

Abstract
The convergent synthesis of candidate stereoisomers of the natural product arenolide was accomplished using recently developed catalytic boron-based reactions. Comparison of the spectral data for candidate structures with that reported for the authentic natural product revealed the likely stereostructure of the natural compound.
Graphical Abstract
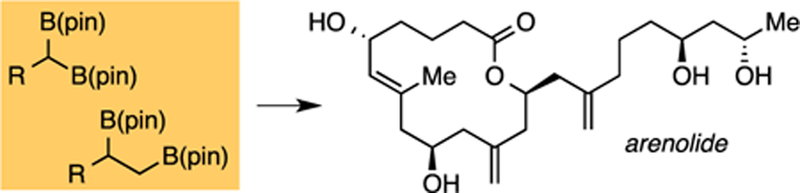
The natural product arenolide (Scheme 1a), together with three other diterpenes, were isolated from a specimen of Dysidea sp. by Lu and Faulkner in 1998.1 While arenolide demonstrated only modest cytotoxicity against HCT human colon carcinoma cells (IC50: 21 mM) and A2780 human ovarian carcinoma cells (IC50: 9.8 mM), it was noted that much of the compound may have decomposed prior to the assay, leaving its true biological activity in question. Of further interest, it was noted that macrolides had yet to be isolated from the Dysidea species, suggesting that arenolide may have been secreted by another producing organism and then absorbed by the sponge. In terms of structure, the macrocyclic core of arenolide is reported to be a fourteen-membered ring bearing an attached hydroxylated sidechain. While the relative configuration of the macrocycle was assigned by NOESY analysis, the relative configuration at C19 and C21 was left unassigned. Moreover, the absolute configuration of arenolide was not established. For these reasons, and to study the utility of recently-developed borylation methods under development in our laboratory, we undertook the synthesis of candidate stereoisomers of arenolide with the goal of establishing its overall stereostructure.
Scheme 1.
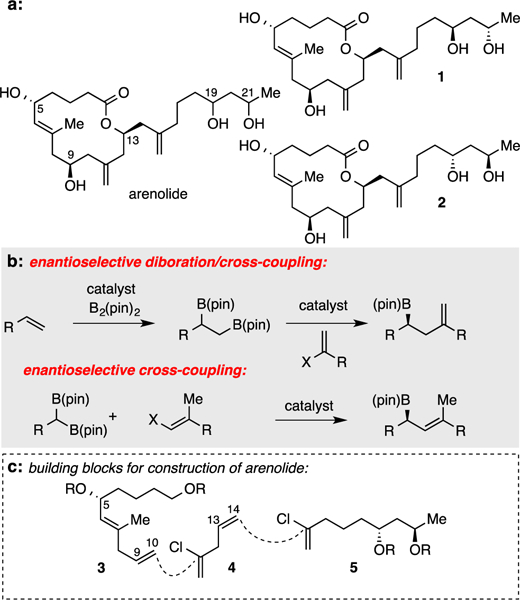
Structure of Arenolide Candidates and a Plan for Constructing them Using Boron-Based Reactions.
To streamline our efforts toward arenolide, we examined the spectral data reported for the natural product. Importantly, Lu and Faulkner employed COSY, HMQC and HMBC to completely assign the 1H and 13C NMR spectra and provided the observation that the 13C resonances for C-19 and C-21 reside at δ 69.1 and 65.5 ppm. Employing a correlation first established by Hoffmann2 and recently employed by Bruckner3, the chemical shifts reported for the C-19 and C-21 13C resonances are strongly supportive of a 1,3-anti relative configuration (see Supporting Information (SI) for details) and thus, compounds 1 and 2, were targeted for synthesis.
Recently, a number of stereochemically-complex, densely functionalized natural products have been prepared using chiral organoboron based synthesis methods.4 Along these lines, our group has developed convergent catalytic methods to efficiently construct chiral organoboronic esters (Scheme 1b), and we envisioned these might facilitate the construction of arenolide. Critical synthetic connections for arenolide assembly are illustrated in Scheme 1c. We considered that the macrolactone would be closed by intramolecular esterification, and an advanced intermediate might be derived from alkene diboration/cross-coupling sequences5 involving an alkene at C9/C10 of 3 and alkenyl chloride 4; a second diboration/cross-coupling sequence involving an alkene at C13/C14 and alkenyl chloride 5 would complete the carbon skelton. Importantly, we anticipated ready access to substrate 3 through asymmetric cross-coupling of a 1,1-geminal bis(boronic) ester to establish the carbinol at C5 in compound 3.6
Synthesis of fragment 3 commenced with readily available bis(boryl) methane7, which was deprotonated by treatment with lithium tetramethylpiperidide (LiTMP) and then used in an SN2 alkylation of TBS-protected 4-bromo-1-butanol (Scheme 2).8 The product 1,1-bis(boronate) 6 was then subjected to an enantioselective cross coupling with vinyl bromide 7 in the presence of 1 mol % L1•PdCl26b as catalyst to afford secondary boronate 8 in modest yield (predominant byproduct is protodeboronation) and good enantioselectivity; oxidation and TBS protection furnished 9. The monosubstituted alkene in 9 was an excellent substrate for catalyst-controlled stereoselective diboration and, in the presence of Pt(dba)3/(R,R)-L29, this reaction furnished 1,2-bis(boronate) 10 in good yield and stereoselectivity. The vicinal bis(boronate) 10 engaged in efficient Pd/RuPhos10-catalyzed cross-coupling with readily available alkenyl bromide 11 (see SI) and, after oxidation11 and TBS-protection, delivered 12. Finally, a second Pt-catalyzed enantioselective diboration, this time with (S,S)-L2 as ligand, furnished cross-coupling substrate 13 in excellent yield and stereocontrol.
Scheme 2.
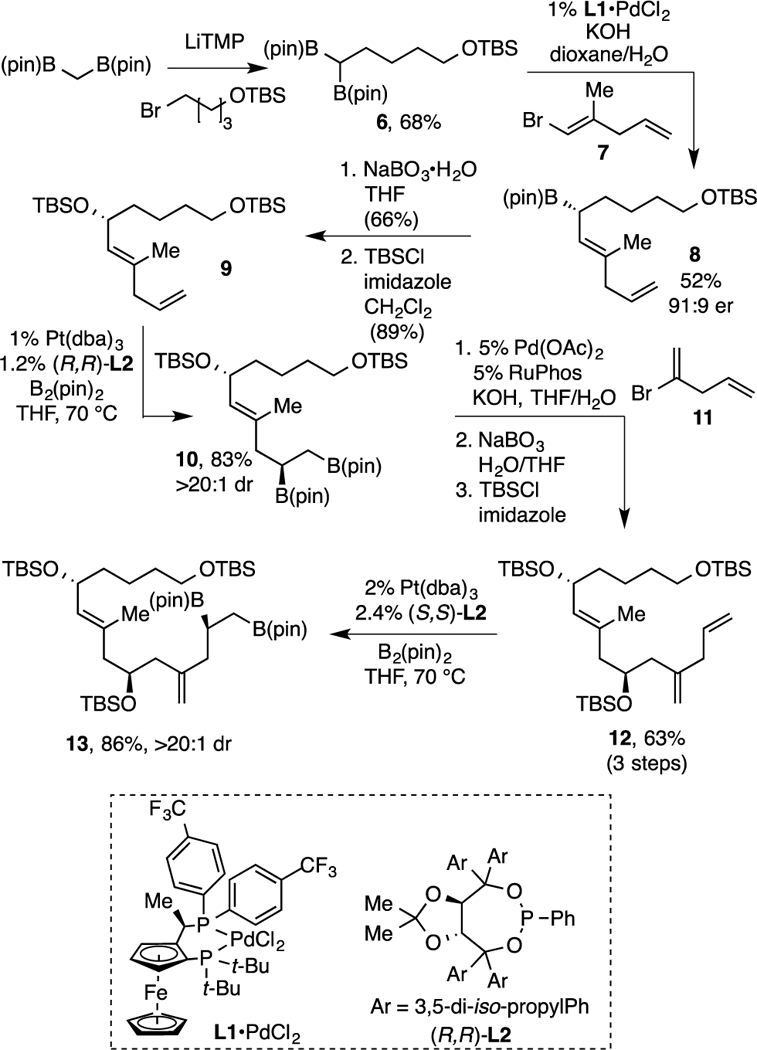
Synthesis of Cross-Coupling Partner 13.
With fragment 13 in hand, the synthesis of the side-chain was then pursued (Scheme 3). Readily available (S)-propylene oxide was treated with vinyl magnesium bromide and the product protected as a silyl ether giving 15.12 While Pt-catalyzed asymmetric diboration is more efficient and slightly more selective than our first-generation carbohydrate-catalyzed diboration13 employing TBS-DHG as catalyst, the later process is less expensive to operate on larger scale and was selected for preparation of 16. Following carbohydrate-catalyzed diboration, Pd/RuPhos-catalyzed cross-coupling and oxidation furnished 16 in modest yield, but high stereoselectivity. Subsequent protection to give 17, followed by sequential 9-BBN hydroboration and B-alkyl Suzuki coupling14 with 1,1-dichloroethylene, proceeded in outstanding yield to furnish cross-coupling partner 18.
Scheme 3.
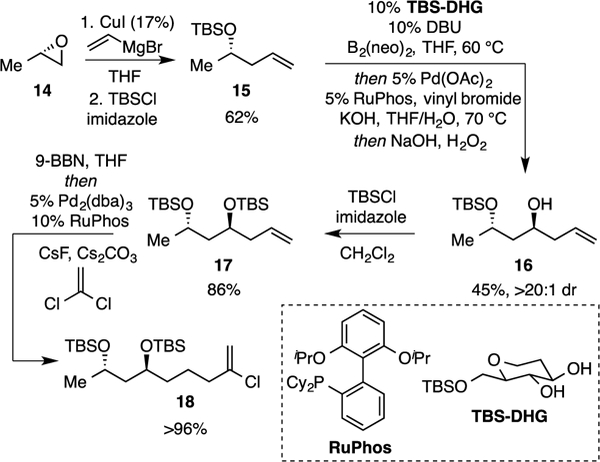
Preparation of Cross-Coupling Alkenyl Chloride 18.
Suzuki-Miyaura cross-coupling of 13 and 18 with Pd/RuPhos furnished 19 in moderate yield (60%, Scheme 4). Selective desilylation of compound 19 delivered primary alcohol 20, which was then subjected to Dess–Martin periodinane15 oxidation, followed by Pinnick oxidation16 to furnish carboxylic acid 21. Importantly, the remaining boronic ester in compound 21 did not appear to be perturbed during this oxidation sequence and, having served its role as a masked hydroxyl group over several steps of synthesis, was then oxidized; the obtained alcohol was subjected to Yamaguchi lactonization17 to afford a fourteen-membered lactone 22. Finally, after global deprotection, compound 1 was isolated.
Scheme 4.
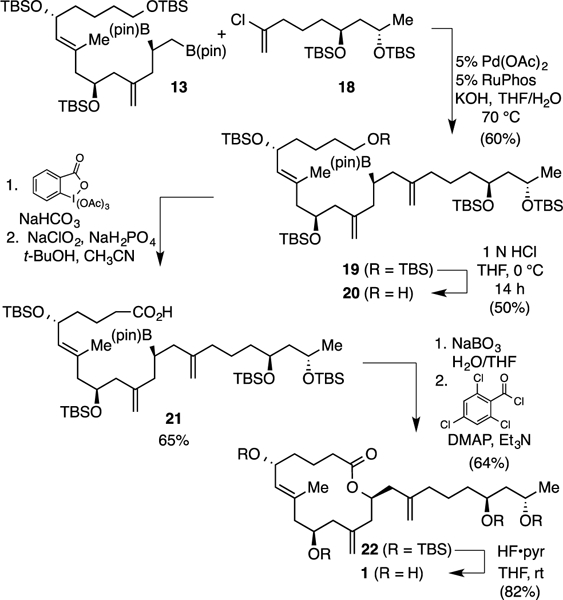
Completion of Structure 1 by Suzuki-Miyaura Fragment Coupling.
Using a similar synthesis strategy, stereoisomer 2 was also prepared (see SI for details). Spectral data for both stereoisomers was collected and compared. While comparison of 1H NMR was not helpful for establishing stereoisomer identity, examination of 13C of NMR spectra was informative. Comparing 13C NMR spectra revealed significant differences between stereoisomer 2 and the natural product (root mean square (rms) deviation = 0.10 ppm) whereas 13C NMR spectra for stereoisomer 1 showed much closer alignment with spectral data reported for arenolide (rms difference = 0.04 ppm). Of note, two resonances for isomer 2 (C16 and C19) exhibited ca. 0.3 ppm difference from the reported values for arenolide (Scheme 5). Similarly, the exocyclic methide (C25) for compound 2 exhibited a 0.14 ppm difference from the natural product. In terms of absolute configuration, the optical rotation measured for compound 1 is [α]D +6.00° (c 0.067, CHCl3), while the value reported for arenolide is [α]D +13.0° (c 0.64, CHCl3). Collectively, the evidence suggests that the structure of arenolide is that depicted for compound 1.
Scheme 5.
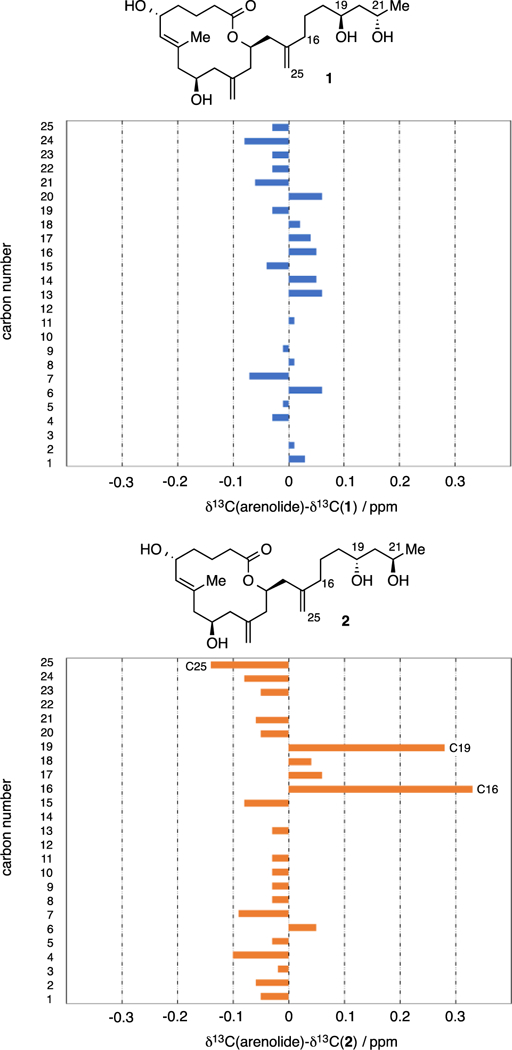
Comparison of 13C NMR for Structures 1 and 2 with Naturally Occurring Arenolide Suggests 1 is the Natural Product.
In summary, we have completed the total synthesis of stereoisomers of the natural product arenolide employing boron-based asymmetric transformations in place of more common carbonyl-based C-C bond constructions. Comparison of physical data suggests that compound 1 is most likely the structure of naturally occurring arenolide.
Supplementary Material
ACKNOWLEDGMENT
This work was supported by the NIH (NIGMS 59417).
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
Supporting Information Placeholder
REFERENCES
- (1).Lu Q; Faulkner DJ J. Nat. Prod. 1998, 61, 1096. [DOI] [PubMed] [Google Scholar]
- (2).Hoffmann RW; Weidmann U Chem. Ber. 1985, 118, 3980. [Google Scholar]
- (3).Diehl J; Brü R Eur. J. Org. Chem. 2017, 278. [Google Scholar]
- (4).For recent total syntheses that employ organoboron intermediates, see: (a) Wu J; Lorenzo P; Zhong S; Ali M; Butts CP; Myers EL; Aggarwal VK Nature 2017, 547, 436. [DOI] [PubMed] [Google Scholar]; (b) Bootwicha T; Feilner JM; Myers EL; Aggarwal VK Nature Chem. 2017, 9, 896. [DOI] [PubMed] [Google Scholar]; (c) Li J; Ballmer SG; Gillis EP; Fujii S; Schmidt MJ; Palazzolo AME; Lehmann JW; Morehouse GF; Burke MD Science 2015, 347, 1221. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Woerly EM; Roy J; Burke MD Nature Chem. 2014, 6, 484. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Feng F; McGrath KP; Hoveyda AH Nature 2014, 513, 367. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Chen L-A; Ashley MA; Leighton JL J. Am. Chem. Soc. 2017, 139, 4568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Mlynarski SN; Schuster CH; Morken JP Nature 2014, 505, 386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).(a) Sun C; Potter B; Morken JP J. Am. Chem. Soc. 2014, 136, 6534. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Potter B; Szymaniak AA; Edelstein EK; Morken JP J. Am. Chem. Soc. 2014, 136, 17918. See also [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Lee JCH; McDonald R; Hall DG Nature Chem. 2011, 3, 894. [DOI] [PubMed] [Google Scholar]; (d) Endo K; Ohkubo T; Hirokami M; Shibata TJ Am. Chem. Soc. 2010, 132, 11033. [DOI] [PubMed] [Google Scholar]
- (7).Commercially available, see: Aldrich 794287. For synthesis, see: Hong K; Liu X; Morken JP J. Am. Chem. Soc. 2014, 136, 10581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).For the alkylation of 6, see: (a) Matteson DS; Moody RJ Organometallics, 1982, 1, 20. [Google Scholar]; (b) Yang C-T; Zhang Z-Q; Tajuddin H; Wu C-C; Liang J; Liu J-H; Fu Y; Czyzewska M; Steel PG; Marder TB; Liu L Angew. Chem. Int. Ed. 2012, 51, 528. [DOI] [PubMed] [Google Scholar]
- (9).For the utility of this catalyst in diboration, see: (a) Coombs JR; Haeffner F; Kliman LT; Morken JP J. Am. Chem. Soc. 2013, 135, 11222. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kliman LT; Mlynarski SN; Morken JP J. Am. Chem. Soc. 2010, 132, 13949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Milne JE; Buchwald SL J. Am. Chem. Soc. 2004, 126, 13028. [DOI] [PubMed] [Google Scholar]
- (11).In our experience, the efficiency of oxidation with NaOH/H2O2 and sodium perborate are comparable. When compounds are anticipated to be base-sensitive, we opt for the later method.
- (12).For the synthesis of 15, see: (a) Kumar P; Gupta P; Naidu SV Chem. Eur. J. 2006, 12, 1397. [DOI] [PubMed] [Google Scholar]; (b) Figueroa R; Hsung RP; Guevarra CG Org. Lett. 2007, 9, 4857. [DOI] [PubMed] [Google Scholar]
- (13).Fang L; Yan L; Haeffner F; Morken JP J. Am. Chem. Soc. 2016, 138, 2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).(a) Liron F; Fosse C; Pernolet A; Roulland E J. Org. Chem. 2007, 72, 2220. For a review, see: [DOI] [PubMed] [Google Scholar]; (b) Chemler SR; Trauner D; Danishefsky SJ Angew. Chem., Int. Ed. 2001, 40, 4544. [DOI] [PubMed] [Google Scholar]
- (15).(a) Dess DB; Martin JC J. Org. Chem. 1983, 48, 4155. [Google Scholar]; (b) Dess DB; Martin JC J. Am. Chem. Soc. 1991, 113, 7277. [Google Scholar]
- (16).(a) Lindgren BO; Hilsson T Acta Chem. Scand. 1973, 27, 888. [Google Scholar]; (b) Kraus GA; Taschner MJ J. Org. Chem. 1980, 45, 1175. [Google Scholar]; (c) Kraus GA; Roth BJ Org. Chem. 1980, 45, 4825. [Google Scholar]; (d) Bal BS; Childers WE Jr.; Pinnick HW Tetrahedron 1981, 37, 2091. [Google Scholar]
- (17).Inanaga J; Hirata K; Saeki H; Katsuki T; Yamaguchi M Bull. Chem. Soc. Jpn. 1979, 52, 1989. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.