

Abstract
Urate is often viewed as an antioxidant. Here, we present an alternative perspective by showing that, when oxidized, urate propagates oxidative stress. Oxidation converts urate to the urate radical and the electrophilic products dehydrourate, 5-hydroxyisourate, and urate hydroperoxide, which eventually break down to allantoin. We investigated whether urate-derived electrophiles are intercepted by nucleophilic amino acid residues to form stable adducts on proteins. When urate was oxidized in the presence of various peptides and proteins, two adducts derived from urate (Mr 167 Da) were detected and had mass additions of 140 and 166 Da, occurring mainly on lysine residues and N-terminal amines. The adduct with a 140-Da mass addition was detected more frequently and was stable. Dehydrourate (Mr 166 Da) also formed transient adducts with cysteine residues. Urate-derived adducts were detected on human serum albumin in plasma of healthy donors. Basal adduct levels increased when neutrophils were added to plasma and stimulated, and relied on the NADPH oxidase, myeloperoxidase, hydrogen peroxide, and superoxide. Adducts of oxidized urate on serum albumin were elevated in plasma and synovial fluid from individuals with gout and rheumatoid arthritis. We propose that rather than acting as an antioxidant, urate's conversion to electrophiles contributes to oxidative stress. The addition of urate-derived electrophiles to nucleophilic amino acid residues, a process we call oxidative uratylation, will leave a footprint on proteins that could alter their function when critical sites are modified.
Keywords: uric acid, oxidative stress, neutrophil, NADPH oxidase, myeloperoxidase, post-translational modification (PTM), biomarker, uratylation, protein adduct, lysine modification
Introduction
Uric acid is an intriguing metabolite. It is a breakdown product of purines and is actively reabsorbed by the kidneys, signifying an essential role in metabolism (1, 2), but its function remains obscure. In vivo, uric acid (pKa 5.4) exits predominantly as urate. This monoanion has a low one-electron reduction potential (E0′ 560 mV), which has led researchers to propose that urate is a powerful antioxidant (3). Thermodynamics dictates that urate will reduce most oxidizing free radicals and react readily with two-electron oxidants such as peroxynitrite (4, 5). Evidence to support its role as an antioxidant is strong in neurological pathologies, most notably Parkinson's disease and ALS, where low levels are associated with greater oxidative stress and worse clinical outcomes (6). However, urate's antioxidant function is less apparent in other pathologies. It is even suggested to act as a pro-oxidant when it amplifies lipid peroxidation promoted by either copper or peroxynitrite or activates NADPH oxidases (7). Hyperuricemia may also be associated with increased oxidative stress when urate is produced by xanthine oxidase, which, unlike xanthine dehydrogenase, generates superoxide and hydrogen peroxide when it oxidizes hypoxanthine and xanthine (8).
The argument that urate acts as a radical scavenger in vivo has an important but often neglected consequence (i.e. when urate is oxidized by radicals, it too is converted to a radical). Unless the urate radical is reduced by ascorbate, it gives rise to several reactive species (Fig. 1) (9–11). Urate radical reacts at diffusion-controlled rates with superoxide to form urate hydroperoxide (12). When it loses a second electron, dehydrourate is formed and slowly hydrolyzes to 5-hydroxyisourate (11). 5-Hydroxyisourate is carcinogenic when not metabolized in mice (13). Consequently, unless the urate radical is intercepted by ascorbate, oxidative stress may be propagated by the ensuing electrophiles.
Figure 1.
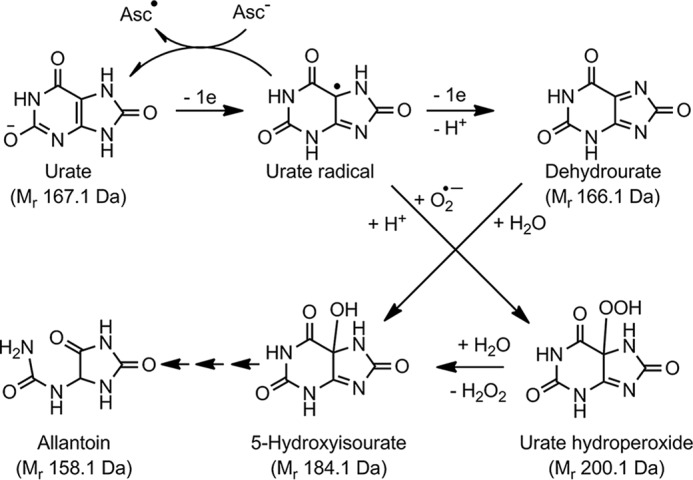
Oxidation of urate to electrophiles and their break down to allantoin. One-electron oxidation of urate produces the urate radical that, unless reduced by ascorbate (Asc), can either be further oxidized to dehydrourate or react with superoxide to produce urate hydroperoxide. Both of these electrophiles hydrolyze to 5-hydroxyisourate that, through a series of reactions (small arrows), breaks down to allantoin. Relative molecular masses (Mr) are given in parentheses.
More recently, urate has been identified as a physiological substrate for myeloperoxidase (MPO),2 a key enzyme of neutrophils, and lactoperoxidase, which is present on epithelial surfaces (14, 15). These enzymes contribute to host defense by producing bactericidal oxidants, including hypochlorous acid and hypothiocyanous acid (16, 17). Urate competes with the main physiological substrates for these enzymes and is converted to several oxidation products. Urate hydroperoxide is the major product when oxidation occurs in the presence of a flux of superoxide, such as with stimulated neutrophils (14). Hence, peroxidases are a source of urate-derived electrophiles, especially during inflammation when myeloperoxidase is abundant.
5-Hydroxyisourate breaks down (Fig. 1) via rearrangement and decarboxylation to form a bicyclic imidazalone (Mr 140 Da), which then hydrolyzes to allantoin (11). Detection of allantoin in numerous inflammatory diseases, including cardiovascular diseases (18), gout (19), rheumatoid arthritis (20), and cystic fibrosis (21), signifies that urate electrophiles are formed during oxidative stress. Previously, urate electrophiles generated by peroxidases and peroxynitrite were shown to alkylate alcohols and ammonia (11, 22, 23). Related reactions have the potential to occur in vivo but have yet to be demonstrated.
In this investigation, we show that oxidized urate forms adducts on amino and thiol residues of peptides and proteins. Stable adducts were present on serum albumin in the plasma of healthy individuals, increased when neutrophils generated oxidants, and were elevated in individuals with neutrophil-driven inflammatory diseases. Modification of amino acid residues by oxidized urate may alter protein function and potentially serve as a biomarker of oxidative stress.
Results
Oxidized urate forms conjugates with peptides
To establish whether oxidized forms of urate react with amino acid residues, urate was oxidized by myeloperoxidase and hydrogen peroxide in the presence of the peptide GGYR. After 30 min, new peptides were detected by MS. They had m/z values of 140 and 166 greater than the parent peptide (Fig. 2A). No new peptides were formed in the absence of either urate, myeloperoxidase or hydrogen peroxide (not shown). The mass spectra of the fragments derived from the new peptides of GGYR (Fig. 2, C and D) both contained the same y2 ([YR + H]+) and y3 ([GYR + H]+) ions of the unmodified parent molecule (Fig. 2B), whereas the b2 ([GG + H]+; 115 m/z) ions had increased by the m/z values of 140 (Fig. 2C) or 166 (Fig. 2D). These data demonstrate that adducts were present on the N-terminal amine. To assess whether oxidized urate could form adducts with other nucleophilic amino acid residues, a range of peptides was selected based on their content of lysine, cysteine, arginine, histidine, and tyrosine residues. (Table S1). Adducts were formed on lysine residues as well as the N-terminal amine (Fig. S1). The adduct with an m/z of 140 was formed in most cases, whereas that of 166 m/z was not always detected. Peptides with a blocked N-terminal amine and without lysine residues did not form adducts. No adducts were detected on cysteine, histidine, or tyrosine residues.
Figure 2.
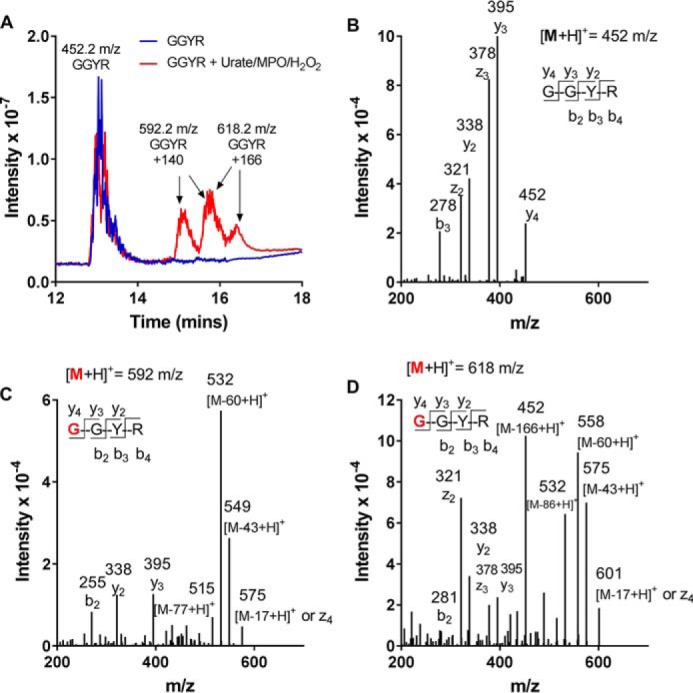
Oxidized urate forms stable covalent adducts with amino residues on peptides. Urate (380 μm) was oxidized by MPO (100 nm) and hydrogen peroxide (100 μm) in the presence of the peptide GGYR (100 μm) in 10 mm phosphate buffer, pH 7.4, for 30 min at 37 °C. The products were separated by LC-MS (A), and the fragmentation spectra for GGYR (B) and GGYR with mass additions of 140 Da (C) and 166 Da (D) were determined. The red residue G indicates the location of the urate-derived adduct.
During inflammation urate is likely to be oxidized in the presence of superoxide, which reacts rapidly with urate radical and myeloperoxidase (12, 24, 25). To determine how superoxide affects adduct formation, urate was oxidized by myeloperoxidase using xanthine oxidase as a source of both hydrogen peroxide and superoxide. The peptides used were MLTELEK or YGGFL. Adducts with an m/z value of 140 were detected on the peptides during the oxidation of urate (see Fig. 3 (A and B) for adduct formation on MLTELEK). No adducts were formed in the absence of myeloperoxidase, xanthine oxidase, or hypoxanthine (not shown). Adduct formation was significantly decreased on both peptides when superoxide dismutase (SOD) was included in the reaction system to scavenge superoxide (Fig. 3C). These results suggest that superoxide enhances the formation of urate-derived adducts on peptides.
Figure 3.
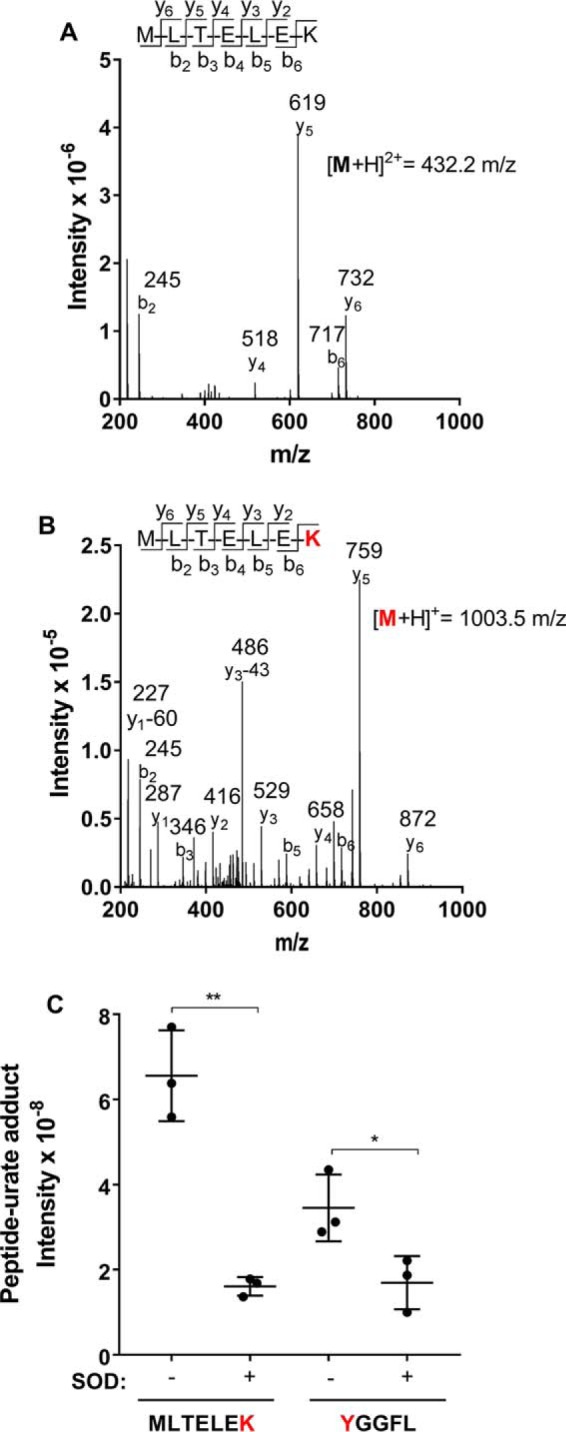
Superoxide enhances formation of urate-derived adducts with amino residues on peptides. The peptide MLTELEK (100 μm) or YGGFL (100 μm) was incubated with urate (380 μm), which was oxidized by MPO (100 nm) with xanthine oxidase (0.025 units/ml) and acetaldehyde (10 mm) to generate low fluxes of superoxide and hydrogen peroxide. Formation of the 140-Da adduct of oxidized urate on MLTELEK was confirmed by comparing fragmentation of the parent peptide (A) and the product identified by LC-MS (B). The red K residue indicates the location of the urate-derived adduct. C, urate-derived adducts on the peptides were generated in the presence and absence of SOD and measured using LC-MS/MS. Data are means ± S.D. (error bars) of triplicate experiments. Statistical differences were determined by Student's t test (**, p < 0.001; *, p = 0.02).
In the above studies, urate-derived adducts were not detected on cysteine residues (Table S1). Rather, when urate was oxidized in the presence of peptides containing cysteine, dimers of the peptides, cross-linked via their cysteine residue, were the predominant products formed (e.g. GGFCD; Table S1). Blocking the cysteine residue with iodoacetamide resulted in formation of the 140-Da adduct on the N-terminal amine group (Table S1). However, when the reaction mixture contained the peptide PFVCG and was analyzed in the first few minutes, a peptide with an increase in m/z of 166 was detected along with the dimer (Fig. 4A). This increase in mass of 166 Da corresponds to the addition of dehydrourate to the peptide, presumably via nucleophilic attack of the thiolate anion at the electron-deficient center of dehydrourate. By comparing the MS/MS spectra of the new peptide with that of the parent peptide, it was apparent that the adduct was located on the cysteine residue (Fig. 4, B and C). Both the parent and modified peptide had the same b3 ion ([PFV + H]+), indicating that the modification was on either the Cys or Gly residues. However, the presence of an ion with an m/z corresponding to the b4 ion with the adduct (Fig. 4C) suggested that dehydrourate was attached to the Cys residue. The adduct was stable for 60 min at 4 °C but decayed rapidly at room temperature (not shown). This result indicates that adducts between oxidized urate and cysteine residues are formed but are unstable.
Figure 4.
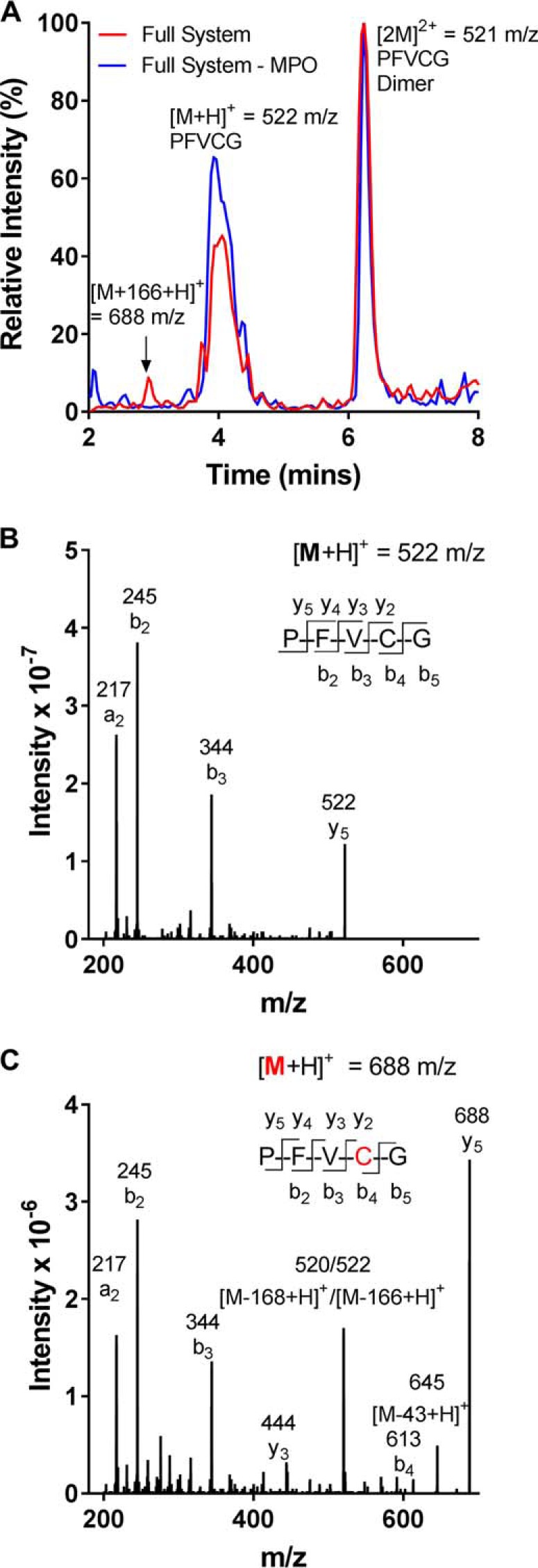
Oxidized urate forms transient adducts with cysteine residues. A, urate (380 μm) was oxidized by MPO (100 nm) and hydrogen peroxide (100 μm) in the presence of the peptide PFVCG (100 μm) in 10 mm phosphate buffer, pH 7.4, at 37 °C for 1 min and kept at 4 °C until the products were identified by LC-MS. The product with an m/z of 688 is equivalent to PFVCG ([M + H]+ = 522 m/z) plus dehydrourate (166 Da). The mass spectra are shown for PFVCG (B) and PFVCG (C) with a mass addition of 166 Da. The red residue C indicates the location of the dehydrourate adduct.
Identification of the urate-derived adducts on amine groups
The addition of 166 Da to a peptide during the oxidation of urate is readily rationalized as the nucleophilic addition of the amine group to dehydrourate (Mr 166 Da). However, the structure of the adduct that results in the addition of 140 Da to a peptide is less obvious. We sought structural information on this adduct by comparing the mass spectra of the adduct obtained from normal urate (Mr 167 Da) with that obtained from urate containing two atoms of the heavy isotope of nitrogen (15N) (Mr 169 Da). Normal or 15N-labeled urate was oxidized in the presence of the amine phenylethylamine (Mr 121.2). Products from normal and heavy urate had positive ions ([M + H]+) with m/z values of 262.2 and 264.2, respectively (Fig. 5A). In other words, the adduct on phenylethylamine had a mass of 140 Da when arising from normal urate or 142 Da from heavy urate. Fragmentation of the product from normal urate and phenylethylamine gave losses of 17, 43, and 60 Da, which can be attributed to the loss of 14NH3, CO14NH, and both, respectively (Fig. 5B). The same losses occur with allantoin (26). The heavy urate isotope had both 15N atoms in its five-membered ring. Its oxidation resulted in marked differences in the mass spectrum for the adduct with phenylethylamine (Fig. 5C). When this species was fragmented, it gave doublet and a triplet sets of ions that resulted from losses of 43 or 44, 17 or 18, and 60, 61, or 62 Da. The doublet and triplet sets suggest that the molecular ion, [M + H]+ was a combination of two structures that differed only in the location of the 15N isotopes.
Figure 5.

Determination of the structure of the 140-Da adduct of oxidized urate with amino groups. Labeled (15N) (Mr 169 Da) or unlabeled urate (Mr 167 Da) (380 μm) was oxidized by MPO (100 nm) and hydrogen peroxide (400 μm) in the presence of phenethylamine (PEA) (Mr 121 Da; 100 μm) in 10 mm phosphate buffer, pH 7.4, for 30 min at 20–22 °C, and products (A) with an addition of 140 Da ([M + 140 + H]+ m/z 262 (blue line) or 264 Da (red line)) were detected by selected ion-monitoring LC-MS. The fragmentation spectra of the unlabeled ([M + H]+ 262 m/z) (B) and labeled ([M + H]+ 264 m/z) (C) products were then determined. D, the proposed reaction sequence for breakdown of 5-hydroxyisourate to the bicylic imidazolone that adds to an amino group (NH2R) to form the stable 140-Da adduct.
The simplest explanation for adduct formation is that urate electrophiles break down to the bicyclic imidazolone (Mr 140 Da) as proposed by Volk et al. (22), which then reacts with amine groups (Fig. 5D). For the isotopically labeled bicylic structure, one ring would contain two 14N, whereas the other would contain both the 15N atoms. Given the symmetrical nature of the structure, either ring could open upon rearrangement, resulting in a single five-membered ring structure that contains only normal or only heavy nitrogen atoms. Consequently, these two structures would be expected to give distinct mass spectra. The fragmentation schemes to support this conclusion are shown in Fig. S2 (A and B).
Inactivation of glyceraldehyde phosphate dehydrogenase (GAPDH) by oxidized urate
Because oxidized urate reacted with thiol residues on peptides, we determined whether urate electrophiles could inactivate the thiol-dependent enzyme GAPDH. Urate electrophiles were generated using xanthine oxidase, hypoxanthine, and lactoperoxidase to oxidize urate. When a solution containing oxidized urate was added to GAPDH, the enzyme lost activity (Fig. 6A). Enzyme inactivation required all components of the reaction system. Omission of exogenous urate had a modest affect because xanthine oxidase generates urate during the oxidation of hypoxanthine. Superoxide dismutase prevented about half of the inactivation of GAPDH. Thus, we conclude that a superoxide-dependent electrophile, presumably urate hydroperoxide, made a major contribution to inactivation of GAPDH. Solutions of oxidized urate containing increasing concentrations of urate hydroperoxide progressively inactivated GAPDH (Fig. 6B). DTT, which reduces disulfides and sulfenic acids, partially reversed inactivation (Fig. 6B). Inactivation, therefore, was partly due to reversible thiol oxidation.
Figure 6.

Urate electrophiles inactivate GAPDH. A, residual enzyme activity (μm/min) was determined after GAPDH (2 μm) was treated for 10 min at pH 7.4 with a solution of urate electrophiles that contained urate hydroperoxide (5 μm). Urate was oxidized using XO, hypoxanthine (HX; 100 μm), urate (400 μm), and lactoperoxidase (LPO; 160 nm) in 50 mm phosphate buffer, pH 7.4, for 20 min. Other additions included SOD (20 μg/ml) or CAT (20 μg/ml). Data are representative of two independent experiments. Error bars, S.D. of triplicate readings for one experiment. A one-way ANOVA followed by Dunnett's multiple-comparison test on triplicate readings was used to identify samples that were significantly different from the full system. All treatments were significantly different from the control (p < 0.001) except catalase. The enzyme GAPDH was treated with oxidized urate containing increasing concentrations of urate hydroperoxide, and its residual activity (B) was determined either before (●) or after (▴) reduction with DTT. C, the content of reduced thiols after treatment with increasing concentrations of urate hydroperoxide.
GAPDH has four subunits each containing four cysteine residues, giving a maximal thiol concentration of 32 μm for 2 μm enzyme. The detectable concentration we measured (21 μm) is similar to that reported previously (27). When treated with oxidized urate containing increasing concentration of urate hydroperoxide, there was increasing loss of thiol residues from the GAPDH (Fig. 6C). After inactivation of GAPDH, the protein was analyzed for the presence of urate-derived adducts, but none were detected with confidence.
Detection of urate-derived adducts on proteins
Next we aimed to determine whether oxidized urate can become attached to amine residues on proteins. We chose the small proteins ubiquitin, β-lactoglobulin, and macrophage migration inhibitory factor (MIF) because changes in their masses can be accurately assessed by MS. We also chose human serum albumin because it is the most abundant plasma protein and, therefore, most likely to be modified by oxidized urate in vivo. Urate was oxidized in the presence of individual proteins, which were then analyzed by whole-protein MS. Urate-derived adducts of 140 Da were detected on all of the small proteins, as shown for ubiquitin (Mr 8,565 Da) (Fig. 7A), β-lactoglobulin (Mr 18,278 Da) (Fig. 7C), and MIF (Mr 12,345 Da) (Fig. S3A). β-Lactoglobulin was also oxidized during the incubation, as seen by the addition of 32 Da, which corresponds to the incorporation of two oxygen atoms. When modified ubiquitin was digested with trypsin and the resulting peptides were identified by LC-MS/MS, adducts with a mass of 140 Da were detected on the N-terminal methionine (1MQIFVK6) (Fig. 7B) as well as Lys-48 (43LIFAGKQLEDGR54) and Lys-63 (55TLSDYNIQKESTLHLVLR72) (Fig. S4). Also, when urate was oxidized in the presence of β-lactoglobulin, a 140-Da adduct was detected on the lysine residue of the tryptic peptide TKIPAVFK (Fig. 7D). MIF was also partially modified by oxidized urate, as judged by the presence of a new species that was 143 ± 5 Da larger than the native protein (Fig. S3A). When the protein was digested with trypsin, the 140-Da adduct of urate was detected on the reactive N-terminal proline residue of MIF (Fig. S3B). No adducts were detected when myeloperoxidase was absent from the oxidizing system (not shown).
Figure 7.

Oxidized urate forms stable adducts on amino residues of proteins. Urate (380 μm) was oxidized by MPO (100 nm) and hydrogen peroxide (100 μm) in the presence of either ubiquitin (A and B; Mr 8,565 Da; 0.1 mg/ml) or β-lactoglobulin (C and D; Mr 18,278 Da; 0.1 mg/ml) in 10 mm phosphate buffer, pH 7.4, for 30 min at 37 °C. The molecular masses of the modified proteins were determined by MS (A and C), and tryptic peptides (B and D) containing the 140-Da adducts from oxidized urate were identified by LC-MS/MS. The red residues M and K indicate the locations of the urate-derived adduct.
After urate was oxidized in the presence of human serum albumin, the 140-Da adduct was detected consistently on 11 of the 59 lysine residues of the protein (Table S2; shown in Fig. 8A). Fragmentation spectra for tryptic peptides of albumin containing the urate-derived adduct with a mass addition of 140 Da are shown in Fig. 8B (NLGK432VGSK) and Fig. S5.
Figure 8.

Oxidized urate forms stable adducts with human serum albumin, which are present in normal plasma and elevated by oxidative stress. A, urate was oxidized by MPO and hydrogen peroxide in the presence of human serum albumin under the conditions listed in Fig. 7. Adducts of urate (140 Da) were detected on the protein's lysine residues indicated in blue on the crystal structure of albumin (yellow) drawn using PyMOl (Protein Data Bank code 1AO6). The red residue is Lys-432. B, the fragmentation spectrum of NLGK432VGSK from albumin containing the urate-derived adduct (140 Da) on lysine 432 (red residue in A). C, using multiple-reaction-monitoring MS, NLGK432VGSK was measured in plasma from healthy donors without (black) or with neutrophils (5 × 106/ml) that had been stimulated with CytB (10 μg/ml) and PMA (100 ng/ml) for 45 min at 37 °C (red). D, urate-derived adducts on serum albumin were measured in plasma from healthy donors alone or with neutrophils. When stimulated, neutrophils were incubated without or with inhibitors of the NADPH oxidase (DPI; 20 μm), myeloperoxidase (TX1; 10 μm), CAT (100 μg/ml), or SOD (20 μg/ml). Data are means ± S.D. (error bars) of three independent experiments with blood from three donors. Significant difference (*, p < 0.05; ***, p < 0.0001) from control plasma (blue line) was determined by ANOVA.
Urate-derived adducts are present on human serum albumin and elevated during inflammation
To identify the 140-Da adduct of oxidized urate on human serum albumin in vivo, we developed an analytical LC-MS/MS method for detection of a modified tryptic peptide of the protein. We chose the peptide NLGK432VGSK containing the 140-Da adduct of the middle lysine residue because it was consistently modified in vitro, was amenable to LC analysis with a characteristic retention time of 7.2 min, and gave specific fragment ions that had good signal/noise ratios (Fig. 8, B and C). For quantification, the intensity of the modified peptide was related to that for the unmodified tryptic peptide NLGK. When plasma from healthy controls was digested with trypsin, the peptide NLGK432VGSK was readily detected (Fig. 8C). To establish that this peptide did indeed result from urate electrophiles, neutrophils were added to plasma and stimulated with a combination of cytochalasin B (CytB) and phorbol 12-myristate 13-acetate (PMA). CytB promotes degranulation of neutrophils, whereas PMA stimulates robust production of superoxide, and in combination, they enable neutrophils to oxidize urate (14). The level of NLGK432VGSK increased substantially when neutrophils were incubated in plasma and stimulated (Fig. 8, C and D). The NADPH oxidase inhibitor diphenyleneiodonium (DPI), the myeloperoxidase inhibitor 2-thioxanthine 3-isobutyl-2-thioxo-7H-purin-6-one (TX1) (28), and catalase all lowered the adduct formation to unstimulated levels (Fig. 8D). These results indicate that formation of NLGK432VGSK depended on hydrogen peroxide production and myeloperoxidase activity of neutrophils. Superoxide dismutase also largely inhibited the neutrophil-dependent increase in NLGK432VGSK. This latter result suggests that superoxide enhanced adduct formation on albumin.
Previously, we found that allantoin, the stable oxidation product of urate, is elevated in plasma of individuals with rheumatoid arthritis or gout (19, 20). Because allantoin is derived from the electrophilic oxidation products of urate, we measured urate-derived adducts on albumin in plasma and synovial fluid from individuals with these inflammatory diseases and compared their levels with those from healthy controls (Fig. 9, A and B). The signal for the urate-derived adduct on the albumin peptide NLGK432VGSK was greater in both plasma and synovial fluid for individuals with rheumatoid arthritis compared with that in plasma from healthy controls (Fig. 9A). When the signal for this peptide was related to that for the unmodified parent peptide NLGK, there was an approximate 3-fold increase in the formation of adduct on plasma albumin in individuals with rheumatoid arthritis (n = 6; p = 0.014) and gout (n = 6; p = 0.005) compared with the healthy controls (n = 8) (Fig. 9B). A similar increase was measured in synovial fluid from the individuals with gout (p = 0.001), whereas there was an even greater increase for the individuals with rheumatoid arthritis (p < 0.0001). Adduct formation was greater in synovial fluid compared with plasma for the individuals with rheumatoid arthritis (p = 0.012) but not those with gout (p = 0.12). These results demonstrate that urate forms adducts with albumin in healthy individuals, and adduct formation is increased during inflammation.
Figure 9.
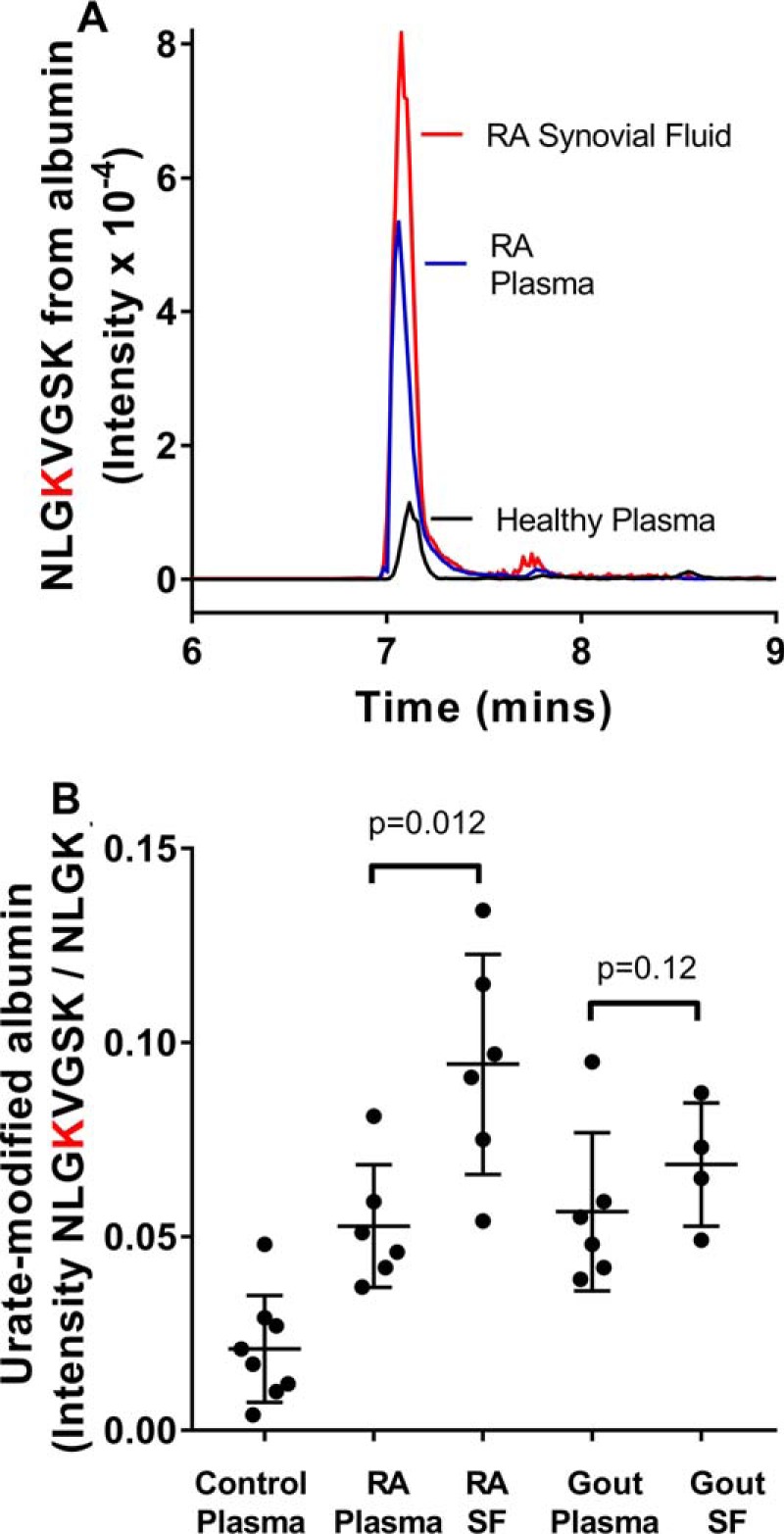
Urate-derived adducts on albumin are elevated during inflammation. A, the LC-MS signal for NLGK432VGSK from albumin was higher in plasma and synovial fluid from a patient with rheumatoid arthritis compared with that from a healthy control. B, the levels of urate-derived adducts on NLGK432VGSK from albumin relative to the normal tryptic peptide NLGK were significantly (p < 0.014) elevated in plasma and synovial fluid from individuals with rheumatoid arthritis (n = 6) and gout (n = 6) compared with healthy controls (n = 8). Plots show individual points, with error bars indicating means and standard deviations. Significant differences were determined by ANOVA after Dunnett's multiple-comparison test.
Discussion
We propose that when urate is oxidized in vivo, it forms reactive electrophiles that add to amino and thiol residues on proteins, a process we have called oxidative uratylation. Furthermore, we contend that these electrophiles are produced during normal metabolism, and their levels become markedly elevated during oxidative stress associated with inflammation. Our findings bring into question the widely held view that urate is an antioxidant that scavenges reactive oxygen species and is itself degraded to safe and soluble allantoin (1, 3, 7). Rather, our work demonstrates that urate is better viewed simply as a versatile reductant of radicals. The fate of the resulting urate radical will determine whether urate oxidation is beneficial or harmful.
Urate will act as an antioxidant under conditions where either its radical is repaired by ascorbate or the reactions of the resulting electrophiles are harmless. Its electrophilic products, however, will be damaging when they are free to react with critical amino or thiol residues on proteins and enzymes. It is possible that post-translational modifications by urate electrophiles may have positive biological outcomes. For example, specific oxidation-dependent modification of extracellular matrix has recently been found to promote migration of macrophages during inflammation (29). Urate-derived adducts on proteins may have similar signaling functions.
Ames et al. (3) first proposed that urate is an antioxidant because it reacted with a variety of oxidants and, at physiological concentrations, protected erythrocyte ghosts from hemolysis and lipid peroxidation. However, further work by Ames's group established that ascorbate is the dominant antioxidant in human blood plasma (30). When neutrophils were stimulated in plasma to produce oxidants, urate was not oxidized until after all of the ascorbate had been consumed (31). Under their reaction conditions, neutrophils release the heme enzyme myeloperoxidase, which converts co-released hydrogen peroxide to hypochlorous acid (16). Urate and ascorbate are both oxidized by hypochlorous acid and are also substrates for myeloperoxidase, which oxidizes them to their respective radicals (32). In plasma, they would be oxidized at similar rates (14). Thus, the lag in urate oxidation when neutrophils were stimulated in plasma suggests that urate was being repaired rather than spared by ascorbate. Consequently, urate is likely to cycle between its reduced and oxidized forms until ascorbate is consumed (see Fig. 1). This concept is important because the urate radical could be gradually diverted to electrophiles, particularly in the presence of superoxide. A low but steady formation of urate electrophiles may account for the urate-derived adducts detected on albumin from healthy individuals replete with ascorbate.
Under our experimental conditions, the main electrophiles produced in the oxidation of urate are expected to be dehydrourate, 5-hydroxyisourate, and urate hydroperoxide (Fig. 1) (11, 14). Urate hydroperoxide is formed predominantly through the rapid reaction of urate radical with superoxide (12, 14). There is no evidence that it is derived from the slow reaction of molecular oxygen with urate radical (10) because it was not detected in the absence of superoxide (14, 15, 22). Dehydrourate formed a 166-Da mass addition product with both amino and cysteine residues (Figs. 2 and 4 and Table S1). This reaction would be a straight forward nucleophilic addition of the amino or sulfur moiety to the electron-deficient carbon on dehydrourate. The low frequency and transient nature of these adducts suggests that they are unstable. The more stable adduct, with a mass addition of 140 Da, was formed under reaction conditions that produce urate hydroperoxide and 5-hydroxisourate, the hydrolysis product of dehydrourate. Based on our analysis using MS (Fig. 5 and Fig. S2), the adduct is a derivative of 2-oxo-4-imino-5-ureido-imidazolidine, where the imino group originates from a lysine residue or an N-terminal amine. This structure is analogous to allantoin (11). We propose that either 5-hydroxyisourate or urate hydroperoxide breaks down to the bicylic imidazolone, which then couples with amine groups (Fig. 5D). Direct reaction of the amines with the individual electrophiles and their subsequent breakdown to the 140-Da adduct is another possible mechanism that cannot be precluded at this time.
We found that superoxide enhanced formation of urate-derived adducts on peptides in the xanthine oxidase system (Fig. 3C) and on albumin with stimulated neutrophils (Fig. 8D). Superoxide-dependent products also played a major part in the inactivation of GAPDH (Fig. 6A). These results suggest that urate hydroperoxide, formed when superoxide reacts with the urate radical (Fig. 1), makes a major contribution to adduct formation. However, the systems we have investigated are complex, and other reactions of superoxide may contribute to adduct formation. For example, superoxide reacts rapidly with myeloperoxidase to form compound III, which may catalyze formation of transient urate electrophiles (33, 34). The precise contribution urate hydroperoxide makes to adduct formation will require purification of this unstable compound from mixtures of urate electrophiles formed when urate is oxidized.
Early evidence that oxidation products of urate could be reactive came when urate was found to substantially enhance radiation-induced inactivation of yeast alcohol dehydrogenase (35, 36). Urate's effects were greatest in the presence of molecular oxygen but substantially decreased by superoxide dismutase. Enzyme inactivation was attributed to a short-lived toxin. Based on our current results and the knowledge that yeast alcohol dehydrogenase is inactivated when its zinc-thiolate active site is oxidized (37), it is likely that urate hydroperoxide or other superoxide-dependent urate electrophiles were generated in the radiation experiments and oxidized critical cysteine residues. In support of this proposal, we showed that urate electrophiles, including a superoxide-dependent species, readily inactivate GAPDH by reversibly and irreversibly oxidizing its cysteine residues (Fig. 6). Also, in recent work, it was found that urate hydroperoxide reacts rapidly with the thiol residues in peroxiredoxins (38). These ubiquitous proteins may protect against the oxidative stress exerted by urate hydroperoxide. Peroxiredoxins may also act as sensors of oxidative stress relayed by urate hydroperoxide.
Methionine residues are also likely to be oxidized by urate hydroperoxide and related electrophiles (39). In an early study that challenged the antioxidant paradigm for urate, it was demonstrated that urate enhanced oxidative inactivation of α1-antiproteinase, a methionine-dependent protease inhibitor (40). In contrast, ascorbate was moderately protective, whereas the combination of ascorbate and urate was even better at preventing inactivation. These findings suggest that when urate radical is converted to a reactive electrophile, it will damage proteins. However, if repaired by ascorbate, it can provide protection over and above that of ascorbate alone.
In the recent work, urate hydroperoxide was shown to react readily with cysteine and methionine (39). Its reactivity with amine groups was considerably slower, but approximately 20% of urate hydroperoxide was lost when it reacted with histidine or lysine over a 10-min period. Given the much slower reactions with amine groups, no attempt was made in that study to look for products with amines.
There is compelling evidence that urate electrophiles are toxic (13). With the exception of humans and other great apes, urate is metabolized to allantoin by three enzymes: urate oxidase, 5-hydroxyisourate hydrolase, and 2-oxo-4-hydroxy-4-carboxy-5-ureidoimidazoline decarboxylase (41). Mice lacking 5-hydroxyisourate hydrolase developed hepatomegaly and hepatocellular cancer (13). This finding suggests that urate metabolism without 5-hydroxyisourate hydrolase liberates toxic metabolites that affect liver growth and transformation. The metabolites of urate oxidase are similar to the electrophiles generated by free radical oxidation of urate (42). The three urate-metabolizing enzymes are not active in humans, which makes humans vulnerable to toxic reactions of urate electrophiles (43). Thus, heightened oxidation of urate, especially in the absence of ascorbate, is likely to contribute to oxidative stress and tissue damage.
There are multiple pathways for urate oxidation in vivo, although heme peroxidases are likely to dominate during inflammation. Given urate's penultimate position in the thermodynamic pecking order of free radical reductants (4), most free radicals will be reduced by urate and potentially lead to urate electrophiles. Oxidizing radicals will include hydroxyl radical, those released by peroxynitrite, tyrosyl radicals, and secondary protein radicals (4, 5, 44). Hence, urate oxidation may reflect overall oxidative damage, and its protein adducts could be valuable biomarkers of oxidative stress. These protein adducts are likely to accumulate and, consequently, will be better indicators of oxidative stress than allantoin, which should be more rapidly cleared from circulation.
Hyperuricemia has been touted as a causative factor in numerous pathologies (45). Much debate has ensued from many contradictory studies on the involvement of serum urate in a range of pathologies, such as nephrolithiasis, metabolic syndrome, hypertension, cardiovascular disease, renal disease, and obesity (46, 47). However, a recent evaluation of serum urate levels in multiple health outcomes found convincing evidence that it plays a clear role in only gout and nephrolithiasis (48). We contend that serum urate levels are a poor indicator of its involvement in diseases, except when urate acts through crystal formation as in gout. Based on our current findings, we propose that urate is likely to contribute to pathologies when it is susceptible to oxidation. This susceptibility will increase when its concentration is high, the ascorbate concentration is low, and there is enhanced oxidative stress, as occurs during inflammation. Therefore, urate-derived adducts on proteins may be a more accurate determinant of urate's involvement in disease than serum urate concentrations.
In conclusion, we have demonstrated that in humans, electrophilic oxidation products of urate are produced during normal metabolism and form adducts with proteins. This process is heightened during inflammation. The challenge now is to decipher how these adducts contribute to the biological chemistry and pathology of urate.
Experimental procedures
All reagents were purchased from Sigma and of the highest grade available unless otherwise stated. All peptides were purchased from GenScript (Piscataway, NJ). Proteins used included rabbit muscle GAPDH (ICN Biomedical Inc., Irvine, CA), bovine β-lactoglobulin, bovine milk lactoperoxidase and xanthine oxidase (XO), human serum albumin (Albumix, CSL Ltd., Victoria, Australia), bovine ubiquitin, bovine erythrocyte SOD, bovine liver catalase (CAT), and trypsin (sequence grade, Promega, Madison, WI). Human MPO (ϵ430 per heme = 89,000 m−1 cm−1) (49) was purchased from Planta Naturstoffe Vertriebs GmbH. Labeled uric acid (15N) was purchased from Icon Isotopes (Summit, NJ). Hydrogen peroxide (ϵ240 43.6 m−1 cm−1) (50) was purchased from Biolab (Victoria, Australia). The TX1 was a gift from AstraZeneca (28).
Reaction of oxidized urate with peptides
Uric acid was dissolved at 10 mm in water at pH 11 and then immediately diluted to 420 μm in phosphate buffer (pH 7.4) to minimize auto-oxidation. Urate (380 μm) was incubated at 37 °C in phosphate buffer (10 mm, pH 7.4) with 100 nm myeloperoxidase and 100 μm of peptide. Oxidation of urate was started by adding 100 μm hydrogen peroxide and continued for 30 min. Controls containing all reagents except hydrogen peroxide or myeloperoxidase were run under conditions identical to those for the full system. Cysteine-containing peptides were either used in their reduced form or alkylated by blocking the reactive thiol with iodoacetamide (50 mm) for 30 min followed by the addition of DTT (50 mm). To minimize breakdown of adducts on some cysteine-containing peptides, reactions were run for approximately 1 min and then kept at 4 °C until analyzed. To determine the effect of superoxide on adduct formation with peptides, superoxide and hydrogen peroxide were generated using xanthine oxidase (0.025 units/ml) and acetaldehyde (10 mm) (51). When used, superoxide dismutase was added at 20 μg/ml. After incubation of peptides with urate, myeloperoxidase, and oxidant, reaction mixtures were analyzed by LC with MS (LC-MS) to detect modified peptides.
Analysis of urate-modified peptides
For identification of modified peptides, samples were dried down, reconstituted in 2% acetonitrile, and then injected onto an Ultimate 3000 HPLC (Thermo, Waltham, MA) fitted with an Aeris Peptide XB-C18 (150 × 2.1 mm, 3.6 μm) column (Phenomenex, Torrance, CA) attached to a Sciex 4000 Q TRAP mass spectrometer (Framingham, MA). The Q TRAP was used in either enhanced mass spectrum mode when identifying peptide masses, or positive enhanced product ion (EPI) mode when fragmenting peptides for sequence identification. A gradient from 2% acetonitrile, 0.1% formic acid (FA) (v/v) up to 50% acetonitrile, 0.1% FA (v/v) over 10 min was applied at a flow rate of 250 μl/min. The ion spray potential was set to 4.0 kV, the nebulizer gas to 20, and the interface heater to 400 °C. For EPI, the collisional gas was nitrogen, and collisional energy was 25% with a 10% collisional energy spread. Peptides and their modifications were identified using Fragment Ion Calculator (Institute for Systems Biology, Seattle, WA). When sequences are given for peptides containing a urate-derived adduct, the conjugated residue is shown in boldface type (red in the figures).
Structural determination of urate-derived adduct
Urate (380 μm), either 15N-labeled (molecular weight 170.1) or unlabeled (molecular weight 168.1), was oxidized by myeloperoxidase (100 nm) and hydrogen peroxide (400 μm) in the presence of phenylethylamine (molecular weight 121.2; 100 μm) in 10 mm phosphate buffer, pH 7.4 at 20–22 °C. Reactions were started by adding hydrogen peroxide and analyzed after 30 min. Conjugates of oxidized urate and phenylethylamine were separated on a Kinetex HILIC 2.6-μm column (75 × 2.1 mm; Phenomenex) and detected on a Sciex 4000 Q TRAP mass spectrometer using selected ion monitoring of [M + H]+ ions. A gradient from 90% acetonitrile, 10% ammonium formate (10 mm, pH 3.6) (v/v) up to 50% acetonitrile, 50% ammonium formate (v/v) over 10 min was applied at a flow rate of 250 μl/min. The ion spray potential was set to 4.0 kV, the nebulizer gas to 20, and the interface heater to 600 °C. For EPI, the collisional gas was nitrogen, and collisional energy was 20% with a 10% collisional energy spread.
Inactivation of GAPDH
Urate hydroperoxide was generated by adding xanthine oxidase (0.025 units/ml) to 100 μm hypoxanthine, 160 nm lactoperoxidase, and 400 μm urate in 50 mm phosphate buffer, pH 7.4. After 20 min, samples were treated with catalase to degrade hydrogen peroxide, and then the concentration of urate hydroperoxide was measured using the ferrous iron xylenol orange assay as described previously (52). Concentrations of urate hydroperoxide were expressed as hydrogen peroxide equivalents. Urate hydroperoxide was then added to GAPDH, and after 10 min, residual enzyme activity was determined by measuring the reduction of NAD+ at 340 nm (53). To establish the degree to which enzyme inhibition was due to reversible oxidation of thiol residues, modified GAPDH was reduced with 2 mm DTT, and its activity was remeasured. Reduced thiols on GAPDH were measured as described previously (27).
Detection of urate-derived adducts on proteins
Urate (380 μm) was incubated in 10 mm phosphate buffer (pH 7.4) with ubiquitin or β-lactoglobulin (0.1 mg/ml), 100 nm myeloperoxidase at 37 °C. Reactions were started by adding 100 μm hydrogen peroxide and analyzed after 30 min. Samples were injected onto a Luna C8 5 μ (20 × 4 mm) HPLC column (Phenomenex) attached to an Agilent (Santa Clara, CA) 1260 binary HPLC and separated using a gradient of 29–45% acetonitrile (both containing 0.1% formic acid) over 3 min. Protein masses were then analyzed using an Agilent 6230 Accurate Mass TOF mass spectrometer, and the resulting m/z spectra were deconvoluted using maximum entropy processing with BioConfirm software.
To identify amino acid residues on proteins that contained adducts of oxidized urate, the proteins were tryptically digested as described previously (54). Tryptic peptides from human serum albumin modified by oxidized urate were separated on a Jupiter Proteo 90A column (150 × 2 mm, 4 μm; Phenomenex) with a solvent gradient of water and acetonitrile (both containing 0.1% formic acid). The gradient started at 98% water and changed to 50% acetonitrile over 45 min at 0.2 ml/min. Peptides in the eluant were detected using a Thermo Velos Pro mass spectrometer. MassMatrix (MassMatrix) and Proteome Discoverer (Thermo) software were used for identification of peptides.
Quantification of urate-derived adducts on a tryptic peptide of albumin
To measure urate-derived adduct formation in plasma, a semiquantitative method was developed to detect modification of Lys-432 of human serum albumin. Plasma proteins were digested with trypsin as described previously (54), and the resulting tryptic peptides NLGK432 (unmodified) and NLGK432(140)VGSK (modified) were detected using MS with multiple reaction monitoring. The peptides were first separated on an Aeris Peptide XB-C18 (150 × 2.1 mm, 3.6 μm) column (Phenomenex) using a gradient from 2% acetonitrile, 0.1% FA (v/v) to 30% acetonitrile, 0.1% FA (v/v) over 8 min at a flow rate of 250 μl/min. Peptides were then detected using a Sciex 4000 Q TRAP mass spectrometer. Collisional energy was 30%, the ion source voltage was 4.5 kV, and the source temperature was 400 °C. The mass spectrometer was set to detect the singly charged b2 and y2 fragment ions of unmodified NLGK432 as well as the singly charged a2, y6, and y4 fragment ions of the doubly charged NLGK432(140)VGSK modified peptide. For quantification, the area under the curve for the a2 fragment ion of NLGK432(140)VGSK was divided by that for the b2 fragment ion of NLGK432. The y fragment ions plus the retention times were used for confirmation of the analytes.
Formation of urate-derived adducts on albumin by isolated neutrophils
Peripheral blood from three healthy male donors (age 45–60 years) was obtained with informed consent as approved by the Southern Health and Disability Ethics Committee of New Zealand. The studies abide by the Declaration of Helsinki principles. Neutrophils were isolated from the heparinized blood by dextran sedimentation followed by Ficoll-Paque centrifugation and hypotonic lysis of contaminating erythrocytes (55). Plasma was also prepared from each donor. Neutrophils were suspended in Hanks' balanced salt solution (10 mm phosphate, pH 7.4, containing 140 mm sodium chloride (PBS), 0.5 mm magnesium chloride, 1 mm calcium chloride, and 5.5 mm glucose) and incubated for 5 min at 37 °C with or without inhibitors including 20 μm DPI, 10 μm TX1, 100 μg/ml catalase, or 20 μg/ml superoxide dismutase. After pretreatment, neutrophils were added to autologous plasma to give a final concentration of 80% plasma with neutrophils at 5 × 106/ml in a 250-μl total volume. Cells were stimulated with a combination of cytochalasin B (10 μg/ml) added for a 5-min incubation prior to adding phorbol 12-myristate 13-acetate (100 ng/ml). After 45 min at 37 °C, cells were pelleted by centrifugation (8,000 × g, 5 min), and supernatant was collected for analysis of urate-derived adducts on tryptic peptides from serum albumin. The supernatant (10 μl) was lyophilized, the protein was denatured by resuspending it in 50 μl of a 100 mm ammonium bicarbonate solution containing 6 mm guanidinium chloride and then made up to 1 ml with more ammonium bicarbonate solution, and trypsin was added to the existing protein at a ratio of 1:50 (w/w). The solution was incubated at 37 °C overnight to liberate tryptic peptides, which were separated on the Aeris Peptide XB C18 column and detected on the Sciex 4000 Q TRAP mass spectrometer as described above. The signal for NLGK432VGSK was expressed relative to that for the unmodified peptide NLGK and normalized to the ratio detected in plasma alone.
Uric acid was determined in the plasma samples using HPLC with UV detection at 293 nm (56). The concentrations of urate in the three donor samples were 370, 400, and 525 μm (62–88 mg/liter).
Analysis of urate-derived adducts on serum albumin from patient samples
Human plasma was obtained from eight healthy donors (five females and three males, 33–59 years old). Plasma and synovial fluids were obtained from individuals with rheumatoid arthritis (n = 6; four females and two males; age 30–69 years) and gout (n = 6; all males age 42–84 years) as defined by American College of Rheumatology Classification Criteria (57, 58). Plasma only was available from two of the individuals with gout. Ethical consent for blood and synovial fluid collection was obtained from the Southern Health and Disability Ethics Committee of New Zealand. The studies abide by the Declaration of Helsinki principles. Proteins in 10 μl of plasma were digested with trypsin, and the tryptic peptide from albumin, NLGK432(140)VGSK, containing the urate-derived adduct was quantified as described above.
Statistical analysis
The effect of superoxide dismutase on the attachment of urate electrophiles to peptides was assessed using unpaired one-tailed Student's t test. For adducts on human serum albumin generated by neutrophils or detected in clinical samples, analysis of variance with Dunnett's post hoc analysis was used to determine differences among groups after Satterthwaite adjustment for unequal variance. For cell and human data, the level of significance was set at p < 0.01.
Author contributions
R. T. performed all of the mass spectrometry experiments and measurements except for the work on MIF. S. O. B. assisted with the mass spectrometry analysis of modified proteins. L. V. A. did the experiments with neutrophils and assisted with writing of the manuscript. N. D. did the experimental work on MIF and assisted with the writing of the manuscript. M. R. H. performed the experimental work on inactivation and modification of GAPDH. J. F. P. advised on the statistical analysis. L. K. S. provided samples from the patients with gout and rheumatoid arthritis and helped write the clinical section of the paper. A. J. K. designed and coordinated the study and wrote the majority of the paper.
Supplementary Material
Acknowledgment
We thank Professor Mark Hampton for helpful suggestions regarding the writing of the manuscript.
This work was supported by grants from the Manatu Hauora Health Research Council of New Zealand. The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Tables S1 and S2 and Figs. S1–S5.
- MPO
- myeloperoxidase
- EPI
- enhanced product ion
- DPI
- diphenyliodonium
- GAPDH
- glyceraldehyde phosphate dehydrogenase
- MIF
- macrophage migration inhbitory factor
- SOD
- superoxide dismutase
- TX1
- 3-isobutyl-2-thioxo-7H-purin-6-one
- XO
- xanthine oxidase
- PMA
- phorbol 12-myristate 13-acetate
- CAT
- catalase
- FA
- formic acid
- ANOVA
- analysis of variance
- CytB
- cytochalasin B.
References
- 1. Becker B. F. (1993) Towards the physiological function of uric-acid. Free Radic. Biol. Med. 14, 615–631 10.1016/0891-5849(93)90143-I [DOI] [PubMed] [Google Scholar]
- 2. Feig D. I., Kang D. H., and Johnson R. J. (2008) Uric acid and cardiovascular risk. New Engl. J. Med. 359, 1811–1821 10.1056/NEJMra0800885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ames B. N., Cathcart R., Schwiers E., and Hochstein P. (1981) Uric-acid provides an antioxidant defense in humans against oxidant-caused and radical-caused aging and cancer: a hypothesis. Proc. Natl. Acad. Sci. U.S.A. 78, 6858–6862 10.1073/pnas.78.11.6858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Buettner G. R. (1993) The pecking order of free-radicals and antioxidants: lipid-peroxidation, α-tocopherol, and ascorbate. Arch. Biochem. Biophys. 300, 535–543 10.1006/abbi.1993.1074 [DOI] [PubMed] [Google Scholar]
- 5. Santos C. X. C., Anjos E. I., and Augusto O. (1999) Uric acid oxidation by peroxynitrite: multiple reactions, free radical formation, and amplification of lipid oxidation. Arch. Biochem. Biophys. 372, 285–294 10.1006/abbi.1999.1491 [DOI] [PubMed] [Google Scholar]
- 6. Paganoni S., and Schwarzschild M. A. (2017) Urate as a marker of risk and progression of neurodegenerative disease. Neurotherapeutics 14, 148–153 10.1007/s13311-016-0497-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sautin Y. Y., and Johnson R. J. (2008) Uric acid: the oxidant-antioxidant paradox. Nucleosides Nucleotides Nucleic Acids 27, 608–619 10.1080/15257770802138558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cantu-Medellin N., and Kelley E. E. (2013) Xanthine oxidoreductase-catalyzed reactive species generation: a process in critical need of reevaluation. Redox Biol. 1, 353–358 10.1016/j.redox.2013.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Maples K. R., and Mason R. P. (1988) Free-radical metabolite of uric acid. J. Biol. Chem. 263, 1709–1712 [PubMed] [Google Scholar]
- 10. Simic M. G., and Jovanovic S. V. (1989) Antioxidation mechanisms of uric acid. J. Am. Chem. Soc. 111, 5778–5782 10.1021/ja00197a042 [DOI] [Google Scholar]
- 11. Volk K. J., Yost R. A., and Brajter-Toth A. (1989) Online electrochemistry thermospray tandem mass spectrometry as a new approach to the study of redox reactions: the oxidation of uric acid. Anal. Chem. 61, 1709–1717 10.1021/ac00190a024 [DOI] [PubMed] [Google Scholar]
- 12. Santus R., Patterson L. K., Filipe P., Morlière P., Hug G. L., Fernandes A., and Mazière J. C. (2001) Redox reactions of the urate radical/urate couple with the superoxide radical anion, the tryptophan neutral radical and selected flavonoids in neutral aqueous solutions. Free Radic. Res. 35, 129–136 10.1080/10715760100300671 [DOI] [PubMed] [Google Scholar]
- 13. Stevenson W. S., Hyland C. D., Zhang J. G., Morgan P. O., Willson T. A., Gill A., Hilton A. A., Viney E. M., Bahlo M., Masters S. L., Hennebry S., Richardson S. J., Nicola N. A., Metcalf D., Hilton D. J., et al. (2010) Deficiency of 5-hydroxyisourate hydrolase causes hepatomegaly and hepatocellular carcinoma in mice. Proc. Natl. Acad. Sci. U.S.A. 107, 16625–16630 10.1073/pnas.1010390107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Meotti F. C., Jameson G. N. L., Turner R., Harwood D. T., Stockwell S., Rees M. D., Thomas S. R., and Kettle A. J. (2011) Urate as a physiological substrate for myeloperoxidase: implications for hyperuricemia and inflammation. J. Biol. Chem. 286, 12901–12911 10.1074/jbc.M110.172460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Seidel A., Parker H., Turner R., Dickerhof N., Khalilova I. S., Wilbanks S. M., Kettle A. J., and Jameson G. N. L. (2014) Uric acid and thiocyanate as competing substrates of lactoperoxidase. J. Biol. Chem. 289, 21937–21949 10.1074/jbc.M113.544957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kettle A. J., and Winterbourn C. C. (1997) Myeloperoxidase: A key regulator of neutrophil oxidant production. Redox Rep. 3, 3–15 10.1080/13510002.1997.11747085 [DOI] [PubMed] [Google Scholar]
- 17. Ashby M. T. (2008) Inorganic chemistry of defensive peroxidases in the human oral cavity. J. Dent. Res. 87, 900–914 10.1177/154405910808701003 [DOI] [PubMed] [Google Scholar]
- 18. Seet R. C. S., Lee C. Y. J., Chan B. P. L., Sharma V. K., Teoh H. L., Venketasubramanian N., Lim E. C. H., Chong W. L., Looi W. F., Huang S. H., Ong B. K. C., and Halliwell B. (2011) Oxidative damage in ischemic stroke revealed using multiple biomarkers. Stroke 42, 2326–2489 10.1161/STROKEAHA.111.618835 [DOI] [PubMed] [Google Scholar]
- 19. Stamp L. K., Turner R., Khalilova I. S., Zhang M., Drake J., Forbes L. V., and Kettle A. J. (2014) Myeloperoxidase and oxidation of uric acid in gout: implications for the clinical consequences of hyperuricaemia. Rheumatology 53, 1958–1965 10.1093/rheumatology/keu218 [DOI] [PubMed] [Google Scholar]
- 20. Stamp L. K., Khalilova I., Tarr J. M., Senthilmohan R., Turner R., Haigh R. C., Winyard P. G., and Kettle A. J. (2012) Myeloperoxidase and oxidative stress in rheumatoid arthritis. Rheumatology 51, 1796–1803 10.1093/rheumatology/kes193 [DOI] [PubMed] [Google Scholar]
- 21. Dickerhof N., Turner R., Khalilova I., Fantino E., Sly P. D., Kettle A. J., and Arest C. F. (2017) Oxidized glutathione and uric acid as biomarkers of early cystic fibrosis lung disease. J. Cyst. Fibros. 16, 214–221 10.1016/j.jcf.2016.10.012 [DOI] [PubMed] [Google Scholar]
- 22. Volk K. J., Yost R. A., and Brajtertoth A. (1990) Online mass spectrometric investigation of the peroxidase-catalyzed oxidation of uric acid. J. Pharmaceut. Biomed. 8, 205–215 10.1016/0731-7085(90)80028-N [DOI] [PubMed] [Google Scholar]
- 23. Gersch C., Palii S. P., Imaram W., Kim K. M., Karumanchi S. A., Angerhofer A., Johnson R. J., and Henderson G. N. (2009) Reactions of peroxynitrite with uric acid: formation of reactive intermediates, alkylated products and triuret, and in vivo production of triuret under conditions of oxidative stress. Nucleosides Nucleotides Nucleic Acids 28, 118–149 10.1080/15257770902736400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kettle A. J., Anderson R. F., Hampton M. B., and Winterbourn C. C. (2007) Reactions of superoxide with myeloperoxidase. Biochemistry 46, 4888–4897 10.1021/bi602587k [DOI] [PubMed] [Google Scholar]
- 25. Kettle A. J., Maroz A., Woodroffe G., Winterbourn C. C., and Anderson R. F. (2011) Spectral and kinetic evidence for reaction of superoxide with compound I of myeloperoxidase. Free Radic. Biol. Med. 51, 2190–2194 10.1016/j.freeradbiomed.2011.09.019 [DOI] [PubMed] [Google Scholar]
- 26. Turner R., Stamp L. K., and Kettle A. J. (2012) Detection of allantoin in clinical samples using hydrophilic liquid chromatography with stable isotope dilution negative ion tandem mass spectrometry. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 891, 85–89 [DOI] [PubMed] [Google Scholar]
- 27. Peskin A. V., and Winterbourn C. C. (2006) Taurine chloramine is more selective than hypochlorous acid at targeting critical cysteines and inactivating creatine kinase and glyceraldehyde-3-phosphate dehydrogenase. Free Radic. Biol. Med. 40, 45–53 10.1016/j.freeradbiomed.2005.08.019 [DOI] [PubMed] [Google Scholar]
- 28. Tidén A. K., Sjögren T., Svensson M., Bernlind A., Senthilmohan R., Auchère F., Norman H., Markgren P. O., Gustavsson S., Schmidt S., Lundquist S., Forbes L. V., Magon N. J., Paton L. N., Jameson G. N. L., et al. (2011) 2-Thioxanthines are mechanism-based inactivators of myeloperoxidase that block oxidative stress during inflammation. J. Biol. Chem. 286, 37578–37589 10.1074/jbc.M111.266981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yakubenko V. P., Cui K., Ardell C. L., Brown K. E., West X. Z., Gao D., Stefl S., Salomon R. G., Podrez E. A., and Byzova T. V. (2018) Oxidative modifications of extracellular matrix promote the second wave of inflammation via β2 integrins. Blood 132, 78–88 10.1182/blood-2017-10-810176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Frei B., England L., and Ames B. N. (1989) Ascorbate is an outstanding antioxidant in human-blood plasma. Proc. Natl. Acad. Sci. U.S.A. 86, 6377–6381 10.1073/pnas.86.16.6377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Frei B., Stocker R., and Ames B. N. (1988) Antioxidant defenses and lipid-peroxidation in human-blood plasma. Proc. Natl. Acad. Sci. U.S.A. 85, 9748–9752 10.1073/pnas.85.24.9748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Winterbourn C. C. (1985) Comparative reactivities of various biological compounds with myeloperoxidase hydrogen peroxide-chloride, and similarity of the oxidant to hypochlorite. Biochim. Biophys. Acta 840, 204–210 10.1016/0304-4165(85)90120-5 [DOI] [PubMed] [Google Scholar]
- 33. Kettle A. J., and Winterbourn C. C. (1992) Oxidation of hydroquinone by myeloperoxidase: mechanism of stimulation by benzoquinone. J. Biol. Chem. 267, 8319–8324 [PubMed] [Google Scholar]
- 34. Ximenes V. F., Silva S. O., Rodrigues M. R., Catalani L. H., Maghzal G. J., Kettle A. J., and Campa A. (2005) Superoxide-dependent oxidation of melatonin by myeloperoxidase. J. Biol. Chem. 280, 38160–38169 10.1074/jbc.M506384200 [DOI] [PubMed] [Google Scholar]
- 35. Kittridge K. J., and Willson R. L. (1984) Uric-acid substantially enhances the free radical-induced inactivation of alcohol-dehydrogenase. FEBS Lett. 170, 162–164 10.1016/0014-5793(84)81391-5 [DOI] [PubMed] [Google Scholar]
- 36. Willson R. L., Dunster C. A., Forni L. G., Gee C. A., and Kittridge K. J. (1985) Organic free-radicals and proteins in biochemical injury: electron-transfer or hydrogen-transfer reactions. Philos. Trans. R. Soc. Lond. B Biol. Sci. 311, 545–563 10.1098/rstb.1985.0163 [DOI] [PubMed] [Google Scholar]
- 37. Crow J. P., Beckman J. S., and McCord J. M. (1995) Sensitivity of the essential zinc-thiolate moiety of yeast alcohol dehydrogenase to hypochlorite and peroxynitrite. Biochemistry 34, 3544–3552 10.1021/bi00011a008 [DOI] [PubMed] [Google Scholar]
- 38. Carvalho L. A. C., Truzzi D. R., Fallani T. S., Alves S. V., Toledo J. C. Jr., Augusto O., Netto L. E. S., and Meotti F. C. (2017) Urate hydroperoxide oxidizes human peroxiredoxin 1 and peroxiredoxin 2. J. Biol. Chem. 292, 8705–8715 10.1074/jbc.M116.767657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Patrício E. S., Prado F. M., da Silva R. P., Carvalho L. A. C., Prates M. V. C., Dadamos T., Bertotti M., Di Mascio P., Kettle A. J., and Meotti F. C. (2015) Chemical characterization of urate hydroperoxide, a pro-oxidant intermediate generated by urate oxidation in inflammatory and photoinduced processes. Chem. Res. Toxicol. 28, 1556–1566 10.1021/acs.chemrestox.5b00132 [DOI] [PubMed] [Google Scholar]
- 40. Aruoma O. I., and Halliwell B. (1989) Inactivation of α1-antiproteinase by hydroxyl radicals: the effect of uric acid. FEBS Lett. 244, 76–80 10.1016/0014-5793(89)81166-4 [DOI] [PubMed] [Google Scholar]
- 41. Tipton P. A. (2006) Urate to allantoin, specifically (S)-allantoin. Nat. Chem. Biol. 2, 124–125 10.1038/nchembio0306-124 [DOI] [PubMed] [Google Scholar]
- 42. Kahn K., and Tipton P. A. (1998) Spectroscopic characterization of intermediates in the urate oxidase reaction. Biochemistry 37, 11651–11659 10.1021/bi980446g [DOI] [PubMed] [Google Scholar]
- 43. Ramazzina I., Folli C., Secchi A., Berni R., and Percudani R. (2006) Completing the uric acid degradation pathway through phylogenetic comparison of whole genomes. Nat. Chem. Biol. 2, 144–148 10.1038/nchembio768 [DOI] [PubMed] [Google Scholar]
- 44. Domazou A. S., Zhu H., and Koppenol W. H. (2012) Fast repair of protein radicals by urate. Free Radic. Biol. Med. 52, 1929–1936 10.1016/j.freeradbiomed.2012.02.045 [DOI] [PubMed] [Google Scholar]
- 45. Kanbay M., Jensen T., Solak Y., Le M., Roncal-Jimenez C., Rivard C., Lanaspa M. A., Nakagawa T., and Johnson R. J. (2016) Uric acid in metabolic syndrome: from an innocent bystander to a central player. Eur. J. Intern. Med. 29, 3–8 10.1016/j.ejim.2015.11.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Li M., Hu X., Fan Y., Li K., Zhang X., Hou W., and Tang Z. (2016) Hyperuricemia and the risk for coronary heart disease morbidity and mortality a systematic review and dose-response meta-analysis. Sci. Rep. 6, 19520 10.1038/srep19520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Soltani Z., Rasheed K., Kapusta D. R., and Reisin E. (2013) Potential role of uric acid in metabolic syndrome, hypertension, kidney injury, and cardiovascular diseases: is it time for reappraisal? Curr. Hypertens. Rep. 15, 175–181 10.1007/s11906-013-0344-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Li X., Meng X., Timofeeva M., Tzoulaki I., Tsilidis K. K., Ioannidis J. P., Campbell H., and Theodoratou E. (2017) Serum uric acid levels and multiple health outcomes: umbrella review of evidence from observational studies, randomised controlled trials, and Mendelian randomisation studies. Brit. Med. J. 357, j2376 10.1136/bmj.j2376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kettle A. J., and Winterbourn C. C. (2016) Myeloperoxidase: structure and function of the green heme peroxidase of neutrophils. in Heme Peroxidases (Raven E., and Dunford H. B., eds) pp. 272–299, Royal Society of Chemistry, Cambridge, UK [Google Scholar]
- 50. Beers R. F., and Sizer I. W. (1952) A spectrophotometric method for measuring the breakdown of hydrogen peroxide by catalase. J. Biol. Chem. 195, 133–140 [PubMed] [Google Scholar]
- 51. Fridovich I. (1985) Xanthine oxidase. in Handbook of Methods for Oxygen Radical Research (Greenwald R. A., ed) pp. 51–53, CRC Press, Inc., Boca Raton, FL [Google Scholar]
- 52. Wolff S. P. (1994) Ferrous ion oxidation in presence of ferric ion indicator xylenol orange for measurement of hydroperoxides. Methods Enzymol. 233, 182–189 10.1016/S0076-6879(94)33021-2 [DOI] [Google Scholar]
- 53. Allison W. S., and Kaplan N. O. (1964) Comparative enzymology of triosephosphate dehydrogenase. J. Biol. Chem. 239, 2140–2152 [PubMed] [Google Scholar]
- 54. Brennan S. O., Potter H. C., Sheen C. R., and George P. M. (2016) Unique albumin with two silent substitutions (540Thr → Ala and 546Ala → Ser): insights into how albumin is recycled. Clin. Chim. Acta 457, 125–129 10.1016/j.cca.2016.04.014 [DOI] [PubMed] [Google Scholar]
- 55. Segal A. W., Dorling J., and Coade S. (1980) Kinetics of fusion of the cytoplasmic granules with phagocytic vacuoles in human polymorphonuclear leukocytes: biochemical and morphological studies. J. Cell Biol. 85, 42–59 10.1083/jcb.85.1.42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kandár R., Drábková P., and Hampl R. (2011) The determination of ascorbic acid and uric acid in human seminal plasma using an HPLC with UV detection. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 879, 2834–2839 10.1016/j.jchromb.2011.08.007 [DOI] [PubMed] [Google Scholar]
- 57. Neogi T., Jansen T. L. T. A., Dalbeth N., Fransen J., Schumacher H. R., Berendsen D., Brown M., Choi H., Edwards N. L., Janssens H. J. E. M., Lioté F., Naden R. P., Nuki G., Ogdie A., Perez-Ruiz F., et al. (2015) 2015 Gout classification criteria an American College of Rheumatology/European League against rheumatism collaborative initiative. Arthritis Rheumatol. 67, 2557–2568 10.1002/art.39254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Neogi T., Aletaha D., Silman A. J., Naden R. L., Felson D. T., Aggarwal R., Bingham C. O. 3rd, Birnbaum N. S., Burmester G. R., Bykerk V. P., Cohen M. D., Combe B., Costenbader K. H., Dougados M., Emery P., et al. (2010) The 2010 American College of Rheumatology/European League against rheumatism classification criteria for rheumatoid arthritis phase 2 methodological report. Arthritis Rheum. 62, 2582–2591 10.1002/art.27580 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.