

Abstract
Small heat-shock proteins (sHsps) are ubiquitous molecular chaperones, and sHsp mutations or altered expression are linked to multiple human disease states. sHsp monomers assemble into large oligomers with dimeric substructure, and the dynamics of sHsp oligomers has led to major questions about the form that captures substrate, a critical aspect of their mechanism of action. We show here that substructural dimers of two plant dodecameric sHsps, Ta16.9 and homologous Ps18.1, are functional units in the initial encounter with unfolding substrate. We introduced inter-polypeptide disulfide bonds at the two dodecameric interfaces, dimeric and nondimeric, to restrict how their assemblies can dissociate. When disulfide-bonded at the nondimeric interface, mutants of Ta16.9 and Ps18.1 (TaCT-ACD and PsCT-ACD) were inactive but, when reduced, had WT-like chaperone activity, demonstrating that dissociation at nondimeric interfaces is essential for sHsp activity. Moreover, the size of the TaCT-ACD and PsCT-ACD covalent unit defined a new tetrahedral geometry for these sHsps, different from that observed in the Ta16.9 X-ray structure. Importantly, oxidized Tadimer (disulfide bonded at the dimeric interface) exhibited greatly enhanced ability to protect substrate, indicating that strengthening the dimeric interface increases chaperone efficiency. Temperature-induced size and secondary structure changes revealed that folded sHsp dimers interact with substrate and that dimer stability affects chaperone efficiency. These results yield a model in which sHsp dimers capture substrate before assembly into larger, heterogeneous sHsp–substrate complexes for substrate refolding or degradation, and suggest that tuning the strength of the dimer interface can be used to engineer sHsp chaperone efficiency.
Keywords: chaperone, small heat shock protein (sHsp), small-angle X-ray scattering (SAXS), oligomerization, protein stability, protein design, protein engineering, chaperone efficiency, disulfides, dynamic light scattering (DLS), native mass spectrometry, substrate recognition, thermal stability, stress response, protein folding
Introduction
Small heat-shock proteins (sHsps)5 are a class of ATP-independent chaperones, expressed across all kingdoms of life, that are proposed to act as a cell's “first-responders” under stress conditions (1–6). Mutations or misexpression of specific human sHsps are associated with myopathies, neuropathies, and cancers (1, 7–9). The canonical function of sHsps is that they capture substrate proteins that are partially denatured by heat or other stresses in large soluble complexes, which are acted upon by ATP-dependent molecular machines to promote either substrate refolding or degradation (1, 10). sHsps are characterized by a 90–amino acid, β-sheet–rich α-crystallin domain (ACD, or Hsp20 domain, PF00011), flanked by sequences of variable length and composition (N-terminal (NT) sequence; C-terminal (CT) sequence) (11, 12). The majority form large oligomers containing 12 to >40 units, with a dimeric substructure (1, 3, 5), although there are a few dimeric sHsps, some of which are active chaperones (5, 13–15). Although vertebrate sHsps assemble principally as polydisperse oligomers, some bacterial, yeast, and plant sHsps are monodisperse. In all oligomeric forms, sHsp dimers interact through a conserved interface between an I-X-I motif in the CT and a groove formed by β4 and β8 in the ACD of a monomer in another dimer (hereafter the CT-ACD interface) (1, 5, 16, 17). Notably, sHsp oligomers are also highly dynamic, exchanging dimers (and monomers) on a timescale of minutes, as fast or faster than their rate of substrate capture. The large size, dynamic nature, and inherent polydispersity of most sHsps have resulted in few high-resolution structures (17–22) and differing hypotheses on sHsp mode of action. Interaction with substrates has been proposed to require either conformational changes in the oligomer or dissociation of oligomers into active units (3–5). A mechanism involving structural rearrangements of the oligomers is supported by presence of multiple oligomer conformations (23–25), with a shift in equilibria proposed to activate the sHsp. This model is also supported by experiments indicating that sHsp chaperone activity is unaffected by mutations or cross-linking that alter rates of subunit exchange (26–28). In contrast, multiple studies have measured temperature-dependent subunit exchange of several sHsps, consistent with a model in which sHsp dissociation releases suboligomers that interact with unfolding proteins (29–38). In this model, the oligomeric form sequesters the major substrate-binding regions of the sHsp, which is further supported by analysis of cross-linking interactions with substrates (39).
Higher plants express large numbers of sHsps belonging to 11 gene families (40), likely because of their “immobile” life that exposes them to environmental changes. Understanding sHsp function has implications for engineering plants to withstand temperature changes, as well as for defining their role in multiple human disease states. We have aimed to ascertain the mechanism and substrate-binding unit of sHsps through studies of cytosolic class-I sHsps from plants, which have proved to be exceptional models for the study of sHsp chaperone mechanism. Their largely monodisperse, dodecameric form under ambient conditions (41–43) and the availability of a high-resolution structure for wheat (Triticum aestivum) sHsp Ta16.9 (PDB ID: 1GME) (17) have made them amenable to functional and mechanistic studies. It has been suggested that these dodecamers, as well as oligomers of other sHsps, are reservoirs of dimers that are released under stress conditions to interact with unfolding substrates (6, 44). In this study, we used Ta16.9 and its close homologue from Pisum sativum, Ps18.1 (5, 17, 41), to address models of sHsp substrate interaction. These sHsp dodecamers display temperature-dependent dynamics in subunit exchange studies, with exchange mediated primarily, but not exclusively, by dimers (33–35, 45). Analysis of stoichiometries of substrate–sHsp complexes for Ps18.1 has shown that there is a bias for an even number of sHsp subunits in sHsp–substrate complexes (44, 46). Although the above studies suggest that substructural dimers of these sHsps interact with substrates, there is no direct evidence for sHsp dissociation being essential for activity, or for dimers being important for substrate capture. To address the mechanism of substrate protection by sHsps, we engineered disulfide bridges that prevent dissociation at either of the two major oligomeric interfaces, dimeric and nondimeric, in the Ta16.9 and Ps18.1 dodecamers, and tested their activity in protecting a heat-sensitive protein, malate dehydrogenase (MDH). The mutants led us to re-examine the quaternary structures of Ta16.9 and Ps18.1 and support the conclusions that sHsps need to dissociate at specific interfaces to protect substrate, and that free, folded dimers are the active substrate-capturing units.
Results
Protein design, expression, and purification
To test the functional importance of sHsp oligomer dynamics, we made cysteine mutant pairs in Ta16.9 and Ps18.1 to prevent dissociation at the nondimeric or dimeric interfaces. To link the nondimeric interfaces, cysteine residues were introduced to create a disulfide bridge between the CT of one monomer and β4 of the ACD from a monomer in another dimer (Fig. 1A). Of cysteine pairs tested, the mutants E74C V144C Ta16.9 (TaCT-ACD) and E81C V151C Ps18.1 (PsCT-ACD) were soluble and purified as dodecamers (Table S1, Fig. S1). To prevent dissociation at the dimeric interface, cysteines were introduced in a nonhydrogen-bonded registered pair between β6 on one monomer and β2 of the partner subunit such that the resulting disulfide-bonded dodecamer would dissociate into six covalent dimers when oxidized (Fig. 1A). The mutant K49C W96C Ta16.9 (Tadimer) was soluble and purified as a dodecamer; we were unable to create an analogous, dodecameric Psdimer (Table S1), and studies on this interface are confined to the Tadimer protein. This interface, which involves strand-swapping between ACDs, is characteristic of plant, bacterial, and yeast sHsps (3, 5, 17, 19).
Figure 1.

Disulfide bonds introduced within the sHsp dimer or between sHsp dimers, the latter of which defines a new geometry of the sHsp dodecamer. A, positions of the Cys substitutions linking dimers (Inter-dimer; top inset) with the CTD of one dimer shown in red, and a segment of the β-sandwich of the other dimer in gray. Positions of the Cys substitutions linking monomers within a dimer (Intra-dimer; bottom inset) with segments of one monomer in dark gray and the other monomer in light gray. B, SDS-PAGE separation of the indicated wheat WT or mutant proteins either oxidized (left) or reduced (right). C, mass spectra of E74C V144C (TaCT-ACD) in the oxidized state before (black) and after activation (MS2) of the 32+ ion (gray), showing dissociation to covalent trimers. D, the possible theoretical geometries of dodecamers comprising dimers with each edge corresponding to a dimer. The only geometry consistent with a three-point linkage is a tetrahedron (middle). E, tetrahedral models of Ta16.9 (see text and supporting figures and methods for details). Three dimers are colored (blue, orange, red) and three are rendered in gray. F, the best-fit model highlighting positions of the disulfides in the dodecamer. Enlargement shows detail of an interdimer disulfide (between the blue and orange dimers) (left) and the intradimer disulfide (between the light blue and dark blue monomers within the dimer) (right).
The formation of disulfide bonds and absence of free cysteines in all the disulfide mutants after oxidation were confirmed by DTNB assays (Table S2). To ascertain if the desired disulfide bonds were formed, the wheat proteins were examined by SDS-PAGE in the presence or absence of a reducing agent (Fig. 1B). The reduced proteins migrated at the expected monomeric size, whereas without reducing agent, TaCT-ACD and Tadimer migrated at sizes close to that of a covalent trimer (51 kDa) and covalent dimer (34 kDa), respectively. The apparent size of TaCT-ACD was surprising, as it was expected to form covalent tetramers (68 kDa) based on the two hexagonal ring structure in the Ta16.9 crystal structure (PDB ID:1GME) (17). To verify this observation, we obtained an accurate mass of the noncovalent and covalent units in TaCT-ACD by using native MS with collisional activation. This confirmed the unexpected result that disulfide linkage at the CT-ACD interface produced covalent trimers rather than tetramers (Fig. 1C). These observations can be explained only if the sHsp quaternary arrangement is tetrahedral, in contrast to the double disk of the Ta16.9 crystal structure (17). In fact, the only arrangement featuring dimers connected exclusively in groups of three is the tetrahedron (Fig. 1, D and E) (4). Notably, native MS of PsCT-ACD also revealed a covalent trimer building block (Fig. S2A) (47), indicating it is a tetrahedral dodecamer like TaCT-ACD. This tetrahedral architecture is equivalent to that observed for the sHsp Acr1 from Mycobacterium tuberculosis (48). To validate the structural similarity of Ta16.9 and Ps18.1 in the context of the WT forms, we performed small angle X-ray scattering (SAXS) of each (Fig. S2B). Their overlapping SAXS profiles demonstrate them to have indistinguishable native architectures.
A model for the tetrahedral geometry of the sHsp dodecamer
It was important to ensure that TaCT-ACD had retained the same quaternary structure as the WT Ta16.9. Therefore, we performed native ion mobility MS (IM-MS) experiments. Both proteins were dodecameric, and TaCT-ACD had a very similar collisional cross section (CCS) to the WT, whether oxidized or reduced (Fig. S3), indicating that they are architecturally equivalent. We conclude that both Ta16.9 and Ps18.1 are tetrahedral dodecamers in solution, and that the cysteine mutations and disulfide bonds do not disrupt this dodecameric architecture.
We capitalized on the IM-MS data to build models for Ta16.9 dodecamers consistent with overall tetrahedral architecture and linkages between ACDs and CTs. An exhaustive search of the roto-translational space, assuming all ACD dimers to be in equivalent environments, and filtering of the models according to the CCS and linkage constraints, resulted in four models that represent the data well (Fig. 1E). All four models have proteins in the tetrahedral architecture with minor differences because of uncertainty in the CCS measurements. The positions of the disulfide bonds in the tetrahedral model are further illustrated in Fig. 1F.
Note that the NTs were not included in the modeling because of lack of experimental restraints on this domain, but evidence from the Ta16.9 crystal structure and other sHsps indicates that they reside on the interior of the ACD cage (4). We measured the volume inside the tetrahedral cavity of our models (130395 ± 3060 Å3) and calculated the density that would be required in order for 12 NT domains to fit inside. We obtained a density of 0.42 Da/Å3, well below the average density of proteins (0.81 Da/Å3) (49), revealing that the cavity is readily able to accommodate 12 NT sequences.
Disulfide-bonded sHsps have stabilized secondary structure
We considered that, in addition to restricting the mode of dodecamer dissociation, introducing cysteine residues and disulfide bonds could affect the stability of sHsp secondary and quaternary structure, which might alter chaperone activity. Therefore, we first assessed secondary structure of the sHsps at different temperatures by obtaining far-UV CD spectra (Fig. 2A). Although CD spectra for all the sHsps in the oxidized state and the reduced Tadimer were recorded, it was not possible to obtain fully reduced TaCT-ACD or PsCT-ACD at concentrations of reducing agent compatible with CD (Table S2). Therefore, we created the single cysteine mutants TaV144C (V144C Ta16.9) and PsV151C (V151C Ps18.1) to approximate reduced TaCT-ACD and PsCT-ACD, respectively. The CD spectra for native sHsps are similar to those characteristic of β-sheets, indicating that the CD signal predominantly arises from the ACD β-sandwich (Fig. 2A) (36, 39). At 25 °C, spectra from all proteins are basically superimposable, but differences are observed already at 45 °C (Fig. 2A). Based on the temperature at which the different proteins (at 10 μm) show loss of secondary structure, the relative stability is: oxidized PsCT-ACD > oxidized TaCT-ACD > oxidized Tadimer > Ps18.1, PsV151C > TaV144C, reduced Tadimer, Ta16.9. Clearly, stabilizing the two CT-ACD interfaces or the dimeric interface of each monomer in the oligomer with disulfide bonds stabilizes sHsp secondary structure.
Figure 2.
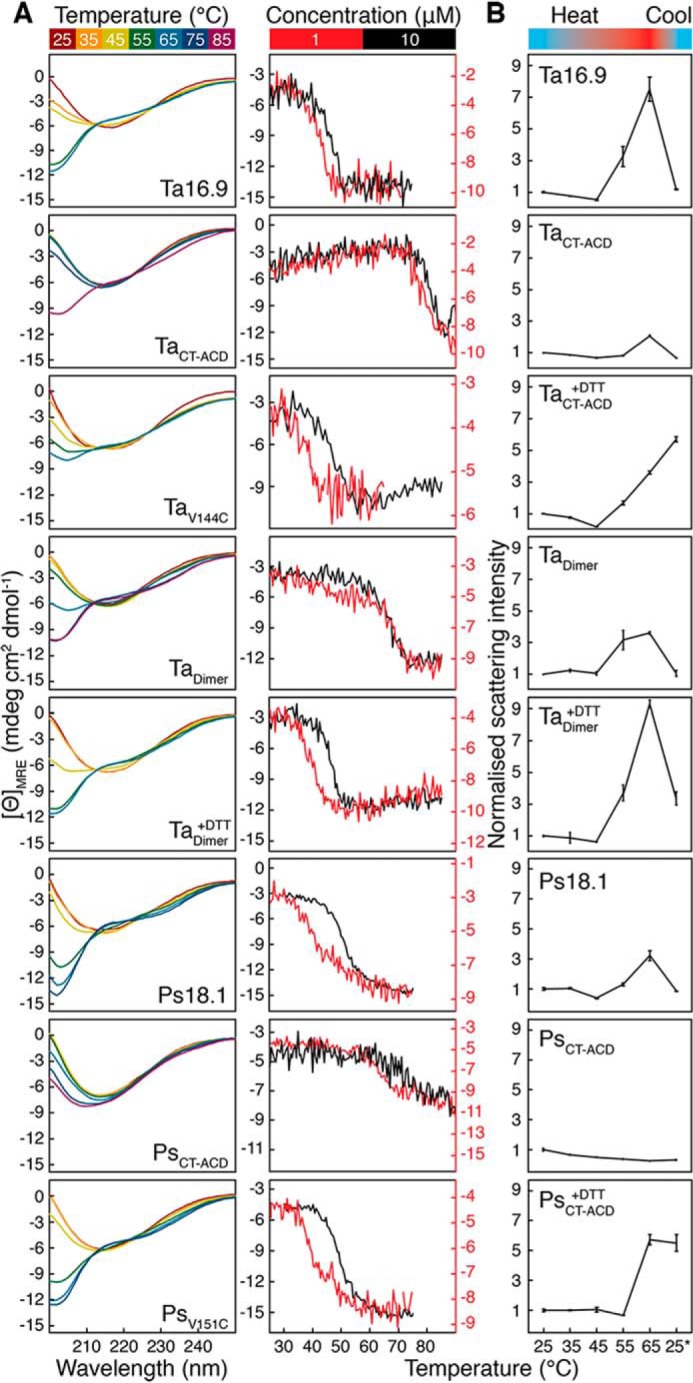
Secondary structure, thermal stability, and size of the sHsps. A, left, far-UV CD spectra for Ta16.9, oxidized TaCT-ACD, TaV144C, oxidized Tadimer, reduced Tadimer, Ps18.1, oxidized PsCT-ACD, and PsV151C (all 10 μm) collected at temperatures from 25 to 65 °C. Spectra were also collected at 75 and 85 °C for oxidized TaCT-ACD, Tadimer, and PsCT-ACD. Right, mean residue ellipticity at 205 nm as a function of temperature for 10 μm (black) and 1 μm (red) sHsp. B, average values with standard deviations for the TSI of proteins from three experiments carried out at each temperature. Measurements were normalized with respect to 25 °C, which was assigned a value of 1.0.
We also observed that unfolding of TaCT-ACD, Tadimer, and reduced Tadimer at 65 °C was irreversible (Fig. S4). These data, along with poor expression and solubility of certain interface mutants (Table S1), indicate that residues on the interfaces are likely important for folding and that the introduced cysteines and disulfide bonds disrupt the normal refolding pathway.
Oligomeric state changes with temperature
To relate sHsp activity to oligomeric state, we assessed oligomeric transitions in the WT and mutant proteins at different temperatures. If unfolding is accompanied by changes in sHsp oligomeric state, the unfolding transition temperature will depend on protein concentration. We therefore carried out thermal melts monitored by CD at 10 and 1 μm sHsp (Fig. 2A). For Ta16.9, TaV144C, and reduced Tadimer, cooperative unfolding occurs in the range of 30 to 50 °C, with the transition shifting to a lower temperature at the lower protein concentration. Ps18.1 and PsV151C behaved similarly, although cooperative unfolding occurred at a higher temperature (40 to 55 °C) (Fig. 2A). These data indicate that the oligomeric state of the unfolded and folded species of these proteins is different and proceeds without an intermediate well-folded monomeric state. In contrast, for the oxidized, disulfide-bonded species, Tadimer and TaCT-ACD (PsCT-ACD is highly stabilized, and an unfolded baseline could not be obtained), the temperature ranges of the unfolding transition, 60 and 75 °C, respectively, were the same at both protein concentrations (Fig. 2A), indicating that their unfolding transitions are for the covalent dimer/trimer.
To better understand the oligomeric states involved in substrate protection we examined the temperature dependence of sHsp size by dynamic light scattering (DLS) (Fig. 2B, Table S3). When heated to 45 °C, all proteins decreased in total scattering intensity (TSI), consistent with dodecamer dissociation, except oxidized Tadimer and reduced PsCT-ACD, which showed little change from 25 °C (Fig. 2B, Table S3). To relate the TSI to particle sizes, we deconvoluted the DLS correlation curves and broadly classified the particles into dodecameric (7–20 nm diameter), suboligomeric (<7 nm diameter), and aggregates (>30 nm diameter). At 25 °C, the major species were dodecameric, whereas at 45 °C, except for reduced Tadimer, the major species for all proteins are smaller. Temperature-dependent formation of both smaller particles and large self-aggregates is consistent with previous studies of sHsps (32, 38, 44, 45). We suggest that the presence of multiple particle sizes explains the unchanged TSI in oxidized Tadimer and reduced PsCT-ACD and conclude that all these sHsps undergo dissociation at 45 °C. DLS measurements were also made after cooling the proteins back to 25 °C from 65 °C. Ta16.9, Ps18.1, oxidized TaCT-ACD, PsCT-ACD, and Tadimer reverted primarily to the dodecameric form, whereas reduced TaCT-ACD, Tadimer, and PsCT-ACD were unable to reform dodecamers, but rather formed even larger aggregates. It should be noted that TaCT-ACD, PsCT-ACD, and Tadimer in their oxidized states, as shown by CD, have native-like secondary structure and are largely folded at 65 °C. The inability of the reduced mutants to revert to dodecamers on cooling (Fig. S5) suggests that interfaces are critical to refolding.
Constraining oligomer dissociation alters sHsp chaperone activity
Ta16.9 and Ps18.1 have been well-characterized for their ability to protect the heat-sensitive substrate MDH (17, 41, 50, 51). Therefore, to determine how restricting the ability to dissociate at either the nondimeric (TaCT-ACD and PsCT-ACD) or dimeric interfaces (Tadimer) affected sHsp chaperone activity, we assayed the reduced and oxidized sHsps for protection of MDH heated to 45 °C. We first tested to see if the high levels of DTT required to fully reduce the disulfide mutants (so that they would be free to dissociate like WT) did not interfere with the chaperone activity of the WT proteins. Both Ta16.9 and Ps18.1 (neither of which have cysteine residues) protected MDH (which has seven cysteine residues) in the presence and absence of reducing agent, as determined by changes in light scattering following heating (Fig. 3). As expected, Ps18.1 was more effective in preventing light scattering than Ta16.9, with complete protection of 3 μm MDH at a monomeric molar ratio of sHsp:MDH of 1:1, compared with 6:1 for Ta16.9 (Fig. 3) (41, 51). Thus, the WT proteins functioned to protect MDH under both reducing and oxidizing conditions.
Figure 3.

Restricting dodecamer dissociation alters sHsp chaperone activity. Light scattering by aggregated MDH mixed with Ta16.9, TaCT-ACD, or Tadimer under reducing (top left) or oxidizing (bottom left) conditions, and Ps18.1 and PsCT-ACD under reducing (top right) or oxidizing (bottom right) conditions. The sHsp monomer: MDH monomer ratio is indicated on the x axis. Scattering values were normalized with respect to that for MDH heated alone, which was assigned a value of 1.0. Means (filled circles) from three replicate experiments (open circles) are plotted with standard deviation. Unheated MDH (4 °C) served as a control for absence of aggregation.
Comparing the chaperone activity of the reduced and oxidized TaCT-ACD and PsCT-ACD revealed that, under reducing conditions, both proteins were essentially as effective as their corresponding WT in preventing MDH light scattering (Fig. 3). However, strikingly, under oxidizing conditions, which limits these dodecamers to dissociating into trimers, these sHsps afforded no protection to MDH (Fig. 3). Even at a molar ratio of three times the ratio that afforded full protection by Ps18.1, oxidized PsCT-ACD had no ability to limit MDH light scattering (Fig. 3). These data imply that for the sHsp to protect MDH, it is necessary for the CT-ACD (nondimeric) interfaces to dissociate, and the complete absence of TaCT-ACD and PsCT-ACD activity is most readily explained by the inability to dissociate into dimers like WT.
Assays with Tadimer, which can dissociate into dimers under both the reduced and oxidized conditions, yielded dramatically different results. Reduced Tadimer was somewhat less efficient than Ta16.9 at suppressing MDH aggregation, although still highly effective (Fig. 3). In contrast, oxidized Tadimer protected MDH much more efficiently than Ta16.9, with complete protection at a ratio of Tadimer:MDH of 1:1 (Fig. 3). Notably, the CD measurements showed that oxidized TaCT-ACD, PsCT-ACD, and Tadimer all have highly stable secondary structure, but only oxidized Tadimer is an effective chaperone. Thus, stable ACD secondary structure alone does not account for the enhanced chaperone activity of oxidized Tadimer, but rather stabilizing the dimeric interface, while still allowing dissociation of dimers from the oligomer, results in a highly effective chaperone.
To examine in more detail how these sHsps interacted with MDH compared with the WT proteins, these same samples were analyzed for the presence of MDH–sHsp complexes by size-exclusion chromatography (SEC) (Fig. 4). As expected, at the sHsp:MDH molar ratios where protection was observed by light scattering, WT Ta16.9 and Ps18.1 formed complexes with MDH under reducing or oxidizing conditions, with complexes eluting similarly, ahead of the 670 kDa marker. In contrast, MDH complexes with TaCT-ACD and PsCT-ACD were only observed under reducing conditions, when the sHsps are free to dissociate into dimers like WT; no complexes were formed under oxidizing conditions (Fig. 4).
Figure 4.

sHsp–MDH complex formation correlates with aggregation protection. A and B, SEC profiles were generated on a TSKgel SWxl Ultra SW column for soluble fractions of reaction mixtures of sHsp–MDH under reducing (top two rows) or oxidizing conditions (bottom two rows) for samples prepared as in Fig. 3 with (A) Ta16.9 and mutants or (B) Ps18.1 and PsCT-ACD. The dashed and solid lines represent unheated and heated reaction mixtures, respectively. The sHsp monomer: MDH monomer molar ratios are indicated at the top right corner of each plot. Calibration standards above the top panels correspond from left to right: Void volume, 670, 158, 44, and 17 kDa. Peaks seen at 7 ml and 8.5 ml correspond to sHsp and MDH, respectively. * indicates absorbance from protein with spurious disulfide linkages. Peaks between 4.7 and 6.5 ml correspond to sHsp–MDH complexes.
Formation of complexes between Tadimer and MDH also paralleled results of the light scattering assays. The lower efficiency of reduced Tadimer is reflected in formation of more variable sized Tadimer–MDH complexes compared with Ta16.9–MDH complexes (Fig. 4). Conversely, the MDH–sHsp complexes that formed with oxidized Tadimer include smaller complexes than those formed with WT Ta16.9, consistent with Tadimer being a more efficient chaperone (30).
Discussion
Outstanding questions about the mechanism of sHsp action concern the mode of substrate capture and the functional importance of sHsp oligomer dynamics. To address these questions, we engineered the structurally defined, dodecameric sHsps Ta16.9 and Ps18.1 to restrict dissociation at each of two major oligomeric interfaces, dimeric and CT-ACD. A surprise on analysis of the proteins linked at the CT-ACD interface, TaCT-ACD and PsCT-ACD, was that, when oxidized, the substructure of these sHsps proved to be covalent trimers, as opposed to covalent tetramers, as predicted from the available X-ray structure (1GME) (17). This observation, along with MS data, leads us to propose a tetrahedral structure for these dodecameric sHsps. Importantly, our analysis of the chaperone activity of Ta16.9 and Ps18.1 with fixed interfaces provides strong evidence that dissociation of the dodecamer at the CT-ACD interfaces is essential for optimal chaperone activity, and that stabilized dimers are more effective chaperones and likely to mediate the first encounter with denaturing substrate.
A new geometry for the sHsp dodecamer
Our mutant studies, MS, and modeling data support a new geometry for Ta16.9 and Ps18.1 dodecamers that is highly populated in solution. This dodecamer form has dimers arranged in a tetrahedral structure rather than in the double disk of the Ta16.9 X-ray structure, and this tetrahedron is maintained in the disulfide mutants. The tetrahedral arrangement requires no changes in the sHsp monomer or dimer compared with the X-ray structure, other than altering the angle of the CT relative to the ACD at a flexible hinge. In the double disk structure, the CT must adopt two different angles to form CT-ACD interfaces. In contrast, for tetrahedral geometry the CT in each monomer can have the same angle with respect to the ACD. Attempts to reduce and reoxidize TaCT-ACD under different conditions, including those used for crystallography, also only yielded covalent trimers. Thus, we suggest that the double disk geometry is a minor form in solution that is trapped by crystallization. The ability of sHsps to adopt different geometries accommodated by varying the angle of the CT while maintaining the CT-ACD interface intact is amply illustrated by different known sHsp oligomeric structures (2, 5, 52, 53). The ability to reassemble into different geometries, potentially using this same CT-ACD interface, may also contribute to the variable stoichiometries and morphologies of sHsp–substrate complexes (44, 54).
sHsp temperature transitions and chaperone activity
Our thermal unfolding data indicate that stability of sHsp secondary structure is greatly enhanced when either of the dodecameric interfaces is linked by disulfides. Each TaCT-ACD and PsCT-ACD monomer has two interfaces with other monomers that are stabilized by disulfides, as opposed to Tadimer, which has only one stabilized interface with its partner in the dimer. As a result, TaCT-ACD and PsCT-ACD are stabilized to a greater extent than Tadimer. Unfolding profiles for different concentrations of the same protein will overlay when loss of secondary structure is independent of oligomeric state, whereas a shift to a higher melting temperature at higher concentrations indicates the protein is stabilized by association in a higher-order structure. A cooperative transition in secondary structure is independent of concentration for the disulfide-linked sHsps (TaCT-ACD and Tadimer), implying that TaCT-ACD and Tadimer dissociate to well-folded covalent trimer and dimer, respectively, before unfolding. In contrast, unfolding of nondisulfide-linked sHsps occurs with deoligomerization. In summary, DLS studies show that the sHsp molecules undergo dissociation at both dimeric and CT-ACD interfaces upon heating. CD melt studies show that suboligomers (trimers and dimers) are folded at activity assay temperatures. Because stabilization of the dimeric interface enhances activity and dissociation of CT-ACD interfaces is essential for activity, we infer that the folded dimers from sHsps are primary substrate capturing units.
The two sHsps, Ta16.9 versus Ps18.1, differ in stability
Ta16.9 and Ps18.1 share 69% sequence identity (Fig. S1), but differ significantly in ability to protect MDH, with Ps18.1 being more efficient (Fig. 3) (41). Previous molecular dynamics studies of Ps18.1 and Ta16.9 dimers suggest that Ps18.1 has larger exposed hydrophobic patches and that the Ps18.1 NT makes fewer contacts to itself compared with Ta16.9 (55). The surface area buried in the ACD dimers of Ta16.9 and Ps18.1 were calculated to be 2945 and 3059 Å2, using PDB files, 1GME (17) and 5DS2 (47), respectively. The somewhat larger buried surface area of the Ps18.1 dimer indicates a stronger dimeric interface, which would make it more stable than the Ta16.9 dimer. This is substantiated by the thermal melt data, from which the apparent Ta16.9 and Ps18.1 melting temperatures are estimated (from the first derivative plots at 10 μm) to be 48.3 and 50.6 °C, respectively (Fig. 2A). The greater stability of Ps18.1 is also seen in DLS studies; Ps18.1 has a lesser tendency to form larger self-aggregates than Ta16.9, although both display dissociation to smaller species, including dimers. This difference in stability is likely a significant factor in the more efficient substrate protection by Ps18.1 compared with Ta16.9, and should be considered when assessing chaperone activity of other sHsps.
sHsp dimers as the substrate capture unit
Our data support a model for sHsp activity as depicted in Fig. 5, in which the sHsp dimer makes the effective first encounter with denaturing substrate before assembly into higher mass sHsp–substrate complexes. By cross-linking the dimer interface in Tadimer, we shifted the equilibrium of these dynamic dodecamers to the dimer form and increased chaperone activity. In contrast, by preventing dissociation at the CT-ACD interface we essentially eliminated the dimer and monomer species and also the chaperone activity (Fig. 5A). At the assay temperature of 45 °C, the equilibrium species of all sHsps studied was shifted to suboligomers as observed by DLS; however, only suboligomers not linked at CT-ACD interfaces protected substrate. Our model reflects the dynamics of the dodecamer and importance of the dimer as the “substrate-capture unit.” sHsp oligomers exchange dimers at a rate that is temperature dependent (25), which tunes the availability of the dimer capture unit to the increase in unfolding proteins with elevated temperature.
Figure 5.

The sHsp dimer is the major substrate encounter species. A, table summarizing how the disulfide constrained sHsps (CT-ACD and Dimer) alter the population of dodecamer, dimer, or monomer compared with WT and resultant changes in chaperone activity. Check mark indicates presence and abundance of a specific protein form or extent of chaperone activity; × indicates absence of that protein form or activity. B, schematic showing how the dimer, which is the favored form in the dimer mutant and disfavored form in the CT-ACD mutants (as in A) is the major substrate capture form of the sHsp, followed by subsequent assembly of multiple dimers and substrates into heterogeneous complexes.
It is possible that linking the CT-ACD interface blocks substrate binding in the β4-β8 groove (44, 46). However, our data, along with previous studies of Ta16.9 and Ps18.1 show that the NT is a major region involved in substrate (including MDH) binding (39, 41, 51). Studies of mammalian, yeast, archaeal, and bacterial sHsps also have concluded that the NT is essential for substrate binding (56–59). Our data imply that the combined conformation of NT sites in the dimer are more effective in binding substrates than NT site conformations available in three linked monomers. Further, all the active sHsps retained complete (i.e. oxidized Tadimer) or partial native-like secondary structure (i.e. Ta16.9, TaV144C, reduced Tadimer, PsV151C) at 45 °C. Because the CD characteristics are primarily contributed by the ACD, and Tadimer is the most efficient chaperone (Fig. 5A), we infer that folded dimers are the primary substrate-capturing units. The recognition of substrates by oligomer dissociation products is an elegant mechanism of action, because each dodecamer comprises six dimers which diffuse much more rapidly. This allows for an efficient chaperone response, with subsequent assembly into larger complexes conferring stability over a longer lifetime. The subunit dynamics and evidence that the NT of many sHsps is involved in substrate binding are consistent with our model being generally applicable to other sHsps interacting with diverse substrates.
Conclusions
Our data clearly support the importance of a specific mode of oligomer dissociation and the critical role of dimers in the mechanism of sHsp action. In addition to leading to a reassessment of the quaternary structure of these plant sHsps, analysis of the disulfide mutants shows that increasing stability of the dimeric interface enhances the sHsp holdase activity. Notably, it has also been observed that when stabilized with an interface disulfide, the core ACD of human αB-crystallin dimers was better at protecting substrates than WT protein (60), and stabilizing crystallins has pharmacological potential in treating cataracts (61, 62). Thus, strategies to modulate the strength of the sHsp dimeric interface could be an approach to producing designer holdases active at different temperatures.
Experimental procedures
Design and purification of disulfide mutants
The coordinates from the PDB file 1GME (17) for Ta16.9 were used for selecting positions to introduce cysteine residues for inter-polypeptide disulfide formation. The structure was analyzed using Naccess (63) for identifying residues at the interaction surfaces. Pairs of residues on neighboring monomers, among which at least one residue was accessible (>7% accessibility) and were positioned such that Cα-Cα and Cβ-Cβ distances were <7.0 and <6.0 Å, respectively, were chosen (64, 65) to introduce disulfides at dimeric and nonpartner interfaces (Table S1).
Ta16.9 and Ps18.1 had been previously cloned under the control of the IPTG-inducible T7 promoter (41). Cysteines were introduced by site-directed mutagenesis at the (i) dimeric interfaces K49C W96C Ta16.9 and W48C H97C Ta16.9 and (ii) nonpartner interfaces E74C V144C Ta16.9 and V73C K145C Ta16.9. K49C W96C Ta16.9 and E74C V144C Ta16.9 were chosen for further study compared with Ta16.9 (Fig. 1). Single cysteine mutations E74C Ta16.9, V144C Ta16.9, K49C Ta16.9, W96C Ta16.9 were also made. Corresponding mutations (K56C W103C Ps18.1, W55C R105C Ps18.1, D54C R105C Ps18.1, E81C V151C Ps18.1, E81C Ps18.1, V151C Ps18.1) were introduced in a homologue from Pisum sativum, Ps18.1, which shares 69% identity with Ta16.9 (Fig. S1). The residues in Ps18.1 for cysteine mutations were identified by sequence alignment of Ps18.1 and Ta16.9. BL21-DE3 Escherichia coli cells transformed with the required plasmid were grown in terrific broth at 37 °C to an outer diameter of 0.6 before induction with 1 mm IPTG.
Following induction, cells were grown at 25 °C for 12–16 h. The cells were pelleted and then lysed by sonication in 20 mm Tris, pH 8.0, 20 mm NaCl. The supernatant following lysis and centrifugation (5000 × g, 15 min) was loaded on a 40 ml Q Sepharose ion exchange column and separated with a gradient of 20–250 mm NaCl in 20 mm Tris, pH 8.0, spanning a total volume of 400 ml collecting 6-ml fractions. Fractions containing sHsp, which eluted between ∼100 and 150 mm NaCl, were concentrated to 0.5 ml and separated on an S-200 Superdex GE gel filtration column equilibrated with 20 mm Tris, pH 8.0, 150 mm NaCl. All buffers used in purification contained 1 mm EDTA, 10 μm PMSF, 1 mm ϵ-aminocaproic acid and 1 mm benzamidine. For purification of single cysteine sHsps, all buffers included 1 mm β-mercaptoethanol. Fractions containing purified protein were used for further analysis. Purity of the proteins and formation of disulfide bonds was ascertained by SDS-PAGE in the presence and absence of 10 mm DTT.
Dithionitrobenzoic acid (DTNB) assay
The number of free cysteine residues in each of the proteins was determined by reaction with DTNB. Each of the proteins at 15 μm was incubated with 5 mm DTNB in 100 mm Tris, pH 8.0, and 4 m urea for 30 min at room temperature in the dark. 5 and 10 μm DTT samples were used as positive controls. Following incubation, absorbance of samples was measured at 412 nm to determine the concentration of thionitrobenzoic acid (TNB) using the extinction coefficient of 13,700 M−1 cm−1. The amount of thionitrobenzoic acid formed corresponds to the concentration of free thiols in the protein sample.
IM-MS of Ta16.9, Ps18.1, and cysteine mutants
Proteins were prepared for native MS at concentrations of 1–5 μm oligomer, in 200 mm ammonium acetate, pH 6.9. Mass spectra of all proteins were obtained on instruments (Q-ToF2, SYNAPT G1 High Definition MS; Waters Corp.) modified for the analysis of high mass species, according to methods described previously (66).Collision-induced dissociation was performed by acceleration into a dedicated collision cell within the instrument, either with or without selection of a particular ion population using the quadrupole analyzer (as specified). CCSs were obtained as described previously (67). Data were calibrated and analyzed using MassLynx software and presented with minimal smoothing and the absence of background subtraction.
Far-UV CD spectroscopy
Protein samples for CD spectra collection were buffer-exchanged into 5 mm sodium phosphate, pH 7.5, 5 mm NaCl using 5 ml HiTrap desalting columns (GE Healthcare). The buffer included 0.5 mm tris(2-carboxyethyl)phosphine as reducing agent for proteins TaV144C and PsV151C and for reduced Tadimer. Spectra were collected with 10 μm proteins on a Jasco J-1500 spectrometer. Spectra were collected at different temperatures using the temperature control program software. In the range of 25–85 °C, spectra were collected at data intervals of 10 °C. The rate for temperature increase was set to 2 °C min−1, with the temperature held constant during data collection. The scan rate, data pitch, response time, and bandwidth parameters were set to 100 nm min−1, 1 nm, 2 s and 1 nm, respectively. An average of three spectra collected for each sample in the range of 250–200 nm are reported after subtraction of corresponding buffer spectra and smoothing. All samples after data collection were incubated at room temperature for 15 min, and then spectra were collected with the parameters as above to check for protein refolding.
Thermal stability of sHsp secondary structure
Thermal stability of sHsp secondary structure was assessed on the Jasco J-1500 CD spectrometer by monitoring the sample ellipticity at 205 nm as a function of temperature. Measurements were made for 1.0 and 10.0 μm protein in 5 mm sodium phosphate, pH 7.5, 5 mm NaCl, in 1 cm and 1 mm path length cuvettes, respectively. A scan rate of 1 °C min−1 with a data pitch of 0.5 °C was used.
Dynamic light scattering (DLS)
DLS profiles were collected for protein samples in 25 mm HEPES, pH 7.5, 150 mm KCL, 5 mm MgCl2 on a Malvern Zetasizer Nano instrument. For reduced protein samples the buffer included 20 mm DTT. Data were collected for 50 μl, 10 μm protein samples in a microquartz cuvette (Malvern-ZEN2112). Protein samples were filtered through a 0.22-μm filter prior to measurement. Each sample was incubated at 25 °C for 10 min in the cuvette in the instrument set to 25 °C. Following incubation, three measurements were made, each consisting of 15 runs of 15 s each. The cuvette holder temperature was then increased by 10 °C. The same sample was then incubated at the increased temperature for 10 min prior to data collection as before. This was repeated until data collection at 65 °C was complete. The sample was then cooled to 25 °C, incubated for 10 min, and the DLS measurements repeated. Data were analyzed using the software Zetasizer 7.11. Corrections for viscosity and refractive index of water with respect to temperature were taken into account for calculating size distributions. The derived count rates were used for generating scatter intensity plots as a function of temperature.
Activity assays for protection of substrate protein by sHsps
The substrate protein used was porcine heart MDH. All protein concentrations are cited for protein monomers. 3 μm MDH was incubated with different concentrations of sHsp in 25 mm HEPES, pH 7.5, 150 mm KCl, 5 mm MgCl2. For assays of sHsps in the reduced state, 20 mm DTT was included in the buffer. Reaction mixtures were incubated at 45 °C for 2 h, and then cooled on ice for 10 min, as described previously (41). To ascertain the extent of MDH precipitation, reaction mixtures were diluted 8-fold with assay buffer, and sample scattering intensity was measured on a Photon Technology International fluorometer. Excitation and emission wavelengths were set to 500 nm with both slit widths set to 2 nm. Scattering intensity was measured over 20 s, with a data pitch of 1 s. Measurements were averaged over the 20 s period. For analysis of sHsp–MDH complex formation by analytical SEC, 20 μl of supernatant obtained after centrifugation (20 min at 16,000 × g) for each reaction mixture was injected on a TSKgel UltraSW Aggregate column (Tosoh Bioscience). The column was run at 1 ml−1 in 5 mm sodium phosphate pH 7.5, 150 mm KCl, 5 mm MgCl2, and absorbance at 220 nm of the eluent was recorded.
SAXS
Data were collected at the B21 bending-magnet instrument at the Diamond Light Source (Harwell, UK). Samples were prepared in 200 mm ammonium acetate to a concentration of 5 mg ml−1 and two successive 2-fold dilutions. Protein and corresponding buffer solutions were exposed to the beam in a 1.6 mm diameter quartz capillary at 15 °C. The sample capillary was held in vacuum and subjected to a cleaning cycle between each measurement. A Pilatus 2 m two-dimensional detector was used to collect 180-frame exposures of 1 s from each sample and the corresponding buffer. The detector was placed at 3.9 m from the sample, giving a useful Q-range from 0.012 to 0.4 Å−1. Two-dimensional data reduction consisted of normalization for beam current and sample transmission, radial sector integration, background buffer subtraction, and averaging. Each frame was inspected manually and discarded if signs of radiation damage were apparent. Data scaling, merging, and Guinier analysis were performed in PRIMUS (68).
Author contributions
I. S. conceptualization; I. S., M. T. D., D. A. S., and G. K. A. H. formal analysis; I. S., M. T. D., D. A. S., and G. K. A. H. methodology; I. S., M. T. D., J. L. P. B., and E. V. writing-original draft; I. S., M. T. D., D. A. S., G. K. A. H., J. L. P. B., and E. V. writing-review and editing; M. T. D. software; J. L. P. B. and E. V. resources; J. L. P. B. and E. V. supervision; J. L. P. B. and E. V. funding acquisition; J. L. P. B. and E. V. investigation; E. V. project administration.
Supplementary Material
Acknowledgments
The authors thank Dr. Lizz Bartlett for assistance with the UMass Amherst Institute for Applied Life Sciences Core Facility for Biophysical Characterization, Dr. Eugenia Clerico for useful discussions, and Umaru Barrie for help with preparation of the single cysteine mutants. We acknowledge access to B21 and help from Mark Tully and James Doutch at the Diamond Synchrotron (SM9384-2) (to J. L. P. B.).
This work was supported by Engineering and Physical Sciences Research Council Grants EP/J01835X/1 (to J. L. P. B.) and EP/P016499/1 (to M. T. D.) and National Institutes of Health Grant RO1 GM42762 (to E. V.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S5 and Tables S1–S3.
- sHsp
- small heat shock protein
- ACD
- α-crystallin domain
- NT
- N-terminal
- CT
- C-terminal
- MDH
- malate dehydrogenase
- DTNB
- dithionitrobenzoic acid
- SAXS
- small angle x-ray scattering
- IM-MS
- ion mobility mass spectrometry
- CCS
- collisional cross section
- DLS
- dynamic light scattering
- TSI
- total scattering intensity
- IPTG
- isopropyl 1-thio-β-d-galactopyranoside
- CD
- circular dichroism
- SEC
- size exclusion chromatography.
References
- 1. Basha E., O'Neill H., and Vierling E. (2012) Small heat shock proteins and α-crystallins: Dynamic proteins with flexible functions. Trends Biochem. Sci. 37, 106–117 10.1016/j.tibs.2011.11.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Richter K., Haslbeck M., and Buchner J. (2010) The heat shock response: Life on the verge of death. Mol. Cell 40, 253–266 10.1016/j.molcel.2010.10.006 [DOI] [PubMed] [Google Scholar]
- 3. Haslbeck M., and Vierling E. (2015) A first line of stress defense: Small heat shock proteins and their function in protein homeostasis. J. Mol. Biol. 427, 1537–1548 10.1016/j.jmb.2015.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hilton G. R., Lioe H., Stengel F., Baldwin A. J., and Benesch J. L. P. (2013) Small heat-shock proteins: Paramedics of the cell. Top. Curr. Chem. 328, 69–98 10.1007/128_2012_324 [DOI] [PubMed] [Google Scholar]
- 5. Santhanagopalan I., Basha E., Ballard K. N., Bopp N. E., and Vierling E. (2015) Model chaperones: Small heat shock proteins from plants. in The Big Book on Small Heat Shock Proteins (Tanguay R. M., and Hightower L. E., eds), pp. 119–153, Springer International Publishing, Cham, Switzerland: 10.1007/978-3-319-16077-1 [DOI] [Google Scholar]
- 6. Van Montfort R., Slingsby C., and Vierling E. (2001) Structure and function of the small heat shock protein/α-crystallin family of molecular chaperones. Adv. Protein Chem. 59, 105–156 10.1016/S0065-3233(01)59004-X [DOI] [PubMed] [Google Scholar]
- 7. Arrigo A. P. (2013) Human small heat shock proteins: Protein interactomes of homo- and hetero-oligomeric complexes: An update. FEBS Lett. 587, 1959–1969 10.1016/j.febslet.2013.05.011 [DOI] [PubMed] [Google Scholar]
- 8. Charmpilas N., Kyriakakis E., and Tavernarakis N. (2017) Small heat shock proteins in ageing and age-related diseases. Cell Stress Chaperones 22, 481–492 10.1007/s12192-016-0761-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mymrikov E. V., Seit-Nebi A. S., and Gusev N. B. (2011) Large potentials of small heat shock proteins. Physiol. Rev. 91, 1123–1159 10.1152/physrev.00023.2010 [DOI] [PubMed] [Google Scholar]
- 10. Tyedmers J., Mogk A., and Bukau B. (2010) Cellular strategies for controlling protein aggregation. Nat. Rev. Mol. Cell Biol. 11, 777–788 10.1038/nrm2993 [DOI] [PubMed] [Google Scholar]
- 11. Caspers G. J., Leunissen J. A., and de Jong W. W. (1995) The expanding small heat-shock protein family, and structure predictions of the conserved “α-crystallin domain.” J. Mol. Evol. 40, 238–248 10.1007/BF00163229 [DOI] [PubMed] [Google Scholar]
- 12. de Jong W. W., Leunissen J. A., and Voorter C. E. (1993) Evolution of the α-crystallin/small heat-shock protein family. Mol. Biol. Evol. 10, 103–126 10.1093/oxfordjournals.molbev.a039992 [DOI] [PubMed] [Google Scholar]
- 13. Basha E., Jones C., Blackwell A. E., Cheng G., Waters E. R., Samsel K. A., Siddique M., Pett V., Wysocki V., and Vierling E. (2013) An unusual dimeric small heat shock protein provides insight into the mechanism of this class of chaperones. J. Mol. Biol. 425, 1683–1696 10.1016/j.jmb.2013.02.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bepperling A., Alte F., Kriehuber T., Braun N., Weinkauf S., Groll M., Haslbeck M., and Buchner J. (2012) Alternative bacterial two-component small heat shock protein systems. Proc. Natl. Acad. Sci. U.S.A. 109, 20407–20412 10.1073/pnas.1209565109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kim K. P., Yu J. H., Park S. M., Koo H. J., and Hong C. B. (2011) Tobacco mitochondrial small heat shock protein NtHSP24.6 adopts a dimeric configuration and has a broad range of substrates. BMB Rep. 44, 816–820 10.5483/BMBRep.2011.44.12.816 [DOI] [PubMed] [Google Scholar]
- 16. Delbecq S. P., and Klevit R. E. (2013) One size does not fit all: The oligomeric states of αB crystallin. FEBS Lett. 587, 1073–1080 10.1016/j.febslet.2013.01.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. van Montfort R. L., Basha E., Friedrich K. L., Slingsby C., and Vierling E. (2001) Crystal structure and assembly of a eukaryotic small heat shock protein. Nat. Struct. Biol. 8, 1025–1030 10.1038/nsb722 [DOI] [PubMed] [Google Scholar]
- 18. Hanazono Y., Takeda K., Oka T., Abe T., Tomonari T., Akiyama N., Aikawa Y., Yohda M., and Miki K. (2013) Nonequivalence observed for the 16-meric structure of a small heat shock protein, SpHsp16.0, from Schizosaccharomyces pombe. Structure 21, 220–228 10.1016/j.str.2012.11.015 [DOI] [PubMed] [Google Scholar]
- 19. Hanazono Y., Takeda K., Yohda M., and Miki K. (2012) Structural studies on the oligomeric transition of a small heat shock protein, StHsp14.0. J. Mol. Biol. 422, 100–108 10.1016/j.jmb.2012.05.017 [DOI] [PubMed] [Google Scholar]
- 20. Kim K. K., Kim R., and Kim S. H. (1998) Crystal structure of a small heat-shock protein. Nature 394, 595–599 10.1038/29106 [DOI] [PubMed] [Google Scholar]
- 21. Fleckenstein T., Kastenmüller A., Stein M. L., Peters C., Daake M., Krause M., Weinfurtner D., Haslbeck M., Weinkauf S., Groll M., and Buchner J. (2015) The chaperone activity of the developmental small heat shock protein Sip1 is regulated by pH-dependent conformational changes. Mol. Cell 58, 1067–1078 10.1016/j.molcel.2015.04.019 [DOI] [PubMed] [Google Scholar]
- 22. Mani N., Bhandari S., Moreno R., Hu L., Prasad B. V., and Suguna K. (2016) Multiple oligomeric structures of a bacterial small heat shock protein. Sci. Rep. 6, 24019 10.1038/srep24019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Shi J., Koteiche H. A., McDonald E. T., Fox T. L., Stewart P. L., and McHaourab H. S. (2013) Cryoelectron microscopy analysis of small heat shock protein 16.5 (Hsp16.5) complexes with T4 lysozyme reveals the structural basis of multimode binding. J. Biol. Chem. 288, 4819–4830 10.1074/jbc.M112.388132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. White H. E., Orlova E. V., Chen S., Wang L., Ignatiou A., Gowen B., Stromer T., Franzmann T. M., Haslbeck M., Buchner J., and Saibil H. R. (2006) Multiple distinct assemblies reveal conformational flexibility in the small heat shock protein Hsp26. Structure 14, 1197–1204 10.1016/j.str.2006.05.021 [DOI] [PubMed] [Google Scholar]
- 25. White H. E., Saibil H. R., Ignatiou A., and Orlova E. V. (2004) Recognition and separation of single particles with size variation by statistical analysis of their images. J. Mol. Biol. 336, 453–460 10.1016/j.jmb.2003.12.015 [DOI] [PubMed] [Google Scholar]
- 26. Aquilina J. A., Benesch J. L. P., Ding L. L., Yaron O., Horwitz J., and Robinson C. V. (2005) Subunit exchange of polydisperse proteins: Mass spectrometry reveals consequences of αA-crystallin truncation. J. Biol. Chem. 280, 14485–14491 10.1074/jbc.M500135200 [DOI] [PubMed] [Google Scholar]
- 27. Augusteyn R. C. (2004) Dissociation is not required for α-crystallin's chaperone function. Exp. Eye Res. 79, 781–784 10.1016/j.exer.2004.08.010 [DOI] [PubMed] [Google Scholar]
- 28. Franzmann T. M., Wühr M., Richter K., Walter S., and Buchner J. (2005) The activation mechanism of Hsp26 does not require dissociation of the oligomer. J. Mol. Biol. 350, 1083–1093 10.1016/j.jmb.2005.05.034 [DOI] [PubMed] [Google Scholar]
- 29. Shashidharamurthy R., Koteiche H. A., Dong J., and McHaourab H. S. (2005) Mechanism of chaperone function in small heat shock proteins: Dissociation of the HSP27 oligomer is required for recognition and binding of destabilized T4 lysozyme. J. Biol. Chem. 280, 5281–5289 10.1074/jbc.M407236200 [DOI] [PubMed] [Google Scholar]
- 30. Giese K. C., and Vierling E. (2002) Changes in oligomerization are essential for the chaperone activity of a small heat shock protein in vivo and in vitro. J. Biol. Chem. 277, 46310–46318 10.1074/jbc.M208926200 [DOI] [PubMed] [Google Scholar]
- 31. Haslbeck M., Walke S., Stromer T., Ehrnsperger M., White H. E., Chen S., Saibil H. R., and Buchner J. (1999) Hsp26: A temperature-regulated chaperone. EMBO J. 18, 6744–6751 10.1093/emboj/18.23.6744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Benesch J. L. P., Aquilina J. A., Baldwin A. J., Rekas A., Stengel F., Lindner R. A., Basha E., Devlin G. L., Horwitz J., Vierling E., Carver J. A., and Robinson C. V. (2010) The quaternary organization and dynamics of the molecular chaperone HSP26 are thermally regulated. Chem. Biol. 17, 1008–1017 10.1016/j.chembiol.2010.06.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Friedrich K. L., Giese K. C., Buan N. R., and Vierling E. (2004) Interactions between small heat shock protein subunits and substrate in small heat shock protein-substrate complexes. J. Biol. Chem. 279, 1080–1089 10.1074/jbc.M311104200 [DOI] [PubMed] [Google Scholar]
- 34. Painter A. J., Jaya N., Basha E., Vierling E., Robinson C. V., and Benesch J. L. P. (2008) Real-time monitoring of protein complexes reveals their quaternary organization and dynamics. Chem. Biol. 15, 246–253 10.1016/j.chembiol.2008.01.009 [DOI] [PubMed] [Google Scholar]
- 35. Sobott F., Benesch J. L. P., Vierling E., and Robinson C. V. (2002) Subunit exchange of multimeric protein complexes. Real-time monitoring of subunit exchange between small heat shock proteins by using electrospray mass spectrometry. J. Biol. Chem. 277, 38921–38929 10.1074/jbc.M206060200 [DOI] [PubMed] [Google Scholar]
- 36. Bova M. P., McHaourab H. S., Han Y., and Fung B. K. (2000) Subunit exchange of small heat shock proteins. Analysis of oligomer formation of αA-crystallin and Hsp27 by fluorescence resonance energy transfer and site-directed truncations. J. Biol. Chem. 275, 1035–1042 10.1074/jbc.275.2.1035 [DOI] [PubMed] [Google Scholar]
- 37. Baldwin A. J., Lioe H., Robinson C. V., Kay L. E., and Benesch J. L. P. (2011) αB-crystallin polydispersity is a consequence of unbiased quaternary dynamics. J. Mol. Biol. 413, 297–309 10.1016/j.jmb.2011.07.016 [DOI] [PubMed] [Google Scholar]
- 38. Zhang K., Ezemaduka A. N., Wang Z., Hu H., Shi X., Liu C., Lu X., Fu X., Chang Z., and Yin C. C. (2015) A novel mechanism for small heat shock proteins to function as molecular chaperones. Sci. Rep. 5, 8811 10.1038/srep08811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Jaya N., Garcia V., and Vierling E. (2009) Substrate binding site flexibility of the small heat shock protein molecular chaperones. Proc. Natl. Acad. Sci. U.S.A. 106, 15604–15609 10.1073/pnas.0902177106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Waters E. R. (2013) The evolution, function, structure, and expression of the plant sHSPs. J. Exp. Bot. 64, 391–403 10.1093/jxb/ers355 [DOI] [PubMed] [Google Scholar]
- 41. Basha E., Jones C., Wysocki V., and Vierling E. (2010) Mechanistic differences between two conserved classes of small heat shock proteins found in the plant cytosol. J. Biol. Chem. 285, 11489–11497 10.1074/jbc.M109.074088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kirschner M., Winkelhaus S., Thierfelder J. M., and Nover L. (2000) Transient expression and heat-stress-induced co-aggregation of endogenous and heterologous small heat-stress proteins in tobacco protoplasts. Plant J. 24, 397–411 10.1046/j.1365-313x.2000.00887.x [DOI] [PubMed] [Google Scholar]
- 43. Tiroli A. O., and Ramos C. H. (2007) Biochemical and biophysical characterization of small heat shock proteins from sugarcane. Involvement of a specific region located at the N-terminus with substrate specificity. Int. J. Biochem. Cell Biol. 39, 818–831 10.1016/j.biocel.2007.01.014 [DOI] [PubMed] [Google Scholar]
- 44. Stengel F., Baldwin A. J., Painter A. J., Jaya N., Basha E., Kay L. E., Vierling E., Robinson C. V., and Benesch J. L. P. (2010) Quaternary dynamics and plasticity underlie small heat shock protein chaperone function. Proc. Natl. Acad. Sci. U.S.A. 107, 2007–2012 10.1073/pnas.0910126107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Benesch J. L. P., Sobott F., and Robinson C. V. (2003) Thermal dissociation of multimeric protein complexes by using nanoelectrospray mass spectrometry. Anal. Chem. 75, 2208–2214 10.1021/ac034132x [DOI] [PubMed] [Google Scholar]
- 46. Stengel F., Baldwin A. J., Bush M. F., Hilton G. R., Lioe H., Basha E., Jaya N., Vierling E., and Benesch J. L. P. (2012) Dissecting heterogeneous molecular chaperone complexes using a mass spectrum deconvolution approach. Chem. Biol. 19, 599–607 10.1016/j.chembiol.2012.04.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hochberg G. K. A., Shepherd D. A., Marklund E. G., Santhanagoplan I., Degiacomi M. T., Laganowsky A., Allison T. M., Basha E., Marty M. T., Galpin M. R., Struwe W. B., Baldwin A. J., Vierling E., and Benesch J. L. P. (2018) Structural principles that enable oligomeric small heat-shock protein paralogs to evolve distinct functions. Science 359, 930–935 10.1126/science.aam7229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kennaway C. K., Benesch J. L. P., Gohlke U., Wang L., Robinson C. V., Orlova E. V., Saibi H. R., and Keep N. H. (2005) Dodecameric structure of the small heat shock protein Acr1 from Mycobacterium tuberculosis. J. Biol. Chem. 280, 33419–33425 10.1074/jbc.M504263200 [DOI] [PubMed] [Google Scholar]
- 49. Fischer H., Polikarpov I., and Craievich A. F. (2004) Average protein density is a molecular-weight-dependent function. Protein Sci. 13, 2825–2828 10.1110/ps.04688204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Cheng G., Basha E., Wysocki V. H., and Vierling E. (2008) Insights into small heat shock protein and substrate structure during chaperone action derived from hydrogen/deuterium exchange and mass spectrometry. J. Biol. Chem. 283, 26634–26642 10.1074/jbc.M802946200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Basha E., Friedrich K. L., and Vierling E. (2006) The N-terminal arm of small heat shock proteins is important for both chaperone activity and substrate specificity. J. Biol. Chem. 281, 39943–39952 10.1074/jbc.M607677200 [DOI] [PubMed] [Google Scholar]
- 52. Baldwin A. J., Lioe H., Hilton G. R., Baker L. A., Rubinstein J. L., Kay L. E., and Benesch J. L. P. (2011) The polydispersity of αB-crystallin is rationalized by an interconverting polyhedral architecture. Structure 19, 1855–1863 10.1016/j.str.2011.09.015 [DOI] [PubMed] [Google Scholar]
- 53. Baldwin A. J., Hilton G. R., Lioe H., Bagnéris C., Benesch J. L. P., and Kay L. E. (2011) Quaternary dynamics of αB-crystallin as a direct consequence of localised tertiary fluctuations in the C-terminus. J. Mol. Biol. 413, 310–320 10.1016/j.jmb.2011.07.017 [DOI] [PubMed] [Google Scholar]
- 54. Stromer T., Ehrnsperger M., Gaestel M., and Buchner J. (2003) Analysis of the interaction of small heat shock proteins with unfolding proteins. J. Biol. Chem. 278, 18015–18021 10.1074/jbc.M301640200 [DOI] [PubMed] [Google Scholar]
- 55. Patel S., Vierling E., and Tama F. (2014) Replica exchange molecular dynamics simulations provide insight into substrate recognition by small heat shock proteins. Biophys. J. 106, 2644–2655 10.1016/j.bpj.2014.04.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. McHaourab H. S., Godar J. A., and Stewart P. L. (2009) Structure and mechanism of protein stability sensors: Chaperone activity of small heat shock proteins. Biochemistry 48, 3828–3837 10.1021/bi900212j [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Fu X., Zhang H., Zhang X., Cao Y., Jiao W., Liu C., Song Y., Abulimiti A., and Chang Z. (2005) A dual role for the N-terminal region of Mycobacterium tuberculosis Hsp16.3 in self-oligomerization and binding denaturing substrate proteins. J. Biol. Chem. 280, 6337–6348 10.1074/jbc.M406319200 [DOI] [PubMed] [Google Scholar]
- 58. Haslbeck M., Ignatiou A., Saibil H., Helmich S., Frenzl E., Stromer T., and Buchner J. (2004) A domain in the N-terminal part of Hsp26 is essential for chaperone function and oligomerization. J. Mol. Biol. 343, 445–455 10.1016/j.jmb.2004.08.048 [DOI] [PubMed] [Google Scholar]
- 59. Plater M. L., Goode D., and Crabbe M. J. (1996) Effects of site-directed mutations on the chaperone-like activity of αB-crystallin. J. Biol. Chem. 271, 28558–28566 10.1074/jbc.271.45.28558 [DOI] [PubMed] [Google Scholar]
- 60. Hochberg G. K., Ecroyd H., Liu C., Cox D., Cascio D., Sawaya M. R., Collier M. P., Stroud J., Carver J. A., Baldwin A. J., Robinson C. V., Eisenberg D. S., Benesch J. L. P., and Laganowsky A. (2014) The structured core domain of αB-crystallin can prevent amyloid fibrillation and associated toxicity. Proc. Natl. Acad. Sci. U.S.A. 111, E1562–E1570 10.1073/pnas.1322673111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Zhao L., Chen X. J., Zhu J., Xi Y. B., Yang X., Hu L. D., Ouyang H., Patel S. H., Jin X., Lin D., Wu F., Flagg K., Cai H., Li G., Cao G., et al. (2015) Lanosterol reverses protein aggregation in cataracts. Nature 523, 607–611 10.1038/nature14650 [DOI] [PubMed] [Google Scholar]
- 62. Makley L. N., McMenimen K. A., DeVree B. T., Goldman J. W., McGlasson B. N., Rajagopal P., Dunyak B. M., McQuade T. J., Thompson A. D., Sunahara R., Klevit R. E., Andley U. P., and Gestwicki J. E. (2015) Pharmacological chaperone for α-crystallin partially restores transparency in cataract models. Science 350, 674–677 10.1126/science.aac9145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Tina K. G., Bhadra R., and Srinivasan N. (2007) PIC: Protein Interactions Calculator. Nucleic Acids Res. 35, W473–W476 10.1093/nar/gkm423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Dani V. S., Ramakrishnan C., and Varadarajan R. (2003) MODIP revisited: Re-evaluation and refinement of an automated procedure for modeling of disulfide bonds in proteins. Protein Eng. 16, 187–193 10.1093/proeng/gzg024 [DOI] [PubMed] [Google Scholar]
- 65. Indu S., Kochat V., Thakurela S., Ramakrishnan C., and Varadarajan R. (2011) Conformational analysis and design of cross-strand disulfides in antiparallel β-sheets. Proteins 79, 244–260 10.1002/prot.22878 [DOI] [PubMed] [Google Scholar]
- 66. Kondrat F. D., Struwe W. B., and Benesch J. L. P. (2015) Native mass spectrometry: Towards high-throughput structural proteomics. Methods Mol. Biol. 1261, 349–371 10.1007/978-1-4939-2230-7_18 [DOI] [PubMed] [Google Scholar]
- 67. Bush M. F., Hall Z., Giles K., Hoyes J., Robinson C. V., and Ruotolo B. T. (2010) Collision cross sections of proteins and their complexes: A calibration framework and database for gas-phase structural biology. Anal. Chem. 82, 9557–9565 10.1021/ac1022953 [DOI] [PubMed] [Google Scholar]
- 68. Petoukhov M. V., Franke D., Shkumatov A. V., Tria G., Kikhney A. G., Gajda M., Gorba C., Mertens H. D., Konarev P. V., and Svergun D. I. (2012) New developments in the ATSAS program package for small-angle scattering data analysis. J. Appl. Crystallogr. 45, 342–350 10.1107/S0021889812007662 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.