

Abstract
Erythropoietin (EPO) signaling is critical to many processes essential to terminal erythropoiesis. Despite the centrality of iron metabolism to erythropoiesis, the mechanisms by which EPO regulates iron status are not well-understood. To this end, here we profiled gene expression in EPO-treated 32D pro-B cells and developing fetal liver erythroid cells to identify additional iron regulatory genes. We determined that FAM210B, a mitochondrial inner-membrane protein, is essential for hemoglobinization, proliferation, and enucleation during terminal erythroid maturation. Fam210b deficiency led to defects in mitochondrial iron uptake, heme synthesis, and iron–sulfur cluster formation. These defects were corrected with a lipid-soluble, small-molecule iron transporter, hinokitiol, in Fam210b-deficient murine erythroid cells and zebrafish morphants. Genetic complementation experiments revealed that FAM210B is not a mitochondrial iron transporter but is required for adequate mitochondrial iron import to sustain heme synthesis and iron–sulfur cluster formation during erythroid differentiation. FAM210B was also required for maximal ferrochelatase activity in differentiating erythroid cells. We propose that FAM210B functions as an adaptor protein that facilitates the formation of an oligomeric mitochondrial iron transport complex, required for the increase in iron acquisition for heme synthesis during terminal erythropoiesis. Collectively, our results reveal a critical mechanism by which EPO signaling regulates terminal erythropoiesis and iron metabolism.
Keywords: heme, iron metabolism, cell metabolism, erythrocyte, erythropoiesis, red blood cell
Introduction
The glycoprotein cytokine erythropoietin (EPO)6 plays a major role in regulation of erythroid survival and differentiation by activation of signaling pathways via EPO receptor binding (1). Among its roles is the up-regulation of erythroferrone to suppress hepcidin to increase availability of iron during stress erythropoiesis (2, 3). However, the physiological functions of EPO in the regulation of iron metabolism are largely uncharacterized.
Iron, as an ion, and in the forms of iron–sulfur [Fe-S] clusters and heme, is an essential cofactor for redox reactions in diverse cellular processes, such as dopamine synthesis, oxygen transport, mitochondrial respiration, xenobiotic metabolism, and maintenance of the circadian rhythm (4–9). However, the majority of iron in the body is used by erythroid cells to synthesize hemoglobin (10, 11). Free iron is highly toxic because of its ability to generate free radicals through the Fenton reaction. Although iron is obtained from outside the cell and is required for reactions that take place in membrane-bound organelles, cellular membranes are not permeable to iron. Organisms have therefore evolved sophisticated mechanisms to efficiently transport iron across membranes (11, 12). In the majority of vertebrate cells, iron is mainly obtained through the transferrin-mediated pathway (13, 14), which is SNX3- and SEC15L1-dependent for efficient transferrin receptor recycling in erythroid cells (15, 16). In the plasma, each transferrin molecule binds two Fe3+ ions. Iron-bound transferrin interacts with transferrin receptor on the cell surface. This complex is internalized via clathrin-mediated endocytosis. Within the acidified endosome, Fe3+ is reduced to Fe2+ by STEAP proteins (17). Fe2+ is exported from the endosome by endosomal DMT1 (18–20) and may enter the mitochondrial intermembrane space via DMT1, which is localized on the mitochondrial outer membrane (21). Iron that is used for heme synthesis or iron–sulfur [Fe-S] clusters is imported into the mitochondrial matrix by the mitochondrial iron importer mitoferrins 1 and/or 2 (MFRN1/SLC25A37 or MFRN2/SLC25A28) (22–24). In developing red cells, the stabilization of MFRN1 by ABCB10 (25) is a mechanism by which the cell ensures efficient iron import for its utilization in hemoglobin synthesis (26). However, the Δmrs3Δmrs4 yeast strain that is deficient in the mitoferrins only exhibited a partial defect in mitochondrial iron import. This suggests the existence of other mitochondrial iron importers (27, 28) or of auxiliary mechanisms that augment iron import to compensate for the genetic insufficiency of an iron transporter.
We identified potential EPO-regulated iron transport genes by microarray analysis of the EPO-treated, EpoR-expressing pro-B cell line, 32D, and we compared EPO-responsive genes to genes that were up-regulated during terminal erythropoiesis. One candidate gene, Fam210b (C20orf120), encoded a protein containing an N-terminal mitochondrial-targeting sequence. Fam210b was previously described to be a target of the erythroid transcription factor, GATA1, which encoded a mitochondrial-localized protein (29) that regulates mitochondrial metabolism in nonerythroid cells (30). In this study, we decided to interrogate the role of Fam210b in erythroid physiology. Fam210b expression is highly up-regulated during terminal erythropoiesis, which requires large quantities of iron. Loss-of-function studies in zebrafish embryos, primary murine fetal liver cells, and Friend murine erythroleukemia (MEL) cell lines show that FAM210B is required specifically to maintain the massive quantities of mitochondrial iron necessary for heme synthesis during terminal erythropoiesis. Although FAM210B is not an iron transporter per se, it interacts with terminal heme synthesis enzymes to form an oligomeric complex. Collectively, our data reveal that FAM210B plays a critical role as an adaptor protein in a mitochondrial iron import oligomeric complex. FAM210B is essential for integration of the rapid iron import with a high rate of porphyrin synthesis that supports hemoglobinization during terminal erythropoiesis. This study describes a novel mechanism by EPO governs enucleation and cellular proliferation, processes underlying terminal erythropoiesis, by its control of mitochondrial iron uptake and heme biosynthesis.
Results
Fam210b is an immediate early target of EPO during terminal erythroid differentiation
Maturing erythroid cells acquire large quantities of exogenous iron to keep pace with the enormous demands of hemoglobin production (31, 32). To determine how EPO regulates iron transport, we identified genes up-regulated by EPO in the EpoR-expressing pro-B cell line, 32D (33). Cells were cultured in media without erythropoietin for 6 h prior to treatment with Epo (0.5 units/ml) for 2 h before harvesting. We analyzed global gene expression in control and EPO-treated murine 32D cells by microarray analysis. To determine which EPO targets could also be required for erythroid terminal differentiation, we compared the EPO-responsive genes in 32D cells with genes that were enriched in TER119+ cells (34), which comprise the terminally differentiating erythroid cell population. We found that Fam210b expression was enriched in both datasets (Fig. 1A). To determine whether Fam210b was an EPO early response gene, a parallel experiment was set up in which 32D cells were treated with cycloheximide (10 μg/ml) 30 min prior to EPO stimulation to inhibit protein translation (35, 36). We observed that Fam210b expression was induced by EPO treatment, suggesting that Fam210b is an EPO target. The up-regulation of Fam210b expression persisted during cycloheximide treatment, indicating that Fam210b is an EPO early-response gene (Fig. 1B). The increase in Fam210b mRNA levels in response to EPO stimulation and during terminal differentiation translated to increases in the FAM210B protein. Interestingly, FAM210B protein was also up-regulated in the presence of cycloheximide, indicating that EPO increases the post-translational stability of FAM210B (Fig. 1C).
Figure 1.

Fam210b is an EPO early response gene and is induced in terminally differentiating erythroid cells. A, microarray analysis of EPO-treated 32D pro-B cells shows that Fam210b is an EPO-responsive gene that is also highly enriched in the terminally differentiating TER119+ population of fetal liver erythroid cells (34). B, qRT-PCR demonstrates that Epo treatment of the EpoR-expressing pro-B cell line, 32D, up-regulated expression of Fam210b mRNA. This up-regulation persists in the presence of cycloheximide (CHX), an inhibitor of protein translation, demonstrating that Fam210b is an EPO early response gene n = 6. *, p < 0.05, Student's t test. C, FAM210B protein levels are up-regulated in response to EPO treatment of the EpoR-expressing 32D pro-B cell line. The increase in protein levels persist with cycloheximide treatment, indicating increased stability. Changes in FAM210B protein expression normalized to GAPDH are quantitated relative to control levels. D, RNAseq analysis of primary murine fetal liver cells sorted according to TER119 and CD71 (R1–R5) expression demonstrates up-regulation of Fam210b during the R2–R3 transition. E, this up-regulation is recapitulated by Western blot analysis of FAM210B protein expression. F, FAM210B protein expression is up-regulated during in vitro differentiation of primary fetal liver cells. G, FAM210B protein is induced upon terminal differentiation of MEL cells in parallel with genes required for heme synthesis, TFRC and FECH. *, p < 0.05, Student t test.
To determine how Fam210b is developmentally regulated, we performed RNA sequencing (RNAseq) on transcripts from murine fetal liver erythroid cells (34) that were sorted into fractions corresponding to their stage of maturation (R1–R5) (15, 37–39). Fam210b expression was up-regulated during maturation from R2 to R3, which corresponds to the developmental shift from TER119− to TER119+ expression. This expression pattern was similar to that of genes involved in heme synthesis and iron import, such as Fech and Tfrc (Fig. 1D), and it is consistent with the enrichment of Fam210b in the TER119+ population (Fig. 1A). FAM210B protein expression in the R2–R3 transition was recapitulated at the protein level (Fig. 1E). Consistent with this, FAM210B protein levels increase during in vitro differentiation of primary fetal liver erythroid cells (Fig. 1F) and in DMSO-induced terminal differentiation of MEL cells (Fig. 1G). The increases in protein expression are coordinated with α-globin (HBA) (Fig. 1F), TFRC, and FECH (Fig. 1G), which are critical to erythroid differentiation, heme, and hemoglobin synthesis, suggesting that FAM210B plays an essential role in erythroid development. Although the expression of iron homeostasis genes is sometimes regulated by intracellular iron status via iron-regulatory proteins, IRP1 and IRP2 (13), we found that this was not the case for FAM210B (Fig. S1).
In situ hybridization of E12.5 murine embryos (performed as described (40)) indicated that Fam210b mRNA is enriched in the murine fetal liver at E12.5, the site of definitive erythropoiesis (Fig. 2A). In the adult mouse, Fam210b is enriched in the bone marrow, liver, and skeletal muscle, which are tissues that require large amounts of heme for synthesis of hemoproteins, and the testis, where iron plays an essential role in spermatogenesis (Fig. 2B) (41). In contrast to gata1, fam210b mRNA is maternally expressed in the developing zebrafish embryo. At 24 hpf, expression of fam210b mRNA is restricted to neuronal tissue and the intermediate cell mass (ICM), which is the site of primitive erythropoiesis in teleosts. fam210b is not expressed in cloche embryos (42, 43), which have defective blood and vascular progenitors. Conversely, fam210b expression is expanded in dino embryos. In dino embryos, the tail is expanded at the expense of the head (44). The tail is also the site of the ICM, which is the site of primitive hematopoiesis in the zebrafish. These zebrafish therefore have expanded blood due to their ventralized phenotype. The enrichment of fam210b in early hematopoietic tissues in zebrafish embryos, its absence in mutants that do not make hematopoietic progenitors, and the expansion of its expression in the dino mutants, which have an expanded ICM, indicate that fam210b expression depends on hematopoietic specification (Fig. 2C). At 72 hpf, fam210b expression persists in neural tissue and the liver (Fig. 2D), which are tissues that require large amounts of iron for their function (8, 13).
Figure 2.

Expression of Fam210b is enriched in vertebrate tissues with high heme content. A, Fam210b expression (red pseudocolor) is enriched in the murine fetal liver (FL) at E12.5. B, in murine tissues, Fam210b expression is enriched in the bone marrow and fetal liver (erythroid), adult liver, skeletal muscle, and testis, all of which require high levels of iron for their function. C, zebrafish fam210b is maternally expressed during early embryonic development. At 24 hpf, fam210b expression is enriched in neural tissue and the intermediate cell mass, the site of primitive hematopoiesis (top). The expression of fam210b in the ICM is abolished in cloche embryos, which do not form hematopoietic or vascular tissue. In contrast, fam210b expression in the intermediate cell mass is expanded in dino embryos, which exhibit a ventralized phenotype (bottom), paralleling the increased expression of erythroid gata1. D, at 72 hpf, fam210b expression persists in the neural tissue and is enriched in the liver, delineated by sid4 expression. n = 3.
Fam210b is required for erythropoiesis in vivo
To determine the biological function of fam210b, we injected antisense morpholinos to knock down its expression in developing zebrafish embryos. As fam210b is highly expressed in terminally differentiating erythroid cells (Fig. 1, A and B), we assayed heme synthesis in morphant embryos by o-dianisidine staining as a measure of terminal erythropoiesis. Compared with the controls, fam210b morphants have significantly decreased heme staining, indicative of their anemia (Fig. 3A). We quantitated the erythropoietic defect by flow cytometry analysis of GFP+ cells in morphant Tg(globin-LCR:eGFP) transgenic zebrafish, which express GFP under the control of a globin locus control region (LCR) enhancer, restricting GFP expression to the erythroid lineage (45). Morphant embryos exhibited decreased erythropoiesis at 24- and 48-hpf, highlighting the importance of fam210b in erythropoiesis (Fig. 3B).
Figure 3.
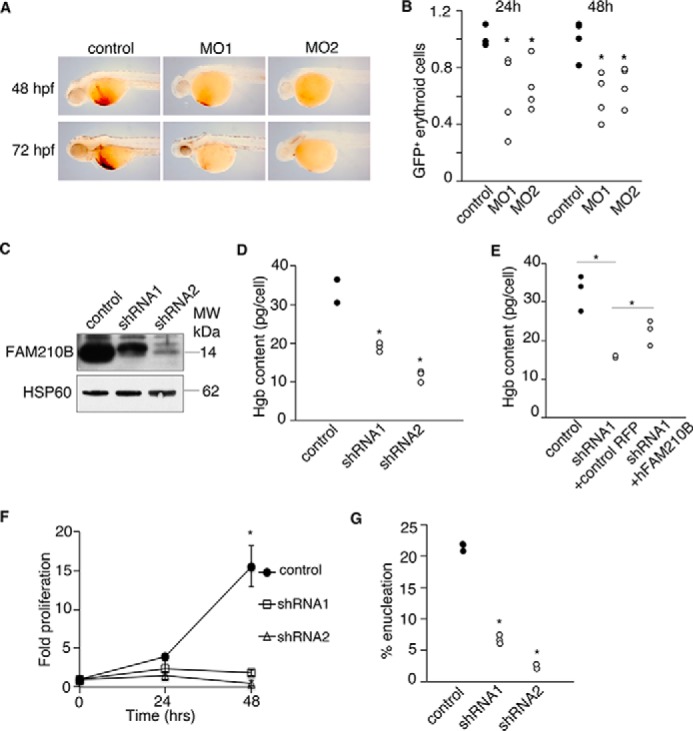
Fam210b is continuously required for terminal erythroid differentiation. A, knockdown of fam210b in zebrafish embryos with two independent antisense morpholinos caused a defect in erythroid hemoglobinization. B, Tg(globin LCR:eGFP), fam210b morphant zebrafish exhibited a decrease in GFP+ erythroid cells, indicating defective erythropoiesis, n = 4; *, p value < 0.05 Student's t test. C, shRNA-mediated knockdown of Fam210b in primary fetal liver cells decreased FAM210B protein expression. D, Fam210b knockdown primary fetal liver cells had a decrease in hemoglobin content. E, hemoglobinization defect in murine Fam210b knockdown cells is complemented by human FAM210B. F, Fam210b knockdown in primary fetal liver cells caused a cell proliferation defect. G, Fam210b knockdown caused an enucleation defect, indicative of erythroid maturation arrest. n = 3, *, p value < 0.05 Student's t test.
To determine whether Fam210b was required for mammalian erythropoiesis, we knocked down Fam210b in primary fetal liver erythroid cells with shRNA (Fig. 3C). Fam210b knockdown caused a reduction in hemoglobinization (Fig. 3D), which was partially ameliorated by co-expression of human FAM210B (Fig. 3E), indicating the functional orthology between the human and mouse FAM210B genes. Knockdown of FAM210B caused defects both in cell proliferation (Fig. 3F) and in enucleation (Fig. 3G), indicating its additional function in erythroid maturation. The independent validations of the erythroid defect in Fam210b knockdown zebrafish and murine models indicate that the role of FAM210B in terminal erythroid maturation is highly conserved in vertebrates. In higher vertebrates, continuous FAM210B expression in the erythron is required for optimal erythroid cell proliferation, maturation, hemoglobinization, and enucleation.
Fam210b is localized to the mitochondrial inner membrane
To delineate the cellular mechanism by which FAM210B regulates erythropoiesis, we transfected fam210b-GFP into HEK293T cells and examined its subcellular localization. Fam210b contains three hydrophobic stretches of ∼21 amino acids, suggesting that FAM210B is an integral membrane protein. Confocal immunofluorescence microscopy revealed that FAM210B-GFP co-localized with COX4, a mitochondrial resident protein (Fig. 4A). Subcellular fractionation and Western blot analysis of primary fetal liver cells confirmed that FAM210B specifically co-localized with HSP60, a mitochondrial resident protein (Fig. 4B).
Figure 4.
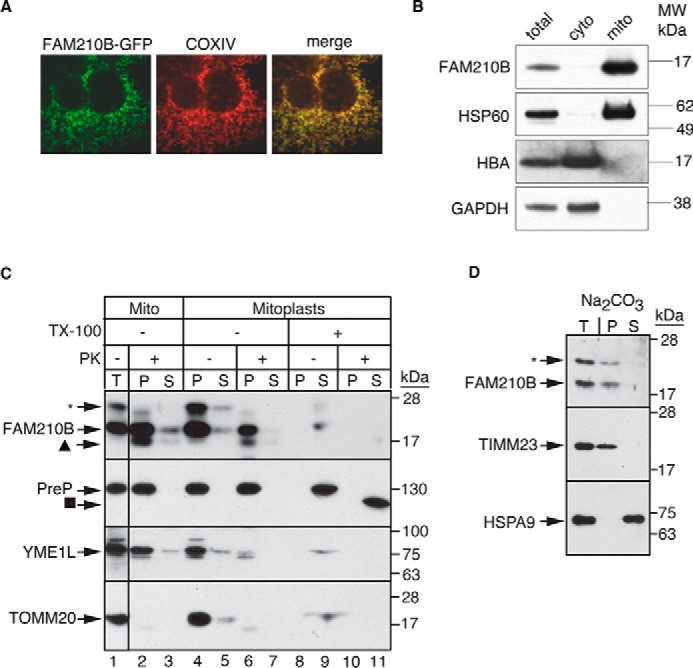
FAM210B localizes to the mitochondrial inner membrane. A, confocal fluorescence microscopy of exogenously expressed FAM210B-GFP (green) in HEK293T cells indicated that FAM210B co-localized with COXIV (red), a mitochondrial resident protein. The overlap is indicated in the merge panel (yellow). B, subcellular fractionation of primary fetal liver cells showed that FAM210B co-sedimented with HSP60 (mitochondrial) but not with HBA and GAPDH (cytoplasmic), indicating its mitochondrial localization. C, isolated mitochondria (Mito, T; lanes 1–3) purified from a MEL cell line stably expressing mouse Fam210b, was treated with proteinase K, which degraded outer membrane proteins (TOMM20) but not intermembrane space proteins (YME1L) or matrix (PreP) proteins (lane 2). Inner mitochondrial proteins, which were impervious to proteinase K treatment, (P) were separated from S proteins by centrifugation. FAM210B co-sedimented with the pellet fraction (lane 2). Mitoplasts (lanes 4–11) were generated by subjecting intact mitochondria to osmotic shock, exposing inner membrane and intermembrane space proteins to proteinase K digestion. Mitoplasts were centrifuged to separate the soluble proteins from the mitoplast pellet. Proteinase K treatment degraded a fraction of FAM210B (lane 6) and completely degraded YME1L but not PreP, suggesting that at least a fraction of FAM210B was situated in the mitochondrial inner membrane or intermembrane space. Triton X-100 (TX-100) treatment liberated FAM210B, PreP, and YME1L from the pellet into the soluble fraction (lane 9). These solubilized proteins were digested by proteinase K, demonstrating specificity of assay (lane 10). The asterisk marks a nonspecific band that was detected with the mouse FAM210B antibody; the triangle marks a likely FAM210B degradation product; and the square marks a core of PreP that is tightly folded and resistant to protease treatment. D, to determine whether FAM210B was an integral inner-membrane protein, isolated mitochondria (T) were analyzed by carbonate extraction. P and S fractions were analyzed by immunoblotting for FAM210B. The blot was probed for TIMM23 as an integral membrane protein control and mortalin as a soluble protein control. An asterisk marks nonspecific bands detected by the FAM210B antibody.
We carried out further localization studies in MEL cells stably expressing Fam210b to determine the sub-mitochondrial localization of FAM210B. To determine the localization of FAM210B in the mitochondrial outer membrane, purified mitochondria were treated with proteinase K, followed by centrifugation to separate the intact mitochondria (P) from soluble proteins (S) (Fig. 4C). Immunoblotting showed that TOMM20, an outer membrane protein with a large cytosolic domain, was degraded upon protease treatment, whereas FAM210B and control proteins YME1 (intermembrane space) and PreP (matrix) were intact. FAM210B is therefore not an outer mitochondrial membrane protein.
The mitochondria were then subjected to osmotic shock to determine whether FAM210B was in the intermembrane space or matrix (Fig. 4C). Mitoplasts, which contain the inner membrane and matrix components and intermembrane space/outer membrane components that stick to the mitoplasts, were recovered by centrifugation (P), whereas soluble proteins (S) remained in the supernatant. The mitoplasts were subsequently treated with a low dose of proteinase K to degrade proteins that were in the intermembrane space. FAM210B and PreP were resistant to protease digestion, whereas intermembrane space protein YME1 was degraded as expected. As a control for proteinase K activity, the mitoplasts were treated with Triton X-100, thereby releasing membrane-bound proteins into the supernatant and exposing digestion sites to enzymatic digestion. The addition of proteinase K to the Triton X-100 detergent-treated samples resulted in complete degradation, with the exception of PreP that has a tightly folded domain that is resistant to the low doses of proteinase K. These data suggested that FAM210B was bound to the mitochondrial inner membrane.
To rigorously confirm that this was the case, mitochondria were subjected to sodium carbonate extraction (46), followed by centrifugation, to separate membrane proteins (pellet, P) from soluble (S) proteins (Fig. 4D). FAM210B and membrane-bound TIMM23 were recovered in the pellet, whereas soluble matrix-localized HSPA9 (mortalin) was released in the supernatant. Thus, FAM210B is an integral membrane protein residing in the inner membrane. The collective data indicate that FAM210B is integrated in the inner membrane, and any soluble domains are likely facing the matrix, because protease addition during osmotic shock did not markedly degrade the full-length protein. A smaller fragment, which was sub-stoichiometric, was detected in the pellet (Fig. 4C, marked with a triangle), which may indicate that the N or C terminus is facing the intermembrane space and is digested by protease that may have leaked into the intermembrane space. The smaller fragment sediments with the pellet, suggesting that the segment of FAM210B that is exposed to the protease is tightly associated with the membrane.
FAM210B facilitates mitochondrial iron import during terminal erythropoiesis
To determine the biochemical function of FAM210B in heme synthesis, we generated Fam210b-deficient MEL cell lines by CRISPR/Cas9 genome editing (Fig. 5A). Metabolic 55Fe-labeling revealed that undifferentiated Fam210b−/− MEL cells had normal rates of heme synthesis and iron uptake compared with WT controls (Fig. 5, B and C). However, when MEL cells were chemically induced to differentiate, heme synthesis and iron uptake in Fam210b−/− MEL cells was significantly impaired (Fig. 5, D and E). The heme synthesis defect in Fam210b−/− MEL cells was due to a deficiency in mitochondrial iron acquisition (Fig. 5, F and G). As FAM210B is required for optimal mitochondrial iron uptake, we assayed the activity of mitochondrial aconitase (EC 4.2.1.3) in FAM210B−/− cells. Mitochondrial aconitase activity is sensitive to mitochondrial iron deficiency (47) caused by defects in mitochondrial iron transport, and this is reflected by the aconitase defect in Mfrn1−/− MEL cells (Fig. 5H) (23, 28). Fam210b−/− cells had a decrease in mitochondrial aconitase activity that is comparable with Mfrn1−/− cells, indicating that it plays a critical role in mitochondrial iron metabolism.
Figure 5.

Fam210b is required for heme synthesis by facilitating mitochondrial iron import. A, Fam210b−/− MEL cell lines generated by CRISPR/Cas9 expressed no detectable FAM210B protein. B, 55Fe metabolic labeling showed that undifferentiated Fam210b−/− MEL cell clones did not cause changes in heme synthesis (n = 3) or iron uptake (n = 3) (C). D, differentiated Fam210b knockout MEL cells exhibited defective heme synthesis (n = 6). E, iron uptake (n = 5). F, mitochondria from differentiated Fam210b knockout cells had decreased 55Fe-transferrin uptake (n = 5). G, ICP-MS demonstrated that mitochondria from Fam210b knockout cells had a decrease in total iron (n = 6). H, mitochondrial aconitase activity, which depends on [Fe-S] cluster synthesis, is decreased in Fam210b−/− cells indicating a defect in mitochondrial iron acquisition. Mfrn1−/− MEL clones served as controls for mitochondrial iron deficiency (n = 4). *, p < 0.05; §, p < 0.1 Student's t test. All data are normalized to WT controls.
The heme synthesis (Fig. 6A) and iron transport (Fig. 6B) defects in Fam210b−/− MEL cells were functionally complemented to the level of WT cells by hinokitiol (CAS 499-44-5; ChEBI: 10447), a natural product that binds iron and autonomously transports both Fe(II) and Fe(III) across lipid membranes to bypass defects in iron transport (48). Iron-hinokitiol functionally complemented the erythropoietic defect in fam210b morphant zebrafish embryos (Fig. 6C), demonstrating that the primary role of FAM210B is to facilitate mitochondrial delivery of iron for the synthesis of heme and [Fe-S] clusters in the developing erythroid cell. C2-deoxyhinokitiol, which does not bind or transport iron, did not chemically complement the anemia in Fam210b−/− cells (Fig. 6, A and B), indicating that the ability of the iron-hinokitiol to rescue heme synthesis was directly dependent on the ability of hinokitiol to chelate and transport iron across membranes (48). The specificity of the functional complementation is further demonstrated by the restoration of both 55Fe and 55Fe-heme only in cells defective for iron transport, such as the Dmt1 knockdown cell line (19, 20), but not in cells with a porphyrin transport defect, such as the Tmem14c CRISPR cell line (38).
Figure 6.

Fam210B increases the kinetics of mitochondrial iron import in differentiating MEL cells by functioning as an auxiliary factor. A, 55Fe metabolic labeling confirmed a decrease in heme synthesis in differentiating Fam210b (CRISPR1–3), Tmem14c, and Dmt1 deficient MEL cells (black bars). The addition of hinokitiol, a lipid-soluble iron carrier (white bars), restored heme synthesis in Fam210b- and Dmt1-deficient cells, but not Tmem14c-deficient cells. C2-deoxyhinokitiol, which does not chelate iron, did not complement heme synthesis in the Fam210b knockout cells (gray bars) (n = 6). B, 55Fe metabolic labeling confirmed an iron uptake defect in differentiating Fam210b-, Tmem14c-, and Dmt1-deficient cells (black bars). The iron uptake defect in Fam210b- and Dmt1-deficient cells was chemically complemented by hinokitiol (white bars). Hinokitiol did not rescue iron uptake in Tmem14c-deficient cells, which have a porphyrin synthesis defect. C2-deoxyhinokitiol did not rescue the iron deficiency in Fam210b−/− cells (gray bars) (n = 6). C, treatment of fam210b morphant zebrafish embryos (MO) with iron citrate and hinokitiol (MO + Hino/Fe) rescued their anemia (n = 8). D, FECH activity was measured in intact mitochondria isolated from WT and Fam210b−/− MEL cells, using 55Fe and DP as substrates. FECH activity in Fam210b−/− mitochondria was ∼1/3 that of WT controls. Both WT and Fam210b−/− mitochondria had very little measurable FECH activity when NMMP was used as a substrate (n = 4). E, FECH activity was measured in isolated mitochondria from WT and Fam210b−/− MEL cells that were treated with detergent and disrupted by sonication, allowing access of reaction substrates to FECH in the absence of intact membranes. FECH activity was decreased in Fam210b−/− mitochondria, but not to the extent of intact mitochondria (n = 3). F, FLAG-tagged MFRN1 and FAM210B co-localized with porin in Δmrs3/4 yeast, indicating correct mitochondrial localization. G, Mfrn1 expression complemented the growth defect of Δmrs3/4 yeast in low-iron media, whereas Fam210b expression did not. H, survival of Mfrn2−/− fibroblasts in DFO is significantly complemented by expression of Mfrn2-GFP, but less so by expression of Fam210b-HA (n = 3). I, expression of Fam210b-HA and Mfrn2-GFP in WT fibroblasts increase cell survival in the presence of DFO, with Fam210b overexpression possessing a protective effect over a larger dose range than Mfrn2-GFP. Mean ± S.E., n = 3, *, p < 0.05; §, p < 0.1 Student's t test. N.S., not significant.
HPLC analysis of protoporphyrinogen IX (PPIX) content in differentiating Fam210b−/− cells indicated that FAM210B was not required for synthesis of the tetrapyrrole ring in the mitochondria (Fig. S2A). This was further confirmed by the inability of deuteroporphyrin IX (DP), a PPIX analog that can chemically complement porphyrin synthesis defects (38), to rescue heme synthesis in Fam210b−/− cells (Fig. S2B).
To exclude the possibility that the heme defect in FAM210B deficiency was caused by a lesion in mitochondrial biogenesis or physiology, we stained control and Fam210b−/− cells with Mitotracker dye and analyzed mitochondrial staining by flow cytometry. The percentage of Mitotracker-positive cells and average Mitotracker dye staining did not differ between control and Fam210b−/− cells, indicating that Fam210b deficiency did not cause defects in cell survival (Fig. S2C), mitochondrial membrane potential, and mitochondrial mass (Fig. S2D).
FAM210b is not a direct mitochondrial iron carrier in vertebrate erythroblasts
To determine whether FAM210B functions as a mitochondrial iron importer or whether it played a role in regulation of FECH, we carried out an in vitro heme synthesis assay to compare the amount of iron transported across the mitochondrial membrane to synthesize deuteroheme (49). 55Fe and DP were added to isolated mitochondria from WT and Fam210b−/− MEL cells. DP was added in excess. As DP is lipid-soluble, any differences in the synthesis of 55Fe-DP are caused by alterations in FECH activity or iron transport across the mitochondrial membrane. In the absence of FAM210B, in vitro heme synthesis decreased to background levels comparable with cells treated with NMMP, a PPIX analog that inhibits FECH and cannot be metallated (Fig. 6D) (50). These data suggested that FAM210B is required for either iron import or heme synthesis. To determine which was the case, we performed a FECH activity assay as described (81). Using this method, cells are disrupted by sonication and treatment with Tween 20 detergent, allowing the substrates, mesoporphyrin IX, and zinc access to FECH without the need to cross membranes. FECH activity is measured by quantification of the product zinc-mesoporphyrin IX by HPLC. In the absence of a mitochondrial membrane, we observed that Fam210b−/− cells exhibited a decrease in FECH activity (Fig. 6E, left). This was not attributable to a decrease in FECH protein levels (Fig. 6E, right). Based on Fig. 6, D and E, FAM210B is required for optimal FECH activity in erythroid cells. However, we observed that the decrease in FECH activity was far less significant when membranes were disrupted (Fig. 6E) compared with when the mitochondrial membrane was intact (Fig. 6D). We concluded that FAM210B is required both for full activation of FECH but also plays a role in facilitating iron transport across the mitochondrial membrane for heme synthesis.
To determine whether FAM210B is a bona fide iron transporter, we expressed epitope-tagged Fam210 or Mfrn in the Δmrs3Δmrs4 yeast strain (denoted as Δmrs3/4), deficient in the Mfrn orthologs (23), or in Mfrn2−/− primary murine fibroblasts,7 which are deficient in mitochondrial iron transporter Slc25a28 (22). Mouse FAM210B, with the N-terminal mitochondrial targeting propiece from Mrs3, properly targeted to the mitochondrial compartment in yeast (Fig. 6F and Fig. S3). Δmrs3/4 yeast exhibited iron auxotrophy when grown on iron-deficient media, which was genetically complemented by vertebrate Mfrn1 but not Fam210b (Fig. 6G), suggesting that FAM210B does not directly function as a iron-solute carrier.
We next treated mouse embryonic fibroblasts from Mfrn2−/− with varying concentrations of desferrioxamine (DFO), an iron chelator, for 48 h. As cells require iron for basic cellular processes such as respiration, defects in cellular iron uptake and utilization would decrease cellular viability with DFO treatment. Conversely, increasing the expression of iron transporters would increase the efficiency of iron uptake and utilization in cells, resulting in increased survival. As expected, the expression of Mfrn2-GFP in Mfrn2−/− fibroblasts significantly improved cellular viability in DFO-treated cells (Fig. 6H). In contrast, Fam210b co-expression did not significantly enhance fibroblast viability above the basal control, demonstrating that Fam210b could not genetically complement Mfrn2 deficiency (Fig. 6H). However, FAM210B significantly increases the survival of WT fibroblasts in a wide range of DFO concentrations beyond that of Mfrn2-GFP expression (Fig. 6I). As FAM210B augments iron import in WT but not in Mfrn2−/− MEFs, we propose that FAM210B increases the efficiency of mitochondrial iron import by an MFRN-dependent mechanism.
FAM210b is in an iron-import oligomeric complex to enhance heme synthesis
To further characterize the biochemical function of FAM210B in an oligomeric complex, we examined MFRN1 levels in Fam210b−/− MEL cells to determine whether Fam210b plays a role in Mfrn1 stability analogous to ABCB10, as MFRN1 is the predominant erythroid mitochondrial iron transporter (22, 23, 25). MFRN1 protein levels were similar in control and Fam210b−/− mitochondria, indicating that FAM210B does not play a role in the maintenance of MFRN1 stability (Fig. S2E). Immunoprecipitation of FAM210B that was ectopically co-expressed with MFRN1 in HEK293T cells showed that FAM210B and MFRN1 do not interact (Fig. 7A). In contrast, we co-immunoprecipitated ectopically expressed FAM210B with PPOX and FECH (Fig. 7B), which are terminal enzymes in the heme synthesis pathway. As the ectopically expressed proteins are expressed at levels far higher than endogenous FAM210B, PPOX, and FECH or potential bridging proteins that can mediate indirect interactions, it is likely that the physical interactions between FAM210B, PPOX, and FECH are direct. Notably, PPOX and FECH are in the same complex as MFRN1 (Chen et al. (26) and Medlock et al. (79)). The specificity of these protein–protein interactions is ensured by the lack of CPOX co-immunoprecipitation with FAM210B (Fig. 7B). To further determine whether FAM210B can interact with FECH at endogenous levels, we generated a MEL cell line that stably expressed FAM210B–FLAG. We immunoprecipitated FAM210B with FLAG antibodies in differentiating cells. Western blot analysis of the immunoprecipitated protein showed that FAM210B–FLAG interacted with endogenous FECH in vivo (Fig. 7C). FAM210B does not homooligomerize, as would be expected if FAM210B was a membrane-embedded solute carrier (Fig. 7C). Collectively, these results are consistent with a role for FAM210B as an auxiliary factor in an iron transport oligomeric complex that includes the terminal heme biosynthesis enzymes in the mitochondria. The intact oligomeric complex is essential for maintaining the high rate of mitochondrial iron import that is required to fulfill the demands for heme synthesis in maturing red cells.
Figure 7.

FAM210B directly interacts with terminal heme synthesis enzymes but does not with MFRN1 (Slc25a37). A, immunoprecipitation of co-expressed FAM210B-HA and FLAG-MFRN1 demonstrates FAM210B does not directly interact with MFRN1. B, FAM210B-HA interacts with the terminal enzymes of the heme synthesis pathway, PPOX and FECH, but not CPOX. MFRN1, CPOX, PPOX, and FECH are all localized to the mitochondrial inner membrane. C, immunoprecipitation of FAM-210B FLAG stably expressed in differentiating MEL cells shows that ectopically expressed FAM210B interacts with endogenous FECH. D, FAM210B-HA and FAM210B-FLAG do not interact, demonstrating that FAM210B does not homooligomerize. E, model of the role of FAM210B as an adaptor protein in an oligomeric complex with terminal heme enzymes, required for terminal erythropoiesis and mitochondrial iron importation.
Discussion
Our study demonstrates that FAM210B, a protein of previously unknown function, is required for mitochondrial iron import to support sustained synthesis of heme, iron–sulfur cluster, and hemoglobin during erythroid maturation. In addition, FAM210B regulates FECH enzyme activity. Our results are consistent with and extend the findings of a recent study identifying Fam210b as a GATA1 transcriptional target that is required for erythroid differentiation (29). Mitochondrial mass, membrane potential, and porphyrin levels are normal in Fam210b−/− cells, precluding a role for FAM210B in housekeeping mitochondrial homeostasis or tetrapyrrole synthesis, respectively. Although heme synthesis and iron import was decreased in Fam210b-deficient cells, we did not observe an accumulation in porphyrin, similar to the phenotype in Mfrn1 deficiency (51, 52). This is attributable to impaired [Fe-S] cluster biosynthesis in Fam210b-deficient cells (Fig. 5H), which elevates IRP1 RNA binding and suppresses Alas2 mRNA translation and porphyrin accumulation (52, 53).
FAM210B deficiency can be functionally complemented by the addition of hinokitiol, which acts as a small-molecule mimetic of iron transporters (48). However, Fam210b overexpression cannot genetically complement the iron transport defect in Δmrs3/4 yeast or Mfrn2−/− fibroblasts. We therefore conclude that the primary function of FAM210B is to facilitate mitochondrial iron import in differentiating erythroid cells by functioning as an auxiliary factor in promoting formation of an oligomeric iron–transport complex with terminal heme synthesis enzymes, thereby increasing the kinetics of mitochondrial iron import to keep pace with its cellular demands for heme. Our studies on FAM210B are the first to describe an EPO-induced mitochondrial chaperone that is tissue- and differentiation stage-specific, revealing a novel mechanism by which cells regulate intracellular iron metabolism. We previously identified a mitochondrial chaperone, CLPX, which facilitates the delivery of the vitamin B6 cofactor required for the catalytic activity of ALAS, the enzyme that catalyzes the committed step of protoporphyrin synthesis (54). The oligomerization of MFRN1, ABCB10, and FECH integrates the efficient import of iron with the metallation of PPIX to form heme during erythroid maturation (26). We further note that the use of small molecules that autonomously perform protein-like functions as a distinct type of biological probe, as demonstrated herein with the transmembrane iron transporter hinokitiol, may be more widely applicable and have complementary strengths to other experimental approaches (48, 55).
Fam210b is transcriptionally activated by GATA-1 (29, 56), consistent with its expression in erythropoietic tissues. Iron chelation by DFO or iron supplementation by iron citrate did not cause changes in its steady-state protein expression, indicating that FAM210B is not regulated by cellular iron status (Fig. S1).
EPO regulates FAM210B expression both at the levels of mRNA transcription and protein stability, suggesting that FAM210B is an essential component of EPO's function and signaling in erythropoiesis. The up-regulation of Fam210b by EPO in EpoR-expressing 32D pro-B cells suggests that Fam210b is direct target of EPO. Although FAM210B is localized in the mitochondria, it is required for cellular processes that take place during terminal erythroid differentiation, such as proliferation, and finally enucleation. These data suggest a link between nuclear processes in terminal erythroid differentiation and mitochondrial iron metabolism and also demonstrate a novel mechanism by which EPO regulates the biology of terminal erythropoiesis via its regulation of cellular iron uptake.
Although published GWAS studies did not reveal any extant association between FAM210B mutations and hematologic traits (57–61), the profoundly anemic phenotype of the fam210b morphant embryos and the heme defects in primary murine fetal liver and in MEL cells predict that mutations in FAM210B may lead to hematologic and iron metabolism defects in humans. As MFRN1 genetic or splicing defects have been shown to be modifiers of porphyria (62) or sideroblastic anemia (63), we predict that detailed genetic studies will reveal the function of FAM210B mutations in patients suffering from anemias and porphyrias of unknown etiology. As Fam210b−/− cells only exhibited heme and iron metabolism defects in differentiating erythroid cells, it is expected that FAM210B mutations will exhibit a phenotype specifically in the terminally differentiating erythroid cell population.
To adapt to their role as oxygen carriers in vertebrates, erythroid cells have evolved several mechanisms to efficiently transport and utilize iron within the cell for adequate heme synthesis. This is critical as mitochondrial iron import is tightly coupled with heme synthesis, and cells cannot preload iron in the mitochondria (64). In addition to the housekeeping mitochondrial iron importer MFRN2, erythroid cells express MFRN1 during their terminal maturation (22, 23). The tissue-restricted regulation at the transcriptional level for MFRN1 at “erythroid super-enhancer” by GATA-factors (65, 66), is reminiscent of the transcriptional regulation of ALAS, the first and rate-limiting enzyme in the heme biosynthetic pathway (67). Like the MFRN genes, these ALAS genes are encoded by two separate genes, encoding distinct ALAS isoenzymes. The ubiquitous ALAS1 is regulated by heme via the peroxisome proliferator-activated 1α (PGC-1α) (68). The erythroid-enriched ALAS2 and MFRN1 genes are not regulated by heme. The post-translational regulation of ALAS2 is mediated by the intracellular levels of iron and iron-responsive protein 1 (IRP1) binding to its cognate iron-responsive element (IRE) in the 5′-untranslated region (UTR) of the ALAS2 mRNA (69); however, no such regulation by iron on IRE have been reported and ascribed to the MFRN genes.
During terminal erythropoiesis, MFRN1 protein stability is also enhanced by oligomerization with ABCB10 (25, 26). MFRN1 and ABCB10 are in a physical complex with FECH, which kinetically favors the coupling of iron transport and heme synthesis. The addition of FAM210B, which forms an oligomer with PPOX and FECH to enhance mitochondrial iron import and heme synthesis, fits into this experimental model where FAM210B can nucleate or stabilize the formation of a complex between PPOX and FECH. In this manner, it can facilitate the transfer of PPIX to FECH and promote iron import coupled with heme synthesis (Fig. 7D). While PPOX and FECH physically interact, they do not form a tight complex. This supports the idea that bridging molecules, such as FAM210B, may mediate the strength of the interaction, hence modulating the rate of iron import and heme synthesis (70).
FAM210B plays a second role in heme synthesis by activating FECH enzymatic activity (Fig. 6, D and E). It is well-established that red cells increase the expression of ferrochelatase protein during erythropoiesis. This occurs either by increase in FECH mRNA transcription (71) or stabilization of FECH protein by binding of iron–sulfur clusters (72). In contrast, FAM210B deficiency does not alter FECH protein levels (Fig. 6E), suggesting that its effects on FECH activity may be mediated by the requirement for FAM210B for efficient iron delivery to FECH, and/or by playing a role in allosteric activation of the FECH enzyme. These data add to our understanding of the mechanisms by which Epo signaling regulates heme synthesis by post-translational regulation of FECH (49) and suggest mechanisms by which the activity of heme synthetic enzymes, in this case, FECH, may be rapidly modulated in response to metabolic requirements.
As Fam210b is also expressed in nondifferentiating erythroid cells and neural and hepatic tissues, it may also play a role in these tissues that is independent of heme synthesis and iron metabolism. Because FAM210B is a mitochondrial protein, detailed sequencing studies may reveal mutations in FAM210B that play a role in modifying the severity of mitochondriopathy in these tissues. Our identification of Fam210b as a novel mitochondrial protein that is required for optimal mitochondrial iron uptake during terminal erythropoiesis thus provides a novel genetic tool for further studies on vertebrate erythropoiesis and mitochondrial biology.
Materials and methods
Cell lines
MEL DS19 subclone cells were obtained from Arthur Skoultchi (Albert Einstein College of Medicine, New York). Human HEK293T cells were used for transient transfections.
GenBankTM accession numbers
Zebrafish fam210b (c20orf108) is GenBankTM accession number NM_001033751; mouse Fam210b is GenBankTM accession number NM_025912.
Knockdown of Dmt1 and Fam210b by shRNA hairpins in mouse cells
Dmt1 (GenBankTM accession number NM_008732.2) stable knockdown MEL clones were obtained by stable transfection of an shRNA hairpin, TRCN0000079535. Fam210b (GenBankTM accession number NM_025912) stable knockdown MEL clones were obtained by transfection of shRNA hairpins: shFam210b-1 and TRCN0000103732; shFam210b-2 TRCN0000103734. shRNA hairpins were obtained from Sigma. MEL clones were electroporated with shRNA plasmids by electroporation, and stable clones were obtained by limiting dilutions and selection with G418 (Dmt1).
CRISPR design and cloning
CRISPR guide sequences were designed to direct cleavages at genetic loci to generate chromosomal deletions (73, 74). CRISPR guide sequences were designed to have a unique 12-bp seed sequence 5′-NNNNNNNNNNNNNGG-3′ in the mouse genome to minimize off-target cleavages. Exons 1 and 3 of the Fam210b locus were targeted. The exon 1 tgt1 targeting sequence was as follows: 5′-GCTCGCGGCGCGCCATGGCC-3′; exon 1 tgt2 targeting sequence was 5′-TCGGGCGTCCGGAACCGGAT-3′, and the exon 3 targeting sequence was 5′-TACCGGGAGCAAACAGCTTATGGA-3′. Exons 2 and 4 of the Mfrn1 locus were targeted. The exon 2 targeting sequence was as follows: 5′-GATGCTTGTATACCGGGCTT-3′; the exon 4 targeting sequence was 5′-GAAGAACTCATAAACGGACC-3′. CRISPR guides were cloned into pX330 plasmid (Addgene) with BbsI ligation as described previously (75).
CRISPR/Cas9 genome editing in MEL cells
CRISPR/Cas9 constructs were delivered to MEL DS19 cells by electroporation. CRISPR/Cas9 constructs were co-electroporated with pEF1α at a 9:1 ratio. Two days later, cells were resuspended in puromycin media (5 μg/ml). Cells were plated by limiting dilution at 0.3 cells per well in 96-well plates and cultured at 37 °C. Clones were screened by gDNA isolation (QuickExtract, Epicenter), and PCR for the deletion allele (CRISPR 1), or PCR for the WT allele (CRISPR 2 and CRISPR 3). Clones were validated by Western blot analysis for FAM210B protein; Mfrn1 gene expression was assayed by qRT-PCR (48).
The PCR primers to genotype Fam210b CRISPR 1 were as follows: mFam210b_F 5′-CTATAGAGCCCCGCCTATCC-3′ and mFam210b_R 5′-TCGGTCAGGTGCAATGTAAA-3′. The PCR primers used to genotype Fam210b CRISPR 2 and 3 were as follows: E3F 5′-GCATAGACATGTCTGCAATCC-3′ and E3R 5′-AGCCACCUUGCACACAGCCC-3′.
Iron radiolabeling and radio iron–heme measurements
55FeCl3 (specific activity: 1.28 Ci/mmol) (PerkinElmer Life Sciences) were loaded onto transferrin as described previously (80). Metabolic labeling with 55Fe-transferrin and quantitation of the incorporation of 55Fe into heme were carried out as described previously (38).
ICP analysis of mitochondrial iron content
Mitochondrial iron was measured as described previously (76). Isolated mitochondria were treated with nitric acid, and sample iron content was determined by using an ICP optical emission spectrometer (PerkinElmer Life Sciences). The results were normalized to mitochondrial protein content.
HPLC analysis of heme and porphyrins
HPLC analysis was carried out with MEL cells that were differentiated for 72 h with 1.5% DMSO. A cell pellet spun down from a 30–50-ml culture was mixed with water to about 200 μl in a microcentrifuge tube and sonicated for 12 cycles with 5-s intervals at 50% duty (about 2.5 s on, 2.5 s off) using a microtip. A 50-μl aliquot was mixed vigorously with 200 μl of an extraction mixture of ethyl acetate (4 volumes) and glacial acetic acid (1 volume). The phases were separated by microcentrifugation for 1 min at maximum speed. The upper organic layer was immediately analyzed simultaneously for protoporphyrin IX and heme in the HPLC (38, 51).
Silencing of Fam210b in mouse primary fetal liver cells
Silencing of Fam210b and hemoglobin quantification in mouse primary fetal liver cells was carried out as described (77). The retroviral plasmids expressing shRNAs for Fam210b were transfected in a packaging HEK293T cell line. The collected retroviral supernatants were added to erythroid precursor cells purified from E14.5 mouse fetal liver (C57BL/6J). The retrovirally-transduced cells were sorted for GFP expression using a FACSAria machine (BD Biosciences), followed by ex vivo erythroid terminal development in Epo-only medium for 48 h. After culture, cells were collected for analysis on enucleation and hemoglobin content. The hemoglobin content was quantified with Drabkin's reagent used human hemoglobin as standards.
Fetal liver proliferation and enucleation assays
To measure proliferation rates, GFP-positive cells were plated in 24-well plates with a density at 100,000 cells/per ml, cultured in Epo-only erythroid differentiation medium. The number of cells was counted at the 24 and 48 h using a hemocytometer. In each time point, the cell numbers were the average of three biological samples, which were the average of two technical duplicates. To measure the rate of enucleations, after 48 h of culture, one million cells were spun down and resuspended with 100 μl of FACS buffer (PBS + 2% fetal bovine serum + 100 μm EDTA). Anti-Ter119 antibodies conjugated with APC (eBioscience) and Hoechst 33342 (Sigma) were added to stain the cells for 10 min at room temperature following manufacturer protocols. After staining, cells were washed with cold PBS twice and resuspended with the FACS buffer containing propidium iodide (Sigma) to distinguish between live and dead cells and were further analyzed on a FACS Fortessa machine. The details of protocols were previously described (78).
Functional complementation assays with chemicals in cultured cells
MEL cells were differentiated chemically with 2% DMSO for 72 h. For functional complementation assays with hinokitiol, cells were concurrently treated with 10 μm iron citrate and hinokitiol (1 μm) or C2dOHino (1 μm) (48). Chemical complementation with DP was carried out by treating cells with 5 μm DP (Frontier Scientific) as described (38).
Sub-mitochondrial localization studies
Crude mitochondria were isolated from MEL or primary fetal liver cells using a mitochondrial isolation kit (Pierce).
For mitochondrial sublocalization studies, MEL cells stably expressing mouse Fam210b were harvested at confluency and lysed in homogenization buffer (20 mm HEPES, pH 7.4, 220 mm mannitol, 70 mm sucrose) supplemented with 2 mg/ml BSA and 0.5 mm PMSF using a Teflon–Dounce homogenizer to release mitochondria. The lysates were centrifuged at 100 × g at 4 °C for 5 min. The post-nuclear supernatant was centrifuged at 10,000 × g for 10 min to obtain mitochondria pellets. The pellets were subsequently washed with homogenization buffer without BSA and PMSF, and mitochondria were collected by centrifugation. The protein concentration was measured using BCA assay according to the vendor's protocols (ThermoFisher Scientific).
For mitochondrial fractionation, mitochondria (equivalent of 50 μg of mitochondrial protein per reaction) were suspended in homogenization buffer, 20 mm HEPES (for osmotic shock) or 20 mm HEPES with 1% Triton X-100 in the presence or absence of 5 μg/ml proteinase K for 30 min at 4 °C. Proteinase K was inactivated with the addition of 1 mm PMSF. Samples were centrifuged at 10,000 × g at 4 °C to obtain the pellet that contained mitoplasts (the matrix and inner membrane). The supernatant was collected and precipitated with trichloroacetic acid (TCA, final concentration of 15% TCA) to obtain soluble proteins. The proteins (50 μg per lane) were separated by SDS-PAGE followed by immunoblot analysis.
Mitochondria (equivalent of 50 μg of mitochondrial protein per reaction) were suspended in homogenization buffer or 0.1 m sodium carbonate, pH 11.5, for 30 min at 4 °C. Then, the samples were centrifuged at 28,000 × g for 30 min at 4 °C to obtain the pellet of precipitated membrane proteins. The supernatant with soluble proteins was TCA-precipitated. The proteins (50 μg per lane) were separated by SDS-PAGE followed by immunoblot analysis.
Western blot analysis
The following antibodies were used in this study: TIMM23 (BD Biosciences catalog no. 611222); TOMM20 (Santa Cruz Biotechnology clone FL-145); Mortalin (Proteintech catalog no. 1488-14-AP).PreP and Yme1L polyclonal antibodies were raised against recombinant proteins (Pacific Immunology). Immunoblotting of FAM210B was performed using a custom anti-mouse FAM210B polyclonal antibody generated against three peptides (C-LSHPVPDARLLRTARGDC, C-TGTEKKLSRTQQLKKVC, and C-KLGFKESLVQSKMAC), and immunoaffinity-purified against these antigenic peptides (Genemed Synthesis, Inc., San Antonio, TX). Anti-FECH polyclonal (C-20), HA (Y-11), hemoglobin a (H-80), and HSP60 (K-19) antibodies were obtained from Santa Cruz Biotechnology (Athens, GA). Anti-TFRC (H68.4) was obtained from Invitrogen. Anti-GAPDH was obtained from Pierce (GA1R). Anti-FLAG M2-HRP was obtained from Sigma (catalog no. A8592). HSP90 antibody was obtained from Cell Signaling (C45G5). For Fig. 7C, we used anti-FECH polyclonal antibodies from Kerafast (EGA191).
qRT-PCR
qRT-PCR probes were obtained from Invitrogen. Murine Fam210b probe is Mm00508881_m1; Hprt probe is Mm01545399_m1. Zebrafish fam210b probe is Dr03426307_m1; hprt probe is Dr03138604_m1. The mouse tissue cDNA array was purchased from Clontech.
Chemical hinokitiol and functional complementation in zebrafish
24-hpf morphant embryos in the transgenic Tg(globinLCR:eGFP) line were dechorionated with Pronase as described (54) and incubated in the presence of 1 μm hinokitiol + 10 μm iron citrate for another 48 h (48). For specificity, the control, inactive C2 deoxyhinokitiol was 1 μm final concentration with iron citrate. Vehicle-treated embryos were exposed to 0.01 mm DMSO. Control or morphant embryos at roughly 72 hpf were mechanically homogenized as described previously for flow cytometry (45, 54).
Genetic complementation assays
Genetic complementation of the Δmrs3/4 yeast strain with chimeric vertebrate Fam210b and Mfrn1, containing the first N-terminal 23 amino acid residues from yeast Mrs3 subcloned in the pRS426-ADH vector, was performed as described previously (23). Western blot analysis of the total, mitochondrial, and cytosolic fraction was described previously with anti-FLAG and anti-porin antibodies.
Control and Mfrn2−/− mouse embryonic fibroblasts (MEF) were isolated from the skin of 3-month-old mice and immortalized by forcing through a senescence crisis. Mfrn2-GFP or Fam210b-HA was transiently expressed in these MEF cells. Transfected MEF cells were treated with DFO (10–50 μm). Cellular viability was then assayed using a cell titer blue assay, which measures conversion of resazurin to resorufin on a 96-well format (Promega).
Immunoprecipitation of FAM210B and its interacting proteins
CPOX-FLAG, PPOX-FLAG, FECH-FLAG, and FAM210B-HA were cloned into the pEF1 vector and expressed under the control of the EF1 promoter. pEF1/FAM210B-HA was co-transfected with pEF1 empty vector, pEF1/CPOX-FLAG, pEF1/PPOX-FLAG, or pEF1/FECH-FLAG into 10-cm3 dishes of HEK293T cells using a Lipofectamine 2000 reagent-based method (Invitrogen). Cells were harvested 48 h post-transfection and lysed in a buffer containing 1% Nonidet P-40, 300 mm NaCl, 50 mm Tris, pH 8.0, and protease inhibitor mixture (Pierce). Cell lysates were precleared with mouse IgG-agarose (Sigma) for 60 min at 4 °C. Precleared lysates were then incubated with anti-FLAG or anti-HA–agarose beads (Sigma) for 2 h at 4 °C. Beads were washed three times for 15 min in lysis buffer. Immunoprecipitated proteins were subsequently eluted with Laemmli buffer and resolved by SDS-PAGE.
To determine whether FAM210B interacted with endogenous FECH, we electroporated pEF1/FAM210B-FLAG into MEL cells and selected FAM210B-FLAG expressing single cell clones by limiting dilution. pEF1/FAM210B-HA and pEF1 empty vector expressing cell lines were differentiated with 1.5% DMSO for 72 h. For each immunoprecipitation experiment, we lysed 100 million differentiated cells with 1 ml of lysis buffer (77). 2 mg of protein was used as the starting material for each immunoprecipitation sample. Samples were precleared with mouse IgG beads and immunoprecipitated with anti-FLAG M2–agarose for 4 h at 4 °C. Beads were washed three times for 15 min at 4 °C. Proteins were eluted with Laemmli buffer, resolved by SDS-PAGE, and analyzed by Western blotting.
Statistics
Statistical analysis was carried out using two-tailed paired or unpaired Student's t test. Significance was set at p < 0.05. All data points were plotted as scatter plots, representing all replicates of each experiment.
Study approval
All zebrafish experiments were performed in accordance with the Institutional Animal Care and Use Committee (IACUC) regulations of Brigham and Women's Hospital. WT AB zebrafish were used for all zebrafish experiments. All mouse experiments were performed in accordance with IACUC regulations at the Massachusetts Institute of Technology and University of Utah.
Author contributions
Y. Y. Y., J. S., C. C., D. E. B., C. M. K., D. M. W., H. F. L., and B. H. P. conceptualization; Y. Y. Y. and B. H. P. resources; Y. Y. Y., J. S., C. C., J. T. C., A. S. G., R. S., L. L., M. D. K., P. D. K., M. J. K., J. P., D. E. B., C. M. K., J. D. P., J. K., D. M. W., and B. H. P. data curation; Y. Y. Y., J. S., C. C., J. T. C., A. S. G., R. S., L. L., M. D. K., P. D. K., M. J. K., M. D. B., D. E. B., S. H. O., C. M. K., J. D. P., J. K., D. M. W., H. F. L., and B. H. P. formal analysis; Y. Y. Y., C. C., J. P., M. D. B., D. E. B., S. H. O., C. M. K., J. D. P., J. K., D. M. W., H. F. L., and B. H. P. supervision; Y. Y. Y., C. C., L. Z., J. P., M. D. B., D. E. B., S. H. O., C. M. K., J. D. P., J. K., D. M. W., H. F. L., and B. H. P. funding acquisition; Y. Y. Y., J. S., C. C., C. M. K., and D. M. W. validation; Y. Y. Y., J. S., C. C., J. T. C., A. S. G., R. S., L. L., X. Z., M. D. B., C. M. K., J. D. P., J. K., D. M. W., H. F. L., and B. H. P. investigation; Y. Y. Y., J. S., C. C., A. S. G., R. S., L. L., X. Z., J. A., H. L., L. Z., D. E. B., S. H. O., C. M. K., D. M. W., and B. H. P. methodology; Y. Y. Y., J. S., P. D. K., C. M. K., D. M. W., and B. H. P. writing-original draft; Y. Y. Y., M. D. B., D. E. B., C. M. K., J. D. P., J. K., D. M. W., H. F. L., and B. H. P. project administration; Y. Y. Y., J. S., C. C., J. T. C., A. S. G., R. S., L. L., M. D. K., M. J. K., J. A., L. Z., J. P., M. D. B., D. E. B., S. H. O., C. M. K., J. D. P., J. K., D. M. W., H. F. L., and B. H. P. writing-review and editing.
Supplementary Material
Acknowledgments
We thank Iman Schultz for assistance on bioinformatics analysis, Johannes Wittig for assistance with model figure preparation, and Nicholas Huston for assistance with initial morpholino injections. We thank Mahnaz Paktinat for training on the use of the BD FACSCanto cell sorter. We also thank Bingbing Yuan for help with processing and analyzing the microarray data. The DS19 MEL clones were obtained from Arthur Skoultchi (Albert Einstein College of Medicine). We thank Eva Buys and her crew for the zebrafish animal husbandry. We also thank Harry Dailey and Amy Medlock for their critical feedback on the manuscript and Stashauna Carter for technical assistance. Fluorescence confocal microscopy was performed at the Harvard Digestive Disease Center Imaging Facility (Boston Children's Hospital, supported by National Institutes of Health Grant P30 DK34854) with assistance from Ramiro Massol. HPLC analyses of porphyrins were performed at the University of Utah Center for Iron and Heme Disorders supported by National Institutes of Health Grant U54 DK110858.
This work was supported by March of Dimes Foundation Grant 6-FY09-289 (to B. H. P.); the Cooley's Anemia Foundation (to C. C. and Y. Y. Y.); the National Science Foundation (to A. S. G.); National Institutes of Health Grants F32 DK098866, R03 DK118307, and K01 DK106156 (to Y. Y. Y.), K08 DK093705 (to D. E. B.), K99CA20146 (to J. A.), R01 DK020503 and U54 DK083909 (to J. D. P.), R01 GM073981 and R01 GM61721 (to C. M. K.), R01 DK052830 and DK030534 (to D. M. W.), R01 HL116364 (to J. P.), R01 GM118185 (to M. D. B.), R01 DK070838 (to B. H. P.), P01 HL032262 (to H. F. L., S. H. O., L. I. Z., and B. H. P.); and the University of Delaware Biological Sciences Undergraduate Summer Scholar award (to X. Z.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
The microarray data for this study were deposited to the NCBI Gene Expression Omnibus (GEO) database under GEO accession number GSE113657.
This article contains Figs. S1–S3.
D. M. Ward and J. Kaplan, unpublished data.
- EPO
- erythropoietin
- ICM
- intermediate cell mass
- PMSF
- phenylmethylsulfonyl fluoride
- LCR
- locus control region
- PPIX
- protoporphyrinogen IX
- DP
- deuteroporphyrin IX
- qRT
- quantitative RT
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- P
- pellet
- S
- soluble
- MEL
- murine erythroleukemia
- DFO
- desferrioxamine
- ALAS
- 5-aminolevulinate synthase
- IRE
- iron-responsive element
- MEF
- mouse embryonic fibroblast
- BSA
- bovine serum albumin
- ICP
- inductively coupled plasma
- hpf
- hours post-fertilization.
References
- 1. Richmond T. D., Chohan M., and Barber D. L. (2005) Turning cells red: signal transduction mediated by erythropoietin. Trends Cell Biol. 15, 146–155 10.1016/j.tcb.2005.01.007 [DOI] [PubMed] [Google Scholar]
- 2. Gammella E., Diaz V., Recalcati S., Buratti P., Samaja M., Dey S., Noguchi C. T., Gassmann M., and Cairo G. (2015) Erythropoietin's inhibiting impact on hepcidin expression occurs indirectly. Am. J. Physiol. Regul. Integr. Comp. Physiol. 308, R330–R335 10.1152/ajpregu.00410.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Nai A., Rubio A., Campanella A., Gourbeyre O., Artuso I., Bordini J., Gineste A., Latour C., Besson-Fournier C., Lin H. Y., Coppin H., Roth M.-P., Camaschella C., Silvestri L., Meynard D., Franke K., et al. (2016) Limiting hepatic Bmp-Smad signaling by matriptase-2 is required for erythropoietin-mediated hepcidin suppression in mice. Blood 127, 99–101 10.1182/blood-2015-11-681494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Feng D., and Lazar M. A. (2012) Clocks, metabolism, and the epigenome. Mol. Cell 47, 158–167 10.1016/j.molcel.2012.06.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Girvan H. M., and Munro A. W. (2013) Heme sensor proteins. J. Biol. Chem. 288, 13194–13203 10.1074/jbc.R112.422642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Maio N., and Rouault T. A. (2015) Iron–sulfur cluster biogenesis in mammalian cells: new insights into the molecular mechanisms of cluster delivery. Biochim. Biophys. Acta 1853, 1493–512 10.1016/j.bbamcr.2014.09.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Martínková M., Kitanishi K., and Shimizu T. (2013) Heme-based globin-coupled oxygen sensors: linking oxygen binding to functional regulation of diguanylate cyclase, histidine kinase, and methyl-accepting chemotaxis. J. Biol. Chem. 288, 27702–27711 10.1074/jbc.R113.473249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Matak P., Matak A., Moustafa S., Aryal D. K., Benner E. J., Wetsel W., and Andrews N. C. (2016) Disrupted iron homeostasis causes dopaminergic neurodegeneration in mice. Proc. Natl. Acad. Sci. U.S.A. 113, 3428–3435 10.1073/pnas.1519473113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ramsey A. J., Hillas P. J., and Fitzpatrick P. F. (1996) Characterization of the active site iron in tyrosine hydroxylase. J. Biol. Chem. 271, 24395–24400 10.1074/jbc.271.40.24395 [DOI] [PubMed] [Google Scholar]
- 10. Andrews N. C. (2008) Forging a field: the golden age of iron biology. Blood 112, 219–230 10.1182/blood-2007-12-077388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chen C., and Paw B. H. (2012) Cellular and mitochondrial iron homeostasis in vertebrates. Biochim. Biophys. Acta 1823, 1459–1467 10.1016/j.bbamcr.2012.01.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Philpott C. C., Leidgens S., and Frey A. G. (2012) Metabolic remodeling in iron-deficient fungi. Biochim. Biophys. Acta 1823, 1509–1520 10.1016/j.bbamcr.2012.01.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hentze M. W., Muckenthaler M. U., and Andrews N. C. (2004) Balancing acts. Cell 117, 285–297 10.1016/S0092-8674(04)00343-5 [DOI] [PubMed] [Google Scholar]
- 14. Levy J. E., Jin O., Fujiwara Y., Kuo F., and Andrews N. C. (1999) Transferrin receptor is necessary for development of erythrocytes and the nervous system. Nat. Genet. 21, 396–399 10.1038/7727 [DOI] [PubMed] [Google Scholar]
- 15. Chen C., Garcia-Santos D., Ishikawa Y., Seguin A., Li L., Fegan K. H., Hildick-Smith G. J., Shah D. I., Cooney J. D., Chen W., King M. J., Yien Y. Y., Schultz I. J., Anderson H., Dalton A. J., et al. (2013) Snx3 regulates recycling of the transferrin receptor and iron assimilation. Cell Metab. 17, 343–352 10.1016/j.cmet.2013.01.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lim J. E., Jin O., Bennett C., Morgan K., Wang F., Trenor C. C. 3rd., Fleming M. D., and Andrews N. C. (2005) A mutation in Sec15l1 causes anemia in hemoglobin deficit (hbd) mice. Nat. Genet. 37, 1270–1273 10.1038/ng1659 [DOI] [PubMed] [Google Scholar]
- 17. Ohgami R. S., Campagna D. R., Greer E. L., Antiochos B., McDonald A., Chen J., Sharp J. J., Fujiwara Y., Barker J. E., and Fleming M. D. (2005) Identification of a ferrireductase required for efficient transferrin-dependent iron uptake in erythroid cells. Nat. Genet. 37, 1264–1269 10.1038/ng1658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fleming M. D., Romano M. A., Su M. A., Garrick L. M., Garrick M. D., and Andrews N. C. (1998) Nramp2 is mutated in the anemic Belgrade (b) rat: evidence of a role for Nramp2 in endosomal iron transport. Proc. Natl. Acad. Sci. U.S.A. 95, 1148–1153 10.1073/pnas.95.3.1148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Fleming M. D., Trenor C. C. 3rd., Su M. A., Foernzler D., Beier D. R., Dietrich W. F., and Andrews N. C. (1997) Microcytic anaemia mice have a mutation in Nramp2, a candidate iron transporter gene. Nat. Genet. 16, 383–386 10.1038/ng0897-383 [DOI] [PubMed] [Google Scholar]
- 20. Gunshin H., Mackenzie B., Berger U. V., Gunshin Y., Romero M. F., Boron W. F., Nussberger S., Gollan J. L., and Hediger M. A. (1997) Cloning and characterization of a mammalian proton-coupled metal-ion transporter. Nature 388, 482–488 10.1038/41343 [DOI] [PubMed] [Google Scholar]
- 21. Wolff N. A., Ghio A. J., Garrick L. M., Garrick M. D., Zhao L., Fenton R. A., and Thévenod F. (2014) Evidence for mitochondrial localization of divalent metal transporter 1 (DMT1). FASEB J. 28, 2134–2145 10.1096/fj.13-240564 [DOI] [PubMed] [Google Scholar]
- 22. Paradkar P. N., Zumbrennen K. B., Paw B. H., Ward D. M., and Kaplan J. (2009) Regulation of mitochondrial iron import through differential turnover of mitoferrin 1 and mitoferrin 2. Mol. Cell. Biol. 29, 1007–1016 10.1128/MCB.01685-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Shaw G. C., Cope J. J., Li L., Corson K., Hersey C., Ackermann G. E., Gwynn B., Lambert A. J., Wingert R. A., Traver D., Trede N. S., Barut B. A., Zhou Y., Minet E., Donovan A., et al. (2006) Mitoferrin is essential for erythroid iron assimilation. Nature 440, 96–100 10.1038/nature04512 [DOI] [PubMed] [Google Scholar]
- 24. Christenson E. T., Gallegos A. S., and Banerjee A. (2018) In vitro reconstitution, functional dissection, and mutational analysis of metal ion transport by mitoferrin-1. J. Biol. Chem. 293, 3819–3828 10.1074/jbc.M117.817478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chen W., Paradkar P. N., Li L., Pierce E. L., Langer N. B., Takahashi-Makise N., Hyde B. B., Shirihai O. S., Ward D. M., Kaplan J., and Paw B. H. (2009) Abcb10 physically interacts with mitoferrin-1 (Slc25a37) to enhance its stability and function in the erythroid mitochondria. Proc. Natl. Acad. Sci. U.S.A. 106, 16263–16268 10.1073/pnas.0904519106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chen W., Dailey H. A., and Paw B. H. (2010) Ferrochelatase forms an oligomeric complex with mitoferrin-1 and Abcb10 for erythroid heme biosynthesis. Blood 116, 628–630 10.1182/blood-2009-12-259614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Froschauer E. M., Rietzschel N., Hassler M. R., Binder M., Schweyen R. J., Lill R., Mühlenhoff U., and Wiesenberger G. (2013) The mitochondrial carrier Rim2 co-imports pyrimidine nucleotides and iron. Biochem. J. 455, 57–65 10.1042/BJ20130144 [DOI] [PubMed] [Google Scholar]
- 28. Mühlenhoff U., Stadler J. A., Richhardt N., Seubert A., Eickhorst T., Schweyen R. J., Lill R., and Wiesenberger G. (2003) A specific role of the yeast mitochondrial carriers Mrs3/4p in mitochondrial iron acquisition under iron-limiting conditions. J. Biol. Chem. 278, 40612–40620 10.1074/jbc.M307847200 [DOI] [PubMed] [Google Scholar]
- 29. Kondo A., Fujiwara T., Okitsu Y., Fukuhara N., Onishi Y., Nakamura Y., Sawada K., and Harigae H. (2016) Identification of a novel putative mitochondrial protein FAM210B associated with erythroid differentiation. Int. J. Hematol. 103, 387–395 10.1007/s12185-016-1968-4 [DOI] [PubMed] [Google Scholar]
- 30. Sun S., Liu J., Zhao M., Han Y., Chen P., Mo Q., Wang B., Chen G., Fang Y., Tian Y., Zhou J., Ma D., Gao Q., and Wu P. (2017) Loss of the novel mitochondrial protein FAM210B promotes metastasis via PDK4-dependent metabolic reprogramming. Cell Death Dis. 8, e2870 10.1038/cddis.2017.273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Napier I., Ponka P., and Richardson D. R. (2005) Iron trafficking in the mitochondrion: novel pathways revealed by disease. Blood 105, 1867–1874 10.1182/blood-2004-10-3856 [DOI] [PubMed] [Google Scholar]
- 32. Richardson D. R., Lane D. J., Becker E. M., Huang M. L., Whitnall M., Suryo Rahmanto Y., Sheftel A. D., and Ponka P. (2010) Mitochondrial iron trafficking and the integration of iron metabolism between the mitochondrion and cytosol. Proc. Natl. Acad. Sci. U.S.A. 107, 10775–10782 10.1073/pnas.0912925107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lu X., Huang L. J., and Lodish H. F. (2008) Dimerization by a cytokine receptor is necessary for constitutive activation of JAK2V617F. J. Biol. Chem. 283, 5258–5266 10.1074/jbc.M707125200 [DOI] [PubMed] [Google Scholar]
- 34. Wong P., Hattangadi S. M., Cheng A. W., Frampton G. M., Young R. A., and Lodish H. F. (2011) Gene induction and repression during terminal erythropoiesis are mediated by distinct epigenetic changes. Blood 118, e128–38 10.1182/blood-2011-03-341404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Socolovsky M., Fallon A. E., Wang S., Brugnara C., and Lodish H. F. (1999) Fetal anemia and apoptosis of red cell progenitors in Stat5a(−/−)5b(−/−) mice: a direct role for stat5 in Bcl-X(L) induction. Cell 98, 181–191 10.1016/S0092-8674(00)81013-2 [DOI] [PubMed] [Google Scholar]
- 36. Schneider-Poetsch T., Ju J., Eyler D. E., Dang Y., Bhat S., Merrick W. C., Green R., Shen B., and Liu J. O. (2010) Inhibition of eukaryotic translation elongation by cycloheximide and lactimidomycin. Nat. Chem. Biol. 6, 209–217 10.1038/nchembio.304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Chung J., Bauer D. E., Ghamari A., Nizzi C. P., Deck K. M., Kingsley P. D., Yien Y. Y., Huston N. C., Chen C., Schultz I. J., Dalton A. J., Wittig J. G., Palis J., Orkin S. H., Lodish H. F., et al. (2015) The mTORC1/4E-BP pathway coordinates hemoglobin production with l-leucine availability. Sci. Signal. 8, ra34 10.1126/scisignal.aaa5903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yien Y. Y., Robledo R. F., Schultz I. J., Takahashi-Makise N., Gwynn B., Bauer D. E., Dass A., Yi G., Li L., Hildick-Smith G. J., Cooney J. D., Pierce E. L., Mohler K., Dailey T. A., Miyata N., et al. (2014) TMEM14C is required for erythroid mitochondrial heme metabolism. J. Clin. Invest. 124, 4294–4304 10.1172/JCI76979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zhang J., Socolovsky M., Gross A. W., and Lodish H. F. (2003) Role of Ras signaling in erythroid differentiation of mouse fetal liver cells: functional analysis by a flow cytometry-based novel culture system. Blood 102, 3938–3946 10.1182/blood-2003-05-1479 [DOI] [PubMed] [Google Scholar]
- 40. Kingsley P. D., Greenfest-Allen E., Frame J. M., Bushnell T. P., Malik J., McGrath K. E., Stoeckert C. J., and Palis J. (2013) Ontogeny of erythroid gene expression. Blood 121, e5–e13 10.1182/blood-2012-04-422394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Metzendorf C., and Lind M. I. (2010) Drosophila mitoferrin is essential for male fertility: evidence for a role of mitochondrial iron metabolism during spermatogenesis. BMC Dev. Biol. 10, 68 10.1186/1471-213X-10-68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Stainier D. Y., Weinstein B. M., Detrich H. W. 3rd., Zon L. I., and Fishman M. C. (1995) Cloche, an early acting zebrafish gene, is required by both the endothelial and hematopoietic lineages. Development 121, 3141–3150 [DOI] [PubMed] [Google Scholar]
- 43. Reischauer S., Stone O. A., Villasenor A., Chi N., Jin S.-W., Martin M., Lee M. T., Fukuda N., Marass M., Witty A., Fiddes I., Kuo T., Chung W.-S., Salek S., Lerrigo R., et al. (2016) Cloche is a bHLH-PAS transcription factor that drives haemato-vascular specification. Nature 535, 294–298 10.1038/nature18614 [DOI] [PubMed] [Google Scholar]
- 44. Hammerschmidt M., Pelegri F., Mullins M. C., Kane D. A., van Eeden F. J., Granato M., Brand M., Furutani-Seiki M., Haffter P., Heisenberg C. P., Jiang Y. J., Kelsh R. N., Odenthal J., Warga R. M., and Nüsslein-Volhard C. (1996) Dino and mercedes, two genes regulating dorsal development in the zebrafish embryo. Development 123, 95–102 [DOI] [PubMed] [Google Scholar]
- 45. Ganis J. J., Hsia N., Trompouki E., de Jong J. L., DiBiase A., Lambert J. S., Jia Z., Sabo P. J., Weaver M., Sandstrom R., Stamatoyannopoulos J. A., Zhou Y., and Zon L. I. (2012) Zebrafish globin switching occurs in two developmental stages and is controlled by the LCR. Dev. Biol. 366, 185–194 10.1016/j.ydbio.2012.03.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Fujiki Y., Hubbard A. L., Fowler S., and Lazarow P. B. (1982) Isolation of intracellular membranes by means of sodium carbonate treatment: application to endoplasmic reticulum. J. Cell Biol. 93, 97–102 10.1083/jcb.93.1.97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ye H., and Rouault T. A. (2010) Human iron–sulfur cluster assembly, cellular iron homeostasis, and disease. Biochemistry 49, 4945–4956 10.1021/bi1004798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Grillo A. S., SantaMaria A. M., Kafina M. D., Cioffi A. G., Huston N. C., Han M., Seo Y. A., Yien Y. Y., Nardone C., Menon A. V., Fan J., Svoboda D. C., Anderson J. B., Hong J. D., Nicolau B. G., et al. (2017) A small-molecule iron transporter restores hemoglobinization in protein-deficient animals. Science 356, 608–616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Chung J., Wittig J. G., Ghamari A., Maeda M., Dailey T. A., Bergonia H., Kafina M. D., Coughlin E. E., Minogue C. E., Hebert A. S., Li L., Kaplan J., Lodish H. F., Bauer D. E., Orkin S. H., et al. (2017) Erythropoietin signaling regulates heme biosynthesis. Elife 10.7554/eLife.24767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Dailey H. A., and Fleming J. E. (1983) Bovine ferrochelatase. Kinetic analysis of inhibition by N-methylprotoporphyrin, manganese, and heme. J. Biol. Chem. 258, 11453–11459 [PubMed] [Google Scholar]
- 51. Troadec M. B., Warner D., Wallace J., Thomas K., Spangrude G. J., Phillips J., Khalimonchuk O., Paw B. H., Ward D. M., and Kaplan J. (2011) Targeted deletion of the mouse Mitoferrin1 gene: From anemia to protoporphyria. Blood 117, 5494–5502 10.1182/blood-2010-11-319483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Chung J., Anderson S. A., Gwynn B., Deck K. M., Chen M. J., Langer N. B., Shaw G. C., Huston N. C., Boyer L. F., Datta S., Paradkar P. N., Li L., Wei Z., Lambert A. J., Sahr K., et al. (2014) Iron regulatory protein-1 protects against mitoferrin-1-deficient porphyria. J. Biol. Chem. 289, 7835–7843 10.1074/jbc.M114.547778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wingert R. A., Galloway J. L., Barut B., Foott H., Fraenkel P., Axe J. L., Weber G. J., Dooley K., Davidson A. J., Schmidt B., Paw B. H., Shaw G. C., Kingsley P., Palis J., Schubert H., et al. (2005) Deficiency of glutaredoxin 5 reveals Fe-S clusters are required for vertebrate haem synthesis. Nature 436, 1035–1039 10.1038/nature03887 [DOI] [PubMed] [Google Scholar]
- 54. Kardon J. R., Yien Y. Y., Huston N. C., Branco D. S., Hildick-Smith G. J., Rhee K. Y., Paw B. H., and Baker T. A. (2015) Mitochondrial ClpX activates a key enzyme for heme biosynthesis and erythropoiesis. Cell 161, 858–867 10.1016/j.cell.2015.04.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Cioffi A. G., Hou J., Grillo A. S., Diaz K. A., Burke M. D. (2015) Restored physiology in protein-deficient yeast by a small-molecule channel. J. Am. Chem. Soc. 137, 10096–10099 10.1021/jacs.5b05765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Yu M., Riva L., Xie H., Schindler Y., Moran T. B., Cheng Y., Yu D., Hardison R., Weiss M. J., Orkin S. H., Bernstein B. E., Fraenkel E., and Cantor A. B. (2009) Insights into GATA-1-mediated gene activation versus repression via genome-wide chromatin occupancy analysis. Mol. Cell 36, 682–695 10.1016/j.molcel.2009.11.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Chami N., Chen M.-H., Slater A. J., Eicher J. D., Evangelou E., Tajuddin S. M., Love-Gregory L., Kacprowski T., Schick U. M., Nomura A., Giri A., Lessard S., Brody J. A., Schurmann C., Pankratz N., et al. (2016) Exome genotyping identifies pleiotropic variants associated with red blood cell traits. Am. J. Hum. Genet. 99, 8–21 10.1016/j.ajhg.2016.05.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ganesh S. K., Zakai N. A., van Rooij F. J., Soranzo N., Smith A. V., Nalls M. A., Chen M.-H., Kottgen A., Glazer N. L., Dehghan A., Kuhnel B., Aspelund T., Yang Q., Tanaka T., Jaffe A., et al. (2009) Multiple loci influence erythrocyte phenotypes in the CHARGE Consortium. Nat. Genet. 41, 1191–1198 10.1038/ng.466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Hindorff L. A., Sethupathy P., Junkins H. A., Ramos E. M., Mehta J. P., Collins F. S., and Manolio T. A. (2009) Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc. Natl. Acad. Sci. U.S.A. 106, 9362–9367 10.1073/pnas.0903103106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Soranzo N., Spector T. D., Mangino M., Kühnel B., Rendon A., Teumer A., Willenborg C., Wright B., Chen L., Li M., Salo P., Voight B. F., Burns P., Laskowski R. A., Xue Y., et al. (2009) A genome-wide meta-analysis identifies 22 loci associated with eight hematological parameters in the HaemGen consortium. Nat. Genet. 41, 1182–1190 10.1038/ng.467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Ulirsch J. C., Nandakumar S. K., Wang L., Giani F. C., Zhang X., Rogov P., Melnikov A., McDonel P., Do R., Mikkelsen T. S., and Sankaran V. G. (2016) Systematic functional dissection of common genetic variation affecting red blood cell traits. Cell 165, 1530–1545 10.1016/j.cell.2016.04.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Wang Y., Langer N. B., Shaw G. C., Yang G., Li L., Kaplan J., Paw B. H., and Bloomer J. R. (2011) Abnormal mitoferrin-1 expression in patients with erythropoietic protoporphyria. Exp. Hematol. 39, 784–794 10.1016/j.exphem.2011.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Visconte V., Avishai N., Mahfouz R., Tabarroki A., Cowen J., Sharghi-Moshtaghin R., Hitomi M., Rogers H. J., Hasrouni E., Phillips J., Sekeres M. A., Heuer A. H., Saunthararajah Y., Barnard J., and Tiu R. V. (2015) Distinct iron architecture in SF3B1 mutant myelodysplastic syndromes patients is linked to an SLC25A37 splice variant with a retained intron. Leukemia 29, 188–195 [DOI] [PubMed] [Google Scholar]
- 64. Lange H., Kispal G., and Lill R. (1999) Mechanism of iron transport to the site of heme synthesis inside yeast mitochondria. J. Biol. Chem. 274, 18989–18996 10.1074/jbc.274.27.18989 [DOI] [PubMed] [Google Scholar]
- 65. Amigo J. D., Yu M., Troadec M.-B., Gwynn B., Cooney J. D., Lambert A. J., Chi N. C., Weiss M. J., Peters L. L., Kaplan J., Cantor A. B., and Paw B. H. (2011) Identification of distal cis-regulatory elements at mouse mitoferrin loci using zebrafish transgenesis. Mol. Cell Biol. 31, 1344–1356 10.1128/MCB.01010-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Huang J., Liu X., Li D., Shao Z., Cao H., Zhang Y., Trompouki E., Bowman T. V., Zon L. I., Yuan G.-C., Orkin S. H., and Xu J. (2016) Dynamic control of enhancer repertoires drives lineage and stage-specific transcription during hematopoiesis. Dev. Cell 36, 9–23 10.1016/j.devcel.2015.12.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Surinya K. H., Cox T. C., and May B. K. (1997) Transcriptional regulation of the human erythroid 5-aminolevulinate synthase gene. Identification of promoter elements and role of regulatory proteins. J. Biol. Chem. 272, 26585–26594 10.1074/jbc.272.42.26585 [DOI] [PubMed] [Google Scholar]
- 68. Handschin C., Lin J., Rhee J., Peyer A. K., Chin S., Wu P. H., Meyer U. A., and Spiegelman B. M. (2005) Nutritional regulation of hepatic heme biosynthesis and porphyria through PGC-1α. Cell 122, 505–515 10.1016/j.cell.2005.06.040 [DOI] [PubMed] [Google Scholar]
- 69. Cox T. C., Bawden M. J., Martin A., and May B. K. (1991) Human erythroid 5-aminolevulinate synthase: promoter analysis and identification of an iron-responsive element in the mRNA. EMBO J. 10, 1891–1902 10.1002/j.1460-2075.1991.tb07715.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Proulx K. L., Woodard S. I., and Dailey H. A. (1993) In situ conversion of coproporphyrinogen to heme by murine mitochondria: terminal steps of the heme biosynthetic pathway. Protein Sci. 2, 1092–1098 10.1002/pro.5560020703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Ning B., Liu G., Liu Y., Su X., Anderson G. J., Zheng X., Chang Y., Guo M., Liu Y., Zhao Y., and Nie G. (2011) 5-Aza-2′-deoxycytidine activates iron uptake and heme biosynthesis by increasing c-Myc nuclear localization and binding to the E-boxes of transferrin receptor 1 (TfR1) and ferrochelatase (Fech) genes. J. Biol. Chem. 286, 37196–37206 10.1074/jbc.M111.258129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Crooks D. R., Ghosh M. C., Haller R. G., Tong W. H., and Rouault T. A. (2010) Posttranslational stability of the heme biosynthetic enzyme ferrochelatase is dependent on iron availability and intact iron–sulfur cluster assembly machinery. Blood 115, 860–869 10.1182/blood-2009-09-243105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Canver M. C., Bauer D. E., Dass A., Yien Y. Y., Chung J., Masuda T., Maeda T., Paw B. H., and Orkin S. H. (2014) Characterization of genomic deletion efficiency mediated by CRISPR/Cas9 in mammalian cells. J. Biol. Chem. 289, 21312–21324 10.1074/jbc.M114.564625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Cong L., Ran F. A., Cox D., Lin S., Barretto R., Habib N., Hsu P. D., Wu X., Jiang W., Marraffini L. A., and Zhang F. (2013) Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823 10.1126/science.1231143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Ran F. A., Hsu P. D., Wright J., Agarwala V., Scott D. A., and Zhang F. (2013) Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 8, 2281–2308 10.1038/nprot.2013.143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Li L., Chen O. S., McVey Ward D., and Kaplan J. (2001) CCC1 is a transporter that mediates vacuolar iron storage in yeast. J. Biol. Chem. 276, 29515–29519 10.1074/jbc.M103944200 [DOI] [PubMed] [Google Scholar]
- 77. Hattangadi S. M., Burke K. A., and Lodish H. F. (2010) Homeodomain-interacting protein kinase 2 plays an important role in normal terminal erythroid differentiation. Blood 115, 4853–4861 10.1182/blood-2009-07-235093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Cheng A. W., Shi J., Wong P., Luo K. L., Trepman P., Wang E. T., Choi H., Burge C. B., and Lodish H. F. (2014) Muscleblind-like 1 (Mbnl1) regulates pre-mRNA alternative splicing during terminal erythropoiesis. Blood 124, 598–610 10.1182/blood-2013-12-542209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Medlock A. E., Shiferaw M. T., Marcero J. R., Vashisht A. A., Wohlschlegel J. A., Phillips J. D., and Dailey H. A. (2015) Identification of the mitochondrial heme metabolism complex. PLoS One e0135896 10.1371/journal.pone.0135896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Roy C. N., Penny D. M., Feder J. N., and Enns C. A. (1999) The hereditary hemochromatosis protein, HFE, specifically regulates transferrin-mediated iron uptake in HeLa cells. J. Biol. Chem. 274, 9022–9028 10.1074/jbc.274.13.9022 [DOI] [PubMed] [Google Scholar]
- 81. Rossi E., Costin K.A., and Garcia-Webb P. (1988) Ferrochelatase activity in human lymphocytes, as quantified by a new high-performance liquid-chromatographic method. Clin. Chem. 34, 2481–2485 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.