

Abstract
The apolipoprotein (apo) E ε4 allele is the primary genetic risk factor for late-onset Alzheimer’s disease (AD). ApoE in the brain is produced primarily by astrocytes; once secreted from these cells, apoE binds lipids and forms high-density lipoprotein (HDL)-like particles. Accumulation of amyloid-β protein (Aβ) in the brain is a key hallmark of AD, and is thought to initiate a pathogenic cascade leading to neurodegeneration and dementia. The level and lipidation state of apoE affect Aβ aggregation and clearance pathways. Elevated levels of plasma HDL are associated with lower risk and severity of AD; the underlying mechanisms, however, have not been fully elucidated. This study was designed to investigate the impact of an HDL-mimetic peptide, 4F, on the secretion and lipidation of apoE. We found that 4F significantly increases apoE secretion and lipidation in primary human astrocytes as well as in primary mouse astrocytes and microglia. Aggregated Aβ inhibits glial apoE secretion and lipidation, causing accumulation of intracellular apoE, an effect that is counteracted by co-treatment with 4F. Pharmacological and gene editing approaches show that 4F mediates its effects partially through the secretory pathway from the endoplasmic reticulum to the Golgi apparatus and requires the lipid transporter ABCA1. We conclude that the HDL-mimetic peptide 4F promotes glial apoE secretion and lipidation and mitigates the detrimental effects of Aβ on proper cellular trafficking and functionality of apoE. These findings suggest that treatment with such an HDL mimetic peptide may provide therapeutic benefit in AD.
Keywords: Alzheimer’s disease, apolipoprotein E, lipidation, HDL mimetic peptide, amyloid-β, astrocytes and microglia
The level and lipidation state of apoE affect the pathogenesis of Alzheimer’s disease (AD). In this study, an HDL-mimetic peptide, 4F, was found to promote the secretion and lipidation of apoE and to counteract Aβ-induced inhibition of apoE secretion and lipidation in primary human astrocytes, as well as in primary mouse astrocytes and microglia. Pharmacological and gene editing approaches show that 4F mediates its effects partially through the secretory pathway from the endoplasmic reticulum (ER) to the Golgi apparatus and requires the lipid transporter ABCA1. These findings suggest the therapeutic potential of HDL mimetic peptides in AD.
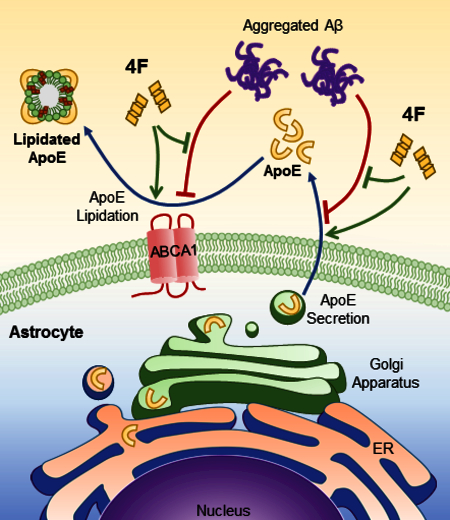
Introduction:
Alzheimer’s disease (AD) is the leading cause of dementia in the elderly; the risk of developing AD doubles every five years after the age of 65, and over one-third of Americans over 85 are affected (Hebert et al. 2013). Although the pathogenesis of AD is not fully understood, it is widely accepted that accumulation of amyloid-β protein (Aβ) in the brain initiates a pathogenic cascade, ultimately leading to neurodegeneration and dementia (Hardy & Selkoe 2002, Mucke & Selkoe 2012). There is currently no effective therapeutic that can halt, or even slow, the progression of AD pathology.
The apolipoprotein (apo) E ε4 allele is the strongest genetic risk factor identified to date for late-onset AD, associated with an earlier age of onset (Corder et al. 1993, Saunders et al. 1993, Strittmatter et al. 1993). In the brain, apoE is primarily secreted by astrocytes while microglia also produce apoE (Xu et al. 2006). Once secreted, apoE binds lipids and forms high-density lipoprotein (HDL)-like particles in the interstitial and cerebrospinal fluids through interactions with the ATP-binding cassette transporter A1 (ABCA1). The human apoE gene has three common alleles: ε2, ε3, and ε4 (designated apoE2, E3, and E4). The three resulting isoforms differ only at two amino acid sites in the protein. ApoE3 is the most common isoform and has a Cysteine residue at position 112 and an Arginine residue at 158. ApoE2, the rarest isoform, has Cysteine at both positions 112 and 158, whereas apoE4 has Arginine residues at both positions (Weisgraber et al. 1981).
ApoE has been extensively studied in the context of AD. It has been shown that apoE interacts with Aβ and that the level and lipidation state of apoE affect Aβ aggregation and clearance (Bu 2009, Holtzman et al. 2012, Liao et al. 2017).
ApoE4 has been shown to be poorly lipidated compared with apoE2 and apoE3 (Kanekiyo et al. 2014, Tai et al. 2014b, Hu et al. 2015, Hanson et al. 2013, Heinsinger et al. 2016), which is thought to be the cause of many of its deleterious effects (Bu 2009, Holtzman et al. 2012). Indeed, the receptor binding ability of apoE is dependent on its lipidation state; key residues involved in receptor binding become unburied when apoE is in a lipidated state (Chen et al. 2011). ApoE2, which reduces the risk for AD, has increased total levels of apoE compared to apoE3 or apoE4 (Cruchaga et al. 2012). This indicates that apoE level and lipidation state may be of crucial importance in combating the toxic Aβ pathway in AD pathogenesis.
Aggregated Aβ reduces the secretion of apoE and causes its cellular accumulation in astrocytes (LaDu et al. 2000, Handattu et al. 2013). The mechanism of this effect has not been fully described, although cAMP and β-adrenergic receptors have been shown to play a role (Igbavboa et al. 2006, Rossello et al. 2012). In addition, apoE binds to cell surface heparin sulfate proteoglycan (HSPG) and cell membrane associated receptors and competes with Aβ for uptake (Huang & Mahley 2014, Fu et al. 2016). Recent studies have also demonstrated that apoE binds to the triggering receptor expressed on myeloid cells 2 (TREM2) and regulates microglial function (Atagi et al. 2015, Bailey et al. 2015, Krasemann et al. 2017, Yeh et al. 2016). It has been shown that the LDL receptor (LDLR), but not HSPG, mediates the effects of Aβ on apoE in astrocytes (LaDu et al. 2000). This highlights the direct role of the lipid metabolism pathway in AD pathogenesis. Further, the formation of apoE/Aβ complexes is observed (Wisniewski et al. 1993, LaDu et al. 1994, Tai et al. 2014b), and may increase the ability of enzymes such as insulin degrading enzyme (IDE) to degrade Aβ in the extracellular space (Russo et al. 1998). Highly lipidated apoE promotes IDE degradation of Aβ to a greater extent than less lipidated forms (Jiang et al. 2008). Interestingly, apoE4 carriers produce less apoE/Aβ, and the complexes they do form are less stable (Tai et al. 2013). The reduced sequestration of Aβ into these complexes may hinder its degradation and lead to increased levels of toxic oligomers in the brain. However, a recent study showed that apoE minimally associates with Aβ but retains its significant influence on Aβ clearance, possibly through competing for binding to cell surface receptors (Verghese et al. 2013).
Multiple large-scale human clinical studies have found HDL levels to be highly correlated with cognitive performance late in life, and to be inversely correlated with both AD risk and severity (Hottman et al. 2014). A number of animal studies also support the role of apoA-I/HDL in AD (Lewis et al. 2010)(Lefterov et al. 2010)(Robert et al. 2016)(Song et al. 2014). These data led to the hypothesis that apoA-I/HDL may be an attractive target for AD therapy. However, the potential of full-length apoA-I protein as a therapeutic is greatly limited by its high cost of production and lack of oral bioavailability. A series of small peptides that mimic HDL function have been developed as potential therapeutics for cardiovascular disease (Osei-Hwedieh et al. 2011, Leman et al. 2014, White et al. 2014, Getz & Reardon 2014). The most notable of these is the peptide known as 4F, which is an 18 amino acid peptide containing 4 phenylalanine (F) residues (Ac-DWFKAFYDKVAEKFKEAF-NH2) (Segrest et al. 1983). 4F does not share sequence homology with any natural proteins but mimics the class A amphipathic helixes contained in HDL associated apolipoproteins such as apoA-I and apoE (Segrest et al. 1992). Animal studies have shown that treatment with 4F enhances HDL function and inhibits atherosclerosis (Navab et al. 2008). Intriguingly, animal experiments have also demonstrated that treatment with 4F reduces neuroinflammation and promotes cognitive function in atherosclerotic and AD mice (Buga et al. 2006, Handattu et al. 2009); the underlying mechanisms, however, have not been elucidated. As apoE is the primary HDL-associated protein in the brain, and 4F promotes cellular lipid efflux in the periphery (Xie et al. 2010, Liu et al. 2010, Tang et al. 2006), the beneficial effects of 4F on brain function may be mediated through apoE secretion and lipidation in the brain, which has not been explored previously.
Thus, the present study aimed to determine the impact of 4F on the secretion and lipidation of apoE in primary astrocytes and microglia, and whether 4F could mitigate the effects of Aβ therein. Our results demonstrate that 4F increases the secretion and lipidation of apoE from both mouse and human astrocytes, as well as mouse microglia, and ameliorates Aβ-induced inhibition in apoE secretion and lipidation. We also show that the 4F-mediated effects depend in part on the secretory transport pathway, from the endoplasmic reticulum (ER) to the Golgi apparatus, and the function of cell surface associated receptors. Specifically, we show that 4F-mediated enhancement of apoE secretion is abolished in the absence of ABCA1 expression. These findings suggest that 4F may reduce the detrimental effects of poorly lipidated apoE in the brain and serve as a potential therapeutic agent for AD.
Methods:
Primary astrocyte and microglial culture:
This study was not pre-registered. Primary glial cells were collected and cultured as previously described (Fagan et al. 1999). Briefly, neonatal pups (total n=84) of wild-type C57BL/6J mice (Stock No: 000664, The Jackson Laboratory, Bar Harbor, ME; RRID:IMSR_JAX:000664) were sacrificed within the first 3 days post-natal. Both sexes of the pups were used and no randomization was performed to allocate animals in the study. The animals were anesthetized by isoflurane inhalation prior to decapitation. Brains were dissected out, then cortex and hippocampal tissue were triturated into a single-cell suspension and cultured for 14 days in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum, 16mM HEPES buffer, 0.1mM non-essential amino acids, 2mM GlutaMAX, 2.5μg/ml amphotericin B and 50μg/ml gentamicin, at which point microglia were removed by shaking. Medium was replaced, and microglia were allowed to grow up an additional week, at which point they were shaken loose and removed again. Astrocytes were then passaged two times to increase purity. The purity of astrocyte and microglial cultures was determined by immunofluorescence analysis of markers specific for astrocytes and microglia (Fig. S1 and S2). Finally, cells were plated for treatment at 2×105 cells per well on poly-D-lysine (PDL) coated 12-well tissue culture plates and allowed to attach and grow for 2 days prior to experimental treatment. Human primary astrocytes were purchased from ScienCell (Carlsbad, CA; Cat# 1800) and cultured following the manufacturer’s protocols. The peptides (with purity > 95%), 4F (Ac-DWFKAFYDKVAEKFKEAF-NH2), D-4F (same sequence as 4F but with D-amino acids), and Scrambled 4F (S. 4F) (Ac-DWFAKDYFKKAFVEEFAK-NH2), were purchased from American Peptide Company (now Bachem; Sunnyvale, CA). During treatment, cells were washed twice with sterile phosphate buffered saline (PBS) and treatments were performed in serum-free OPTI-MEM supplemented with 50μg/ml gentamicin for various durations, as defined for each experiment. No blinding was performed during the experiment. All animal procedures were prospectively reviewed and approved by the Institutional Animal Care and Use Committee (IACUC protocol # 1607–33963A) of the University of Minnesota.
Gel electrophoresis and Western blot analysis:
After treatments were performed, media was collected and cells were lysed in ice-cold RIPA buffer. Media and cell lysates were then subjected to 12% SDS-PAGE. Proteins were transferred to PVDF membranes and probed with anti-mouse apoE (Santa Cruz Biotechnology, Dallas, TX; Cat# sc-6384; RRID:AB_634036) and tubulin (Sigma-Aldrich, St. Louis, MO; Cat# T5168; RRID:AB_477579) antibodies, followed by HRP-conjugated secondary antibody and chemiluminescence detection using Western Lighting Plus-ECL reagents (PerkimElmer, Waltham, MA; Cat# NEL103001EA). ApoE from immortalized apoE3 targeted-replacement astrocytes was probed with an anti-human apoE antibody (Millipore, Burlington, MA; Cat# 178479; RRID:AB_564230). Other primary antibodies used include: anti-glial fibrillary acidic protein (GFAP) (Millipore; Burlington, MA; Cat# MAB3402; RRID:AB_94844), anti-ionized calcium-binding adapter molecule 1 (Iba1) (Wako; Richmond, VA; Cat# 019–19741; RRID:AB_839504), anti-apoJ (clusterin) (R&D; Minneapolis, MN; Cat# AF2747; RRID:AB_2083314), anti-complement C3 (MP Biomedical; Santa Ana, CA; Cat# 0855444; RRID:AB_2334469), anti-ABCA1 (Novus; Littleton, CO; Cat# NB400–105; RRID:AB_10000630), anti-LDLR (Abnova; Taipei, Taiwan, Cat# PAB8804; RRID:AB_1676510), and anti-LRP1 (Millipore Cat# MABN1796, clone 6F8; generously provided by Dr. Guojun Bu, Mayo Clinic, Jacksonville, FL).
Non-denaturing gradient gel electrophoresis (NDGGE) was used to assess apoE lipidation. Fresh media was run on 4–20% polyacrylamide tris-glycine gels (ThermoFisher; Waltham, MA; Cat# EC6021BOX) in the absence of SDS, reducing agents or sample boiling at 125V for 5 hours. Proteins were transferred to PVDF membranes at 100V for 90 minutes and probed for apoE, followed by HRP-conjugated secondary antibody and chemiluminescence detection using ECL reagents. Poorly lipidated apoE was defined as complexes smaller than 8.2 nm as measured by NDGGE based on the high molecular weight marker (GE Healthcare; Buckinghamshire, UK; Cat# 17044501).
Preparation of aggregated Aβ:
Aggregated Aβ42 was prepared as previously described (Stine et al. 2011). Briefly, hexafluoroisopropanol (HFIP) treated Aβ42 film, kindly provided by Dr. Mary Jo LaDu and Dr. Leon Tai (University of Illinois at Chicago), was resuspended in fresh dry DMSO, diluted in phenol-free F12 cell culture media, and incubated at 37°C for 24 hours for aggregation. Aliquots of the Aβ preparation at 0 and 24 hours were examined by LDS-NuPAGE (4–12% bis-tris gels; ThermoFisher; Waltham, MA; Cat# NP0322BOX) followed by immunoblot analysis with the anti-Aβ antibody, 6E10 (Covance (now BioLegend); San Diego, CA; Cat# SIG-39340; RRID:AB_662806), and transmission electron microscopy (TEM) as previously described (Fitz et al. 2017) (Fig. 3S). This aggregated Aβ preparation was used to treat cells.
Figure 3:

Scrambled 4F has no effect on apoE Secretion or Lipidation, while D-4F is equally effective as 4F. (A,B) Primary mouse astrocytes were treated for 6 hours with 5μM 4F or Scrambled 4F (S. 4F) in serum-free OPTI-MEM. (A) SDS-PAGE and (B) NDGGE were performed to determine the relative secretion and lipidation state of apoE, respectively. (C,D) Primary mouse astrocytes were treated for 6 hours with 5 μM 4F or D-4F in serum-free OPTI-MEM. (C) SDS-PAGE and (D) NDGGE were performed to determine the relative secretion and lipidation state of apoE, respectively. Tubulin was used as a loading control. Results were obtained from n=3 independent experiments with each treatment in duplicate. * = p <0.05, ** = p < 0.01, *** = p < 0.001.
Pharmacological manipulation:
Primary mouse astrocytes were treated with various pharmacological agents in order to understand potential mechanisms of the effects seen. 4F was prepared in sterile PBS and used at a range of concentrations from 0.1μM to 5μM. Actinomycin D (Sigma-Aldrich; St. Louis, MO; Cat# A1410) was prepared in DMSO and used at 1μg/ml. Cycloheximide (Sigma-Aldrich; Cat# C7698) and brefeldin A (Sigma-Aldrich; Cat# B7651) were prepared in ethanol and used at 2μg/ml and 1μg/ml, respectively. Heparinase I (Sigma-Aldrich; Cat# H2519) and pronase (Sigma-Aldrich; Cat# P8811) were prepared in PBS and used at 5 units/ml and 10μg/ml, respectively.
Targeted deletion of ABCA1 in astrocytes using CRISPR/Cas9:
Immortalized mouse astrocytes derived from human apoE3 targeted-replacement mice (Morikawa et al. 2005), generously provided by Dr. Guojun Bu (Mayo Clinic, Jacksonville, FL), were cultured in DMEM supplemented with 10% FBS, 2mM GlutaMAX, 50μg/ml gentamicin, and 10ng/ml epidermal growth factor (EGF). The cells were co-transfected with a CRISPR/Cas9 vector designed to disrupt/knock out (KO) ABCA1 gene expression, and a homology directed repair (HDR) vector, designed to incorporate genes encoding puromycin resistance as well as red fluorescence protein (RFP) into the genome in place of ABCA1. Both vectors were purchased from Santa Cruz biotechnology (Cat# sc-401086 and sc-401086-HDR, respectively). Transfected cells were visually confirmed by RFP expression, and un-transfected cells were eliminated by titration of puromycin concentration up to a final concentration of 9 μg/ml. Absence of ABCA1 protein expression was confirmed by Western blot analysis, and these cells were plated for experiments and treated with or without 4F as previously described.
Statistical Analysis:
Western blot results were quantified using Image J software. The amount of secreted apoE was analyzed as the ratio of apoE in medium to total apoE, where total apoE = apoE in medium + apoE in cell lysate, and expressed as relative percent in media with the amount in the vehicle treatment set as 100%. The total amount of apoE was normalized by tubulin when it was compared between different treatments. The amount of lipidated apoE was analyzed as the ratio of lipidated apoE to total apoE in medium, where total apoE = lipidated apoE + poorly lipidated apoE, and expressed as relative percent in lipidated form with the amount in the vehicle treatment set as 100%. Data were expressed as mean ± standard error (SE) from at least three independent experiments with each treatment in duplicate or triplicate. No sample size calculation was performed. Comparison of different treatments was performed by Student’s t-test or analysis of variance (ANOVA) (for normally distributed data), or the Mann-Whitney rank sum test (for non-normally distributed data). SigmaPlot v13.0 (Systat Software, San Jose, CA) was used for statistical analysis. p < 0.05 was considered statistically significant.
Results:
4F increases apoE secretion and lipidation in primary astrocytes
Due to the role of apoE in HDL-like particle formation and function, the secretion and lipidation state of apoE is highly important to its ability to perform its functions in the brain. ApoA-I has been shown to increase the secretion of apoE from peripheral macrophages (Rees et al. 1999) and primary mixed glia (Fan et al. 2011). We therefore hypothesized that the HDL-mimetic peptide 4F may mediate apoE secretion and lipidation in astrocytes. Using primary mouse astrocytes, we found that 4F induced a robust concentration- and time-dependent increase in both secretion and lipidation of apoE from these cells (Fig. 1; Fig. 2), without affecting cell survival (Fig. S4). We initially measured the levels of secreted apoE at 24 hours of treatment. We found that apoE secretion was significantly increased at 1, 2, and 5 μM 4F. At 5μM 4F, the secretion of apoE was increased to approximately 300% of control (Fig. 1A). Similarly, 4F produced a dose dependent increase in apoE lipidation as well, over 400% increase at 5μM (Fig 1B). We then used 5uM 4F to test the time-dependent effects from 2 to 24 hours, finding that both secretion and lipidation of apoE increased with time (Fig. 2). Interestingly, the 4F effect appears to begin plateauing between 6 and 12 hours of treatment (Fig. 2), although it is possible that such phenomenon was caused partly by the intrinsic saturation of the in vitro cell culture system or Western blotting. Despite the limitations and some variations observed, these results demonstrated that 4F treatment consistently increased apoE secretion and lipidation in these cells.
Figure 1:

Dose dependent effects of 4F on apoE secretion and lipidation. Primary mouse astrocytes were treated for 24 hours with 0–5μM 4F in serum-free OPTI-MEM. (A) SDS-PAGE and (B) NDGGE were performed to determine the relative secretion and lipidation state of apoE, respectively. Tubulin was used as a loading control. Results were obtained from n=3 independent experiments with each treatment in duplicate. ** = p < 0.01.
Figure 2:

Time course for the effects of 4F on apoE secretion and lipidation. Primary mouse astrocytes were treated for 2–24 hours with 5μM 4F in serum-free OPTI-MEM. (A) SDS-PAGE and (B) NDGGE were performed on samples to determine apoE secretion and lipidation. Tubulin was used as a loading control. As the level of secreted/lipidated apoE at Veh-2 h was not detectable, the level at 4F-2 h was set as 100%. Results were obtained from n=2–4 independent experiments with each treatment in duplicate. * = p <0.05, ** = p <0.01, *** = p < 0.001.
To test the specificity of 4F-mediated effects, we utilized a scrambled version of 4F. Scrambled 4F contains the same 18 amino acids as 4F, but in an altered sequence, which precludes the formation of the amphipathic alpha helical structure that is critical to the function of the peptide (Handattu et al. 2009). We found that scrambled 4F had no effect on apoE secretion or lipidation in comparison to 4F (Fig. 3A and3B), confirming the specificity of 4F-mediated effects and further indicating the necessity of the particular amphipathic structure of 4F to mediate such effects. In addition, we found that D-4F, which has the same amino acid sequence as 4F but is composed entirely of the D-enantiomers of each amino acid, produced the same effect as 4F on astrocyte apoE secretion and lipidation (Fig. 3C and3D). These results have significant therapeutic implications because peptides D-amino acids are less susceptible to degradation of digestive/proteolytic enzymes in vivo.
To determine whether the effect of 4F is selective to apoE secretion, we measured the levels of two other proteins, well known to be secreted by astrocytes, apoJ (aka clusterin) and complement C3, in the medium and cell lysate. The results showed that 4F treatment did not change the secretion of these proteins (Fig. S5), indicating the selective effect of 4F on apoE secretion. Furthermore, 4F did not affect the levels of ABCA1, LDLR, or LRP1 in treated cells (Fig. S6).
Next, considering that the regulation of apoE expression differs between mouse and human cells and that mouse apoE is structurally and functionally distinct from human apoE (Fagan et al. 1999, Zhu et al. 2012, Liao et al. 2015), we investigated whether the effects of 4F on apoE secretion and lipidation extend to human cells. To accomplish this, primary human astrocytes were cultured and treated with 4F for 6 and 24 hours, respectively, followed by apoE immunoblot analysis in the medium and cell lysate. The results showed that 4F treatment significantly increased the endogenous apoE secretion and lipidation in human astrocytes (Fig. 4), consistent with the findings in mouse astrocytes described above. These data demonstrate that 4F promotes apoE secretion and lipidation in both mouse and human astrocytes.
Figure 4:

4F enhances apoE secretion and lipidation in primary human astrocytes. Primary human astrocytes were treated for 6 hours (A, B) or 24 hours (C,D) with 5μM 4F in serum-free OPTI-MEM. (A,C) SDS-PAGE and (B,D) NDGGE were performed to determine the relative secretion and lipidation state of apoE, respectively. Tubulin was used as a loading control. Results were obtained from n=3 independent experiments with each treatment in triplicate. * = p < 0.05, ** = p < 0.01, *** = p < 0.001.
4F mitigates aggregated Aβ42-induced inhibition of apoE secretion and lipidation
To investigate the effects of Aβ on apoE secretion and lipidation, we treated primary mouse astrocytes with different concentrations of aggregated Aβ42, followed by immunoblot analysis. We found that aggregated Aβ42 caused a dose-dependent decrease in apoE secretion, accompanied by accumulation of intracellular apoE in astrocytes (Fig. 5A), without affecting cell survival (Fig. S4). Furthermore, we found that apoE secreted from Aβ-treated astrocytes was less lipidated than that released by control cells (Fig. 5B). This indicates that Aβ not only suppresses the secretion of apoE, but also inhibits the lipidation of secreted apoE, thus potentially diminishing the function and stability of apoE as well. Intriguingly, co-treatment with 4F counteracts the inhibitory effects of Aβ on both apoE secretion and lipidation in primary mouse astrocytes (Fig. 5A and5B). The Aβ-induced inhibition of apoE secretion was also observed in primary human astrocytes, and as in mouse astrocytes, co-treatment with 4F overcame the inhibitory effect of Aβ on human apoE secretion (Fig. 5C).
Figure 5:

Aggregated Aβ inhibits apoE secretion and lipidation and 4F counteracts the inhibitory effects of Aβ in astrocytes and microglia. (A,B) Primary mouse astrocytes were cultured for 20 hours with 1–10 μM Aβ42 aggregates in absence or presence of 5 μM 4F in serum-free OPTI-MEM. (C) Primary human astrocytes were treated for 24 hours with 5 μM Aβ42 aggregates and/or 2 μM 4F in serum-free OPTI-MEM. (D) Primary mouse microglia were treated for 24 hours with 2.5 μM Aβ42 aggregates and/or 5 μM 4F. (A, C, D) SDS-PAGE and (B) NDGGE were performed to determine the relative secretion and lipidation state of apoE, respectively. Tubulin was used as a loading control. Results were obtained from n=3 independent experiments with each treatment in duplicate or triplicate. * = p <0.05, ** = p <0.01, *** = p < 0.001.
In addition, microglia also produce apoE, and the importance of these cells in the context of AD is highlighted by findings that apoE interacts with TREM2 and regulates microglial function (Atagi et al. 2015, Bailey et al. 2015, Krasemann et al. 2017, Yeh et al. 2016). Therefore, we tested the effects of 4F and Aβ in primary mouse microglia. Similar results as in astrocytes were observed. 4F treatment increased, whereas Aβ treatment decreased, apoE secretion, and co-treatment with 4F mitigated the inhibitory effect of Aβ on apoE secretion in primary mouse microglia (Fig. 5D).
The effect of 4F on apoE secretion relies on the production of intracellular apoE but is not influenced by the process of transcription and translation per se
To determine whether new protein production is necessary for the 4F-inducted effects on apoE, we treated primary mouse astrocytes with actinomycin D (actD) and cycloheximide (CHX) to inhibit transcription and translation, respectively. ActD treatment alone did not affect the apoE protein levels (Fig. 6A), and the effects of 4F on apoE secretion were unaffected by the presence of actD, suggesting that pre-existing apoE mRNA was stable and sufficient to produce the new protein. In contrast, CHX treatment alone drastically reduced apoE levels in both the media and cell lysates (Fig. 6A). Interestingly, the residual amount of apoE produced in the presence of CHX was efficiently secreted into the medium with the treatment of 4F (Fig. 6A). Together, these results showed that the secretion of apoE promoted by 4F depended on the availability of intracellular apoE but the mechanism was not influenced by the processes of transcription and translation per se.
Figure 6:

4F depends on protein production, partly the canonical secretory pathway, and cell-surface receptors for its effect in primary mouse astrocytes. (A) Effects of protein transcription and translation inhibition on apoE secretion. Primary mouse astrocytes were treated for 12 hours with 1 μg/mL actinomycin D (actD) or 2 μg/mL cycloheximide (CHX) in serum-free OPTI-MEM with or without 4F. (B) Effect of inhibiting the protein secretory pathway on apoE secretion. Primary mouse astrocytes were treated for 2 hours with 5 μM Brefeldin A (BFA) in serum-free OPTI-MEM in the presence or absence of 5 μM 4F. (C) Effects of degrading heparin sulfate proteoglycan or all cell surface proteins on apoE secretion. Primary mouse astrocytes were pre-treated for 1 hour with 5 units/mL heparinase I (Hep) or 10 μg/mL Pronase E (Pro) in serum-free OPTI-MEM, respectively. Cells were then washed twice with PBS and treated with 5μM 4F for 2 hours. SDS-PAGE was performed on samples to determine the relative amount of apoE secreted. Tubulin was used as a loading control. Results were obtained from n=3 independent experiments with each treatment in duplicate. * = p < 0.05, ** = p < 0.01, *** = p < 0.001; NS, not significant (p = 0.052).
4F promotes apoE secretion in part through the protein transport pathway from the endoplasmic reticulum to the Golgi apparatus
To further study the mechanisms by which 4F enhances apoE secretion, we employed the use of brefeldin A (BFA), which interferes with the anterograde transport from the endoplasmic reticulum (ER) to the Golgi apparatus (Klausner et al. 1992). Treatment of primary mouse astrocytes with BFA drastically inhibited basal apoE secretion. Further, BFA treatment significantly reduced the 4F-induced effect on apoE secretion (Fig. 6B). Interestingly, in the presence of BFA, 4F treatment showed a trend increase in apoE secretion compared to BFA alone, although this effect did not reach statistical significance (Fig. 6B). This indicates that there might be a delay in the effects of BFA on the secretory pathway during the co-treatment with 4F or there might be other pathways involved in mediating the effects of 4F on apoE secretion.
4F-stimulated apoE secretion does not involve heparin sulfate proteoglycan but requires cell surface receptors
ApoE is known to bind to heparin sulfate proteoglycan (HSPG) and other receptors on the cell surface to produce a surface-bound pool of apoE, which can be released into the media (Huang & Mahley 2014). Therefore, we aimed to determine if 4F-stimulated apoE secretion involves the release of this extracellular matrix or receptor-bound pool of apoE. To cleave cell surface HSPG and receptors, primary mouse astrocytes were pre-treated with heparinase or pronase for 1 hour, respectively. Following a thorough wash to remove apoE released by this treatment, the cells were treated with 4F or vehicle for 2 hours in the absence of the proteolytic agents. We found that heparinase treatment had no effects on 4F-stimulated apoE secretion. 4F produced an equally significant increase in apoE secretion after heparinase treatment (Fig. 6C), indicating that 4F does not induce its effect by releasing HSPG-bound pools of apoE. Interestingly, while pronase treatment did not affect the basal level of apoE secretion, it abolished 4F-stimulated apoE secretion (Fig. 6C). Together, these results show that 4F-stimulated apoE secretion does not involve cell surface HSPG but relies on the presence of other cell surface receptors.
4F-mediated apoE secretion requires ABCA1
The inhibition of 4F-mediated apoE secretion by pronase treatment led us to question which specific cell-surface protein(s) are required for this phenomenon to occur. ABCA1 is the primary cell surface receptor responsible for the transfer of lipids onto nascent HDL particles (Oram & Heinecke 2005). As such, we aimed to determine whether ABCA1 is required for the 4F-mediated effects on apoE to occur. We utilized immortalized mouse astrocytes with a targeted replacement of mouse apoE for human apoE3 (Morikawa et al. 2005). Using commercially available vectors based on the CRISPR/Cas9 technology (Santa Cruz biotechnology), the ABCA1 gene was disrupted/knocked out in these cells. Transfection and loss of ABCA1 protein expression were confirmed by fluorescent imaging and Western blot analysis (Fig. 7A and7B). We found that total levels (secreted and cellular) of apoE were markedly reduced in ABCA1 KO cells (Fig. 7B). This finding indicates that cellular production of apoE is downregulated in the absence of ABCA1, consistent with findings in animal models (Hirsch-Reinshagen et al. 2005, Koldamova et al. 2005, Wahrle et al. 2005). Compared with ABCA1-intact wild type (WT) astrocytes, 4F failed to induce apoE secretion in ABCA1 KO cells (Fig. 7B). Importantly, 4F-mediated enhancement of apoE secretion could be detected under the condition of reduced cellular apoE levels, as observed in our experiments using the translational inhibitor CHX (Fig. 6A). Thus, the results indicate that the presence of ABCA1 is required for 4F-mediated effects on apoE secretion.
Figure 7:

4F-induced elevation of apoE secretion requires ABCA1. Immortalized apoE3 TR astrocytes were transfected with an ABCA1-specific CRISPR/Cas9 vector and a homology directed repair (HDR) vector for puromycin resistance and red fluorescence protein (RFP) to establish a stably transfected ABCA1 KO cell line. (A) Transfection is confirmed by continuous expression of RFP. Un-transfected apoE3 TR cells (WT, normal ABCA1 expression) show no RFP signal. (B) ABCA1 WT and ABCA1 KO apoE3 TR astrocytes were treated for 6 hours with or without 5μM 4F in serum-free OPTI-MEM. SDS-PAGE was performed on samples to determine the relative amount of apoE secreted, as well as ABCA1 protein expression levels. Tubulin was used as a loading control. Results were obtained from n=3 independent experiments with each treatment in duplicate. * = p < 0.05; NS, not significant.
Discussion:
In the current study, we described the ability of 4F, an 18 amino acid HDL-mimetic peptide, to increase the secretion and lipidation of apoE in primary human astrocytes as well as primary mouse astrocytes and microglia. The primary functions of apoE require it to be part of an HDL-like particle in the brain, and mounting evidence suggests that many of its critical roles are influenced by the degree to which apoE is lipidated. ApoE4, the primary genetic risk factor for late onset AD, has been shown to be poorly lipidated compared with apoE2 and apoE3 (Kanekiyo et al. 2014, Tai et al. 2014b, Hu et al. 2015, Hanson et al. 2013, Heinsinger et al. 2016). As the lipidation status of apoE affects its function and receptor binding capacity (Bu 2009, Koldamova et al. 2014), deficits in lipidation may underlie the pathogenic effects of apoE4. Therefore, increasing the lipidation of apoE may be a viable therapeutic avenue for AD treatment.
A number of studies have shown that oligomeric species of Aβ correlate better with cognitive impairment than Aβ plaque burden in AD (Haass & Selkoe 2007, Lesne et al. 2013, Selkoe & Hardy 2016). Aβ has also been previously shown to inhibit apoE secretion from astrocytes (LaDu et al. 2000, Igbavboa et al. 2006, Handattu et al. 2013). Our results corroborate these previous findings on the inhibition of apoE secretion by Aβ in both mouse and human astrocytes as well as in mouse microglia, and further demonstrate that lipidation of secreted apoE from primary mouse astrocytes is also inhibited by Aβ, which may reduce apoE function, stability, and receptor binding capacity. The interaction between Aβ and apoE may be a crucial part of early pathogenesis, in which early Aβ insults reduce apoE secretion and lipidation, leading to reduced Aβ clearance and thus elevated levels of aggregated Aβ in the brain. This in turn may create a vicious feed-forward cycle in which these elevated levels of aggregated Aβ cause a further decline in apoE secretion and lipidation. Blocking this circular pathway with a pharmacological agent that increases apoE secretion and lipidation may slow or even halt the progression of AD. Interestingly, Verghese and colleagues showed that the association between apoE and Aβ was minimal in the cerebrospinal fluids in human apoE targeted-replacement mice and that apoE-deficient astrocytes cleared Aβ from the medium more efficiently than apoE-expressing astrocytes (Verghese et al. 2013). They further showed that apoE and Aβ compete for the same LRP1-mediated clearance pathway, suggesting that reducing apoE levels would enhance Aβ clearance (Verghese et al. 2013). We examined the effect of co-treatment of 4F on Aβ uptake in primary mouse astrocytes by confocal microscopy and flow cytometry as described previously (Omtri et al. 2012). The results showed that 4F did not interfere with Aβ uptake (Fig. S7). Importantly, chronic treatment with 4F resulted in a significant reduction of amyloid deposition in the APP/PS1 model of AD (Handattu et al. 2009), indicating that 4F may facilitate brain Aβ clearance through multiple mechanisms in vivo.
When the effect of Aβ on reducing secretion of apoE was described initially, it was postulated that the Aβ-induced effect might be protective, based on the assumption that an increase in the cellular production of apoE counteracts the detrimental effects of Aβ (LaDu et al. 2000, Igbavboa et al. 2006). However, more recent studies indicate that apoE must be secreted and properly lipidated in order to perform its canonical functions, including lipid/cholesterol transport, synapse regeneration, immune modulation, and clearance/degradation of Aβ (Kanekiyo et al. 2014, Tai et al. 2014b, Hu et al. 2015, Hanson et al. 2013). As such, Aβ-induced accumulation of intracellular apoE and simultaneous inhibition of secretion is considered detrimental. We have found that co-treatment of astrocytes with 4F mitigates the Aβ-induced inhibitory effects on apoE secretion and lipidation. In the presence of 4F, astrocytes exposed to Aβ secrete similar amounts of apoE as control cells, and the secreted apoE is lipidated as efficiently as in the controls. Consistent with our findings, it has been shown that a hybrid apoE/apoA-I-mimetic peptide, Ac-hE18A-NH2, mitigated the inhibitory effect of Aβ in the U251 astrocyte cell line (Handattu et al. 2013). These results suggest that 4F or related apo mimetic peptides can counteract the detrimental effects of Aβ on astrocytes, potentially serving as a protective agent against AD. These findings in vitro have significant implications in vivo. Although it remains a topic of debate whether an increase or decrease in total apoE levels is protective against AD, improving the lipidation state of apoE ameliorates cognitive deficits in the presence or absence of amyloid pathology regardless of apoE genotype (Cramer et al. 2012, Fitz et al. 2013, Boehm-Cagan & Michaelson 2014).
In the present study, we showed that 4F treatment consistently produced a robust increase in apoE secretion and lipidation in astrocytes throughout the experiments. The mechanisms by which 4F promotes apoE secretion were explored with the use of pharmacological agents and CRISPR/Cas9-mediated gene editing approaches. The effect of 4F occurs in part through the classical ER-Golgi secretion pathway, as inhibition of this pathway by BFA partially blocks the 4F-induced enhancement of apoE secretion. Our experiments with heparinase demonstrate that cell surface HSPG-associated apoE does not contribute to the effect of 4F on apoE secretion, which is in line with a previous study showing that the Aβ-induced elevation of intracellular apoE does not depend on interactions with HSPG (LaDu et al. 2000). However, removal of other cell surface receptors with pronase abolishes the increase of apoE secretion by 4F in primary astrocytes. Furthermore, using ABCA1 KO astrocytes generated by using CRISPR/Cas9 technology, we show that the effect of 4F on apoE secretion requires the presence of ABCA1. Notably, it was reported recently that an apoE-mimetic peptide ameliorated apoE4-driven cognitive and brain pathologies through activation of ABCA1 (Boehm-Cagan et al. 2016). These findings corroborate the importance of ABCA1 in apoE-targeted therapeutic development.
Highlighting the importance of ABCA1 in the context of AD, several studies have shown that ABCA1 deficiency exacerbates amyloid pathology in AD mice (Hirsch-Reinshagen et al. 2005, Koldamova et al. 2005, Wahrle et al. 2005). ABCA1 KO mice exhibit drastically reduced apoE levels and lipidation. In contrast, apoE KO AD mice have reduced amyloid deposition (Bales et al. 1997, Irizarry et al. 2000) and tau pathology (Shi et al. 2017). Furthermore, immunotherapy against apoE was associated with reduced Aβ deposition in AD mice (Kim et al. 2012), an effect that has recently been recapitulated using an antibody targeting only non-lipidated forms of apoE (Liao et al. 2018). These findings, together, indicate that the lipidation state of apoE may influence AD pathology more so than the absolute level of apoE.
In line with these findings, ABCA1 overexpression is associated with elevated apoE levels and lipidation, and attenuates AD pathology (Wahrle et al. 2008). Bexarotene and other nuclear receptor agonists, which upregulate genes including ABCA1, have been shown to increase apoE level and lipidation, while reducing amyloid pathology in mice (Cramer et al. 2012, Zelcer et al. 2007, Skerrett et al. 2014, Donkin et al. 2010, Corona et al. 2016), although the effect on amyloid pathology was not observed in all studies (Veeraraghavalu et al. 2013, Tesseur et al. 2013). Importantly, these agents produce deleterious peripheral effects on triglyceride production and liver health (Hong & Tontonoz 2014, Tousi 2015, Tai et al. 2014a), as well as off-target effects inherent when targeting promiscuous transcription factors, indicating that alternative methods of increasing apoE lipidation may provide safer and greater benefits to those suffering from AD.
The potential for development of full-length HDL-associated apolipoproteins such as apoA-I or apoE as therapeutics is limited by their size, structure and post-translational modifications, making small HDL-mimetic peptides, such as 4F, attractive candidates in this regard. Notably, unlike other HDL-mimetic peptides (Leman et al. 2014, White et al. 2014), 4F has been tested in three human clinical trials with 50, 152 and 62 individuals, respectively, for cardiovascular disease; 4F was found to be safe and well-tolerated when administered orally or by injections, and improved HDL anti-inflammatory properties (Bloedon et al. 2008, Watson et al. 2011, Dunbar et al. 2017). Further, our finding that D-4F improves apoE secretion and lipidation to the same extent as 4F has significant implications for therapeutic development, because D-4F is orally bioavailable with a longer half-life than L-4F in vivo (Navab et al. 2005).
Our studies indicate that the small peptide, 4F, may harbor the beneficial effects of apoA-I/HDL in a form that is both more economically viable and much easier to deliver to the brain. Notably, D-4F, in the presence of a low/ineffective dose of a statin drug, has been shown to mitigate memory deficits and amyloid pathology in the APP/PS1 model of AD (Handattu et al. 2009). In that study, D-4F with pravastatin was administered orally in drinking water to 4–5 month old male APP/PS1 mice for 3 months, using scrambled D-4F with pravastatin and drinking water alone as controls, followed by behavioral assessment and biochemical analyses. The results showed that treatment with D-4F+pravastatin improved the learning and memory performance of APP/PS1 mice in the Morris water maze test and led to a >50% reduction in Aβ load compared with the controls (Handattu et al. 2009). Further analysis showed that D-4F+pravastatin treatment did not alter APP expression or proteolytic processing but was associated with a decrease in glial activation and inflammatory markers in the brain. Thus, it was concluded that D-4F+pravastatin treatment inhibits Aβ deposition and improves cognitive function through exerting anti-inflammatory actions in the brain (Handattu et al. 2009). These findings strongly support the beneficial effects of 4F treatment in vivo, although a study with 4F in the absence of any statin drug will be needed for confirmation. In addition, the role of apoE was not investigated in that study. As apoE plays a pivotal role in Aβ metabolism and immune modulation, and based on the findings in the present study, we hypothesize that the enhancement of apoE secretion and lipidation may underlie the beneficial effects of 4F treatment in AD mice. Further studies will be required to test this hypothesis.
Targeting apoE is a promising approach for AD therapy. However, simply altering the level of apoE may not be sufficient, particularly in individuals carrying the apoE4 allele. Mounting evidence shows that the lipidation status of apoE dictates its function and relative contribution to AD risk. The present study has demonstrated that the HDL-mimetic peptide, 4F, promotes secretion and lipidation of apoE from astrocytes and microglia, in both the presence and absence of Aβ, suggesting that HDL-mimetic peptides such as 4F may serve as effective apoE-modulating agents against AD.
Supplementary Material
Acknowledgments and conflict of interest disclosure:
This work was supported in part by grants from the National Institute on Aging of the National Institutes of Health (AG056025, AG056976, and AG058081), the National Institute of Neurological Disorders and Stroke (NS100704), and the Center on Aging and the Academic Health Center of the University of Minnesota. DC is supported by a pre-doctoral training fellowship in the PharmacoNeuroImmunology Program from the National Institute on Drug Abuse of the National Institutes of Health (T32DA007097) and a 3M Science and Technology Training Fellowship.
We would like to thank Drs. Shuxian Hu and James Lokensgard for their training in the collection and culturing of primary mouse glial cells. We also would like to thank Drs. Mary Jo LaDu and Leon Tai for providing the synthetic Aβ and Dr. Guojun Bu for providing human apoE targeted-replacement immortalized mouse astrocytes and the antibody against LRP1. In addition, we thank Dr. Wei Zhang for her assistance in characterization of Aβ preparations by transmission electron microscopy in the Characterization Facility, University of Minnesota, which receives partial support from NSF through the MRSEC program.
Abbreviations used:
- ActD
actinomycin D
- AD
Alzheimer’s disease
- Aβ
amyloid-β
- ANOVA
analysis of variance
- apoA-I
apolipoprotein A-I
- apoE
apolipoprotein E
- apoJ
apolipoprotein J
- ABCA1
ATP-binding cassette transporter A1
- BFA
brefeldin A
- C3
complement component 3
- CAA
cerebral amyloid angiopathy
- CHX
cycloheximide
- DAPI
4’,6- diamidino-2-phenylindole
- DMEM
Dulbecco’s Modified Eagle Medium
- EGF
epidermal growth factor
- ER
endoplasmic reticulum
- FITC
fluorescein isothiocyanate
- GFAP
glial fibrillary acidic protein
- HDL
high-density lipoprotein
- HDR
homology directed repair
- Hep
heparinase I
- HRP
horseradish peroxidase
- HSPG
heparin sulfate proteoglycan
- Hep
heparinase I
- Iba1
ionized calcium- binding adapter molecule 1
- IDE
insulin degrading enzyme
- KO
knock out
- NDGGE
non-denaturing gradient gel electrophoresis
- LDLR
low density lipoprotein receptor
- LPS
lipopolysaccharide
- LRP1
low density lipoprotein receptor-related protein 1
- MFI
mean fluorescence intensity
- PBS
phosphate buffered saline
- PDL
poly-D-lysine
- pro
pronase E
- RFP
red fluorescent protein
- SDS-PAGE
sodium dodecyl sulfate-polyacrylamide gel electrophoresis
- TEM
transmission electron microscopy
- TR
targeted replacement
- TxRed
Texas Red
- Veh
vehicle
- WT
wild-type
Footnotes
The authors have no conflicts of interest to disclose.
References:
- Atagi Y, Liu CC, Painter MM et al. (2015) Apolipoprotein E Is a Ligand for Triggering Receptor Expressed on Myeloid Cells 2 (TREM2). J Biol Chem, 290, 26043–26050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey CC, DeVaux LB and Farzan M (2015) The Triggering Receptor Expressed on Myeloid Cells 2 Binds Apolipoprotein E. J Biol Chem, 290, 26033–26042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bales KR, Verina T, Dodel RC et al. (1997) Lack of apolipoprotein E dramatically reduces amyloid beta-peptide deposition. Nat Genet, 17, 263–264. [DOI] [PubMed] [Google Scholar]
- Bloedon LT, Dunbar R, Duffy D et al. (2008) Safety, pharmacokinetics, and pharmacodynamics of oral apoA-I mimetic peptide D-4F in high-risk cardiovascular patients. J Lipid Res, 49, 1344–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehm-Cagan A, Bar R, Liraz O, Bielicki JK, Johansson JO and Michaelson DM (2016) ABCA1 Agonist Reverses the ApoE4-Driven Cognitive and Brain Pathologies. J Alzheimers Dis, 54, 1219–1233. [DOI] [PubMed] [Google Scholar]
- Boehm-Cagan A and Michaelson DM (2014) Reversal of apoE4-driven brain pathology and behavioral deficits by bexarotene. J Neurosci, 34, 7293–7301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bu G (2009) Apolipoprotein E and its receptors in Alzheimer’s disease: pathways, pathogenesis and therapy. Nat Rev Neurosci, 10, 333–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buga GM, Frank JS, Mottino GA et al. (2006) D-4F decreases brain arteriole inflammation and improves cognitive performance in LDL receptor-null mice on a Western diet. J Lipid Res, 47, 2148–2160. [DOI] [PubMed] [Google Scholar]
- Chen J, Li Q and Wang J (2011) Topology of human apolipoprotein E3 uniquely regulates its diverse biological functions. Proc Natl Acad Sci U S A, 108, 14813–14818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, Roses AD, Haines JL and Pericak-Vance MA (1993) Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science, 261, 921–923. [DOI] [PubMed] [Google Scholar]
- Corona AW, Kodoma N, Casali BT and Landreth GE (2016) ABCA1 is Necessary for Bexarotene-Mediated Clearance of Soluble Amyloid Beta from the Hippocampus of APP/PS1 Mice. J Neuroimmune Pharmacol, 11, 61–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cramer PE, Cirrito JR, Wesson DW et al. (2012) ApoE-directed therapeutics rapidly clear beta-amyloid and reverse deficits in AD mouse models. Science, 335, 1503–1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruchaga C, Kauwe JS, Nowotny P et al. (2012) Cerebrospinal fluid APOE levels: an endophenotype for genetic studies for Alzheimer’s disease. Hum Mol Genet, 21, 4558–4571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donkin JJ, Stukas S, Hirsch-Reinshagen V et al. (2010) ATP-binding cassette transporter A1 mediates the beneficial effects of the liver X receptor agonist GW3965 on object recognition memory and amyloid burden in amyloid precursor protein/presenilin 1 mice. J Biol Chem, 285, 34144–34154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunbar RL, Movva R, Bloedon LT, Duffy D, Norris RB, Navab M, Fogelman AM and Rader DJ (2017) Oral Apolipoprotein A-I Mimetic D-4F Lowers HDL-Inflammatory Index in High-Risk Patients: A First-in-Human Multiple-Dose, Randomized Controlled Trial. Clinical and translational science, 10, 455–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagan AM, Holtzman DM, Munson G et al. (1999) Unique lipoproteins secreted by primary astrocytes from wild type, apoE (−/−), and human apoE transgenic mice. J Biol Chem, 274, 30001–30007. [DOI] [PubMed] [Google Scholar]
- Fan J, Stukas S, Wong C et al. (2011) An ABCA1-independent pathway for recycling a poorly lipidated 8.1 nm apolipoprotein E particle from glia. J Lipid Res, 52, 1605–1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitz NF, Carter AY, Tapias V, Castranio EL, Kodali R, Lefterov I and Koldamova R (2017) ABCA1 Deficiency Affects Basal Cognitive Deficits and Dendritic Density in Mice. J Alzheimers Dis, 56, 1075–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitz NF, Cronican AA, Lefterov I and Koldamova R (2013) Comment on “ApoE-directed therapeutics rapidly clear beta-amyloid and reverse deficits in AD mouse models”. Science, 340, 924-c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y, Zhao J, Atagi Y, Nielsen HM, Liu CC, Zheng H, Shinohara M, Kanekiyo T and Bu G (2016) Apolipoprotein E lipoprotein particles inhibit amyloid-beta uptake through cell surface heparan sulphate proteoglycan. Mol Neurodegener, 11, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Getz GS and Reardon CA (2014) The structure/function of apoprotein A-I mimetic peptides: an update. Current opinion in endocrinology, diabetes, and obesity, 21, 129–133. [DOI] [PubMed] [Google Scholar]
- Haass C and Selkoe DJ (2007) Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid beta-peptide. Nat Rev Mol Cell Biol, 8, 101–112. [DOI] [PubMed] [Google Scholar]
- Handattu SP, Garber DW, Monroe CE et al. (2009) Oral apolipoprotein A-I mimetic peptide improves cognitive function and reduces amyloid burden in a mouse model of Alzheimer’s disease. Neurobiol Dis, 34, 525–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handattu SP, Monroe CE, Nayyar G, Palgunachari MN, Kadish I, van Groen T, Anantharamaiah GM and Garber DW (2013) In vivo and in vitro effects of an apolipoprotein e mimetic peptide on amyloid-beta pathology. J Alzheimers Dis, 36, 335–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson AJ, Bayer-Carter JL, Green PS et al. (2013) Effect of apolipoprotein E genotype and diet on apolipoprotein E lipidation and amyloid peptides: randomized clinical trial. JAMA neurology, 70, 972–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy J and Selkoe DJ (2002) The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science, 297, 353–356. [DOI] [PubMed] [Google Scholar]
- Hebert LE, Weuve J, Scherr PA and Evans DA (2013) Alzheimer disease in the United States (2010–2050) estimated using the 2010 census. Neurology, 80, 1778–1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinsinger NM, Gachechiladze MA and Rebeck GW (2016) Apolipoprotein E Genotype Affects Size of ApoE Complexes in Cerebrospinal Fluid. J Neuropathol Exp Neurol, 75, 918–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsch-Reinshagen V, Maia LF, Burgess BL et al. (2005) The absence of ABCA1 decreases soluble ApoE levels but does not diminish amyloid deposition in two murine models of Alzheimer disease. J Biol Chem, 280, 43243–43256. [DOI] [PubMed] [Google Scholar]
- Holtzman DM, Herz J and Bu G (2012) Apolipoprotein E and apolipoprotein E receptors: normal biology and roles in Alzheimer disease. Cold Spring Harbor perspectives in medicine, 2, a006312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong C and Tontonoz P (2014) Liver X receptors in lipid metabolism: opportunities for drug discovery. Nat Rev Drug Discov, 13, 433–444. [DOI] [PubMed] [Google Scholar]
- Hottman DA, Chernick D, Cheng S, Wang Z and Li L (2014) HDL and cognition in neurodegenerative disorders. Neurobiol Dis, 72 Pt A, 22–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J, Liu CC, Chen XF, Zhang YW, Xu H and Bu G (2015) Opposing effects of viral mediated brain expression of apolipoprotein E2 (apoE2) and apoE4 on apoE lipidation and Abeta metabolism in apoE4-targeted replacement mice. Mol Neurodegener, 10, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y and Mahley RW (2014) Apolipoprotein E: structure and function in lipid metabolism, neurobiology, and Alzheimer’s diseases. Neurobiol Dis, 72 Pt A, 3–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igbavboa U, Johnson-Anuna LN, Rossello X, Butterick TA, Sun GY and Wood WG (2006) Amyloid beta-protein1–42 increases cAMP and apolipoprotein E levels which are inhibited by beta1 and beta2-adrenergic receptor antagonists in mouse primary astrocytes. Neuroscience, 142, 655–660. [DOI] [PubMed] [Google Scholar]
- Irizarry MC, Rebeck GW, Cheung B, Bales K, Paul SM, Holzman D and Hyman BT (2000) Modulation of A beta deposition in APP transgenic mice by an apolipoprotein E null background. Ann N Y Acad Sci, 920, 171–178. [DOI] [PubMed] [Google Scholar]
- Jiang Q, Lee CY, Mandrekar S et al. (2008) ApoE promotes the proteolytic degradation of Abeta. Neuron, 58, 681–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanekiyo T, Xu H and Bu G (2014) ApoE and Abeta in Alzheimer’s Disease: Accidental Encounters or Partners? Neuron, 81, 740–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Eltorai AE, Jiang H, Liao F, Verghese PB, Kim J, Stewart FR, Basak JM and Holtzman DM (2012) Anti-apoE immunotherapy inhibits amyloid accumulation in a transgenic mouse model of Abeta amyloidosis. J Exp Med, 209, 2149–2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klausner RD, Donaldson JG and Lippincott-Schwartz J (1992) Brefeldin A: insights into the control of membrane traffic and organelle structure. J Cell Biol, 116, 1071–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koldamova R, Fitz NF and Lefterov I (2014) ATP-Binding Cassette Transporter A1: From metabolism to neurodegeneration. Neurobiol Dis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koldamova R, Staufenbiel M and Lefterov I (2005) Lack of ABCA1 considerably decreases brain ApoE level and increases amyloid deposition in APP23 mice. J Biol Chem, 280, 43224–43235. [DOI] [PubMed] [Google Scholar]
- Krasemann S, Madore C, Cialic R et al. (2017) The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity, 47, 566–581 e569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaDu MJ, Falduto MT, Manelli AM, Reardon CA, Getz GS and Frail DE (1994) Isoform-specific binding of apolipoprotein E to beta-amyloid. J Biol Chem, 269, 23403–23406. [PubMed] [Google Scholar]
- LaDu MJ, Shah JA, Reardon CA, Getz GS, Bu G, Hu J, Guo L and van Eldik LJ (2000) Apolipoprotein E receptors mediate the effects of beta-amyloid on astrocyte cultures. J Biol Chem, 275, 33974–33980. [DOI] [PubMed] [Google Scholar]
- Lefterov I, Fitz NF, Cronican AA, Fogg A, Lefterov P, Kodali R, Wetzel R and Koldamova R (2010) Apolipoprotein A-I deficiency increases cerebral amyloid angiopathy and cognitive deficits in APP/PS1DeltaE9 mice. J Biol Chem, 285, 36945–36957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leman LJ, Maryanoff BE and Ghadiri MR (2014) Molecules that mimic apolipoprotein A-I: potential agents for treating atherosclerosis. J Med Chem, 57, 2169–2196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesne SE, Sherman MA, Grant M, Kuskowski M, Schneider JA, Bennett DA and Ashe KH (2013) Brain amyloid-beta oligomers in ageing and Alzheimer’s disease. Brain, 136, 1383–1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis TL, Cao D, Lu H et al. (2010) Overexpression of human apolipoprotein A-I preserves cognitive function and attenuates neuroinflammation and cerebral amyloid angiopathy in a mouse model of Alzheimer disease. J Biol Chem, 285, 36958–36968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao F, Li A, Xiong M et al. (2018) Targeting of nonlipidated, aggregated apoE with antibodies inhibits amyloid accumulation. J Clin Invest, 128, 2144–2155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao F, Yoon H and Kim J (2017) Apolipoprotein E metabolism and functions in brain and its role in Alzheimer’s disease. Curr Opin Lipidol, 28, 60–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao F, Zhang TJ, Jiang H, Lefton KB, Robinson GO, Vassar R, Sullivan PM and Holtzman DM (2015) Murine versus human apolipoprotein E4: differential facilitation of and co-localization in cerebral amyloid angiopathy and amyloid plaques in APP transgenic mouse models. Acta neuropathologica communications, 3, 70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu XH, Xiao J, Mo ZC et al. (2010) Contribution of D4-F to ABCA1 expression and cholesterol efflux in THP-1 macrophage-derived foam cells. J Cardiovasc Pharmacol, 56, 309–319. [DOI] [PubMed] [Google Scholar]
- Morikawa M, Fryer JD, Sullivan PM et al. (2005) Production and characterization of astrocyte-derived human apolipoprotein E isoforms from immortalized astrocytes and their interactions with amyloid-beta. Neurobiol Dis, 19, 66–76. [DOI] [PubMed] [Google Scholar]
- Mucke L and Selkoe DJ (2012) Neurotoxicity of amyloid beta-protein: synaptic and network dysfunction. Cold Spring Harbor perspectives in medicine, 2, a006338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navab M, Anantharamaiah GM, Reddy ST et al. (2005) Apolipoprotein A-I mimetic peptides. Arterioscler Thromb Vasc Biol, 25, 1325–1331. [DOI] [PubMed] [Google Scholar]
- Navab M, Anantharamaiah GM, Reddy ST, Van Lenten BJ and Fogelman AM (2008) Apo A-1 mimetic peptides as atheroprotective agents in murine models. Curr Drug Targets, 9, 204–209. [DOI] [PubMed] [Google Scholar]
- Omtri RS, Davidson MW, Arumugam B, Poduslo JF and Kandimalla KK (2012) Differences in the cellular uptake and intracellular itineraries of amyloid beta proteins 40 and 42: ramifications for the Alzheimer’s drug discovery. Molecular pharmaceutics, 9, 1887–1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oram JF and Heinecke JW (2005) ATP-binding cassette transporter A1: a cell cholesterol exporter that protects against cardiovascular disease. Physiol Rev, 85, 1343–1372. [DOI] [PubMed] [Google Scholar]
- Osei-Hwedieh DO, Amar M, Sviridov D and Remaley AT (2011) Apolipoprotein mimetic peptides: Mechanisms of action as anti-atherogenic agents. Pharmacol Ther, 130, 83–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rees D, Sloane T, Jessup W, Dean RT and Kritharides L (1999) Apolipoprotein A-I stimulates secretion of apolipoprotein E by foam cell macrophages. J Biol Chem, 274, 27925–27933. [DOI] [PubMed] [Google Scholar]
- Robert J, Stukas S, Button E et al. (2016) Reconstituted high-density lipoproteins acutely reduce soluble brain Abeta levels in symptomatic APP/PS1 mice. Biochim Biophys Acta, 1862, 1027–1036. [DOI] [PubMed] [Google Scholar]
- Rossello XS, Igbavboa U, Weisman GA, Sun GY and Wood WG (2012) AP-2beta regulates amyloid beta-protein stimulation of apolipoprotein E transcription in astrocytes. Brain Res, 1444, 87–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo C, Angelini G, Dapino D et al. (1998) Opposite roles of apolipoprotein E in normal brains and in Alzheimer’s disease. Proc Natl Acad Sci U S A, 95, 15598–15602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders AM, Strittmatter WJ, Schmechel D et al. (1993) Association of apolipoprotein E allele epsilon 4 with late-onset familial and sporadic Alzheimer’s disease. Neurology, 43, 1467–1472. [DOI] [PubMed] [Google Scholar]
- Segrest JP, Chung BH, Brouillette CG, Kanellis P and McGahan R (1983) Studies of synthetic peptide analogs of the amphipathic helix. Competitive displacement of exchangeable apolipoproteins from native lipoproteins. J Biol Chem, 258, 2290–2295. [PubMed] [Google Scholar]
- Segrest JP, Jones MK, De Loof H, Brouillette CG, Venkatachalapathi YV and Anantharamaiah GM (1992) The amphipathic helix in the exchangeable apolipoproteins: a review of secondary structure and function. J Lipid Res, 33, 141–166. [PubMed] [Google Scholar]
- Selkoe DJ and Hardy J (2016) The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO molecular medicine, 8, 595–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Yamada K, Liddelow SA et al. (2017) ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature, 549, 523–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skerrett R, Malm T and Landreth G (2014) Nuclear receptors in neurodegenerative diseases. Neurobiol Dis, 72 Pt A, 104–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Q, Huang M, Yao L et al. (2014) Lipoprotein-based nanoparticles rescue the memory loss of mice with Alzheimer’s disease by accelerating the clearance of amyloid-beta. ACS nano, 8, 2345–2359. [DOI] [PubMed] [Google Scholar]
- Stine WB, Jungbauer L, Yu C and LaDu MJ (2011) Preparing synthetic Abeta in different aggregation states. Methods Mol Biol, 670, 13–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strittmatter WJ, Saunders AM, Schmechel D, Pericak-Vance M, Enghild J, Salvesen GS and Roses AD (1993) Apolipoprotein E: high-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc Natl Acad Sci U S A, 90, 1977–1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai LM, Bilousova T, Jungbauer L et al. (2013) Levels of soluble apolipoprotein E/amyloid-beta (Abeta) complex are reduced and oligomeric Abeta increased with APOE4 and Alzheimer disease in a transgenic mouse model and human samples. J Biol Chem, 288, 5914–5926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai LM, Koster KP, Luo J, Lee SH, Wang YT, Collins NC, Ben Aissa M, Thatcher GR and LaDu MJ (2014a) Amyloid-beta pathology and APOE genotype modulate retinoid X receptor agonist activity in vivo. J Biol Chem, 289, 30538–30555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai LM, Mehra S, Shete V, Estus S, Rebeck GW, Bu G and LaDu MJ (2014b) Soluble apoE/Abeta complex: mechanism and therapeutic target for APOE4-induced AD risk. Mol Neurodegener, 9, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang C, Vaughan AM, Anantharamaiah GM and Oram JF (2006) Janus kinase 2 modulates the lipid-removing but not protein-stabilizing interactions of amphipathic helices with ABCA1. J Lipid Res, 47, 107–114. [DOI] [PubMed] [Google Scholar]
- Tesseur I, Lo AC, Roberfroid A et al. (2013) Comment on “ApoE-directed therapeutics rapidly clear beta-amyloid and reverse deficits in AD mouse models”. Science, 340, 924-e. [DOI] [PubMed] [Google Scholar]
- Tousi B (2015) The emerging role of bexarotene in the treatment of Alzheimer’s disease: current evidence. Neuropsychiatric disease and treatment, 11, 311–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veeraraghavalu K, Zhang C, Miller S et al. (2013) Comment on “ApoE-directed therapeutics rapidly clear beta-amyloid and reverse deficits in AD mouse models”. Science, 340, 924-f. [DOI] [PubMed] [Google Scholar]
- Verghese PB, Castellano JM, Garai K, Wang Y, Jiang H, Shah A, Bu G, Frieden C and Holtzman DM (2013) ApoE influences amyloid-beta (Abeta) clearance despite minimal apoE/Abeta association in physiological conditions. Proc Natl Acad Sci U S A, 110, E1807–1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahrle SE, Jiang H, Parsadanian M, Hartman RE, Bales KR, Paul SM and Holtzman DM (2005) Deletion of Abca1 increases Abeta deposition in the PDAPP transgenic mouse model of Alzheimer disease. J Biol Chem, 280, 43236–43242. [DOI] [PubMed] [Google Scholar]
- Wahrle SE, Jiang H, Parsadanian M et al. (2008) Overexpression of ABCA1 reduces amyloid deposition in the PDAPP mouse model of Alzheimer disease. J Clin Invest, 118, 671–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson CE, Weissbach N, Kjems L et al. (2011) Treatment of patients with cardiovascular disease with L-4F, an apo-A1 mimetic, did not improve select biomarkers of HDL function. J Lipid Res, 52, 361–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisgraber KH, Rall SC Jr. and Mahley RW (1981) Human E apoprotein heterogeneity. Cysteine-arginine interchanges in the amino acid sequence of the apo-E isoforms. J Biol Chem, 256, 9077–9083. [PubMed] [Google Scholar]
- White CR, Garber DW and Anantharamaiah GM (2014) Anti-inflammatory and cholesterol-reducing properties of apolipoprotein mimetics: a review. J Lipid Res, 55, 2007–2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisniewski T, Golabek A, Matsubara E, Ghiso J and Frangione B (1993) Apolipoprotein E: binding to soluble Alzheimer’s beta-amyloid. Biochem Biophys Res Commun, 192, 359–365. [DOI] [PubMed] [Google Scholar]
- Xie Q, Zhao SP and Li F (2010) D-4F, an apolipoprotein A-I mimetic peptide, promotes cholesterol efflux from macrophages via ATP-binding cassette transporter A1. Tohoku J Exp Med, 220, 223–228. [DOI] [PubMed] [Google Scholar]
- Xu Q, Bernardo A, Walker D, Kanegawa T, Mahley RW and Huang Y (2006) Profile and regulation of apolipoprotein E (ApoE) expression in the CNS in mice with targeting of green fluorescent protein gene to the ApoE locus. J Neurosci, 26, 4985–4994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh FL, Wang Y, Tom I, Gonzalez LC and Sheng M (2016) TREM2 Binds to Apolipoproteins, Including APOE and CLU/APOJ, and Thereby Facilitates Uptake of Amyloid-Beta by Microglia. Neuron, 91, 328–340. [DOI] [PubMed] [Google Scholar]
- Zelcer N, Khanlou N, Clare R, Jiang Q, Reed-Geaghan EG, Landreth GE, Vinters HV and Tontonoz P (2007) Attenuation of neuroinflammation and Alzheimer’s disease pathology by liver x receptors. Proc Natl Acad Sci U S A, 104, 10601–10606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Nwabuisi-Heath E, Dumanis SB, Tai LM, Yu C, Rebeck GW and LaDu MJ (2012) APOE genotype alters glial activation and loss of synaptic markers in mice. Glia, 60, 559–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.