aspartate receptors and scopolamine blocks muscarinic receptors (Li et al., 2010; Voleti et al., 2013; Wohleb et al., 2017). These two initial actions inhibit the inhibitory interneuron, resulting in decreased GABAergic activity that, in turn, leads to disinhibition of pyramidal neurons and increased extracellular glutamate release, activation of the mammalian target of rapamycin complex 1 cascade, elevated brain derived neurotrophic factor (BDNF) concentrations, and an increased number and function of spine synapses in the prefrontal cortex (Li et al., 2010; Voleti et al., 2013; Hare et al., 2017; Wohleb et al., 2017).
We previously suggested that a novel approach to developing biomarkers of antidepressant response would be to contrast interventions with a rapid onset of action, such as sleep deprivation or i.v. drugs (e.g., ketamine or scopolamine) (Zarate et al., 2013; Niciu et al., 2014). Increases in plasma BDNF levels have been shown to be associated with antidepressant response to ketamine (Haile et al., 2014), though other studies have not demonstrated this association (Machado-Vieira et al., 2009). In animal models, BDNF release was recently shown to be necessary for scopolamine’s antidepressant effects (Ghosal et al., 2018). Neuroimaging modalities may also offer an important tool for assessing target engagement and understanding the underlying mechanisms of drug action (Carmichael et al., 2017). A recent study of subjects with treatment-resistant MDD who received i.v. ketamine found that depressed and healthy subjects exhibited robust increases in gamma power in response to ketamine administration (Nugent et al., 2018). The relationship between increased gamma power and antidepressant response was modulated by baseline gamma levels, such that large increases in gamma power were associated with better antidepressant response in MDD subjects with lower baseline gamma; this relationship was inverted in MDD subjects with higher baseline gamma.
This study sought to examine the antidepressant efficacy of the muscarinic antagonist scopolamine in MDD patients across a broad range of severity and treatment resistance (i.e., there were no exclusion criteria for symptom severity or past number of treatment trials). This study also sought to generate preliminary evidence for a shared mechanism of action between ketamine and scopolamine, testing the hypothesis that, like ketamine, scopolamine’s antidepressant effects would be associated with increased plasma BDNF and increased MEG gamma power from baseline to post-treatment.
METHODS
Participants
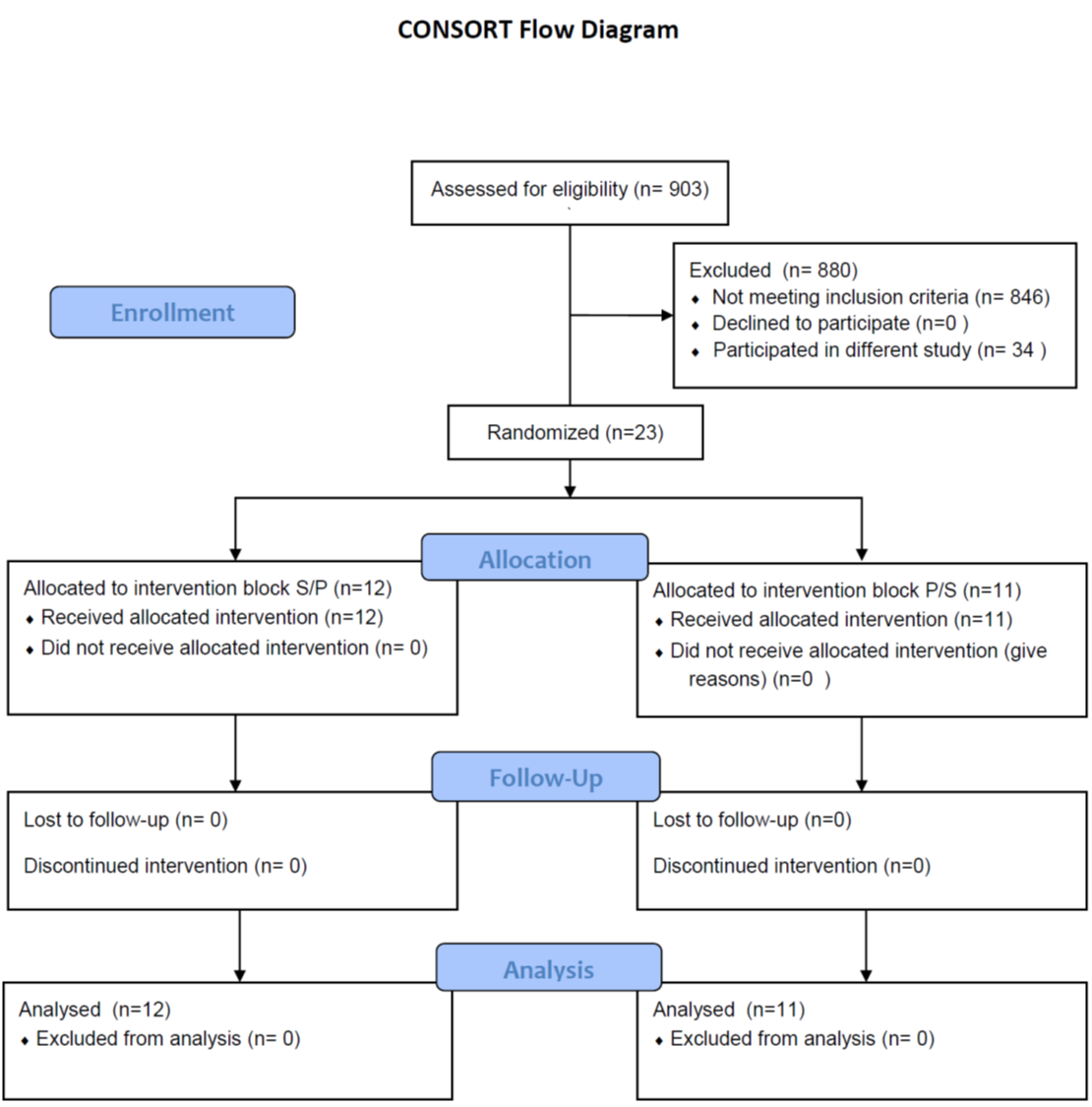
Subjects were recruited from local inpatient psychiatric units, the internet, and local and national physician referrals and were studied as inpatients (n = 7) and outpatients (n = 16) at the National Institute of Mental Health (NIMH) Mood Disorders Research Unit in Bethesda, Maryland. Eligible participants were 11 male and 12 female inpatients, 18 to 55 years old (n = 23); all subjects were diagnosed with MDD, currently depressed without psychotic features, as assessed by the Structured Clinical Interview for Axis I DSM-IV Disorders—Patient Version (First et al., 2001), and were required to have a score ≥20 on the Montgomery-Asberg Depression Rating Scale (MADRS) at screening and at the start of each infusion.
Subjects were judged clinically not to be at serious risk for suicide. Other exclusion criteria included a history of drug or alcohol abuse within 1 year or a lifetime history of alcohol or drug dependence; a current or past history of other Axis I disorders that preceded the onset of MDD; general MRI exclusion criteria; vision and/or hearing problems severe enough to interfere with testing; electrocardiographic evidence of ischemia, arrhythmia, conduction defect, or myocardial infarction; current blood pressure of >140 mm Hg or <90 mm Hg systolic, or >90 mm Hg diastolic; clinically significant cerebrovascular or cardiovascular disease; congestive heart disease; angina pectoris; advanced arteriosclerosis; gross neurological impairment; hyperthyroidism; known hypersensitivity to anticholinergic agents; renal or hepatic impairment; clinical history of glaucoma or narrow angle glaucoma; age of onset of MDD >45 years; exposure within 2 weeks to medications likely to affect cerebral blood flow and metabolism or likely to interact with anti-cholinergic medications (e.g., narcotics or anti-cholinergic agents), as verified by history and urine drug screen; concomitant treatment with psychotropic medications in the two weeks before randomization (6 weeks for fluoxetine); and weight >275 pounds. In addition, female subjects could not be pregnant or nursing.
All subjects were in good physical health, as determined by medical history, physical exam, blood labs, ECG, chest x-ray, urinalysis, and toxicology. The study was approved by the Combined Neuroscience Institutional Review Board at the National Institutes of Health (NIH; NCT00369915). All subjects provided written informed consent before entry into the study and were assigned a Clinical Research Advocate from the NIMH Human Subjects Protection Unit to monitor the consent process and research participation throughout the study.
Study Design
Based on prior studies, an effect size (d = 0.8) was used to estimate the difference between pre-and post-treatment response for the purposes of the power calculation. With 80% power to detect an effect using 2-tailed significance at 0.05, we estimated that a minimum of 15 subjects would be needed.
This was a single-center, randomized, placebo-controlled, crossover design with a single-blind placebo lead-in. After a 2-week drug-free period, seven 15-minute infusions of either placebo or 4 μg/kg of scopolamine were administered (Figure 1). The first of these infusions was the single-blind placebo lead-in, followed by 2 blocks of 3 placebo or scopolamine infusions. Study participants were randomized to block order (placebo/scopolamine (P/S): n = 11; scopolamine/placebo (S/P): n = 12). Blocks were counterbalanced across subjects so that there were equal numbers of P/S and S/P block orders. Both the MADRS and the Hamilton Anxiety Rating Scale (HAM-A) were administered prior to each infusion and at the final follow-up visit.
Figure 1.

Study design. Following a 2-week wash-out and a single-blind placebo lead-in, participants were randomized to receive 2 counterbalanced blocks of 3 i.v. infusions of scopolamine (4 μg/kg) and placebo infusions. Block order was randomized and infusions were administered in a double-blind manner. Please note that the last follow-up visit is not represented in this diagram. Missing data: In placebo/scopolamine (P/S) group, Block 2, visit 2: n = 2 missing Montgomery-Asberg Depression Rating Scale (MADRS), n = 4 missing Hamilton Anxiety Rating Scale (HAM-A); in S/P group, Block 0, visits 1 and 2: n = 1 missing HAM-A, Block 1, visit 1: n = 1 missing HAM-A, Block 2, visit 2: n = 2 missing MADRS, n = 4 missing HAM-A.
Vital signs were monitored during the infusion and for 135 minutes post-infusion. An ECG, complete blood counts, electrolyte panels, and liver function tests were obtained at baseline and at the end of the study.
Data Collection
The primary outcome measure was the MADRS, a 10-item clinician-administered scale of depressive symptoms, and the secondary outcome was the HAM-A. Antidepressant response was defined using existing conventions to classify percent improvement from the final assessment of the placebo lead-in: improvements ≥50% on the MADRS were considered full response and MADRS scores ≤10 indicated remission (Zimmerman et al., 2004). Although the number of prior treatment trial failures was not systematically collected, past treatment history was assessed through clinician interviews of medical and psychiatric history. The maximum number of failed prior antidepressant treatment trials was not an exclusion.
Resting-state MEG recordings were obtained following the single-blind placebo lead-in and on the day of the infusion (SCOP-Day0 and Placebo-Day0). Up to two 250-second resting state recordings per time point were analyzed; in general, one recording occurred at the beginning of the session, and the second was acquired approximately 30 minutes to 1 hour later after a series of tasks (to be reported elsewhere). For the resting state recordings, subjects were instructed to relax with their eyes closed and remain still. All data were acquired on a 275-channel CTF system at 1200 Hz. Background environmental magnetic noise was attenuated by synthetic third gradient balancing. T1 weighted MRI scans were acquired on a 3T GE scanner for co-registration.
Blood samples (for BDNF) were obtained using the vacutainer system prior to and after each treatment phase, as previously described (Machado-Vieira et al., 2009). Briefly, blood samples were centrifuged at 1000 rpm at 4°C for 5 minutes and stored at −80°C until assay. BDNF concentrations were measured using an anti-BDNF sandwich ELISA kit (Chemicon International) according to the manufacturer’s instructions. Plasma was diluted 1:2 with sample buffer and carried out in duplicate blind to clinical information. BDNF standard solution was diluted to concentrations from 7.8 to 500 pg/mL of BDNF in a microplate reader to create the standard curve for BDNF concentrations. After the addition of streptavidin enzyme, substrate, and stop solution, BDNF concentrations were determined by absorbance in 450 nm using optical density values based on the standard curve values.
Statistical Analysis
Clinical Data Analysis
The model was a repeated-measures ANOVA with a compound symmetry covariance structure and restricted maximum likelihood estimation with a random effect of subject. Degrees of freedom were calculated using the Satterthwaite approximation. The first and last on-drug assessments for each period were selected for analysis, such that baseline assessments 1 and 2 formed Block 0, assessments 3 and 5 formed Block 1, and assessments 6 and 8 formed Block 2. The model included effects for randomization, block, assessment, and their interactions. Significant interactions were probed using posthoc tests with Tukey-Kramer adjustment. Effect sizes were quantified using the least square mean estimates. Although mixed models are robust to missing data, which did occur in this study, the missing data at random assumption is not directly testable. For this reason, we carried out 2 sensitivity analyses to assess the robustness of the primary outcome. First, we excluded altogether participants for whom any datapoint was missing. Second, we analyzed the first arm of the trial as a parallel design. Complementary analyses were performed to analyze secondary hypotheses. In these mixed models, baseline values were controlled and fixed effects of block (Block 1 or Block 2), order (randomization group), assessment (first or last within block), and drug (scopolamine versus placebo) were entered. Again, the model was a repeated-measures ANOVA with a compound symmetry covariance structure and restricted maximum likelihood estimation, with a random effect of subject. All data analyses were performed using SAS/STAT Version 9.3.
MEG Data Analysis
MEG data were processed using CTF software (http://www.ctf.com), MNE-python (Gramfort et al., 2013), Analysis of Functional NeuroImages (AFNI) (Cox, 1996), and routines developed in-house. This work used the computational resources of the NIH HPC Biowulf cluster (http://hpc.nih.gov). Each MEG dataset was filtered using a high-pass filter of 2 Hz and visually inspected to identify and mark time periods with significant muscular, ocular, or movement artifacts. Up to 10 segments of 15-second duration outside marked artifacts were identified in an automated fashion. Datasets were discarded if at least five 15-second artifact-free segments could not be defined. All further described imaging analyses and quality control measures were carried out on the clean epochs.
Data were localized to source space on a 5-mm grid using synthetic aperture magnetometry (Robinson and Vrba, 1999), and a multisphere head model was calculated from co-registered MRI scans. MRI and MEG images were coregistered using MRI-visible fiducial markers placed on the head at the time of MRI scanning. Beamformer weights were calculated using a bandpass frequency of 2 to 100 Hz, and power was normalized by the projected noise floor of the virtual sensor. The resulting images represented root-mean-square power in the gamma band (30–50 Hz). All images were warped to Talairach space using AFNI and masked to remove non-brain matter and cerebellum. The final gamma band images were then normalized by the square root of the sum of squared images for 6 canonical bands between 2 and 100 Hz (delta, theta, alpha, beta, gamma, and high gamma). From this point forward, “gamma power” refers to the normalized root-mean-square gamma power.
Images were analyzed using a linear mixed model implemented in the AFNI routine 3dLME (Chen et al., 2013). If more than one usable recording existed for a given subject and session, both were included and coded as having occurred before or after a battery of cognitive tasks (to be reported elsewhere). MEG scans were obtained 60 to 120 minutes following scopolamine or placebo infusion. Session (baseline, Placebo-Day0, SCOP-Day0) and run number (first or second recording) were included in the model. The initial baseline scan was conducted during the placebo lead-in phase. Gender and age were initially included as main effects and removed if nonsignificant. Posthoc tests were performed within the 3dLME routine to assess individual contrasts. False discovery rate (FDR) over the contrast image was used to determine significance, with a threshold set at PFDR < .05.
BDNF
Plasma BDNF concentrations were measured immediately before and 150 minutes after the first infusion of each condition (i.e., Session 2 and Session 5). The effect of scopolamine on natural log-transformed BDNF was assessed using a mixed model, with repeated effect of time (0 minutes vs 150 minutes) and a random subject effect. All BDNF analyses included age, sex, and weight as covariates.
RESULTS
General Characteristics
Of the 23 participants enrolled into the protocol, 11 were assigned to the P/S order group and 12 were assigned to the S/P order group. Participant characteristics are presented in Table 1. There were no premature withdrawals, and all enrolled participants completed the protocol.
Table 1.
Subject Characteristics
| P/S (n = 11) | S/P (n = 12) | Total (N = 23) | ||||
|---|---|---|---|---|---|---|
| M | SD | M | SD | M | SD | |
| Age (y) | 32.91 | 9.08 | 40.42 | 11.32 | 36.83 | 10.78 |
| Age of onset (y) | 17.3 | 6.96 | 22.5 | 10.22 | 20.14 | 9.08 |
| Duration of illness (y) | 14.6 | 10.05 | 17.92 | 13.54 | 16.41 | 11.92 |
| MADRS | 31.64 | 4.2 | 34.08 | 4.25 | 32.91 | 4.32 |
| HAM-A | 19 | 5.67 | 25.73 | 8.33 | 22.36 | 7.76 |
| n | % | n | % | n | % | |
| Male | 7 | 63 | 4 | 33 | 11 | 48 |
| Race | ||||||
| White, non-Hispanic | 9 | 82 | 6 | 50 | 15 | 65 |
| Black or multiracial | 2 | 18 | 3 | 25 | 5 | 22 |
| Unknown | 0 | 3 | 25 | 3 | 13 | |
| Comorbid diagnoses | ||||||
| Anxiety disorder | 4 | 36 | 3 | 25 | 7 | 30 |
| Obsessive compulsive disorder | 0 | 3 | 25 | 3 | 13 | |
| Posttraumatic stress disorder | 1 | 9 | 2 | 17 | 3 | 13 |
| Personal history of alcohol/substance abuse | 1 | 9 | 6 | 50 | 7 | 30 |
| Medication response history | ||||||
| Naïve | 2 | 18 | 4 | 33 | 6 | 26 |
| Resistant | 7 | 63 | 8 | 66 | 15 | 65 |
| Responder | 2 | 18 | 0 | 2 | 9 | |
| Previous medication trials | ||||||
| 0–1 | 3 | 27 | 5 | 42 | 8 | 35 |
| 2–3 | 5 | 45 | 3 | 25 | 8 | 35 |
| 4–7 | 1 | 9 | 4 | 33 | 5 | 22 |
| 8+ | 2 | 18 | 0 | 2 | 9 | |
| Previous ECT trial | 2 | 18 | 3 | 25 | 5 | 22 |
Abbreviations: ECT, electroconvulsive therapy; HAM-A, Hamilton Anxiety Rating Scale; MADRS, Montgomery-Asberg Depression Rating Scale; P/S, randomized to placebo then scopolamine; S/P, randomized to scopolamine then placebo.
Primary and Secondary Outcomes
Mean MADRS scores at all assessments are shown in Figure 2. A significant effect of block was observed (F(2,43.1) = 10.82, P = .0002), explained by moderate improvements across both randomization groups from Block 0 to Block 1 (Cohen’s d = 0.85, 95% CI: 0.22–1.47; t(41.7) = 2.74, Padj = .02) and to Block 2 (Cohen’s d = 1.39, 95% CI: 0.79–2.00; t(43.9) = 4.62, Padj = .0001). The improvement from Block 1 to Block 2 exceeded the threshold for significance (Cohen’s d = 0.58, 95% CI: -0.03–1.19; t(101) = 2.37, Padj = .14). However, no main effect or interaction with randomization was observed, indicating that scopolamine had no effect on the primary outcome (Table 2). Two of the 23 participants (one each in the S/P and P/S group) met criteria as responders during the scopolamine condition (i.e., ≥50% improvement in MADRS from baseline to post-treatment). One of these responders also met criteria for remission (MADRS ≤10). One participant who responded in the scopolamine condition also met criteria for response in the placebo condition. Finally, 2 participants in the S/P group who did not respond in the scopolamine condition demonstrated remission in the placebo condition.
Figure 2.

Montgomery-Asberg Depression Rating Scale (MADRS) and Hamilton Anxiety Rating Scale (HAM-A) scores. Assessments 4 and 7 were excluded from the analysis. Randomization group by block interaction was not significant for either scale. A main effect of block was significant for both scales. Posthoc tests indicated that for MADRS both Block 1 (padj = .02) and Block 2 (padj = .0001) differed from Block 0 but not from one another (Padj = .14). HAM-A scores for Block 1 (Padj = .01) and Block 2 (Padj = .001) differed from Block 0 but not from one another (Padj = .59). P, placebo; S, scopolamine.
Table 2.
Results of Repeated-Measures ANOVA
| Num DF | Den DF | F | P | |
|---|---|---|---|---|
| MADRS | ||||
| Block | 2 | 43.1 | 10.82 | .0002 |
| Visit | 1 | 62 | 0.65 | .42 |
| Group | 2 | 62 | 0.09 | .91 |
| Block*visit | 1 | 21.1 | 0.07 | .79 |
| Block*GROUP | 2 | 43.1 | 0.65 | .53 |
| Visit*group | 1 | 62 | 0.08 | .78 |
| Block*visit*group | 2 | 62 | 0 | 1.00 |
| HAM-A | ||||
| Block | 2 | 42.4 | 8.33 | .0009 |
| Visit | 1 | 56.9 | 4.72 | .03 |
| Group | 1 | 21.3 | 2.29 | .15 |
| Block*visit | 2 | 56.8 | 1.82 | .17 |
| Block*group | 2 | 42.4 | 1.2 | .31 |
| Visit*group | 1 | 56.9 | 2.59 | .11 |
| Block*visit*group | 2 | 56.8 | 1.36 | .26 |
Abbreviations: Den DF, denominator degrees of freedom; HAM-A, Hamilton Anxiety Rating Scale; Num DF, numerator degrees of freedom; MADRS, Montgomery-Asberg Depression Rating Scale.
DFs were calculated using the Satterthwaite approximation. Repeated measures nested within block were modeled with a compound symmetry variance structure and a random subject effect.
Four participants (2 in each of the randomization groups) did not provide data at the final assessment. To assess robustness of the results to these missing data, the 4 participants were excluded and the analysis re-run. The results did not differ (i.e., main effect of block, P = .0017; main effect of randomization, P = .79; block-by-randomization interaction, P = .53). Next, we analyzed the first arm of the trial, controlling for the session 2 (baseline) score. Scopolamine had no effect [F(1,20) = 0.38, P = .54], and no interaction between drug and visit [F(2,42) = 0.01, P = .99] was noted.
Similar results were observed for the secondary outcome measure, the HAM-A (Table 2). The main effect of block was significant (F(2,42.4) = 8.33, P = .0009), driven by significant improvement from Block 0 to Block 1 (Cohen’s d = 0.95, 95% CI: 0.31–1.59; t(39.8) = 3.01, Padj = .005) and to Block 2 (Cohen’s d = 1.17, 95% CI: 0.57–1.78; t(43.9) = 3.89, Padj = .0003). Change from Block 1 to Block 2 was not significant (Cohen’s d = 0.30, 95% CI: -0.31–0.91; t(43.8) = 0.98, Padj = .59).
MEG Results
No significant effects of age or gender on gamma power in MDD subjects were observed, so these effects were removed from the model. There was a significant overall main effect of session (minimum PFDR = .0005). However, this was primarily accounted for by increased gamma power in the baseline placebo-lead in session, primarily in fronto-temporal areas, and presumably due to the novelty of the scanning environment and potentially increased muscular activity. For the contrast of interest—scopolamine vs placebo—no significant effects were observed (minimum PFDR = .195). Maps of this comparison, thresholded at Puncorreced < 0.005, are shown in Figure 3. Gamma power was nominally increased in bilateral precuneus/angular gyrus, but nominally reduced in parahippocampal gyrus and inferior frontal gyrus.
Figure 3.

Z-map of the comparison of gamma power in the post-scopolamine vs post-placebo condition. Red indicates increased gamma in the scopolamine condition, and blue indicates decreased gamma in the scopolamine condition.
BDNF Results
Plasma BDNF levels did not change significantly between 0 and 150 minutes under either condition (placebo, F(1,42) = 1.78, P = .19; scopolamine, F(1,42) = 0.06, P = .80) (placebo vs scopolamine, F(1,42) = 1.26, P = .27). To address the possibility of carry-over effects, data from the first block of the trial were analyzed separately (i.e., Session 2, 0 minutes vs Session 5, 0 minutes). No significant change in BDNF levels was observed following 3 infusions of placebo (F(1,10) = 0.01, P = .94) or scopolamine (F(1,10) = 0.04, P = .84) (placebo vs scopolamine, F(1,20) = 0.04, P = .84). Change in BDNF levels was not related to change in MADRS score during the first block under either condition (placebo, t(15) = -0.35, P = .73; scopolamine, t(15) = -1.55, P = .14), and this did not differ between condition (change in BDNF by condition interaction, F(1,15) = 0.12, P = .73) (see Figure 4).
Figure 4.

Results of a repeated measures model with fixed effects of time, condition, and their interaction. Least square mean estimates (with SE) are plotted. The pre-post change in natural log-transformed brain-derived neurotrophic factor (BDNF) concentrations did not differ between conditions (F(1,42) = 1.26, P = .27).
Adverse Effects
Most participants reported transient minor side effects at the time of the infusions (of both scopolamine and placebo). These included expected anticholinergic side effects such as dry mouth, constipation, blurred vision, drowsiness, and nervousness. Two participants reported worsening of depression during the study; both were closely monitored and were able to complete the protocol. No unexpected or serious adverse events occurred; while a formal assessment of blinding was not conducted, clinical reports suggest that unblinding was not a major concern.
Discussion
In this randomized, placebo-controlled, crossover trial of 23 subjects with MDD, a series of 3 scopolamine infusions did not significantly improve depressive or anxiety symptoms compared with placebo. These negative results are not in line with previous studies of scopolamine for depression and suggest that the current study sample may have differed significantly from samples used in previous studies. One possible explanation for the lack of significant results in this trial may be the increased level of treatment resistance in this subject sample. While previous studies (Furey and Drevets, 2006; Drevets and Furey, 2010) did not report data for past medication trials, the average past number of medication trials in the current study was greater than three. Furthermore, a larger proportion of patients in the previous studies were treatment naive (50% compared with 26% in the current study). Moreover, the previous studies examined wholly outpatient samples while the current study had a mix of inpatients and outpatients (7 inpatients, 16 outpatients). Another potential difference between the current and previous samples may be increased severity of depressive symptoms; specifically, average MADRS score on entering the treatment phases were between 23 and 30 in the previous studies compared with 33 in the current study.
From a translational perspective, the lack of clinical efficacy in this study did not allow us to test the hypothesis that scopolamine and ketamine have a shared mechanism of action. The results do suggest that the neurobiological effects (i.e., drug effects) of scopolamine and ketamine are at least partially distinct. It is important to acknowledge that because the response to scopolamine was small in this sample, it was not possible to assess any neurophysiological correlates of response (i.e., mechanism of action). For instance, while this cohort exhibited no drug-dependent changes in BDNF levels in response to scopolamine, it is possible that significant correlations would have been observed if clinical response had been greater. It should be noted, however, that peripheral BDNF concentrations may be difficult to interpret. For instance, in humans—though not in mice—BDNF blood levels may primarily be influenced by platelet stores, which are typically released during the coagulation process, more than brain BDNF activity (Chacon-Fernandez et al., 2016).
Regarding the neuroimaging biomarker analysis, we found no significant change in MEG gamma power following scopolamine administration in contrast to studies conducted with ketamine. For instance, both animal (Pinault, 2008; Anderson et al., 2014; Jones et al., 2014) and human (Rivolta et al., 2015; Shaw et al., 2015) studies have indicated that acute, subanesthetic doses of ketamine are associated with robust increases in gamma power regardless of clinical response. A recent study by Nugent and colleagues not only found increases in MEG gamma power in response to ketamine administration in both MDD and healthy controls, but also demonstrated a relationship between increased gamma power and antidepressant response in MDD subjects with lower baseline MEG gamma power (Nugent et al., 2018). For ketamine, it is thought that the decreased activity in GABAergic interneurons and the disinhibition of excitatory pyramidal neurons (Homayoun and Moghaddam, 2007) presumably provide the mechanism underlying these increased gamma oscillations (Carlen et al., 2012), which may function as a biomarker of antidepressant response to ketamine. The association between change in gamma power and antidepressant response could not be assessed in the current study given the lack of clinical efficacy in this cohort, and thus scopolamine’s mechanism of antidepressant action could not be evaluated.
Strengths of the study include: the randomized design, the medication-free status of all the participants, the i.v. administration of the study drug (which ensures uniform administration and compliance), the 100% completion rate of the participants, the inclusion of subjects regardless of the number of past medication trials, and the inclusion of 2 distinct neurophysiological modalities obtained in relationship with treatment to scopolamine.
Taken together, these findings suggest that scopolamine may have only modest or no significant antidepressant effects in patients with more severe and treatment-resistant forms of depression. It is possible, however, that subjects who respond more robustly to scopolamine would exhibit similar neurobiological effects to those who respond to ketamine. Future studies with scopolamine in depression should factor in level of symptom severity and treatment resistance. Investigations of physiological markers associated with antidepressant response to scopolamine should be reexamined in a patient cohort that responds to this agent, as reported in previously published trials.
Funding
Funding for this work was supported by the Intramural Research Program at the NIMH, NIH (ZIA MH002927; NCT00369915), by a NARSAD Independent Investigator Award to Dr. Zarate, and by a Brain and Behavior Mood Disorders Research Award to Dr. Zarate.
Supplementary Material
{kind=link}
Acknowledgments
The authors thank the 7SE research unit and staff for their support. Ioline Henter (NIMH) provided invaluable editorial assistance.
Statement of Interest
Dr. Zarate is listed as a co-inventor on a patent for the use of ketamine in major depression and suicidal ideation; as a co-inventor on a patent for the use of (2R,6R)-hydroxynorketamine, (S)-dehydronorketamine, and other stereoisomeric dehydro and hydroxylated metabolites of (R,S)-ketamine metabolites in the treatment of depression and neuropathic pain; and as a co-inventor on a patent application for the use of (2R,6R)-hydroxynorketamine and (2S,6S)-hydroxynorketamine in the treatment of depression, anxiety, anhedonia, suicidal ideation, and posttraumatic stress disorders. He has assigned his patent rights to the US government but will share a percentage of any royalties that may be received by the government. Dr. Furey is identified as a co-inventor on a patent in the US and Europe for the use of scopolamine as an antidepressant agent. Dr. Furey is a full-time employee at Janssen Pharmaceuticals, Neuroscience Research and Development, La Jolla, CA. All other authors have no conflict of interest to disclose, financial or otherwise.
References
- Anderson PM, Pinault D, O’Brien TJ, Jones NC(2014)Chronic administration of antipsychotics attenuates ongoing and ketamine-induced increases in cortical gamma oscillations. Int J Neuropsychopharmacol 17:1895–1904. [DOI] [PubMed] [Google Scholar]
- Betin C, DeFeudis FV, Blavet N, Clostre F(1982)Further characterization of the behavioral despair test in mice: positive effects of convulsants. Physiol Behav 28:307–311. [DOI] [PubMed] [Google Scholar]
- Cannon DM, Klaver JK, Ghandi SK, Solorio G, Peck SA, Erickson K, Akula N, Savitz J, Eckelman WC, Furey ML, Sahakian BJ, McMahon FJ, Drevets WC(2011)Genetic variation in cholinergic muscarinic-2 receptor gene modulates M2 receptor binding in vivo and accounts for reduced binding in bipolar disorder. Mol Psychiatry 16:407–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlen M, Meletis K, Siegle JH, Cardin JA, Futai K, Vierling-Claassen D, Ruhlmann C, Jones SR, Deisseroth K, Sheng M, Moore CI, Tsai LH(2012)A critical role for NMDA receptors in parvalbumin interneurons for gamma rhythm induction and behavior. Mol Psychiatry 17:537–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmichael O, Schwarz AJ, Chatham CH, Scott D, Turner JA, Upadhyay J, Coimbra A, Goodman JA, Baumgartner R, English BA, Apolzan JW, Shankapal P, Hawkins KR(2017)The role of fMRI in drug development. Drug Discov Today Nov 15:pii: S1359-6446(1317)30341-30340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chacon-Fernandez P, Sauberli K, Colzani M, Moreau T, Ghevaert C, Barde YA(2016)Brain-derived neurotrophic factor in megakaryocytes. J Biol Chem 291:9872–9881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Saad ZS, Britton JC, Pine DS, Cox RW(2013)Linear mixed-effects modeling approach to FMRI group analysis. NeuroImage 73:176–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comings DE, Wu S, Rostamkhani M, McGue M, Iacono WG, MacMurray JP(2002)Association of the muscarinic cholinergic 2 receptor (CHRM2) gene with major depression in women. Am J Med Genet 114:527–529. [DOI] [PubMed] [Google Scholar]
- Cox RW.(1996)AFNI: software for analysis and visualization of functional magnetic resonance neuroimages. Comput Biomed Res 29:162–173. [DOI] [PubMed] [Google Scholar]
- Dilsaver SC.(1986)Pathophysiology of “cholinoceptor supersensitivity” in affective disorders. Biol Psychiatry 21:813–829. [DOI] [PubMed] [Google Scholar]
- Drevets WC, Furey ML(2010)Replication of scopolamine’s antidepressant efficacy in major depressive disorder: a randomized, placebo-controlled clinical trial. Biol Psychiatry 67:432–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drevets WC, Zarate CAJ, Furey ML(2013)Antidepressant effects of the muscarinic cholinergic receptor antagonist scopolamine: a review. Biol Psychiatry 73:1156–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- First MB, Spitzer RL, Gibbon M, Williams JBW(2001)Structured clinical interview for DSM-IV-TR axis I disorders research version, patient edition. New York, NY: Biometrics Research, New York State Psychiatric Institute. [Google Scholar]
- Furey ML, Drevets WC(2006)Antidepressant efficacy of the antimuscarinic drug scopolamine: a randomized, placebo-controlled clinical trial. Arch Gen Psychiatry 63:1121–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosal S, Bang E, Yue W, Hare BD, Lepack AE, Girgenti MJ, Duman RS(2018)Activity-dependent brain-derived neurotrophic factor release is required for the rapid antidepressant actions of scopolamine. Biol Psychiatry 83:29–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillin JC, Sutton L, Ruiz C, Darko D, Golshan S, Risch SC, Janowsky DS(1991)The effects of scopolamine on sleep and mood in depressed patients with a history of alcoholism and a normal comparison group. Biol Psychiatry 30:157–169. [DOI] [PubMed] [Google Scholar]
- Gramfort A, Luessi M, Larson E, Engemann DA, Strohmeier D, Brodbeck C, Goj R, Jas M, Brooks T, Parkkonen L, Hamalainen M(2013)MEG and EEG data analysis with MNE-Python. Front Neurosci 7:267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haile CN, Murrough JW, Iosifescu DV, Chang LC, Al Jurdi RK, Foulkes A, Iqbal S, Mahoney J Jr, De La Garza Rn, Charney DS, Newton TF, Mathew SJ(2014)Plasma brain derived neurotrophic factor (BDNF) and response to ketamine in treatment-resistant depression. Int J Neuropsychopharmacol 17:331–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hare BD, Ghosal S, Duman RS(2017)Rapid acting antidepressants in chronic stress models: molecular and cellular mechanisms. Chronic Stress (Thousand Oaks). doi: 10.1177/2470547017697317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homayoun H, Moghaddam B(2007)NMDA receptor hypofunction produces opposite effects on prefrontal cortex interneurons and pyramidal neurons. J Neurosci 27:11496–11500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janowsky DS, el-Yousef MK, Davis JM(1974)Acetylcholine and depression. Psychosom Med 36:248–257. [DOI] [PubMed] [Google Scholar]
- Janowsky DS, el-Yousef MK, Davis JM, Sekerke HJ(1972)A cholinergic-adrenergic hypothesis of mania and depression. Lancet 2:632–635. [DOI] [PubMed] [Google Scholar]
- Janowsky DS, Risch SC, Huey LY, Kennedy B, Ziegler M(1985)Effects of physostigmine on pulse, blood pressure, and serum epinephrine levels. Am J Psychiatry 142:738–740. [DOI] [PubMed] [Google Scholar]
- Jones NC, Anderson P, Rind G, Sullivan C, van den Buuse M, O’Brien TJ(2014)Effects of aberrant gamma frequency oscillations on prepulse inhibition. Int J Neuropsychopharmacol 17:1671–1681. [DOI] [PubMed] [Google Scholar]
- Kessler RC, Petukhova M, Sampson NA, Zaslavsky AM(2012)Twelve-month and lifetime prevalence and lifetime morbid risk of anxiety and mood disorders in the United States. Int J Methods Psychiatr Res 21:169–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishimoto T, Chawla JM, Hagi K, Zarate CA, Kane JM, Bauer M, Correll CU(2016)Single-dose infusion ketamine and non-ketamine N-methyl-d-aspartate receptor antagonists for unipolar and bipolar depression: a meta-analysis of efficacy, safety and time trajectories. Psychol Med 46:1459–1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N, Lee B, Liu RJ, Banasr M, Dwyer JM, Iwata M, Li XY(2010)mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science 329:959–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machado-Vieira R, Yuan P, Brutsche N, DiazGranados N, Luckenbaugh D, Manji HK, Zarate CA Jr(2009)Brain-derived neurotrophic factor and initial antidepressant response to an N-methyl-D-aspartate antagonist. J Clin Psychiatry 70:1662–1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niciu MJ, Mathews DC, Nugent AC, Ionescu DF, Furey ML, Richards EM, Machado-Vieira R, Zarate CA Jr(2014)Developing biomarkers in mood disorders research through the use of rapid-acting antidepressants. Depress Anxiety 31:297–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nugent AC, Ballard E, Gould TD, Park LT, Moaddel R, Brutsche NE, Zarate CAJ(2018)Ketamine has distinct electrophysiological and behavioral effects in depressed and healthy subjects. Mol Psychiatry doi: 10.1038/s41380-018-0028-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nurnberger JI Jr, Jimerson DC, Simmons-Alling S, Tamminga C, Nadi NS, Lawrence D, Sitaram N, Gillin JC, Gershon ES(1983)Behavioral, physiological, and neuroendocrine responses to arecoline in normal twins and “well state” bipolar patients. Psychiatry Res 9:191–200. [DOI] [PubMed] [Google Scholar]
- Olfson M, Blanco C, Marcus SC(2016)Treatment of adult depression in the United States. JAMA Intern Med 176:1482–1491. [DOI] [PubMed] [Google Scholar]
- Overstreet DH, Russell RW, Hay DA, Crocker AD(1992)Selective breeding for increased cholinergic function: biometrical genetic analysis of muscarinic responses. Neuropsychopharmacology 7:197–204. [PubMed] [Google Scholar]
- Pinault D.(2008)N-methyl d-aspartate receptor antagonists ketamine and MK-801 induce wake-related aberrant gamma oscillations in the rat neocortex. Biol Psychiatry 63:730–735. [DOI] [PubMed] [Google Scholar]
- Risch SC, Kalin NH, Janowsky DS(1981)Cholinergic challenges in affective illness: behavioral and neuroendocrine correlates. J Clin Psychopharmacol 1:186–192. [DOI] [PubMed] [Google Scholar]
- Rivolta D, Heidegger T, Scheller B, Sauer A, Schaum M, Birkner K, Singer W, Wibral M, Uhlhaas PJ(2015)Ketamine dysregulates the amplitude and connectivity of high-frequency oscillations in cortical-subcortical networks in humans: evidence from resting-state magnetoencephalography recordings. Schizophr Bull 41:1105–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson SE, Vrba J(1999)Functional neuroimaging by synthetic aperture magnetometry (SAM). In: Recent advances in biomagnetism (Yoshimoto T, Kotani M, Kuriki S, Karibe H, Nakasato N, eds), pp302-305. Sendai: Tohoku University Press. [Google Scholar]
- Rubin RT, O’Toole SM, Rhodes ME, Sekula LK, Czambel RK(1999)Hypothalamo-pituitary-adrenal cortical responses to low-dose physostigmine and arginine vasopressin administration: sex differences between major depressives and matched control subjects. Psychiatry Res 89:1–20. [DOI] [PubMed] [Google Scholar]
- Shaw AD, Saxena N, L EJ, Hall JE, Singh KD, Muthukumaraswamy SD(2015)Ketamine amplifies induced gamma frequency oscillations in the human cerebral cortex. Eur Neuropsychopharmacol 25:1136–1146. [DOI] [PubMed] [Google Scholar]
- Trivedi MH, Fava M, Wisniewski SR, Thase ME, Quitkin F, Warden D, Ritz L, Nierenberg AA, Lebowitz BD, Biggs MM, Luther JF, Shores-Wilson K, Rush AJ (2006a) Medication augmentation after the failure of SSRIs for depression. N Engl J Med 354:1243–1252. [DOI] [PubMed] [Google Scholar]
- Trivedi MH, Rush AJ, Wisniewski SR, Nierenberg AA, Warden D, Ritz L, Norquist G, Howland RH, Lebowitz B, McGrath PJ, Shores-Wilson K, Biggs MM, Balasubramani GK, Fava M (2006b) Evaluation of outcomes with citalopram for depression using measurement-based care in STAR*D: implications for clinical practice. Am J Psychiatry 163:28–40. [DOI] [PubMed] [Google Scholar]
- Voleti B, Navarria A, Liu RJ, Banasr M, Li N, Terwilliger R, Sanacora G, Eid T, Aghajanian G, Duman RS(2013)Scopolamine rapidly increases mammalian target of rapamycin complex 1 signaling, synaptogenesis, and antidepressant behavioral responses. Biol Psychiatry 74:742–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JC, et al. (2004)Evidence of common and specific genetic effects: association of the muscarinic acetylcholine receptor M2 (CHRM2) gene with alcohol dependence and major depressive syndrome. Hum Mol Genet 13:1903–1911. [DOI] [PubMed] [Google Scholar]
- Wohleb ES, Gerhard D, Thomas A, Duman RS(2017)Molecular and cellular mechanisms of rapid-acting antidepressants ketamine and scopolamine. Curr Neuropharmacol 15:11–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarate CA Jr, Mathews DC, Furey ML(2013)Human biomarkers of rapid antidepressant effects. Biol Psychiatry 73:1142–1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarate CA Jr, Singh JB, Carlson PJ, Brutsche NE, Ameli R, Luckenbaugh DA, Charney DS, Manji HK(2006)A randomized trial of an N-methyl-D-aspartate antagonist in treatment-resistant major depression. Arch Gen Psychiatry 63:856–864. [DOI] [PubMed] [Google Scholar]
- Zimmerman M, Posternak MA, Chelminski I(2004)Derivation of a definition of remission on the Montgomery-Asberg depression rating scale corresponding to the definition of remission on the Hamilton rating scale for depression. J Psychiatr Res 38:577–582. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.