

Abstract
Live imaging of stem cells and their support cells can be used to visualize cellular dynamics and fluctuations of intracellular signals, proteins, and organelles in order to better understand stem cell behavior in the niche. We describe a simple protocol for imaging stem cells in the Drosophila ovary that improves on alternative protocols in that flies of any age can be used, dissection is simplified because the epithelial sheath that surrounds each ovariole need not be removed, and ovarioles are imaged in a closed chamber with a large volume of medium that buffers oxygen, pH, and temperature. We also describe how to construct the imaging chamber, which can be easily modified and used to image other tissues and non-adherent cells. Imaging is limited by follicle cells moving out of the germarium in culture around the time of egg chamber budding; however the epithelial sheath delays this abnormal cell migration. This protocol requires an hour to prepare the ovarioles followed by half an hour on the confocal microscope to locate germaria and set z limits. Successful imaging time depends on germarial morphology at the time of dissection, but we suggest 10–11 h to encompass all specimens.
Introduction
The Drosophila ovary is widely used as a model for investigating both germline stem cells and epithelial stem cells (also known as follicle stem cells (FSCs)). These stem cells work together to produce eggs that develop from egg chambers that bud off a structure called the germarium. It has been challenging to image stem cells in the germarium for long time periods because germaria are sensitive to their environment; they move as they develop, thus often moving out of the imaging window, and they lose their integrity during or soon after egg chamber budding after the ovary is separated into individual ovarioles for imaging. To address these problems, we designed a reusable imaging chamber that holds a large volume of medium and allows gas exchange; we embedded ovarioles in Matrigel to reduce movement; and we determined that the presence of the muscle sheath, also called the epithelial sheath, can delay abnormal follicle cell movement out of the germarium in culture. Using this chamber, we visualized FSCs undergoing radial back-and-forth movements in the germarium and moving anteriorly into the region where a population of nondividing somatic escort cells reside1. In this protocol, we provide detailed instructions to facilitate the manufacture and use of the chamber to image Drosophila ovarioles.
Development of the protocol
This protocol arose from a collaboration between cell biologists and engineers specializing in the design and development of innovative bioreactor chambers for stem cell studies1. Before the development of our system, makeshift chambers that require assembly each time were used, and many of these systems were impermeable to gas. Our goal was to construct a reliable reusable chamber to enable prolonged imaging of FSCs in Drosophila ovaries.
Silicone imaging chamber.
The dimensions and appearance of the chamber we developed are shown in Fig. 1a. The construction of the imaging chamber is shown in Supplementary Video 1. The chamber is made of silicone and is bonded to a glass coverslip. The chamber is customizable, is easy to build, and can be used for live imaging of many non-adherent cells or tissues. It can hold a range of media volumes depending on the diameter of the biopsy punch used and the thickness of the silicone slabs. The fundamental role of the chamber is to supply nutrients and allow gas exchange for a prolonged time without media evaporation. In this specific case, the chamber is equipped with three wells: one for imaging the specimen and two as medium reservoirs, connected to the former by small channels. The purpose of the reservoir wells is to increase the medium volume while keeping the sample confined to a small area, and also to allow the medium to be changed in the reservoir wells without disrupting the sample. In the design shown in Fig. 1a, the center imaging well holds a volume of 1 mL. The reservoir wells are not cored all the way through as an additional safeguard against chamber leakage (Supplementary Video 1) and hold ~ 0.7 mL each. A gas-permeable silicone lid prevents evaporation while allowing oxygenation of the medium.
Figure 1. Chamber design and mathematical modeling.
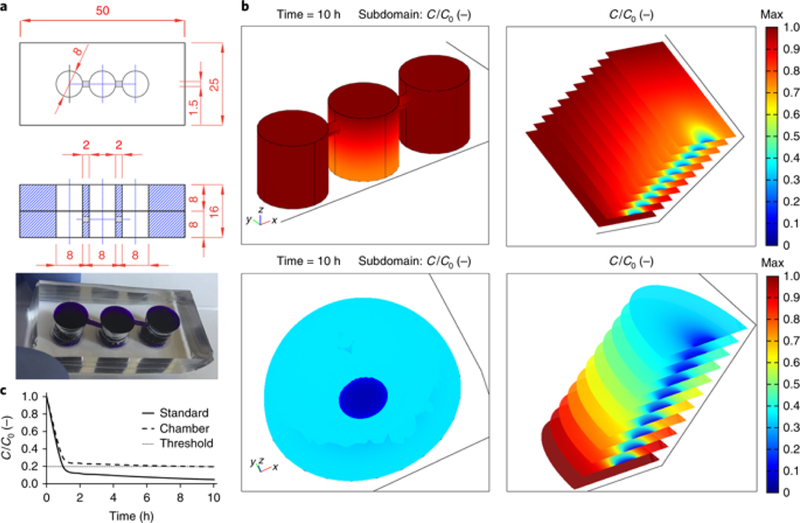
a, Technical drawings and image of a representative chamber composed of two layers of PDMS bonded together to achieve the desired height. Blue dye was added to highlight the wells and connecting channels. All lengths are in millimeters. Dimensions, diameter, and number of wells can be varied. b, Results of mathematical modeling of glucose consumption in a single-layer model chamber (top images) and standard culture (bottom, modeled as a large droplet of medium). Color-coded scale represents glucose concentration relative to its value at time zero. The images on the left give a snapshot of concentration values after 10 h, whereas those on the right show the temporal evolution of concentration values at 1- h intervals (the first and last slices have time stamps of 0 and 10 h, respectively). Whereas our chamber ensured maintenance of physiological levels for prolonged time spans, standard cultures led to rapid formation of highly depleted regions. c, Trace plots of relative glucose concentrations vs. time (in hours). C, concentration.
Maintenance of physiological nutrient levels.
Prolonged imaging is facilitated by the use of the medium formulation initially described by Prasad et al. in which insulin and serum are critical ingredients that keep ovarioles healthy in culture, enabling the imaging of border cell migration in stage 9 egg chambers 2. The chamber maintains a pseudo-steady state of nutrient levels throughout the duration of the experiment and its design allows fresh medium to be added to the reservoir wells without disrupting observation of the sample. Fig. 1b shows representative results of a simplified mathematical analysis of concentration profiles in a single-layer model of the chamber (top) and a conventional setup in which the sample is bathed in a ‘droplet’ of culture medium (bottom). The latter has been overestimated in size and volume to ease the comparison and was modeled as an 8-mm diameter and 4-mm height dome. The chamber well measures 8 × 8mm (diameter × height). The biological sample is modeled as a 2 ×.1-mm (diameter × height) cylinder at the bottom surface. We based our calculations on glucose consumption and assumed a maximum uptake reaction rate expressed by a mass action law for ligand-receptor association kinetics3. To obtain concentration maps, we solved the mass balance equations for a diffusive regime using Comsol Multiphysics modeling software. All parameters used are available in the literature3 and can be easily modified to model smaller or larger molecules that diffuse faster or slower than glucose. We set the initial glucose concentration to Co=11 mol/m3 (0.011 mol/L) equal to its concentration in Schneider’s medium. The threshold ensuring sufficient cell viability was set at 20% of the initial value. The curves in Fig. 1c highlight how the use of the chamber ensures maintenance of nutrient levels compatible with cell survival and correct viability, whereas standard culture quickly leads to the establishment of depleted regions. The color-coded heat maps in Fig. 1b correlate to the relative glucose concentration with respect to the initial value. The slice plots on the right are time sequences (1-h intervals) representative of vertical midsections of the modeled geometries.
Minimization of movement of germaria.
Because germaria move in culture, we embed ovarioles in the minimum gelling concentration of Matrigel in order to prevent them from moving excessively but allow their expansion as they proceed through development.
Applications of the method
Live imaging in the germarium can be used to study the cell cycle and asymmetric segregation of molecules in germline stem cells4–6, interaction of niche support cells with stem cells7,8, and the behavior and movement of FSCs within the niche1. Using this method we have observed divisions and movement of germline and FSCs (Supplementary Videos 2 and 3), stalk formation after egg chamber budding, and egg chamber elongation and rotations (Supplementary Video 4) in the Drosophila ovariole. It could also be applied to imaging other non-adherent tissues.
Comparisons with other methods
The main advantage of using this device, rather than other techniques is that the biological sample can be maintained at physiological nutrient levels during the entire course of the experiment. A large volume of medium ensures a buffered environment and that media components are not limiting although this is a disadvantage if users wish to include expensive or difficult to obtain components. Other protocols for imaging germaria or older egg chambers use 50 to 150μL of imaging medium 2,9–11. For example, a recent protocol for imaging germline stem cells in the germarium encases the sample between a coverslip and glass slide, separated by invisible tape9. The only other published long-term imaging study for FSCs used a small drop of medium together with a wet Kimwipe to reduce evaporation8. One protocol developed for older egg chambers described an improved setup with 5 mL of medium12. However, this protocol immobilized egg chambers using a Kimwipe held in place by brass washers on top, which must be placed with care in order to avoid crushing the egg chambers. The chamber we designed does not carry this risk, as the ovarioles are kept in place with a medium containing a low percentage of Matrigel, and the lid of the chamber is removed from the sample. The smaller specimen area relative to a larger total volume of medium is advantageous because it is more efficient to select germaria to image when they are confined to a smaller area.
Most protocols call for removal of the muscle sheath (also called the epithelial sheath) surrounding each ovariole in order to prevent movement of ovarioles during imaging8,9,11. We do not try to remove the muscle sheath, although it does sometimes come off during dissection. We found that all germaria can be used for imaging and those without the sheath move less in culture; however those that retain the muscle sheath stay intact for longer on average and have a higher rate of budding.
Limitations
In order to track individual cells moving in a germarium, the ovary is dissected and separated into individual ovarioles. The major limitation of this technique is that by dissecting ovarioles apart from one another, germaria lose their integrity during or soon after budding an egg chamber, because of follicle cells moving out of region 3 or the presumptive region 3 (Fig. 2 and Supplementary Video 5). Follicle cells migrating out of the germarium are not dead; they do not take up propidium iodide which only enters dead cells (Supplementary Video 6). We speculate that the germarium may require mechanical tension for integrity because when it is bunched with older egg chambers in the sheath it buds along the larger egg chamber (Supplementary Video 4). Also, the rare cases in which a germarium remained intact for hours after budding occurred when it was bunched against other egg chambers in the muscle sheath. In vivo, a germarium is packed with 14–19 other ovarioles in an ovary, with the entire ovary covered by a muscle layer called the peritoneal sheath that generates contractions around the ovarioles. Both the epithelial and peritoneal muscles sheaths contract several times per minute13, and it has been shown that contractions of the epithelial sheath play a role in the elongation of older egg chambers14.
Fig. 2: The muscle sheath prolongs the useful imaging time.
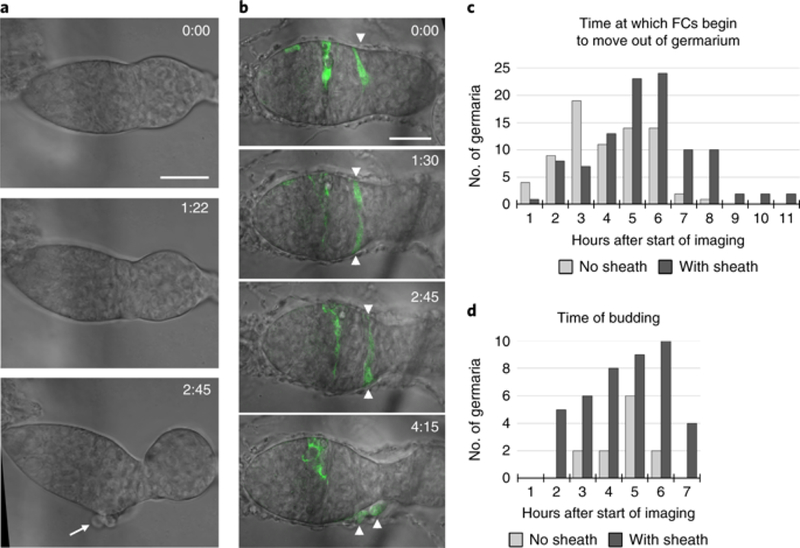
a, A germarium without an epithelial sheath, showing follicle cells (FCs) that abnormally moved out of region 3 (arrow) as the germarium budded. Time is in hours:minutes. See also Supplementary Video 5. b, A germarium with the epithelial sheath containing two GFP-labeled FCs (arrowheads) that abnormally moved out of the germarium at 4:15 (h:min). At time 0, the second FC is on the other side of the germarium and is not visible. c, The times at which FCs began to move out of the germarium after the start of imaging were recorded for germaria without the muscle sheath (light gray bars; n = 74) and for germaria with the muscle sheath (dark gray bars; n = 102). Times were binned into 1-h intervals, and the time at the end of each interval is displayed on the x axis. d, For cases in which budding was successful, the time of budding after the start of imaging is shown for germaria without the epithelial sheath (12 budded out of 74; light gray bars) or with the epithelial sheath (42 budded out of 102; dark gray bars). Scale bars, 20 μm.
Budding from the germarium occurs approximately every 12 h in vivo and can occur at any time after the start of imaging, depending on the initial morphology of the germarium. Because it takes 1.5 h from the start of dissection to the start of imaging, the longest expected time frame for imaging a germarium before it buds is 10.5 h, leaving a median of 5.25 h of useful imaging time before follicle cells begin to abnormally exit the germarium in culture. With the epithelial sheath present, the majority of germaria stayed intact for 5–6 h before follicle cells began to emerge, with a median time of follicle cell exit of 4.75 h and a range up to 10.5 h (Fig. 2c). Without the muscle sheath, follicle cells start to exit germaria with a median time of 3.5 h after the start of imaging in a range up to 7.3 h.
Although germaria without the sheath are more likely to stay in the imaging frame (Table 1), the chances of successful budding are much lower. Budding is observed 40% of the time with the sheath (n=102 germaria) and 16% of the time without (n=74).
Table 1.
Failure rates for imaging germaria with and without the epithelial sheath.
| Overall Failure rate |
Moved out of focus |
Moved out of frame |
Germarium/ first egg chamber damaged |
No development |
Difficult to see due to bunching in sheath |
|
|---|---|---|---|---|---|---|
| No sheath (n=89) |
17% (15) |
3% (3) |
1% (1) |
7% (6) |
6% (5) |
0 (0) |
| With Sheath (n=156) |
35% (54) |
12% (18) |
11% (17) |
8% (12) |
3% (5) |
1% (2) |
The overall failure rate is taken from 89 ovarioles without the muscle sheath (15 failures) and 156 ovarioles with the muscle sheath (54 failures). The specific reasons for those failures are given. The numbers in parentheses are the numbers of ovarioles in each category.
Experimental design
Customizing the imaging chamber (Steps 1–19)
The volume of the imaging and reservoir wells can be customized by altering the height of the polydimethylsiloxane (PDMS) and the diameter of the biopsy punch. We used two stacked PDMS slabs of 8 mm height to image ovarioles. To achieve a height > 8 mm, we recommend stacking two membranes on top on of one another because it is difficult to cleanly punch through a thicker membrane with the biopsy punch. The thickness of the coverslip is also important. We use no. 1 coverslips, rather than the standard no. 1.5, due to the thickness of the tissue (to keep germaria within the focal distance of the 63x objective). The PDMS is bonded to the coverslip with either a plasma cleaner, orsilicone adhesive.
Age and number of flies (Step 20)
An alternative method for imaging the germarium in a droplet of medium required newly-eclosed flies because older ovarioles with mature follicles moved too much8. We were able to limit the movement of the germarium by detaching older egg chambers with forceps and embedding ovarioles in Matrigel. The advantage of being able to use older flies is that we are able to heat shock adult flies and image GFP-labeled cells using the MARCM (mosaic analysis with a repressible cell marker) technique15, for which we have found that we need to wait 6 d after clone induction for GFP to reach a suitable brightness for live imaging. We routinely dissect two flies to screen 40–50 ovarioles for the presence of GFP- marked FSCs (some ovarioles are lost in the preparation).
We dissect only two flies in order to limit the time from dissection to arriving at the microscope; however, the imaging well is not crowded with 50 ovarioles and can accommodate 120 ovarioles, so if two people dissect flies simultaneously, more ovarioles can be screened and used.
Ovary dissection (Steps 21–29)
Other protocols recommend removal of the muscle sheath surrounding each ovariole in order to prevent movement of ovarioles during imaging8,9,11. The muscle sheath contracts at irregular intervals and causes movement of the germarium and egg chambers. We do not try to remove the muscle sheath, although it does sometimes come off during dissection. We found that all germaria are useful for imaging; however those that retain the muscle sheath stay intact for longer as they elongate and bud egg chambers (Fig. 2). An intact muscle sheath causes bunching up of egg chambers in a full ovariole, as well as excessive movement of the germarium, and we found that it is best to rip the muscle sheath with forceps around the stage 7/8 egg chamber, or simply to sever the ovariole above the stage 7egg chamber with forceps.
Embedding in Matrigel (Steps 30–34)
Matrigel reduces but does not eliminate movement of the muscle sheath. We use the minimum gelling concentration for Matrigel (3 mg/mL) in order to allow germaria to move as they develop. To ensure an accurate concentration of Matrigel, refer to the certificate of analysis provided with each batch of Matrigel to determine the lot-specific protein concentration.
Imaging multiple germaria and minimizing photodamage (Steps 37–41)
It is preferable to image multiple germaria per experiment because they rotate and move during imaging, often moving out of the imaging window. By marking positions with the microscope software, we image 2 to 10 germaria per experiment depending on the number of fluorescent colors in the sample and the time interval needed between image captures. Ten germaria can be imaged per experiment (imaging a z series encompassing each germarium) with a time interval of 15 min between image acquisitions and imaging GFP plus transmitted light. We suggest using the longest time interval possible to observe the cells or phenomena being tracked in the germarium, both to minimize photodamage and to be able to image more germaria per experiment.
When every cell in the germarium is labeled, a 6 min interval is sufficient to track movement of all individual cells (Fig. 3 and Supplementary Video 3). When only a few cells are labeled by MARCM a time interval of 15 min suffices to track individual cells. On the confocal microscope, phototoxicity is minimized by reducing the photon budget as much as possible. This is achieved by minimizing laser power and instead using digital gain to increase the signal; scanning at higher speeds and averaging multiple images (using frame or line averaging) to increase the signal to noise ratio instead of scanning at a lower speed because cells respond better to temporally dispersed photons than to a single concentrated dose of photons; imaging at lower number of pixels (e.g. 512 × 512 instead of 1,024 × 1,024); sampling at the longest time intervals possible for the behavior being observed; and taking the minimum number of z slices required to image cell behavior. We perform imaging with a 63×/1.4 numerical aperture (NA) objective at an xyz resolution of 0.2 μm × 0.2 μm × 3 μm at 512 × 512 pixels, frame averaging = 2, scan rate = 7 and pixel dwell time of 3.15 μs. Laser power is ideally kept to ≤2–3%.
Fig. 3: Imaging germline and FSC divisions.
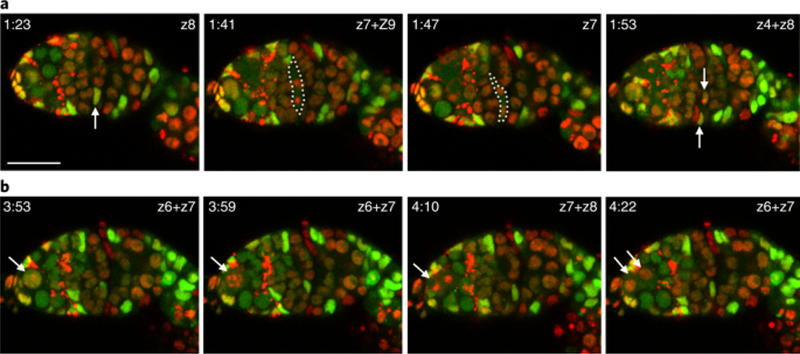
Flies of genotype yw hs-flp/yw; ubi-GFP FRT40A FRT42B ubi-RFP/tub-lacZ FRT40A FRT42B were heat-shocked to generate stem cell lineages that were GFP+RFP+, GFP+, or RFP+, as described1. a, An FSC division occurred in a GFP+RFP+ cell (white arrow). z sections are indicated; two z sections were combined when daughter cell nuclei were located in different sections. The dotted white line highlights the mitotic cell. Arrows indicate the daughter cells in the final panel (an RFP+ cell entering mitosis in z4 overlays the GFP+RFP+ daughter cell in z8 shown by the upper arrow). b, A germline stem cell division (white arrows). Images were collected every 5 min 58 s. Scale bar, 20 μm.
Materials
Biological materials
Flies with fluorescent reporters. To generate GFP-labeled MARCM clones we used flies of genotype FRT42D crossed to hs-Flp, UAS-GFP, tub-GAL4; FRT42D tub-GAL80/ FRT42D; act > CD2 > Gal4 (“>“ indicates a flippase recognition target (FRT)); to label all cells in the ovariole and produce clones of cells that have lost GFP or RFP we used yw hs-flp/yw; ubi-GFP FRT40A FRT42B ubi-RFP/tub-lacZ FRT40A FRT42B. Vasa-mCherry was used to label the germline cells (flies were from R. Lehmann, New York University School of Medicine). 10×STAT-GFP (ten Stat-binding sites in a promoter driving GFP) was used as a JAK/STAT signaling reporter (flies were from E. Bach, New York University School of Medicine). Twinspot-MARCM flies of genotype yw/yw hs-Flp; FRT40A UAS-mCD2-RFP UAS-GFP-miRNA/FRT40A UAS-mCD8-GFP UAS-CD2- miRNA; act > CD2 > Gal4 were heat-shocked to produce surface-labeled GFP+ or RFP+ clones by the twin-spot generator technique16.
Reagents
Polydimethylsiloxane (PDMS) elastomer (Sylgard 184 silicone elastomer; Dow Corning, distributed by Krayden, cat. no. DC4019862) for fabricating the imaging chamber
PDMS membrane for the lid of the imaging chamber (fabricated in-house or purchased; Specialty Silicone Products, cat. no. SSP-M823)
Ethanol (70% (vol/vol) in H2O)
Schneider’s Insect Medium (MilliporeSigma, cat. no. S0146)
Fetal bovine serum (FBS; Thermo Fisher Scientific, cat. no. 10437010)
Penicillin-streptomycin (10,000 U/mL) (Thermo Fisher Scientific, cat. no. 15140148)
Insulin solution I0516 (10 mg/mL, from bovine pancreas; MilliporeSigma)
Matrigel basement membrane matrix (Corning, cat. no. 356234)
Equipment
Balance, covered with aluminum foil to catch any drops of PDMS
Disposable paper cups
Vacuum desiccator (Cole Parmer, cat. no. EW-06514–20)
Petri dishes for casting (10-cm dish or another container compatible with the polymers and temperatures up to 70 °C; Corning, cat. no. 430591)
Convection oven (Thermo Fisher Scientific, model no. Isotemp 500)
Scalpel
Self-healing cutting mat (DAFA A4 professional self-healing mat; Amazon.com)
Plasma cleaner (Herrick Plasma, cat. no. PDC-001) connected to a vacuum pump (Edwards, model no. RV3). Minimum pump speed of 1.4 m3/h and an ultimate total pressure of 0.27 mbar
As an alternative to plasma bonding: Silicone RTV 4500, FDA grade, high-strength silicone sealant, clear (Amazon.com)
Biopsy punches; size depends on the desired imaging well dimension, up to 12 mm (Acuderm, cat. nos. ACKP825 and ACKP1525 for the 8- and 1.5-mm biopsy punches)
Removable tape (3M 811 Scotch Magic Removable Tape, Matte Finish)
50 × 24-mm coverslips, thickness no. 1 (Fisher Scientific, cat. no. 12–545F)
Tissue culture hood
Stereomicroscope (Nikon, model no. SMZ10A)
Inverted confocal microscope (Zeiss, model LSM 700)
Forceps, Dumont no. 5 (Electron Microscopy Sciences, cat. no. 0302–5-P0)
Corning PYREX 9-depression glass spot plates (Corning, cat. no. 7220–85)
Razor blade for cutting off the tips of pipette tips
Cotton-tipped applicators (Puritan, cat. no. 806-WC; http://www.puritanmedproducts.com/)
Reagent setup
Matrigel
Thaw the 5-mL bottle on ice, pipette into 50- to 100-μL aliquots on ice, and freeze at −20 °C. Before dissecting the ovarioles, transfer a frozen aliquot to ice in a small beaker to thaw and keep at 4 °C in a cold cabinet or refrigerator until use. This aliquot can be kept at 4 °C and should be used within a week. Use the lot-specific protein concentration provided on the certificate of analysis to calculate how much Matrigel is needed to obtain a final concentration of mg/mL in a 150-μL volume.
CRITICAL STEP It is very important that Matrigel matrix and all tubes and pipette tips used for aliquoting be prechilled because Matrigel will start to gel at temperatures > 10°C. Keep Matrigel on ice at all times.
Schneider’s medium with 20% FBS (vol/vol)
Make 4-mL aliquots of Schneider’s medium containing 20% FBS and store at 4°C for up to a few weeks.
Procedure
Building the imaging chamber. Timing: ≈4.5 h
-
1
Silicone is supplied as a two-part liquid kit, comprising the elastomer base and the curing agent, which are mixed in a 10:1 (wt/wt) ratio. First, weigh out the base by pouring it slowly into a paper cup; then weigh the curing agent by pouring into the same cup. 100 g of elastomer base (and 10 g of curing agent) will make one or two imaging chambers depending on the desired height of the wells (8 or 16 mm). Stir thoroughly for about a minute with a disposable plastic tissue culture pipette. Many small air bubbles will form upon mixing.
CAUTION It is advisable to cover the balance with foil and to wear a lab coat because drops of PDMS make an oily stain that is difficult to remove.
-
2
Place under vacuum in a vacuum desiccator for 10–15 min to ensure complete removal of air bubbles. Release the vacuum slowly so that the cup does not tip during the return to atmospheric pressure.
-
3
Pour the degassed PDMS mixture into Petri dishes (e.g., with a 100-mm diameter) or other appropriate containers with flat-bottom surfaces. Fill the container to the desired membrane height (1 mm, for making the chamber lid, or 8 mm, for the chamber) by measuring with a ruler and marking with a line, and pouring by eye to the line.
CRITICAL STEP Make sure the bench surface is very flat. Take care not to form air bubbles. If air bubbles form, place the dish in the vacuum desiccator for an additional 5–10 min. Air bubbles on the surface may be popped with a needle or a 10-μL pipette tip. At this point the PDMS should be very transparent.
-
4
Cover the dish with its lid and place it in a convection oven at 65°C for 2–3 h.
CRITICAL STEP The plane of the oven must be horizontal to ensure consistency of membrane thickness.
CRITICAL STEP Polymerization times depend on the thickness of the membrane, with thicker membranes requiring longer polymerization. Polymerization can be verified by visual and/or manual assessment. Avoid touching partially polymerized solutions.
-
5
Carefully peel the polymerized membrane from the casting dish using a scalpel or spatula.
-
6
On a cutting mat with centimeter markings, use a scalpel to cut the membrane to the desired shape and size (50 × 25 mm = length × width of a parallelepiped). Make one or two pieces depending on the desired well height. Bonding two layers together to achieve a taller well is advisable because punching wells through one thick slab is difficult and compromises precision in punching the holes.
-
7
Punch the three main wells (e.g., 8-mm diameter) on both membranes and ensure that the culture well is positioned at the center. The reservoir wells need not be punched all the way through the bottom membrane but may be punched most of the way through and then the biopsy punch can be used to cut the PDMS plug away from the chamber (Supplementary Video 1).
? TROUBLESHOOTING
-
8
Create a hydraulic connection between the outer and central wells by punching a 1.5-mm- diameter channel perpendicular to the wells (Fig. 1), centering it 2 mm from the top. If two membranes are used to increase well height, create the channels in the bottom membrane so that the fresh medium in the reservoir wells is more accessible to the sample.
CRITICAL STEP Take care to not leave any PDMS debris inside the channel.
CRITICAL STEP Take care to not damage the structural integrity of the membrane.
? TROUBLESHOOTING
-
9
Use Scotch removable tape to remove any dust or debris from the membranes and the glass coverslip.
CRITICAL STEP Dust and oils (i.e., from fingerprints) prevent plasma-driven surface activation, so be sure to wear gloves at all times and clean the PDMS surface thoroughly.
Plasma bonding:
Timing: 5 min
CRITICAL If a plasma cleaner is not available, Steps 10–17 can be substituted by spreading a small amount of RTV silicone sealant on the bottom side of the chamber around the perimeter of the chamber and the wells. The chamber can then be attached to the glass coverslip by pressing gently. The chamber should be left to cure for 24 h before using. This glue is UV- resistant.
CRITICAL STEP Most cell types will tolerate this glue very well. In our experience, germaria and ovarioles are healthy and behave normally in glued chambers. In very rare cases, the glue may be toxic to some types of cells (e.g., muscle stem cells).
-
10
Position the coverslip and membranes inside the glass chamber of the plasma cleaner. Plasma is stronger at the back of the chamber, so position as far toward the back as possible. Close the chamber and valves.
CRITICAL STEP The plasma will activate the exposed surfaces, so ensure that clean cuts are made, retaining proper right-angle alignments, and not chipping any surfaces.
-
11
Turn on the pump and allow stabilization at a pressure of ~ 0.3–0.4 mbar.
-
12
Open the needle valve slightly to let a small amount of air into the chamber to form the plasma.
-
13
Switch on the radio frequency coil to drive the formation of the plasma. Expose the surfaces to the plasma for 60 s, and then switch off the coil.
CRITICAL STEP Check the color and stability of the obtained plasma: it should be bright purple and consistent throughout the chamber.
-
14
Open the valve and equilibrate the chamber to atmospheric pressure.
-
15
Open the chamber and extract the glass coverslip and PDMS membranes.
CRITICAL STEP Do not touch any surface with bare hands! Wear gloves, and pinch them from the sides; use forceps, if necessary.
-
16
If combining two membranes, bind the two membranes by putting the activated surfaces in contact. The one with hydraulically connected wells should be at the bottom.
CRITICAL STEP Carefully align the wells before allowing the membranes to touch. Successful bonding should be immediate. The process is irreversible.
-
17
Bind the glass coverslip to the bottom by putting the activated surfaces in contact.
CRITICAL STEP Place the cleaned surfaces together within a minute of removal from the chamber. Do not apply excessive pressure, as the coverslip could break. Successful bonding should be immediate. The process is irreversible.
-
18
Validate the bonding by filling the device with water and checking for leaks. 100% hydraulic sealing must be obtained.
-
19
Cut the PDMS lid for the chamber from a sheet of thin PDMS membrane (0.5- to 1-mm thickness) to a size at which all three wells are covered.
PAUSE POINT Chambers can be stored indefinitely, covered at room temperature (RT; 23 °C).
Preparation of flies for live imaging. Timing: variable
-
20
Raise the desired flies. Flies of any age can be used (we have imaged germaria of flies ranging from 0 to 30 d after eclosion). We raised the flies for which we show illustrative results in this protocol at RT and fed the flies wet yeast paste to ensure continuous division of stem cells in the germarium.
CRITICAL STEP It may be necessary to wait a minimum of 6–7 d after heat shock when imaging GFP-labeled stem cells labeled by the MARCM technique17 in order for the GFP to develop to a brightness in the FSCs that is sufficient for imaging.
Dissection of ovarioles for live imaging. Timing: ≈1 h
-
21
Prepare imaging medium in a tissue culture hood by adding insulin solution to obtain a final concentration of 0.20 mg/mL and penicillin/streptomycin diluted to 0.6× from the 100× stock to 4 mL of Schneider’s medium containing 20% FBS.
-
22
Spray the imaging chamber and silicone lid with 70% ethanol. Keep in a tissue culture hood under UV light for 20 min to sterilize.
-
23
Put an aliquot of frozen Matrigel on ice and keep it in the refrigerator while dissecting ovaries so that the ice does not melt but the Matrigel thaws at 4°C.
-
24
Spray a pair of sharp forceps and a depression glass spot plate with 70% ethanol and dry it with a Kimwipe.
-
25
Pipette 0.75 mL of imaging medium into each of two neighboring wells in the glass spot plate. One well is for the initial dissection and for discarding stage 13/14 egg chambers, and the second is for the final dissected ovarioles. Make sure there is 2.5 mL of imaging medium remaining in the tube for filling the imaging chamber in Step 35.
-
26
Remove the ovaries from one to two females in the first ‘discard’ well and put the ovaries into the neighboring well filled with fresh medium. Females with the largest abdomens will generally have the largest, healthiest ovaries. Healthy ovarioles will have all stages of egg chambers, indicating that they have not arrested egg chamber budding. Remove the ovaries by gently pulling on the tip of the abdomen. The imaging chamber can accommodate ovarioles from more than two females; however, it is time-consuming to remove stage 13/14 egg chambers (at Step 29), hence it may be best to limit each experiment to two females so that each step can be completed in an appropriate time frame.
-
27
Use very sharp forceps to separate all ovarioles from one another. See Supplementary Video 7 for further guidance on how to dissect the ovarioles.
CRITICAL STEP If germaria are stuck together, they will move during imaging.
-
28
Detach stage ≥7/8 egg chambers from the ovariole so that the muscle sheath will be broken and the germaria will not move too much during imaging. We try not to remove the muscle sheath, but it does sometimes come off during dissection.
? TROUBLESHOOTING
CRITICAL STEP If stage ≥7/8 egg chambers are still attached, and the muscle sheath is present, the germarium may become bunched up in the muscle sheath and move excessively during imaging.
-
29
Remove 50–75% of all stage 13/14 egg chambers by grasping the dorsal appendages with forceps to remove them. Transfer to the discard well.
CRITICAL STEP If too many stage 13/14 egg chambers are present, they crowd the chamber and bump into germaria during imaging.
-
30
Bring the glass dissection dish and the beaker of ice containing the thawed aliquot of Matrigel to the hood.
-
31
Cut off the tip of a sterile pipette tip with a razor blade in order to pipette up ovarioles. Coat the pipette tip with protein solution by pipetting up and down a few times in the imaging medium in order to prevent ovarioles from sticking to the pipette tip.
-
32
Transfer the appropriate volume of ovarioles (~100 𝛍.L) to the center well of the imaging chamber, depending on the lot-specific concentration of Matrigel that will be needed for a concentration of 3 mg/mL Matrigel in a final volume of 150 μL. Do not discard the pipette tip; adjust the pipette to 150 μL to use in Step 33.
CRITICAL STEP To pipette out the ovarioles from the glass dissection well, put the cut pipette tip directly on the bottom of the well, holding the pipette vertically, and slowly suction up while swirling around the bottom of the well to get all of the ovarioles. Check the well while the Matrigel is gelling to see how well you removed the ovarioles.
-
33
Use a second pipette with a cut-off pipette tip to add Matrigel to the center well. Use the first pipette that has the non-stick tip and pipette up and down to mix (~4x); make sure that all the pink color of the Matrigel is gone. Avoid creating air bubbles in the mixture.
CRITICAL STEP Work quickly so the Matrigel does not have time to gel before mixing.
-
34
Use forceps to add the silicone lid to cover the chamber. Leave the chamber in the tissue culture hood for 15 min with the UV light off to allow the Matrigel to gel.
-
35
Remove the silicone lid with forceps and slowly pipette the remaining medium first into the side wells until the channels are filled with medium, and then into all three wells, alternating between the wells to keep the level of liquid equal. Add medium until all wells have a positive meniscus in order to seal the silicone lid to the chamber and prevent bubbles from forming when laying the lid on the chamber.
CRITICAL STEP Start with the two edge wells and then carefully add medium to the middle well. The Matrigel forms only a soft gel, so liquid must be pipetted slowly on top.
-
36
Use forceps to cover the chamber with the silicone lid.
Live imaging of ovarioles Timing ≈30 min to choose ovarioles and set individual z positions; imaging will take up to 11 h
-
37
Mount the imaging chamber on the microscope stage.
CRITICAL STEP The coverslip is very fragile; be careful to not break it.
-
38
Use transmitted light to locate germaria, and then examine them for the desired fluorescent marking of cells. Mark the positions of the desired germaria with the microscope software at a magnification of 10× or 20× in order to quickly screen through germaria.
-
39
Move to the 63X objective; first add oil to the objective, taking care not to touch the stage so as to not disturb the marked positions.
-
40
Visit each position at 63X and set the z limits for each germarium to be imaged. On the Zeiss LSM confocal microscope, positions added at low magnification must be visited sequentially, removed and re-added with the correct z limits for each germarium. All germaria must have the same number of z slices. It is advisable to add a few extra z slices because germaria move during imaging.
-
41
Image the sample with the desired time intervals and length of time. We image at 512 × 512 pixels, frame averaging = 2 and scan rate = 7, with a pixel dwell time of 3.15 𝛍s. We use 13 z slices with a spacing of 3 𝛍m. The xy resolution is 0.2 𝛍m. If only a subset of cells are marked, we recommend simultaneously capturing differential interference contrast (DIC) images to outline the shape of the germarium (Figs. 2 and 4).
Fig. 4: Quantifying movement of FSCs.
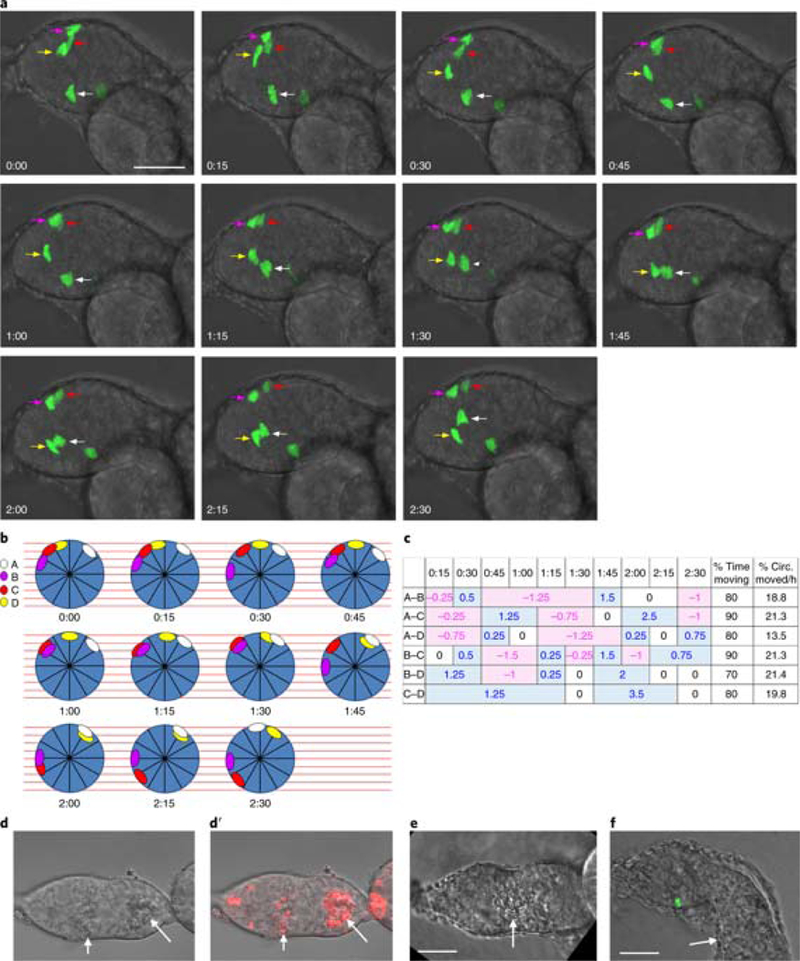
a, Time series showing GFP-labeled FSC movement. Each frame is a maximum projection of z sections to show all four labeled FSCs, indicated by colored arrows. Circumferential movement of FSCs in middle z slices (red and magenta arrows) cannot be seen in maximum projections. The germarium moved during imaging of time point 12, which is therefore not shown. Anterior is to the left. b, FSC positions at the time points in a were plotted around circles representing a cross-section of the FSC region as viewed from the anterior. The colored ovals correspond to the colored arrows, and the first cell after the ‘noon’ position on the circle was designated as cell A, with the lettering proceeding clockwise. The circle was subdivided into 12 sectors and 9 z sections (red lines) to place the cells based on their xyz coordinates. Because the germarium sometimes moved or rotated, the critical parameter is movement of one cell nucleus relative to another. c, For each pair of cells, we estimated relative movement as the fraction of a sector by which two cells separated (positive values in blue) or approached (negative values in pink) over the time interval during which they maintained their approach or separation. The minimum resolvable distance was 0.25 of a sector. When it was too difficult to judge whether the relationship of cell pairs had changed, the movement was scored as 0. The proportion of time during which cell pairs altered their radial separation was summed as ‘% time moving’. The average rate at which a cell pair altered its separation during periods of movement was calculated as the percentage of the circumference moved per hour (‘% circ. moved/h’). d-f, Examples of damaged germaria that should not be imaged. d, Dead cells in the region 3 cyst and scattered dead somatic cells (arrows) can be recognized from their rough appearance. D’ Dead cells have taken up propidium iodide (red; arrows). e, A misshapen germarium with dead cells (arrow; note the rough appearance). Region 3 is too large and should have already budded from the germarium. f, The budding egg chamber appears to have merged with the first budded egg chamber (arrow). We have noticed that if the first egg chamber is not healthy, the germarium will not develop properly. Scale bars, 20 μm.
CRITICAL STEP We use the Autosave function in the imaging software, Zeiss Zen, that runs the LSM700 in order to save images to the disk instead of software memory. The software memory cannot handle the large dataset being acquired. At the end of imaging, save the entire file.
? TROUBLESHOOTING
-
42
Once imaging has started, it is economical to turn off the microscope’s mercury or LED lamp used to visualize fluorescence through the eyepiece. Unused lasers can also be turned off.
-
43
Allow 5 min to save the file when imaging is finished, as file sizes may be large. Remember to turn off Autosaving of files.
Cleaning and storage of the imaging chamber. Timing ≈2 min
-
44
Use several cotton-tipped applicator swabs to clean the imaging well, first with water, and then with 70% ethanol. Rub the swabs gently against the glass coverslip to make sure that all traces of the specimen are removed.
-
45
Spray the imaging chamber and PDMS lid with 70% ethanol, allow them to dry, and store them in a dust-free environment.
Troubleshooting
Troubleshooting advice can be found in Table 2.
Table 2:
Troubleshooting Table
| Step | Problem | Possible reason | Solution |
|---|---|---|---|
| 7 | Wells are not properly positioned and walls are not perpendicular | Biopsy punches were not properly used | Print a schematic of the design and use it as a guide. Pay attention to applying pressure perpendicularly to the surface |
| 8 | The 1.5 mm diameter connecting channel is difficult to punch | There is not sufficient structural integrity to pierce the PDMS | Place the cored 8-mm plug back inside the well to absorb the pressure of the 1.5-mm punch |
| 28 | Germaria are bunched up with egg chambers in the muscle sheath | The muscle sheath was intact. It should be broken/ ripped near the stage 7 egg chamber. | During dissection pinch off stage 7/8 and older egg chambers from the ovariole with forceps. |
| 41 | Images become darker over time in transmitted light channel Imaging software cannot handle the large dataset being acquired |
An air bubble formed just below the chamber lid Images are being saved to memory |
Make two 1-mm holes in the PDMS lid above the imaging well Use settings in imaging software to autosave to hard drive instead |
Timing
Steps 1–19, building the imaging chamber: 4.5 h
Step 20, preparation of flies for imaging 0- to 30-day-old flies
Steps 21–36, preparation of ovarioles for imaging: 1 h for novice, 30–45 min for an expert
Steps 37–40, selection of germaria under the microscope and setting of positions and z limits for each germarium: 30 min
Steps 41–43, imaging: up to 11 h or overnight.
Steps 44 and 45, cleaning and storage of the imaging chamber: ≈2 min
Anticipated results
Healthy germaria in culture are continually expanding or budding off, and they rotate, twist, and move in all directions within the imaging window (Supplementary Videos 2–6), in some cases even twisting 90° and ending up perpendicular to the imaging window (Supplementary Videos 8 and 9). When a germarium is not elongating, pinching in or in the process of budding, cells may appear to be healthy but the germarium is no longer developing properly. As discussed in the ‘Limitations’ section, owing to follicle cells abnormally moving out of the germarium in culture around the time of budding, the useful imaging time depends on the initial morphology of the germarium and ranges up to 10.5 h with a median of 4.75 h with the sheath (n=102) and 3.5 h without the sheath (n=74). Budding occurs in 40% of those with the sheath and 16% of those without.
It is possible to track the movements of follicle stem cells (FSCs)1, and to visualize germline and follicle stem cells and their divisions (Fig. 3). FSCs move around the circumference of the germarium, frequently reversing direction (Fig. 4). FSCs also move anteriorly to become escort cells, a population of somatic cells required for germline development in the anterior half of the germarium1. A caveat to measuring the rates of cell movement is that the germarium can move or rotate during imaging, giving the appearance of cell movement (Supplementary Video 2). Thus, we quantified the movement of cells relative to one another and found that pairs of cells switch between moving toward and apart from one another1. Figure 4 shows an example of a germarium in which only four FSCs are labeled by MARCM; it is therefore relatively easy to track their movement. At the same time, Fig. 4 demonstrates how the germarium can become bunched up with egg chambers in the muscle sheath, which is not optimal because the germarium tends to move more in this situation (Supplementary Video 10). It is better to rip the muscle sheath with forceps near the stage 7 egg chamber.
In rare cases, the germarium bunches against older egg chambers but remains in the imaging window; in these cases we observed that germaria appear to use older egg chambers as guides for budding (Supplementary Video 4). Examples of damaged germaria that should not be imaged are shown in Fig. 4d-f.
Supplementary Material
Acknowledgements
We thank A. Choi for assistance in performing live-imaging experiments and D. Melamed for capturing the video of the RFP- and GFP-labeled germarium in Fig. 3. We are grateful to R. Lehmann for the Vasa-mCherry flies. This work was supported by National Institutes of Health grants to D.K. (RO1GM079351) and to G. V-N. (EB002520). E.C. was supported by the New York Stem Cell Foundation (NYSCF-D-FO2O).
Footnotes
Competing interests The authors declare no competing financial interests.
Contributor Information
Amy Reilein, Department of Biological Sciences Columbia University, New York, NY 10027, USA.
Elisa Cimetta, Department of Industrial Engineering (DII), Padova University Padova 35131, Italy, and, Fondazione Istituto di Ricerca Pediatrica Città della Speranza, Corso Stati Uniti, Padova 35127, Italy.
Nina M. Tandon, EpiBone, Inc 760 Parkside Avenue Brooklyn, NY 11226, and, The Cooper Union for theAdvancement of Science and Art, New York, New York 10003
Daniel Kalderon, Department of Biological Sciences Columbia University, New York, NY 10027, USA.
Gordana Vunjak-Novakovic, Department of Biomedical Engineering Columbia University New York, NY 10032.
References
- 1.Reilein A et al. Alternative direct stem cell derivatives defined by stem cell location and graded Wnt signalling. Nat Cell Biol 19, 433–444, doi: 10.1038/ncb3505 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Prasad M, Jang AC, Starz-Gaiano M, Melani M & Montell DJ A protocol for culturing Drosophila melanogaster stage 9 egg chambers for live imaging. Nat Protoc 2, 2467–2473, doi: 10.1038/nprot.2007.363 (2007). [DOI] [PubMed] [Google Scholar]
- 3.Cimetta E, Figallo E, Cannizzaro C, Elvassore N & Vunjak-Novakovic G Micro-bioreactor arrays for controlling cellular environments: design principles for human embryonic stem cell applications. Methods 47, 81–89, doi: 10.1016/j.ymeth.2008.10.015 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang Q, Shalaby NA & Buszczak M Changes in rRNA transcription influence proliferation and cell fate within a stem cell lineage. Science 343, 298–301, doi: 10.1126/science.1246384 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fichelson P et al. Live-imaging of single stem cells within their niche reveals that a U3snoRNP component segregates asymmetrically and is required for self-renewal in Drosophila. Nat Cell Biol 11, 685–693, doi: 10.1038/ncb1874 (2009). [DOI] [PubMed] [Google Scholar]
- 6.Mathieu J & Huynh JR Monitoring complete and incomplete abscission in the germ line stem cell lineage of Drosophila ovaries. Methods Cell Biol 137, 105–118, doi: 10.1016/bs.mcb.2016.03.033 (2017). [DOI] [PubMed] [Google Scholar]
- 7.Banisch TU, Maimon I, Dadosh T & Gilboa L Escort cells generate a dynamic compartment for germline stem cell differentiation via combined Stat and Erk signalling. Development 144, 1937–1947, doi: 10.1242/dev.143727 (2017). [DOI] [PubMed] [Google Scholar]
- 8.Morris LX & Spradling AC Long-term live imaging provides new insight into stem cell regulation and germline-soma coordination in the Drosophila ovary. Development 138, 2207–2215, doi: 10.1242/dev.065508 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shalaby NA & Buszczak M Live-Cell Imaging of the Adult Drosophila Ovary Using Confocal Microscopy. Methods Mol Biol 1463, 85–91, doi: 10.1007/978-1-4939-4017-2_6 (2017). [DOI] [PubMed] [Google Scholar]
- 10.Cetera M, Lewellyn L & Horne-Badovinac S Cultivation and Live Imaging of Drosophila Ovaries. Methods Mol Biol 1478, 215–226, doi: 10.1007/978-1-4939-6371-3_12 (2016). [DOI] [PubMed] [Google Scholar]
- 11.Prasad M, Wang X, He L, Cai D & Montell DJ Border Cell Migration: A Model System for Live Imaging and Genetic Analysis of Collective Cell Movement. Methods Mol Biol 1328, 89–97, doi: 10.1007/978-1-4939-2851-4_6 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Peters NC & Berg CA In Vitro Culturing and Live Imaging of Drosophila Egg Chambers: A History and Adaptable Method. Methods Mol Biol 1457, 35–68, doi: 10.1007/978-1-4939-3795-0_4 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Middleton CA et al. Neuromuscular organization and aminergic modulation of contractions in the Drosophila ovary. BMC Biol 4, 17, doi: 10.1186/1741-7007-4-17 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Andersen D & Horne-Badovinac S Influence of ovarian muscle contraction and oocyte growth on egg chamber elongation in Drosophila. Development 143, 1375–1387, doi: 10.1242/dev.131276 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee T & Luo L Mosaic analysis with a repressible cell marker (MARCM) for Drosophila neural development. Trends Neurosci 24, 251–254 (2001). [DOI] [PubMed] [Google Scholar]
- 16.Griffin R et al. The twin spot generator for differential Drosophila lineage analysis. Nat Methods 6, 600–602, doi: 10.1038/nmeth.1349 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wu JS & Luo L A protocol for mosaic analysis with a repressible cell marker (MARCM) in Drosophila. Nat Protoc 1, 2583–2589, doi: 10.1038/nprot.2006.320 (2006). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.