

Abstract
Systemic light-chain amyloidosis (AL) is a human disease caused by overexpression of monoclonal immunoglobulin light chains that form pathogenic amyloid fibrils. These amyloid fibrils deposit in tissues and cause organ failure. Proteins form amyloid fibrils when they partly or fully unfold and expose segments capable of stacking into β-sheets that pair and thereby form a tight, dehydrated interface. These structures, termed steric zippers, constitute the spines of amyloid fibrils. Here, using a combination of computational (with ZipperDB and Boston University ALBase), mutational, biochemical, and protein structural analyses, we identified segments within the variable domains of Ig light chains that drive the assembly of amyloid fibrils in AL. We demonstrate that there are at least two such segments and that each one can drive amyloid fibril assembly independently of the other. Our analysis revealed that peptides derived from these segments form steric zippers featuring a typical dry interface with high-surface complementarity and occupy the same spatial location of the Greek-key immunoglobulin fold in both λ and κ variable domains. Of note, some predicted steric-zipper segments did not form amyloid fibrils or assembled into fibrils only when removed from the whole protein. We conclude that steric-zipper propensity must be experimentally validated and that the two segments identified here may represent therapeutic targets. In addition to elucidating the molecular pathogenesis of AL, these findings also provide an experimental approach for identifying segments that drive fibril formation in other amyloid diseases.
Keywords: amyloid, antibody, crystallography, electron microscopy (EM), protein aggregation, light chain (LC), light-chain amyloidosis (AL), steric zipper, thioflavin T (ThT), variable domain (VL)
Introduction
Systemic light-chain amyloidosis (AL)3 is a lethal disease caused by excess immunoglobulin light chains (1). Each year, ∼4,000 patients in the United States and 1,500 in the United Kingdom are diagnosed with AL. However, recent reports indicate systemic amyloidosis is underdiagnosed (2–4). AL is frequently but not always associated with multiple myeloma (5–8). In AL, plasma cells produce excess monoclonal full-length light chains, or their variable domain segments, which circulate in the blood and assemble into pathologic amyloid fibrils. Although the sequences of light chains vary among patients, the molecular progression of the disease is similar: insoluble and degradation-resistant amyloid fibrils deposit in essential tissues and cause organ failure (9–12).
In AL, the amyloid fibrils contain full-length light chains or just their variable domains. Overexpressed light chains (LCs) form dimers of identical LCs rather than the normal pairing of LCs with Ig heavy chains. LCs consist of two domains, variable (VL) and constant (CL), which are covalently connected by a joining segment (J). The amino acid sequences of LCs are determined by somatic gene recombination and are of two classes, λ or κ (13, 14). In AL, the amyloid fibrils include either full-length LCs (VL–J–CL) or VLs, yet the ubiquitous presence of VLs indicates that this domain may be the minimal and essential unit for fibril assembly (15–17).
Amyloid fibrils exhibit common biochemical and structural properties. They are self-propagating, insoluble in aqueous solutions, unusually resistant to degradation, and bind the fluorescent dye thioflavin T (ThT). Molecular structures of amyloid fibrils reveal pairs of tightly mated β-sheets, termed “steric zippers.” Upon exposure to X-rays, the spine gives rise to a cross-β diffraction pattern (8, 9, 18, 19). The interdigitating side chains of segments form mating sheets with a dry, highly complementary interface, which serves as a scaffold for fibril extension (20). Once a steric zipper has formed, hydrogen bonds between the main chains propagate the spine of amyloid fibrils, whereas the dry interface contributes to adhesion of the mating sheets.
Proteins contain putative steric zipper–forming segments. The propensity of a segment to form a steric zipper can be estimated from its sequence (21). In globular proteins, segments predicted to form steric zippers are often buried in the interior and protected from forming fibrils. However, upon full or partial unfolding of the protein, these segments become exposed to solvent and can stack to form the amyloid fibril spine (22). Proteins known to assemble into amyloid fibrils may contain multiple segments capable of forming steric zippers. In the absence of experiments, it is unclear whether every, some, or only one of these segments is responsible for driving fibril formation. If multiple such segments can drive fibril formation independently, the protein could form amyloid fibrils having different spines and thus different structures (Fig. 1). The different fibril structures formed by the same protein are known as amyloid polymorphs (23–26).
Figure 1.
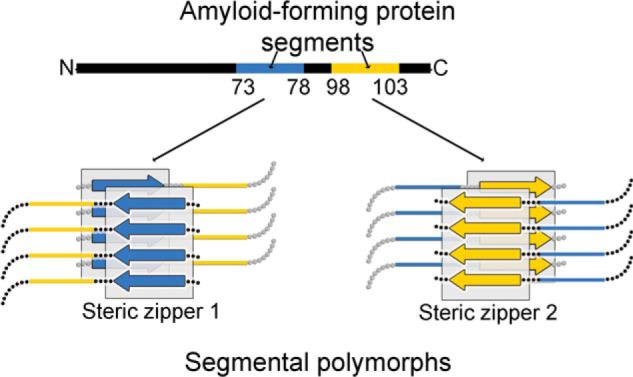
Segmental polymorphism. Amyloid fibrils form through adhesive segments that stack to form steric zippers. Ig VLs contain two segments capable of forming steric-zipper fibril spines, shown here schematically in blue and yellow. Each one of the steric zipper-forming segments can induce assembly of a particular amyloid polymorph with its distinctive spine.
Here, we seek the molecular basis of the formation of amyloid fibrils from Ig light chains of both λ and κ families. The Human Gene Nomenclature Committee lists 33 functional genes for λ and 38 for κ variable domains (27–29). The repertoire of VLs is greatly expanded by the linkage of λ and κ domains to a variety of J-segments and constant domains. Consequently the Amyloid Light Chain Database lists 616 λ and 192 κ VLs (17). Sequence alignments of these VLs involved in AL do not reveal a single residue or segment that exclusively accounts for their amyloidogenic property, and differences in amino acids between VLs within the same family, λ or κ, affect the propensity to form amyloid fibrils (30–32). Hence, the identification of the amyloidogenic segments of VLs is challenging.
We define an amyloidogenic segment by two criteria. 1) Introduction of amyloid-inhibiting residues, such as proline, into the segment by site-directed mutagenesis halts fibril formation of the parent VL. 2) The segment in isolation from the remainder of the VL forms a steric zipper.
Our search for amyloidogenic segments focuses on a human genomic reference VL sequence and two patient-derived VL sequences, one κ and one λ. We found two amyloidogenic segments in VLs and verified that in combination they fulfill criterion 1 by site-directed mutagenesis. Each of the two amyloidogenic segments independently causes amyloid fibril formation. Study of the isolated peptides shows that each segment also fulfills criterion 2 because each can form a steric zipper. By sequence comparison, we find that homologs of these segments are amyloidogenic in both λ and κ families of LCs and are the key propagators of fibril formation, thereby generalizing our model of amyloid formation to other LCs and suggesting these two LC segments as therapeutic targets.
Results
Finding amyloid-driving segments
To identify the amyloidogenic steric zippers within VLs, we employed two complementary approaches: a computational assessment using ZipperDB to identify candidate segments with a high propensity to form steric zippers, and experimental mutagenesis to inhibit the ability of candidates to form amyloid fibrils, as detected by ThT assays (33). We identified a genomic variant of a λ VL2-8 with J1-connecting segment (VL2-8–J1) as a useful reference model for our experiments; upon exposure to destabilizing conditions, VL2-8–J1 forms amyloid fibrils (Fig. 2).
Figure 2.

Two assays of amyloid fibril formation of the reference sequence VL2-8–J1 and its amyloid fibril-inhibiting mutation L75P: ThT fluorescence assay and electron micrographs. The ThT assay shows fluorescence, indicative of formation of amyloid fibrils (A) and electron micrographs that visualize them (B). Upon exposure of VL2-8–J1 to destabilizing conditions, the protein forms amyloid fibrils. In contrast, the mutated variant, VLJ2-8–J1-L75P, under the same conditions does not show an increase in ThT fluorescence or display amyloid fibrils in electron micrographs.
Within VL2-8–J1, ZipperDB (https://services.mbi.ucla.edu/zipperdb/)4 (58) identified five high-propensity steric zipper–forming regions, with each region containing conserved residues (Fig. 3, regions A–E). ZipperDB ranks the relative stabilities of the putative zipper structures; however, the potential of a segment to drive amyloid formation depends on additional factors such as environmental conditions and the shielding of the segment by neighboring parts of the sequence. Hence, we performed site-directed mutagenesis to experimentally determine the degree of involvement of these segments in formation of amyloid fibrils. We replaced conserved amino acids with proline residues. Prolines introduce geometric strain into β-sheets and eliminate a main-chain hydrogen bond, thus destabilizing the amyloid spine and impeding formation of amyloid fibrils (34). We sequentially replaced every amino acid with proline within each of the five predicted steric zipper–rich regions A–E. A single mutation, L75P in segment C, stopped formation of amyloid fibrils by the reference sequence, VL2-8–J1, whereas prolines in other positions did not.
Figure 3.

Regions predicted to contain segments capable of forming steric zippers in a genomic reference sequence VL2-8–J1. Purple and blue colors of the amino acid sequence show conserved and variable amino acids (ConSurf, http://consurf.tau.ac.il). ZipperDB (https://services.mbi.ucla.edu/zipperdb/) (58) identified five major, high-propensity steric-zipper regions that coincide with conserved amino acids. (Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.) The predicted steric zippers were the initial focus of our site-directed mutagenesis experiments. The vertical scale to the left of the histogram shows the calculated energy gain upon formation of a steric zipper. The horizontal gray line shows the energetic gain of −23 kcal/mol. Each histogram bar refers to a 6-residue segment, and orange and red bars represent segments with a high propensity to form steric zippers. Gaps in the calculated propensities arise due to the presence of prolines, which impede ZipperDB calculations. The prediction shows five main regions: A includes residues 4–24; B, 37–50; C, 56–81; D, 84–95; and E, 102–111.
Upon identification of the amyloid-inhibiting proline mutation VL2-8–J1-L75P, we examined whether residue Leu-75 is required for formation of amyloid fibrils by other pathologic VLs: a λ-type VL (Mcg) and a κ-type VL (AL09) (35, 36). Both Mcg and AL09 are specific VL variants isolated from patients afflicted with AL. The same site-directed mutation, L75P, did not stop amyloid fibril formation by Mcg (Fig. 2). The opposite effects of the L75P mutation in two different VL types provide an important insight: a single steric zipper is not responsible for formation of amyloid fibrils by all VL types. This insight led us to hypothesize that VLs may form different polymorphs, and there may be more than a single segment that can independently induce formation of amyloid fibrils.
Finding a second amyloid-driving segment
The inability of the L75P to stop amyloid fibril formation by Mcg led us to perform a “proline-scan” experiment. With the aim of identifying segments required for amyloid fibril formation, we introduced proline mutations in four consecutive residues in each of 28 separate constructs that span the entire polypeptide sequence of Mcg, except that we did not replace either of the two structural cysteines with proline (Fig. 4). Table 1 summarizes the results of the proline-scan experiment: all 28 mutated constructs consistently formed amyloid fibrils, except for construct-25, which contains mutations in region E (residues F99P/V100P/F101P/G102P), in which several segments with a ZipperDB-predicted high propensity for amyloid formation are located. For this construct, several independent batches formed amyloid fibrils, whereas others did not. These data suggest that at least for Mcg, no single steric zipper individually accounts for the amyloid-forming property of VLs, and several different polymorphs may exist. When combined, L75P and F99P/V100P/F101P/G102P mutations abolished the ability of Mcg to form amyloid fibrils (Figs. 5 and 6). To summarize, every Mcg construct with tetra-proline mutations covering the entire VL sequence forms amyloid fibrils. However, the Mcg with L75P and F99P/V100P/F101P/G102P together does not form fibrils. These observations support our conjecture that no single steric zipper fully accounts for formation of amyloid fibrils by VLs; rather, at least two segments drive formation of steric zippers.
Figure 4.

Predictions based on proline scanning of steric-zipper propensity for segments of the Mcg sequence from an AL patient. The Mcg sequence is shown on top of the histogram. Four-residue segments of the proline-scanning, site-directed mutagenesis experiment are shown in black bars below the energy histogram. Peptide-scanning was performed by mutating four consecutive residues in each segment to proline. The mutated residues are identified at the bottom of the histogram and represent individual constructs each containing a tetra-proline replacement. No single tetra-proline mutation inhibited amyloid fibril formation of Mcg, but constructs L75P and 25 together did. Red arrows identify amino acids that were identified as involved in formation of amyloid fibrils.
Table 1.
Effects of site-directed mutations on amyloid fibril formation of three different VLs
The tripartite experiment comprises the following: 1) an amyloid-inhibiting mutation in a reference variant of a λ VL; 2) a λ variant from a patient; and 3) a κ variant from another patient. The main conclusion is that there are two amyloidogenic segments in VLs capable of driving formation of amyloid fibrils.
| Variant or mutant | Increase in ThT fluorescence | Detection of fibrils in electron micrographs | Ability of the site-directed mutation to stop amyloid fibrils |
|---|---|---|---|
| 1) Genomic λ variable domain | |||
| VL2-8–J1 | + | + | |
| VL2-8–J1–L75P | − | − | Positive |
| 2) λ variable domain from an AL patient | |||
| Mcg | + | + | |
| Mcg-L75P | + | + | Negative (1 segment) |
| Mcg-Phe99–Val100–Phe101–Gly102 | +/−a | +/−a | Inconclusive (2 segments) |
| Mcg-L75P–Phe99–Val100–Phe101–Gly102 | − | − | Positive (2 segments) |
| All other constructs | + | + | Negative (2 segments) |
| 3) κ variable domain from an AL patient | |||
| AL09 | + | + | |
| AL09-F73P | + | + | Negative |
| AL09-Y96P/T97P/F98P/G99P | + | + | Negative |
| AL09-F73P/Y96P/T97P/F98P/G99P | − | − | Positive |
a +/− indicates that some batches formed amyloid fibrils, and others did not. Three batches formed fibrils, and two did not. All other mutants were tested in at least three batch-independent repetitions.
Figure 5.

Negative-staining electron micrographs of amyloid fibrils formed by Mcg and AL09 sequences. The top row shows the effect of mutations in Mcg on the ability to form amyloid fibrils. Simultaneous mutations in two different segments abolish the ability to form amyloid fibrils. The bottom row shows the same effect of the corresponding mutations in AL09. The amyloid fibril inhibiting mutations are underlined. Black scale bars represent distances of 0.5 μm.
Figure 6.
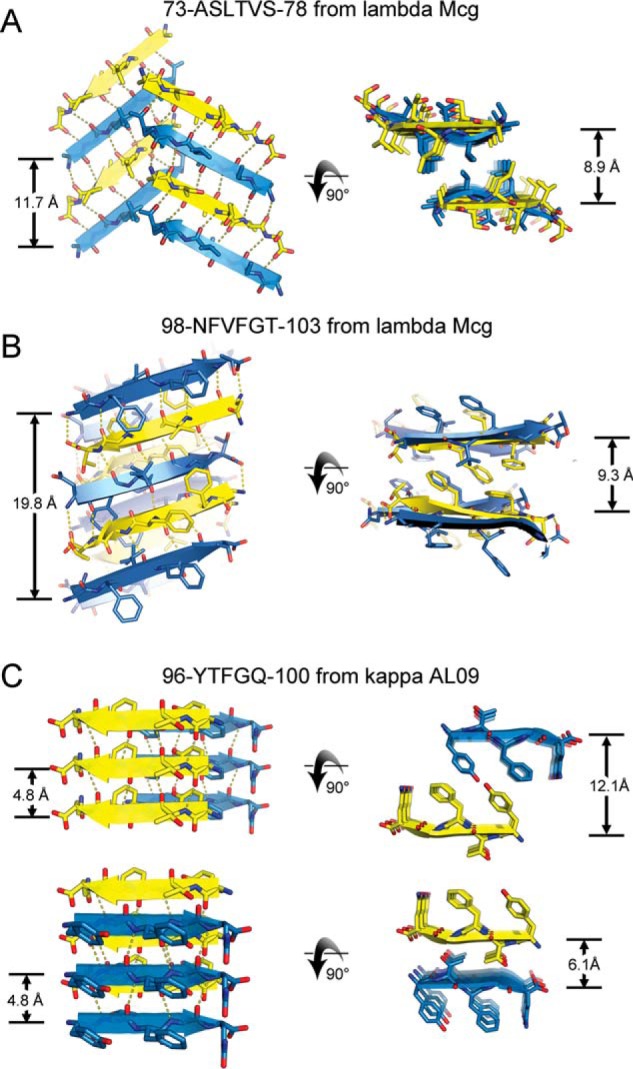
Crystal structures of VL segments that drive the assembly of amyloid fibrils. Each of the three segments adopted a steric-zipper motif, characterized by pairs of sheets, tightly mated by the interdigitation of side chains across the interface and exclusion of water molecules. For each segment, two views are given: fibril axis vertical (left column) and fibril axis normal to the page (right column). Sheet architecture and symmetry differ among the three segments as follows. A, segment 1 of Mcg, ASLTVS. Sheet architecture is anti-parallel, antifacial, and out-of-register (class 5). The translational repeat distance along the fibril axis is 11.7 Å. B, segment 2 of Mcg, NFVFGT. Each sheet contains four orientations of strands in the asymmetric unit, creating a level of complexity beyond any of the 10 previously described steric-zipper symmetry classes. The sheet is out-of-register, with an unusually long translation repeat distance of 19.8 Å. C, segment 2 of AL09, YTFGQ. Sheet architecture is parallel, in-register, with a conventional translational repeat distance of 4.8 Å. Two types of dry interfaces are evident in the crystal packing: face-to-face and back-to-back (both class 1). Distances between sheets are given in the right column.
To verify whether these two segments are essential for formation of amyloid fibrils by another VL type, we introduced the corresponding mutations into a well-studied, patient-derived pathologic κ VL, AL09 (37). Sequence alignment of AL09 and Mcg identified the corresponding mutation sites: Mcg-L75P corresponds to AL09-F73P, and Mcg-F99P/V100P/F101P/G102P corresponds to AL09-Y96P/T97P/F98P/G99P. As with Mcg, only the combined mutant, AL09-F73P/Y96P/T97P/F98P/G99P, stopped amyloid fibril formation; the individual mutations did not affect the amyloidogenic potential of AL09 (Fig. 5). The reproducibility of results in both types of VLs, λ and κ, corroborates the involvement of two segments in formation of VL amyloid fibrils.
Atomic structures of amyloid-driving segments
Crystal structures of individual peptides derived from amyloidogenic segments, two from the λ VL Mcg (ASLTVS and NFVFGT) and one from the κ AL09 (YTFGQ), reveal that these peptides form steric zippers (Fig. 6). Each of these steric zippers features the typical dry interface with high-surface complementarity. The crystals of the fourth segment, EFTFTIS from κ AL09, were of insufficient resolution to resolve its atomic structure. However, cylindrical averaging of the single-crystal diffraction data shows the characteristic reflections of steric zippers (Fig. 7), as do the fibril diffraction patterns calculated from the atomic coordinates of the other three.
Figure 7.

Fibril diffraction patterns (left column) and their radial integration profiles (right column) for amyloidogenic segments. The fibril diffraction patterns are calculated from the crystal structures of the segments (Fig. 6). The radial integration profiles are calculated by cylindrical averaging of these single crystal diffraction patterns. These patterns show reinforcement of reflections in the vicinity of 4.8 and 10 Å, characteristic of steric zippers. In the right column, |Avg(I)| is the average intensity calculated from a Fourier transform of the atomic structures. A, powder diffraction of ASLTVS. The meridional reflection near 4.8 Å is split due to inclined network of hydrogen bonds of the steric-zipper spine. B, powder diffraction of NFVFGT. C, powder diffraction of YTFGQ. D, cylindrical averaging of single-crystal diffraction data of amyloidogenic segment EFTFTIS from κ AL09. The averaged diffraction data of EFTFTIS show strong reflections near 4.8 and 10 Å, perpendicular to each other. These reflections show the amyloid nature of the structure. The meridional reflection near 9.6 Å indicates that the structure contains an anti-parallel amyloid spine.
Our three crystal structures of VL amyloidogenic segments reveal steric zippers belonging to different symmetry classes (20). Symmetry classes define the spatial relationship between identical strands in the zipper. The steric zipper of segment YTFGQ from AL09 is composed of pairs of parallel sheets mated face-to-face, identifying it as class 1 (Fig. 6C). The same crystal structure also reveals a different pair of sheets mated back-to-back, corresponding to a second polymorph of symmetry class 1. ASLTVS from Mcg crystallizes as anti-parallel, anti-facial β-sheets packed face-to-face (Fig. 6). This symmetry pattern is class 5. Furthermore, its β-sheets are out-of-register (Fig. 8A) because its strands are not perpendicular to the fibril axis, but instead are inclined. The third steric zipper, NFVFGT from Mcg, displays a pattern of symmetry more complex than any previously observed steric zipper. The sheets that compose the 10 previously described steric-zipper symmetry classes can be built from translational repeats of one strand along the fibril axis (classes 1–4) or two strands (classes 5–10). Sheets of NFVFGT contain four orientations of the strand, and so they require a minimum of four strands per translational repeat. The symmetry pattern of this sheet is one of six conceivable unique arrangements possible for a sheet with a four-strand translational repeat (Fig. 8B).
Figure 8.
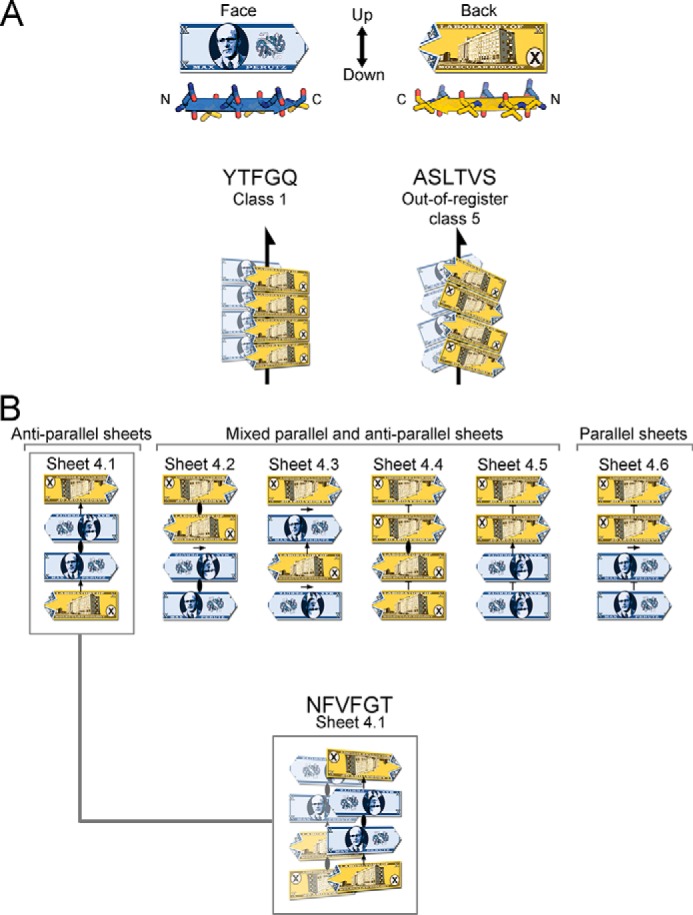
VL steric zipper and sheet geometry of amyloid spines, illustrated schematically with the fictitious Max Perutz banknote. A, Max Perutz banknote represents a protein segment within a steric zipper; it has N and C termini, two distinct faces, and up- and down-direction of hydrogen bonds within a sheet. Arrows represent 21 symmetry axes, meaning that the peptides are related by a 180° rotation about the arrow, and translated one-half of the distance between Max Perutz banknotes along the arrow. The YTFGQ steric zipper belongs to class 1. The ASLTVS steric zipper belongs to class 5. The packing of β-strands is out-of-register with its strands inclined relative to the fibril axis. B, NFVFGT peptide crystallizes in a sheet containing four strands in the asymmetric unit. This sheet belongs to the one of six conceivable arrangements of a sheet with a modulus of four strands and internal symmetry. Arrows represent 2-fold symmetry operators; an ellipse represents a 2-fold symmetry axis perpendicular to the page, and T represents translation.
Genomic analysis of amyloidogenic light chains
Analysis of light chain sequences of AL patients suggests that the segments identified in Mcg and AL09 are the principal amyloid-driving segments in some other individuals with AL. We analyzed protein sequences from individuals with AL, AL/myeloma (597 λ, 186 κ), and a normal repertoire (NR) (158 λ, 401 κ), all obtained from the Boston University ALBase. The protein sequences were aligned with ClustalW. Segments corresponding to segments 1 and 2 of Mcg were isolated from the λ sequences, and segments corresponding to segment 2 of AL09 were isolated from the κ sequences. ZipperDB (https://services.mbi.ucla.edu/zipperdb/)4 (58) was then used to predict the propensity of the segments to form steric zippers. We found distributions of Rosetta energy scores for both λ segments 1 and 2 are significantly different between AL patients and NR individuals (Fig. 9, A and B). Specifically, scores for these segments tend to be lower (i.e. greater propensity to form steric zippers) in AL patients than in NR individuals. This result is based on Wilcoxon rank sum tests (segment 1: U = 26,240, p < 2.2e-16; segment 2: U = 24,574, p = 5.197e-7). We note that the scores are not normally distributed, and the minimum scores are similar for each segment. However, there is a clear shift from a relatively higher frequency of higher scores in NR individuals to a higher frequency of lower scores in AL patients (Fig. 10, A and B). ZipperDB scores for segment 2 also rarely fall below the threshold that predicts a hexa-peptide would form a fibril, suggesting segment 1 may be a more dominant factor in driving the formation of amyloid fibrils of the full-length protein. We found no significant difference in the distribution of κ sequences between AL and NR individuals; however, 227 of the 587 κ sequences were not possible to evaluate with ZipperDB due to the presence of proline residues. In contrast, only 3 of the 753 λ segment 1 sequences and 137 of the 753 λ segment 2 sequences were discarded due to proline residues. Because of the insufficiency of data, analysis of the κ sequences was abandoned. Nevertheless, the λ sequences offer compelling evidence that segments 1 and 2 play an important role in amyloid formation in many AL patients.
Figure 9.

Distribution of Rosetta energy values generated by ZipperDB for λ light chain segment 1 (AL: n = 592, median = −25. 2; NR: n = 158, median = −24.9) (A) and λ light chain segment 2 (AL: n = 474, median = −19.2; NR: n = 142, median = −18.1) for individuals with light-chain AL or NR (B). The dashed red line represents the energetic threshold of −23 kcal/mol. ZipperDB predicts hexa-peptides at or below this threshold to form fibrils. Three data points were omitted from segment 1 AL sequences, zero from segment 1 NR sequences, 121 from segment 2 AL sequences, and 16 from segment 2 NR sequences due to the presence of proline residues or missing sequence data making ZipperDB analysis impossible.
Figure 10.

Rosetta energy values generated by ZipperDB for λ light chain segment 1 (AL: n = 592, median = −25. 2; NR: n = 158, median = −24.9) (A) and λ light chain segment 2 (AL: n = 474, median = −19.2; NR: n = 142, median = −18.1) for individuals with light-chain AL or NR (B). The notches on the boxes represent the 95% confidence interval around the median (± 1.57 × IQR√ of n). Although not a formal test, if the notches of two boxes do not overlap, it is evidence that their medians differ significantly. A Wilcoxon-Mann-Whitney test was performed on each pair, and the median Rosetta energies of the AL group was significantly lower than that of the NR group in both λ segments (A, U = 26240, p < 2.2e-16; B, U = 24574, p = 5.179e-7). Three data points were omitted from segment 1 AL sequences, zero from segment 1 NR sequences, 121 from segment 2 AL sequences, and 16 from segment 2 NR sequences due to the presence of proline residues or missing sequence data making ZipperDB analysis impossible.
Discussion
Our results confirm that computational prediction of steric-zipper propensity must be validated by experiments to correctly identify amyloidogenic segments. Computational prediction suggested several steric zippers within VLs, but site-directed mutagenesis verified only two such segments. We found that both are amyloidogenic independently of each other. Some of the other predicted high-propensity steric-zipper segments do indeed form amyloid fibrils, but only in isolation from the rest of the protein. Specifically, in the context of a full-length protein, not all predicted steric zippers become exposed and available to drive formation of amyloid fibrils. Through a combination of computational, biochemical, and structural work, we have demonstrated the involvement of two distinct amyloidogenic segments in the formation of VL amyloid fibrils.
Although we performed ThT assays under acidic conditions that facilitate a feasible time scale to assess the formation of amyloid fibrils, we show that at least one of the variants, VL2-8–J1, also forms amyloid fibrils at nearly neutral pH (Fig. S1). Other variants, such as AL09, may require a longer time frame, perhaps months or years, to demonstrate formation of amyloid fibril formation at neutral pH. The investigation of variable domain variants, Mcg and AL09, with sequences previously isolated from patients with systemic light-chain amyloidosis, supports the clinical relevance of our findings.
Our conclusions are consistent with previous solid-state nuclear magnetic resonance (ssNMR) experiments. ssNMR experiments identified several segments having immobile conformations in VL amyloid fibrils, suggesting that some of these segments are involved in the formation of amyloid fibril spines. Piehl et al. (38, 39) and Hora et al. (40) each assigned amino acids to these segments in the ssNMR spectra of amyloid fibrils of two VLs, human AL09 and murine MAK33. Two of the ssNMR-assigned segments correlate with our findings: they overlap with the amyloidogenic segments identified by our computational and site-directed mutagenesis experiments (segment 1 in strand E of the Greek-key fold of immunoglobulin; and segment 2 in strands F–G (Fig. 11)). Although AL09 and MAK33 are different in their amino acid sequences and hence their amyloid fibrils are different, both include the amyloidogenic segments 1 and 2. In our work, we have experimentally validated the involvement of segments 1 and 2 in forming the steric zipper spine of the amyloid fibril. Thus, three different experiments, two ssNMR assignments of peptides in amyloid fibrils performed by independent laboratories and our site-directed mutagenesis, identify common regions involved in formation of VL amyloid fibrils (Fig. 11A).
Figure 11.

Sequence alignment of amyloidogenic VL segments 1 and 2. A, alignment of reference sequence VL2-8–J1, Mcg, Mak33S20N, and AL09. Black underlines denote segments assigned in amyloid fibrils by ssNMR experiments. Red bars identify amyloidogenic segments 1 and 2. Residues in which mutations prevent formation of amyloid fibrils are enclosed in red boxes. Both segments 1 and 2 overlap with MAK33S20N VL and AL09. Blue indicates amino acids that vary among of the VLs. Red indicates conserved residues. B, location of the amyloidogenic segments in the Greek-key fold of VLs is indicated in red.
In support of our findings, we succeeded in constructing geometrically-reasonable models of full-length VL amyloid fibrils with minimal deviation from crystallographic coordinates of the steric zippers (Fig. 12). The geometric plausibility of these models in which little unfolding occurs outside of segments 1 and 2 supports our hypothesis that these segments are geometrically capable of driving amyloid formation, even at early time points when unfolding may be incomplete. We envisage that with time, additional segments may unfold and contribute to the cross-β–structure and further stabilize the fibril. As some measure of validation, we find that the diameters of the fibril models are consistent with the measured values for the diameters of amyloid fibrils in negative-stain electron micrographs, ∼10–11 nm.
Figure 12.
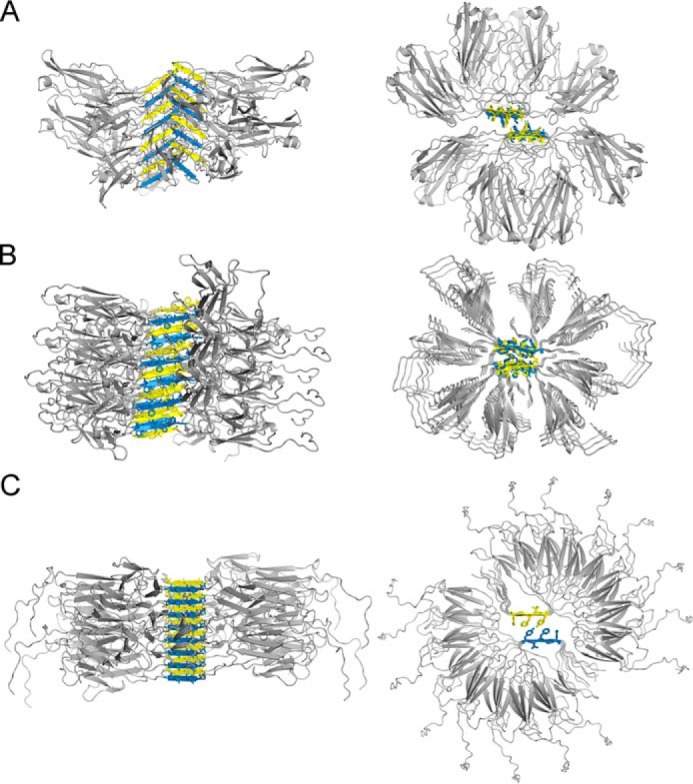
Speculative atomic models for VL amyloid fibrils based on the crystal structure of steric-zipper spines. Each of the steric-zipper spines forms when VLs partly unfold and expose the amyloidogenic segment to solvent. To model the full-length protein around the crystalline structures of the steric-zipper spines, we used 8 or 16 individual VL domains with somewhat different orientations. The left column shows models aligned sideways along the vertical axis of the fibril. The axes of fibrils propagate along the steric-zipper spine through hydrogen bonding between the peptide backbones. The right panel shows the top view of the steric-zipper spine and the interdigitating side chains. A, model of the Mcg VL fibril based on the peptide derived from amyloidogenic segment 1. B, model of the Mcg VL fibril based on the peptide derived from amyloidogenic segment 2. C, model of the AL09 VL fibril based on the peptide derived from amyloidogenic segment 2.
Identification of amyloidogenic segments has translational implications. Our conclusions have direct bearing on strategies for developing peptide-based inhibitors of VL amyloid fibrils. Current approaches to create peptide-based inhibitors generally aim to impede elongation of the amyloid fibril spine; modified peptides are designed to incorporate into the amyloid fibril spine with one face while arresting the propagation of the spine with the opposite face (41). However, in light of our results, it is clear that inhibitor design must simultaneously block at least two independent spines of VL amyloid fibrils. Future therapies may require a mixture of two or more inhibitors of amyloidogenic segments to halt fibril formation.
The structure of NFVFGT from the λ VL Mcg reveals unprecedented complexity in its β-sheet geometry. It contains four distinct orientations of the β-strand within a single out-of-register β-sheet. These features give the β-sheet a translational periodicity of 19.8 Å, which exceeds the more frequently observed in-register parallel sheets (4.8 Å), in-register antiparallel sheets (9.6 Å), and out-of-register antiparallel sheets (11.7 Å). Such lengthy periods, if truly present in biological fibrils, may produce more complex surface patches with affinity for more complex ligands than thioflavin T or Congo red, typically known to bind amyloid fibrils. It remains to be seen whether such complexity will be observed in other amyloid structures.
The VL segments found to form steric zippers through crystal structures were analyzed in 597 λ and 186 κ VLs of patients with AL as well as 158 λ and 401 κ VLs of patients without AL. In the λ sequences, Rosetta energy scores of each of these segments calculated by ZipperDB tend to be lower for segments from patients with AL, indicating a higher propensity to form fibrils. The κ sequences could not be reliably analyzed the same way due to the presence of proline residues in an exceedingly large proportion of the gathered sequences. These results provide further evidence for the segments identified here to be amyloid-driving segments in light-chain amyloidosis.
In summary, we experimentally identified two amyloidogenic segments within VLs. Each of the segments is a key propagator of amyloid fibrils, giving rise to distinct polymorphs. Both amyloidogenic segments occupy the same spatial location of the Greek-key immunoglobulin fold in both λ and κ VLs (segment 1 in strand E of the Greek-key fold; and segment 2 in strands F–G) (42). We found that this pattern of dual steric zippers is found in VLs belonging to both λ and κ classes, suggesting that homologous segments may be responsible for fibril formation in other VLs. Disrupting a single steric zipper is insufficient for blocking amyloid formation because an alternative zipper-forming region remains intact. These results explain previous difficulties in identifying a single residue or segment within VLs responsible for amyloid formation; there can be more than one. Our work offers an experimental framework to identify multiple amyloidogenic segments in VLs and other amyloid-forming proteins.
Experimental procedures
Computational analysis of VL sequences
Genomic and the variant sequences of VL2-8–J1 and Mcg were analyzed with ZipperDB (https://services.mbi.ucla.edu/zipperdb/)4 (58) for steric-zipper propensity, and with ConSurf (http://bental.tau.ac.il/new_ConSurfDB/)4 for amino acid conservation (33, 43).
Preparation of recombinant proteins
Protein samples were prepared as described previously. Site-directed mutagenesis was performed with PCR (44). DNA of every construct was verified twice by an external DNA-sequencing service (Laragen, Inc.). In summary, VLs were expressed in Escherichia coli, purified in denaturing conditions, refolded, and concentrated (35). In the proline-scanning experiment, we found that four is the largest number of sequential prolines that permits formation of the intended constructs. Longer proline segments resulted in degradation of several VL constructs and hampered the systematic experiments.
Amyloid fibril formation assays
ThT assays were performed as described previously (45). Assays were conducted in acidic conditions (50 mm NaAc/HAc (pH 4), and 150 mm NaCl), 0.5 mg/ml VL (40 μm), at 37 °C with constant shaking at 300 rpm with Teflon balls with a 0.125-inch radius as stirrers (46, 47). ThT fluorescence spectra were recorded at 440/480 nm excitation/emission wavelengths. Each experiment was performed with freshly prepared samples in at least three independent repeats on different dates. We refer to an independent repeat as a new preparation of analytes, including production in E. coli, purification, ThT assays, and analysis. Each experiment was performed at least three times on the same batch of analytes. Analysis of the genomic variant and its amyloid-stopping mutation (VL2-8–J1 and VL2-8–J1-L75P) was performed in at least 14 different independent repeats.
The amyloid fibril formation assays were performed in acidic conditions. Although the Mcg and AL09 are pathologic VLs, derived from patients, their behavior in vitro somewhat differs. Although Mcg and VL2-8–J1 readily form amyloid fibrils in both acidic conditions (pH 4) and conditions close to physiologic (pH 7.4) as monitored by ThT fluorescence (Fig. S1), AL09 only forms amyloid fibrils in acidic conditions (pH 4). The application of acidic conditions is common; its purpose is to facilitate and expedite the formation of amyloid fibrils in vitro (48, 49). In conditions other than acidic, amyloid fibril formation by pathologic VLs shows some increased variability; in some experiments, the VLs form amyloid fibrils, although in others they do not. However, in destabilizing conditions the results are consistent, which enables experimental analysis of the capability to form amyloid fibrils.
Electron microscopy (EM)
Samples from the ThT fibril formation assays were diluted with water to 10% v/v and applied onto copper grids with Formvar-carbon coating (Ted Pella, Inc., catalog no. 01810). Negative staining was performed with 2% w/v uranyl acetate, and images were collected by means of a Tecnai T12 electron microscope at 120 kV with a Gatan CCD camera.
Crystal structure determination
Peptides were purchased from Genscript, with TFA salt substitution for HCl. The purity of peptides was greater than 95%. Peptides were dissolved in water to a concentration of 20 mg/ml, and crystallization trials were set up using a Mosquito robot, with a 1:1 ratio of the peptide solution and crystallization screen. Crystals grew in hanging drops and were of needle-like morphology. Solutions that yielded the best-diffracting crystals are listed in Table 2. Crystals were dry-mounted onto pulled glass capillaries, and diffraction data were collected at Advanced Photon Source beamlines 24-ID-C and 24-ID-E (ASLTVS, 24-ID-E; NFVFGT, 24-ID-C; and YTFGQ, 24-ID-C). Phases were determined by molecular replacement with Phaser (ASLTVS) or by direct methods with Shelx (50, 51) (NFVFGT and YTFGQ). Molecular structures were refined with Refmac5 and deposited into the PDB with codes 6DJ0 for ASLTVS, 6DIX for NFVFGT, and 6DIY for YTFGQ (52).
Table 2.
X-ray diffraction data and refinement statistics for the steric zippers that are responsible for amyloid fibril assembly by VLs
Each structure was derived from a single crystal.
| ASLTVS PDB 6DJ0 | NFVFGT PDB 6DIX | YTFGQ PDB 6DIY | |
|---|---|---|---|
| Data collection | |||
| Space group | P21 | P1 | C2 |
| Cell dimensions | |||
| a, b, c (Å) | 15.16, 11.58, 18.8 | 16.03, 11.66, 21.94 | 36.87, 4.81, 18.91 |
| α, β, γ (°) | 90, 97.32, 90 | 90.89, 103.23, 90.26 | 90, 104.95, 90 |
| Resolution (Å) | 18.6–1.3 | 20.0–1.0 | 18.3–0.9 |
| Rsym or Rmerge (%) | 19.2 (27.0)a | 6.6 (12.4) | 6.2 (21.8) |
| I/σI | 3.7 (1.7) | 12.8 (6.7) | 14.1 (3.2) |
| Completeness (%) | 80.5 (62.5) | 81.7 (62.0) | 90.7 (49.8) |
| Redundancy | 2.5 (1.5) | 3.4 (3.2) | 5.1 (3.0) |
| Refinement | |||
| Resolution (Å) | 18.64–1.3 | 21.3–1.0 | 18.3–0.9 |
| No. of reflections | 1215 | 6154 | 2228 |
| Rwork/Rfree | 18.2/21.8 | 10.4/12.5 | 9.7/12.0 |
| No. of atoms | |||
| Peptide | 93 | 388 | 82 |
| Water | 6 | 18 | 1 |
| B-Factors | |||
| Peptide | 6.4 | 7.1 | 5.2 |
| Water | 12.8 | 21.2 | 23.2 |
| Root mean square deviations | |||
| Bond lengths (Å) | 0.019 | 0.015 | 0.013 |
| Bond angles (°) | 2.1 | 2.0 | 1.4 |
| Crystallization conditions | |||
| 0.1 m MMT (dl-malic acid/MES/Tris base; 1:2:2) (pH 5), 25% w/v PEG 1500 | 0.1 m imidazole HCl (pH 8.0), 20% w/v PEG 3000, 0.2 m zinc acetate | 2.8 m sodium acetate, 0.1 m Bistris propane (pH 7.0) | |
a The highest resolution shell is shown in parentheses.
Construction of amyloid fibril models
In constructing our models of full-length VL amyloid fibrils, we strived to make as few assumptions as possible. Therefore, atoms in the VL zipper spine were strongly restrained to maintain the arrangements observed in the crystal structures of their amyloidogenic segments. Because the exact unfolding mechanism of the variable domains is unknown, the remaining atoms of the VL were restrained to maintain the native globular folds (PDB codes 4UNU (35) and 3CDY (53)).
The largest challenge we faced in constructing the models was avoidance of steric clashes among globular domains, because the VL chains are very closely spaced along the fibril axis (4.8 Å for YTFGQ; 5.8 Å for ASLTVS; and 5.0 Å for NFVFGT). We performed a systematic search of orientation space, counting the number of steric clashes incurred for each possible orientation of the globular domains (in 5° increments). To apply the incremental rotation operations and to evaluate steric clashes, we used the programs PDBSET and CONTACT, respectively, from the CCP4 suite (54). We did not impose any twist in our fibril model, because there was no characteristic twist pitch length evident in electron micrographs of the fibrils. As a matter of convenience, we chose to model either 8 or 16 orientations of the globular domains around the fibril axis. We call this collection of unique orientations the asymmetric unit of the fibril. The fibril is then composed of translational repeats of this asymmetric unit (38.4 Å for YTFGQ, 19.8 Å for NFVFGT, and 46.3 Å for ASLTVS) along the fibril axis. For simplicity, the molecules within the asymmetric unit were restrained to have either cyclic or dihedral symmetry. Those models that produced the least number of clashes were manually edited with the program Coot (55) to build connections between the zipper spine and globular domain. The models were geometrically and energetically minimized with the programs CNS (56) and Rosetta (57).
Author contributions
B. B. and D. S. E. conceptualization; B. B., S. R. E., M. R. S., and A. T. L. data curation; B. B., S. R. E., M. R. S., G. R., A. T. L., M. L., and D. S. E. formal analysis; B. B., S. R. E., G. R., and A. T. L. investigation; B. B., S. R. E., M. R. S., G. R., A. T. L., M. L., and D. S. E. methodology; B. B. and D. S. E. writing-original draft; B. B. and M. R. S. project administration; B. B., S. R. E., M. R. S., G. R., A. T. L., M. L., and D. S. E. writing-review and editing; D. S. E. resources; D. S. E. supervision; D. S. E. funding acquisition.
Supplementary Material
Acknowledgments
We thank Duilio Cascio, Daniel Anderson, and Michael Collazo for assistance with the experiments. This work is based upon research conducted at the Northeastern Collaborative Access Team beamlines 24-ID-C and 24-ID-E, which are funded by National Institutes of Health Grant P41 GM103403 from NIGMS. The Pilatus 6M detector on the 24-ID-C beam line is funded by National Institutes of Health ORIP HEI Grant S10 RR029205. This research used resources at the Advanced Photon Source, a United States Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. We acknowledge the use of the Boston University AL-Base, supported by National Institutes of Health Grant HL68705.
This work was supported in part by National Institutes of Health Grant 1R01AG048120-01 from the NIA and the Howard Hughes Medical Institute. D. S. E. is an equity holder of ADRx, Inc. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Fig. S1.
The atomic coordinates and structure factors (codes 6DJ0, 6DIX, and 6DIY) have been deposited in the Protein Data Bank (http://wwpdb.org/).
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
- AL
- light-chain amyloidosis
- LC
- light chain
- NR
- normal repertoire
- VL
- variable light chain
- CL
- constant light chain
- J
- joining segment
- ThT
- thioflavin T
- ssNMR
- solid-state nuclear magnetic resonance
- PDB
- Protein Data Bank
- Bistris propane
- 1,3-bis[tris(hydroxymethyl)methylamino]propane.
References
- 1. Bellotti V., Mangione P., and Merlini G. (2000) Review: immunoglobulin light-chain amyloidosis–the archetype of structural and pathogenic variability. J. Struct. Biol. 130, 280–289 10.1006/jsbi.2000.4248 [DOI] [PubMed] [Google Scholar]
- 2. Adams D. (2013) Recent advances in the treatment of familial amyloid polyneuropathy. Ther. Adv. Neurol. Disord. 6, 129–139 10.1177/1756285612470192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Pinney J. H., Smith C. J., Taube J. B., Lachmann H. J., Venner C. P., Gibbs S. D., Dungu J., Banypersad S. M., Wechalekar A. D., Whelan C. J., Hawkins P. N., and Gillmore J. D. (2013) Systemic amyloidosis in England: an epidemiological study. Br. J. Haematol. 161, 525–532 10.1111/bjh.12286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wechalekar A. D., Gillmore J. D., and Hawkins P. N. (2016) Systemic amyloidosis. Lancet 387, 2641–2654 [DOI] [PubMed] [Google Scholar]
- 5. Kyle R. A. (2011) Amyloidosis: a brief history. Amyloidosis 18, Suppl. 1, 6–7 10.3109/13506129.2011.574354001 [DOI] [PubMed] [Google Scholar]
- 6. Dahlin D. C., and Dockerty M. B. (1950) Amyloid and myeloma. Am. J. Pathol. 26, 581–593 [PMC free article] [PubMed] [Google Scholar]
- 7. Glenner G. G., Terry W., Harada M., Isersky C., and Page D. (1971) Amyloid fibril proteins: proof of homology with immunoglobulin light chains by sequence analyses. Science 172, 1150–1151 10.1126/science.172.3988.1150 [DOI] [PubMed] [Google Scholar]
- 8. Sipe J. D., Benson M. D., Buxbaum J. N., Ikeda S., Merlini G., Saraiva M. J. M., and Westermark P. (2014) Nomenclature 2014: amyloid fibril proteins and clinical classification of the amyloidosis. Amyloidosis 21, 221–224 10.3109/13506129.2014.964858 [DOI] [PubMed] [Google Scholar]
- 9. Eisenberg D., and Jucker M. (2012) The amyloid state of proteins in human diseases. Cell 148, 1188–1203 10.1016/j.cell.2012.02.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Blancas-Mejía L. M., and Ramirez-Alvarado M. (2013) Systemic amyloidoses. Annu. Rev. Biochem. 82, 745–774 10.1146/annurev-biochem-072611-130030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Falk R. H., Comenzo R. L., and Skinner M. (1997) The systemic amyloidoses. N. Engl. J. Med. 337, 898–909 10.1056/NEJM199709253371306 [DOI] [PubMed] [Google Scholar]
- 12. Jones H. B. (1848) On a new substance occurring in the urine of a patient with mollities ossium. Philos. Trans. R. Soc. Lond. 138, 55–62 [Google Scholar]
- 13. Sakano H., Hüppi K., Heinrich G., and Tonegawa S. (1979) Sequences at the somatic recombination sites of immunoglobulin light-chain genes. Nature 280, 288–294 10.1038/280288a0 [DOI] [PubMed] [Google Scholar]
- 14. Marchalonis J. J., and Schluter S. F. (1989) Evolution of variable and constant domains and joining segments of rearranging immunoglobulins. FASEB J. 3, 2469–2479 10.1096/fasebj.3.13.2509274 [DOI] [PubMed] [Google Scholar]
- 15. Buxbaum J. (1986) Aberrant immunoglobulin synthesis in light-chain amyloidosis. Free light chain and light chain fragment production by human bone marrow cells in short-term tissue culture. J. Clin. Invest. 78, 798–806 10.1172/JCI112643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Olsen K. E., Sletten K., and Westermark P. (1998) Fragments of the constant region of immunoglobulin light chains are constituents of AL-amyloid proteins. Biochem. Biophys. Res. Commun. 251, 642–647 10.1006/bbrc.1998.9508 [DOI] [PubMed] [Google Scholar]
- 17. Bodi K., Prokaeva T., Spencer B., Eberhard M., Connors L. H., and Seldin D. C. (2009) AL-Base: a visual platform analysis tool for the study of amyloidogenic immunoglobulin light chain sequences. Amyloid. 16, 1–8 10.1080/13506120802676781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sipe J. D., and Cohen A. S. (2000) Review: history of the amyloid fibril. J. Struct. Biol. 130, 88–98 10.1006/jsbi.2000.4221 [DOI] [PubMed] [Google Scholar]
- 19. Hobbs J. R. (1973) An ABC of amyloid. Proc. R. Soc. Med. 66, 705–710 [PMC free article] [PubMed] [Google Scholar]
- 20. Eisenberg D. S., and Sawaya M. R. (2017) Structural studies of amyloid proteins at the molecular level. Annu. Rev. Biochem. 86, 69–95 10.1146/annurev-biochem-061516-045104 [DOI] [PubMed] [Google Scholar]
- 21. Goldschmidt L., Teng P. K., Riek R., and Eisenberg D. (2010) Identifying the amylome, proteins capable of forming amyloid-like fibrils. Proc. Natl. Acad. Sci. U.S.A. 107, 3487–3492 10.1073/pnas.0915166107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sawaya M. R., Sambashivan S., Nelson R., Ivanova M. I., Sievers S. A., Apostol M. I., Thompson M. J., Balbirnie M., Wiltzius J. J., McFarlane H. T., Madsen A. Ø., Riekel C., and Eisenberg D. (2007) Atomic structures of amyloid cross-β spines reveal varied steric zippers. Nature 447, 453–457 10.1038/nature05695 [DOI] [PubMed] [Google Scholar]
- 23. Fraser P. E., Duffy L. K., O'Malley M. B., Nguyen J., Inouye H., and Kirschner D. A. (1991) Morphology and antibody recognition of synthetic β-amyloid peptides. J. Neurosci. Res. 28, 474–485 10.1002/jnr.490280404 [DOI] [PubMed] [Google Scholar]
- 24. Fraser P. E., Nguyen J. T., Surewicz W. K., and Kirschner D. A. (1991) pH-dependent structural transitions of Alzheimer amyloid peptides. Biophys. J. 60, 1190–1201 10.1016/S0006-3495(91)82154-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Melki R. (2015) Role of different α-synuclein strains in synucleinopathies, similarities with other neurodegenerative diseases. J. Parkinsons Dis. 5, 217–227 10.3233/JPD-150543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Peelaerts W., Bousset L., Van der Perren A., Moskalyuk A., Pulizzi R., Giugliano M., Van den Haute C., Melki R., and Baekelandt V. (2015) α-Synuclein strains cause distinct synucleinopathies after local and systemic administration. Nature 522, 340–344 10.1038/nature14547 [DOI] [PubMed] [Google Scholar]
- 27. Gray K. A., Yates B., Seal R. L., Wright M. W., and Bruford E. A. (2015) Genenames.org: the HGNC resources in 2015. Nucleic Acids Res. 43, D1079–D1085 10.1093/nar/gku1071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lefranc M. P. (2001) Nomenclature of the human immunoglobulin λ (IGL) genes. Exp. Clin. Immunogenet. 18, 242–254 10.1159/000049203 [DOI] [PubMed] [Google Scholar]
- 29. Lefranc M. P. (2001) Nomenclature of the human immunoglobulin κ (IGK) genes. Exp. Clin. Immunogenet. 18, 161–174 10.1159/000049195 [DOI] [PubMed] [Google Scholar]
- 30. Baden E. M., Sikkink L. A., and Ramirez-Alvarado M. (2009) Light-chain amyloidosis–current findings and future prospects. Curr. Protein Pept. Sci. 10, 500–508 10.2174/138920309789351949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Khurana R., Gillespie J. R., Talapatra A., Minert L. J., Ionescu-Zanetti C., Millett I., and Fink A. L. (2001) Partially folded intermediates as critical precursors of light chain amyloid fibrils and amorphous aggregates. Biochemistry 40, 3525–3535 10.1021/bi001782b [DOI] [PubMed] [Google Scholar]
- 32. Wall J., Schell M., Murphy C., Hrncic R., Stevens F. J., and Solomon A. (1999) Thermodynamic instability of human λ6 light chains: correlation with fibrillogenicity. Biochemistry 38, 14101–14108 10.1021/bi991131j [DOI] [PubMed] [Google Scholar]
- 33. Thompson M. J., Sievers S. A., Karanicolas J., Ivanova M. I., Baker D., and Eisenberg D. (2006) The 3D profile method for identifying fibril-forming segments of proteins. Proc. Natl. Acad. Sci. U.S.A. 103, 4074–4078 10.1073/pnas.0511295103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Richardson J. S., and Richardson D. C. (2002) Natural β-sheet proteins use negative design to avoid edge-to-edge aggregation. Proc. Natl. Acad. Sci. U.S.A. 99, 2754–2759 10.1073/pnas.052706099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Brumshtein B., Esswein S. R., Landau M., Ryan C. M., Whitelegge J. P., Phillips M. L., Cascio D., Sawaya M. R., and Eisenberg D. S. (2014) Formation of amyloid fibers by monomeric light chain variable domains. J. Biol. Chem. 289, 27513–27525 10.1074/jbc.M114.585638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Blancas-Mejía L. M., Horn T. J., Marin-Argany M., Auton M., Tischer A., and Ramirez-Alvarado M. (2015) Thermodynamic and fibril formation studies of full-length immunoglobulin light chain AL-09 and its germline protein using scan rate dependent thermal unfolding. Biophys. Chem. 207, 13–20 10.1016/j.bpc.2015.07.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Baden E. M., Owen B. A., Peterson F. C., Volkman B. F., Ramirez-Alvarado M., and Thompson J. R. (2008) Altered dimer interface decreases stability in an amyloidogenic protein. J. Biol. Chem. 283, 15853–15860 10.1074/jbc.M705347200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Piehl D. W., Blancas-Mejía L. M., Ramirez-Alvarado M., and Rienstra C. M. (2017) Solid-state NMR chemical shift assignments for AL-09 VL immunoglobulin light chain fibrils. Biomol. NMR Assign. 11, 45–50 10.1007/s12104-016-9718-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Piehl D. W., Blancas-Mejía L. M., Wall J. S., Kennel S. J., Ramirez-Alvarado M., and Rienstra C. M. (2017) Immunoglobulin light chains form an extensive and highly ordered fibril involving the N- and C-termini. ACS Omega 2, 712–720 10.1021/acsomega.6b00494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hora M., Sarkar R., Morris V., Xue K., Prade E., Harding E., Buchner J., and Reif B. (2017) MAK33 antibody light chain amyloid fibrils are similar to oligomeric precursors. PLoS One 12, e0181799 10.1371/journal.pone.0181799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sievers S. A., Karanicolas J., Chang H. W., Zhao A., Jiang L., Zirafi O., Stevens J. T., Münch J., Baker D., and Eisenberg D. (2011) Structure-based design of non-natural amino-acid inhibitors of amyloid fibril formation. Nature 475, 96–100 10.1038/nature10154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Richardson J. S., Richardson D. C., Tweedy N. B., Gernert K. M., Quinn T. P., Hecht M. H., Erickson B. W., Yan Y., McClain R. D., and Donlan M. E. (1992) Looking at proteins: representations, folding, packing, and design. Biophysical Society National Lecture, 1992. Biophys. J. 63, 1185–1209 10.1016/S0006-3495(92)81695-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ashkenazy H., Abadi S., Martz E., Chay O., Mayrose I., Pupko T., and Ben-Tal N. (2016) ConSurf 2016: an improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res. 44, W344–W350 10.1093/nar/gkw408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kunkel T. A. (1985) Rapid and efficient site-specific mutagenesis without phenotypic selection. Proc. Natl. Acad. Sci. U.S.A. 82, 488–492 10.1073/pnas.82.2.488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wall J., Murphy C. L., and Solomon A. (1999) In vitro immunoglobulin light chain fibrillogenesis. Methods Enzymol. 309, 204–217 10.1016/S0076-6879(99)09016-3 [DOI] [PubMed] [Google Scholar]
- 46. Martin D. J., and Ramirez-Alvarado M. (2010) Comparison of amyloid fibril formation by two closely related immunoglobulin light chain variable domains. Amyloid. 17, 129–136 10.3109/13506129.2010.530081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Andrich K., Hegenbart U., Kimmich C., Kedia N., Bergen H. R. 3rd., Schönland S., Wanker E., and Bieschke J. (2017) Aggregation of full-length immunoglobulin light chains from systemic light-chain amyloidosis (AL) patients is remodeled by epigallocatechin-3-gallate. J. Biol. Chem. 292, 2328–2344 10.1074/jbc.M116.750323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ivanova M. I., Sievers S. A., Sawaya M. R., Wall J. S., and Eisenberg D. (2009) Molecular basis for insulin fibril assembly. Proc. Natl. Acad. Sci. U.S.A. 106, 18990–18995 10.1073/pnas.0910080106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ivanova M. I., Thompson M. J., and Eisenberg D. (2006) A systematic screen of β(2)-microglobulin and insulin for amyloid-like segments. Proc. Natl. Acad. Sci. U.S.A. 103, 4079–4082 10.1073/pnas.0511298103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., and Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 10.1107/S0021889807021206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sheldrick G. M. (2010) Experimental phasing with SHELXC/D/E: combining chain tracing with density modification. Acta Crystallogr. D Biol. Crystallogr. 66, 479–485 10.1107/S0907444909038360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Murshudov G. N., Skubák P., Lebedev A. A., Pannu N. S., Steiner R. A., Nicholls R. A., Winn M. D., Long F., and Vagin A. A. (2011) REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr. 67, 355–367 10.1107/S0907444911001314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Baden E. M., Randles E. G., Aboagye A. K., Thompson J. R., and Ramirez-Alvarado M. (2008) Structural insights into the role of mutations in amyloidogenesis. J. Biol. Chem. 283, 30950–30956 10.1074/jbc.M804822200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., et al. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 10.1107/S0907444910045749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 10.1107/S0907444910007493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Brunger A. T. (2007) Version 1.2 of the crystallography and NMR system. Nat. Protoc. 2, 2728–2733 10.1038/nprot.2007.406 [DOI] [PubMed] [Google Scholar]
- 57. Leaver-Fay A., Tyka M., Lewis S. M., Lange O. F., Thompson J., Jacak R., Kaufman K., Renfrew P. D., Smith C. A., Sheffler W., Davis I. W., Cooper S., Treuille A., Mandell D. J., Richter F., et al. (2011) ROSETTA3: an object-oriented software suite for the simulation and design of macromolecules. Methods Enzymol. 487, 545–574 10.1016/B978-0-12-381270-4.00019-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Tartaglia G. G., and Vendruscolo M. (2008) The zyggregator method for predicting protein aggregation propensities. Chem. Soc. Rev. 37, 1395–1401 10.1039/b706784b [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.