

Abstract
Lysophospholipid transporter (LplT) is a member of the major facilitator superfamily present in many Gram-negative bacteria. LplT catalyzes flipping of lysophospholipids (LPLs) across the bacterial inner membrane, playing an important role in bacterial membrane homeostasis. We previously reported that LplT promotes both uptake of exogenous LPLs and intramembranous LPL flipping across the bilayer. To gain mechanistic insight into this dual LPL-flipping activity, here we implemented a combination of computational approaches and LPL transport analyses to study LPL binding of and translocation by LplT. Our results suggest that LplT translocates LPLs through an elongated cavity exhibiting an extremely asymmetric polarity. We found that two D(E)N motifs form a head group–binding site, in which the carboxylate group of Asp-30 is important for LPL head group recognition. Substitutions of residues in the head group–binding site disrupted both LPL uptake and flipping activities. However, alteration of hydrophobic residues on the interface between the N- and C-terminal domains impaired LPL flipping specifically, resulting in LPLs accumulation in the membrane, but LPL uptake remained active. These results suggest a dual substrate-accessing mechanism, in which LplT recruits LPLs to its substrate-binding site via two routes, either from its extracellular entry or through a membrane-embedded groove between transmembrane helices, and then moves them toward the inner membrane leaflet. This LPL-flipping mechanism is likely conserved in many bacterial species, and our findings illustrate how LplT adjusts the major facilitator superfamily translocation pathway to perform its versatile lipid homeostatic functions.
Keywords: lysophospholipid, transporter, homology modeling, structural model, substrate specificity, protein-lipid interaction, phospholipid turnover, structure-function, lipid transport, lipid flipping, LplT, major facilitator superfamily, structure-function study, transport mechanism
Introduction
The major facilitator superfamily (MFS)4 is the largest known secondary transporter protein family and consists of several hundred members in any cell type (1). MFS proteins transport a great variety of substrates including ions, sugars, amino acids, metabolites, nucleotides, peptides, and lipids. It is believed that MFS proteins share a common alternating-access mechanism, in which transporters undergo conformational changes to create alternating access to a centrally-located substrate-binding site from each side of the membrane (2). A rocker-switch model was supported by the structures of many MFS transporters, including sugar transporters LacY and GLUT1 (3, 4). Of note, these structurally characterized MFS transporters mediate permeation of small polar substrates; i.e. they recruit substrates from the extracellular environment, move them across the hydrophobic membrane bilayer, and then release these solutes into the intracellular space. This translocation pathway is considered as a general working model for all MFS members (2).
Lysophospholipid transporter (LplT) belongs to the MFS family and is found in many Gram-negative bacteria. Distinct from other MFS members, LplT is a lipid flippase. LplT catalyzes flipping of lysophospholipids (LPLs) across the bacterial inner membrane (IM), playing an important role in bacterial membrane phospholipid homeostasis. In bacteria, LplT is functionally linked with de novo biosynthesis of the major outer membrane lipoprotein (Lpp). Generation of matured Lpp requires acyl-transfer from diacyl phospholipids to its N terminus, which releases LPL as a by-product into the outer leaflet of the IM (5) (reaction catalyzed by apolipoprotein N-acyltransferase Lnt, Fig. 1A). LPLs are nonbilayer forming lipids and may destabilize the lamellar membrane structure by inducing positive curvature (6). LplT catalyzes energy-independent flipping of LPLs from the outer leaflet to the inner leaflet of the IM (7) where they are acted upon the peripheral acyltransferase/acyl-acyl carrier protein synthetase Aas on the cytoplasmic surface for acylation to form their respective parent forms (7, 8). Our recent study showed that mutational inactivation of LplT in Escherichia coli impairs the stability of the IM and lipid asymmetry of the outer membrane (OM) mediated by accumulation of LPLs in the IM (9). This intramembranous LPL-flipping activity is apparently different from the common MFS working model, suggesting that LplT utilizes a specific MFS transport mechanism for lipid flipping.
Figure 1.

A, thematic representation of the dual-substrate accessing mechanism of LplT in the bacterial inner membrane. LplT recruits LPL substrates (1) from the outer leaflet of the inner membrane generated by Lnt or (2) from the periplasmic space to flip them across the bilayer to the inner leaflet, which are then acylated by Aas to form diacyl lipids on the cytoplasmic surface. B, TLC image of the total phospholipids extracted from spheroplasts generated from E. coli BL21(DE3) ΔlplT cells expressing LplTKp WT. C, Western blotting of Lpp in E. coli Trp-3110 WT, ΔlplT, and Δaas, and PAP9502 strains using anti-Lpp antibody. The conditional lnt gene knockout strain PAP9502 was grown in the depleted condition (+glucose) or rescuing condition (+arabinose). The same amount of protein was loaded in each lane. D, [32P]LPE transport assays of LplTKp using spheroplasts prepared from E. coli BL21(DE3) Δaas-lplT strain expressing LplTKp WT (black squares) and vector only (open circles) or vector only inside-out vesicles (ISO, open triangles). Radioactivity counts were directly used to calculate the transport activity.
In addition to the intramembrane-flipping activity, our recent biochemical study also found that LplT catalyzes direct uptake of exogenous LPLs into OM-depleted spheroplasts (8, 9) (Fig. 1A). This LPL influx activity appears to be independent of intramembranous flipping because spontaneous incorporation of LPLs into the membrane bilayer is very inefficient (8). How LplT promotes these two distinct transport modes, whether by sharing one translocation pathway or using two individual routes, remains unknown.
Unlike other known flippases, LplT maintains an extreme specificity for lyso versus diacyl forms. We found that spheroplasts expressing LplT from Klebsiella pneumoniae (LplTKp) transports LPLs including both lysophosphatidylethanolamine (LPE), lysophosphatidylglycerol (LPG), but is not inhibited by diacyl-PE or -PG (8). This acyl-chain selectivity may be critical for LplT to perform its specific LPL-flipping activity in the diacyl lipid-rich membrane bilayer.
Lysophospholipid transporter, namely MFSD2A, is also found in animals, although its sequence homology to bacterial LplT is rather poor. MFSD2A has been found to be essential for brain growth and function. MFSD2A transports exogenous lysophosphatidylcholine (LPC) across the brain-blood barrier as the major route for supplying the ω-3 fatty acid docosohexaenoic acid to the brains (10). Interestingly, dual activities have also been reported in MFSD2A. In addition to its LPL influx activity, MFSD2A also maintains the integrity of the brain-blood barrier by suppressing transcytosis in the central nervous system epithelial cells (11). Whether this membrane-maintaining role is mediated by the LPC uptake or other activities of MFSD2A remains to be addressed (12).
No structure of LplT or MFSD2A is available to date. To understand the novel dual LPL uptake/flipping mechanism of LplT, we constructed a three-dimensional structural model of LplTKp, docked the models with different LPL ligands, and studied its LPL uptake and intramembranous flipping activities using mutational analyses. Our results not only provide molecular insights into how LplT recognizes the chemical structure of the LPL substrates, but also reveal a new MFS transport mechanism for LPL translocation across the cell membrane.
Results
Dual substrate transport activities of LplTKp
The intramembranous LPL-flipping activity of LplTKp was assessed by examining LPL contents in the IM. OM-depleted spheroplasts were generated from metabolically 32P-labeled E. coli BL21(DE3) cells and then washed to remove any extracellular components carefully prior to lipid analysis using thin-layer chromatography (TLC). As shown in Fig. 1B and Table 1, no LPLs were detected in WT spheroplasts. In contrast, LPE and LPG were accumulated to 17 and 5% of the total phospholipid compositions, respectively, in ΔlplT spheroplasts. This LPL accumulation was completely diminished in the presence of LplTKp. The Lpp acylation reaction catalyzed by Lpp acyl apolipoprotein N-acyltransferase (Lnt) is the only known source to generate LPLs in the IM (5). To exclude any change of LPL production in the mutant strain, we analyzed Lpp acylation using Western blotting with a specific anti-Lpp antibody. PAP9502 is a conditional lnt-depletion E. coli strain in which endogenous expression of Lnt is controlled by an PBAD promoter (13). In the presence of glucose, the lack of Lnt resulted in an unacylated Lpp precursor in the cells, which migrated faster than its mature form on urea-denaturing gel (Fig. 1C). No Lpp precursor was detected in either ΔlplT or Δaas strains and the matured form of Lpp was present in a similar level compared with WT. Therefore, it is most likely that the accumulation of LPE/LPG in ΔlplT spheroplasts (Fig. 1B) represents the flipping activity of LplTKp.
Table 1.
Phospholipid compositions of spheroplasts generated from E. coli expressing LplTKp WT or mutants
| Cardiolipin | PE | PG | LPE | LPG | |
|---|---|---|---|---|---|
| BL21(DE3) | 9.0 | 70.7 | 20.3 | NDa | ND |
| ΔlplT | 7.0 | 55.5 | 16.2 | 16.6 | 4.7 |
| LplTKp WT | 8.3 | 71.2 | 20.4 | ND | ND |
| D30N | 7.0 | 54.7 | 16.2 | 16.7 | 5.4 |
| K120C | 7.5 | 57.3 | 16.5 | 14.6 | 4.1 |
| R236M | 7.0 | 55.8 | 16.5 | 16.4 | 4.3 |
| E351C | 7.5 | 58.0 | 16.7 | 14.1 | 3.7 |
| N352C | 7.3 | 57.3 | 15.4 | 15.9 | 4.0 |
| N31C | 6.7 | 57.1 | 13.7 | 17.9 | 4.7 |
| N137C | 8.7 | 71.3 | 20.0 | ND | ND |
| L34F | 6.8 | 54.9 | 16.7 | 14.0 | 7.7 |
| F35N | 7.4 | 54.0 | 14.8 | 15.3 | 8.5 |
| L38F | 8.3 | 55.2 | 16.9 | 13.7 | 6.0 |
| I148F | 7.5 | 64.2 | 19.2 | 6.9 | 2.3 |
a ND, not determined.
The extracellular LPL uptake activity of LplTKp was measured by adding [32P]LPE into OM-depleted spheroplasts generated from E. coli cells. Previously, we have demonstrated the LPL-transport activity of LplT in an E. coli aas− strain, indicating that LplT can catalyze LPL transport independent of Aas (9). To better characterize the transport activity of LplTKp, the transport assay was performed in the background of E. coli lplT-aas double deletions. As shown in Fig. 1D, [32P]LPE was imported into spheroplasts in the presence of LplTKp. In contrast, no uptake was detected in the control spheroplasts. Despite the fact that LPLs are detergent-like molecules, spontaneous incorporation of LPE into the bacterial membrane is negligible in the absence of LplTKp at the assay conditions. We further confirmed it using inside-out vesicles (ISO) (Fig. 1D). Therefore, it is unlikely that extraneous LPLs access the translocation pathway of LplTKp through the bilayer. Instead, they may directly access the pathway from its extracellular protein surface. Taken together, these results demonstrate that LplTKp may utilize two distinct routes to perform each individual LPL uptake or flipping activity (Fig. 1A).
Construction of an LplTKp structural model
To gain structural insights into the novel dual LPL-flipping activity of LplT, we took advantage of the conserved membrane topology and protein architecture of MFS proteins to construct a structural homology model of LplTKp. Despite the determination of nearly 100 MFS protein structures, the low sequence identity (<25%) between LplT and other known MFS protein structures is a major hurdle to structural modeling of LplT. Attempts to search for a suitable structural template for LplTKp using the PSI-BLAST program (14) did not yield any hits. Thus, we made use of four automatic structure prediction programs including HHpred (15), Phyre2 (16), RaptorX (17), and I-TASSER (18) to identify remote structural homologs and create initial structural models of LplTKp. We also carried out homology modeling of LplTKp independent of these structure prediction programs using MODELLER v9.14. Sampling of the conformational space was enhanced in our homology modeling protocol by using diverse template-target alignments generated by different sequence alignment methods (Fig. S1, 1–25). By such, a large pool containing 15,000 LplTKp models was created, from which 600 candidates were selected for substrate docking tests using the DOCK 3.6 program (19).
LplTKp has a unique substrate selectivity of the LPL head group; i.e. it transports LPE and LPG, but not LPC and lysophosphatidic acid (LPA) (8), despite the fact that both LPE and LPC are zwitterionic lipids and share a similar quaternary amine head group. We hypothesized that an ideal LplTKp structural model should be able to distinguish its native substrates (LPE and LPG) from nonsubstrates (LPC and LPA). Considering the fact that C16 and C18 acyl chains are present predominantly (>95%) in E. coli membrane phospholipids (20), the docking screening was performed with C18:1 LPL substrates. Both inward- and outward-open templates were used for modeling. However, we found that models generated based on outward-open templates were much better at distinguishing between LPE/LPG and LPC compared with those based on inward-open templates. Of 300 inward-open models, only 51 models were able to distinguish LPE/LPG from LPC, whereas 72 outward-open models of 300 were able to do the same. Models based on outward-open templates showed clearer demarcation between LPE/LPG and LPC (based on docking scores) compared with inward-open template-based models. We found that a MODELLER-generated model using the structure of a putative MFS transporter YajREc protein from E. coli (PDB code 3WDO) as the template and template-target alignment created using PRALINE pairwise alignment exhibited docking scores, which qualitatively matched well with the known substrate selectivity of LplTKp: LPE ≈ LPG > LPA ≫ LPC (Fig. 2A). This model was considered to be the most accurate homology model of LplTKp and used for further mechanistic studies.
Figure 2.

Docking of lysophospholipids to the LplTKp structural models. A, docking scores of 18:1 LPE, LPG, LPA, and LPC to the LplTKp WT structural model. B, comparison of substrate docking 18:1 LPE or 18:1 LPG to the LplTKp WT, D30N, and K120C mutants. C, docking of LPE, LPG, or LPC with different lengths of the acyl chain (14:1, 16:1, and 18:1) to WT LplT model.
Overview of the LplTKp model
The LplTKp model consists of 12 canonical transmembrane helices (TM1–12) and shows a typical MFS protein overall conformation (Fig. 3, A–C). TM1–6 and TM7–12 are arranged into two 6-helix bundles to assemble the N- and C-domains with a 2-fold pseudo symmetry. The two domains are linked by a hydrophilic intracellular loop region.
Figure 3.
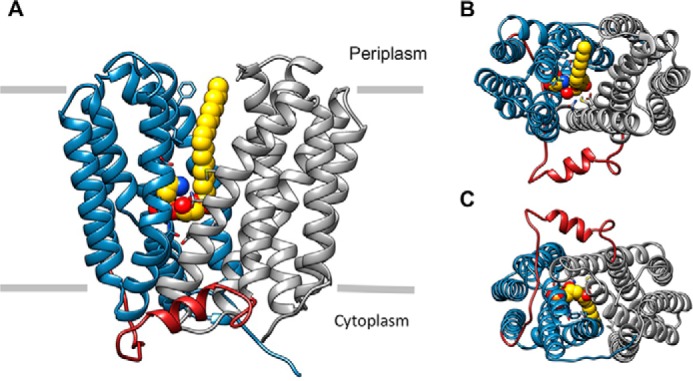
A, overall architecture of the LplTKp structural model in the outward-open conformation with an 18:1 LPE molecule (yellow) docked in the central cavity. The N- and C-domains of LplTKp are shown in blue and gray, respectively, whereas the interdomain linker region is shown in red. B, LplTKp model viewed from the extracellular side (top), and C, intracellular side (bottom).
Despite LplTKp and YajREc sharing a very low protein sequence identity (∼11%) (Fig. S1, 23), the LplTKp model exhibits a similar conformation as that of the YajREc structure with a r.m.s. deviation value of 3.2 Å (Fig. 4B). Both structures also exhibit a remarkable similarity in the distribution of charged residues, particularly those basic residues congregated on the intracellular protein surface (Fig. 4A). Motif A is a fingerprint sequence found in the N-domain of most MFS proteins and assembles a conserved salt-bridge interaction network thought to be important for the outward-open conformation (21). In the YajREc structure, Motif A residues Gly-69TM2, Asp-73TM2, and Arg-77IL2–3, and Arg-77IL2–3 interact with Asp-73TM2 and Asp-126TM4 (Fig. 4B). A similar salt-bridge network is formed by the counterpart residues Gly-70TM2, Asp-74TM2, Lys-78IL2–3, and Glu-126TM4 in the LplTKp model. Charged residues also form conserved salt-bridge interactions in the C-domain of MFS proteins. These interactions were found at the structurally equivalent positions in the LplTKp model, although some interacting residues appear to switch their positions with their partners compared with YajREc (Fig. 4B); i.e. Glu-272TM8 and Lys-329TM10 forming a salt-bridge in YajREc are substituted by Lys-275TM8 and Glu-332TM10 in LplTKp, respectively. Similarly, two acidic residues Glu-211IL6–7 and Glu-330IL10–11 that interact with Arg-213TM7 in YajREc are substituted by Arg-214IL6–7 and Arg-333IL10–11 to interact with Glu-217TM7 (+1) in LplTKp. Taken together, the presence of these conserved salt-bridge networks serves as additional validation for the LplTKp model.
Figure 4.

A, distribution of charged residues: basic/positively-charged (blue) and acidic/negatively-charged (red) residues in LplTKp model. B, superposition of LplTKp model (gray) on YajREc (PDB 3WDO; aquamarine). The conserved motif A in the N-terminal domain of both proteins is highlighted using the dashed square. Other highlighted residues form parts of the C-terminal domain salt-bridge network. Structurally-equivalent residues from LplTKp and YajREc are labeled in gray and aquamarine, respectively.
LPL substrate docking conformations
In the LplTKp model, a large and elongated central cavity is found on the interface between the N- and C-domains, creating a substrate translocation pathway (Fig. 3A). The model has an apparently outward-open conformation, in which the pathway is opened toward the periplasmic surface and ends in the middle of the membrane (Fig. 5). The highest ranked docking poses of LPE and LPG show that the substrates are bound in a similar head-to-tail orientation from the extracellular to the intracellular side in the central cavity (Fig. 5, A and B). Binding of LPE/LPG is likely facilitated by complementation of the asymmetric polarity in the cavity as illustrated by the electrostatic potential map (Fig. 5E). Charged/polar residues are predominantly distributed toward the center of the translocation pathway, whereas hydrophobic residues are found toward the extracellular side. Of note, these cavity-lining residues are conserved in the LplT family (Fig. 6, A and B).
Figure 5.

A–D, sliced section of surface representation showing the orientations of the most favorable docked conformations of (A) 18:1 LPE, (B) 18:1 LPG, (C) 18:1 LPA, and (D) 18:1 LPC within the central cavity of the LplTKp model. E, sliced section of the electrostatic potential surface of LplTKp calculated by the APBS program (40). Regions having positive (blue) and negative (red) potentials surround the negatively-charged phosphate group and positively-charged ethanolamine head group, respectively, whereas neutral (white) regions surround the acyl tail.
Figure 6.

A, conservation of the cavity-lining residues in the LplTKp model. Residues of LplTKp were colored according to conservation scores calculated based on 150 sequence homologs (pairwise sequence identity of 35.2–85.6% with LplTKp) identified from the UniRef database using the ConSurf web server (41). The 10 residues were identified as having critical roles in LplTKp function are shown in sphere representation. B, multiple sequence alignment of 11 LplT sequences from different representative bacterial genera obtained from the UniProt database showing conservation of the 10 critical residues positions (highlighted using same color grades as described for A). The UniProt IDs of the sequences used for producing the MSA are Escherichia (P39196), Shigella (Q32C86), Salmonella (Q8ZMA5), Yersinia (Q1CFA8), Serratia (A8GII5), Enterobacter (A4WE10), Citrobacter (A8AP55), Pectobacterium (C6DE42), Cronobacter (A7MR37), and Photorhabdus (Q7N7A8). C, the binding conformation of 18:1 LPG (blue) in the central cavity. D, 18:1 LPC (magenta) superimposed on the docked conformation of 18:1 LPE (yellow). The residues interacting with the head group are displayed as sticks.
Head group–binding site
Both LPE and LPG were favorably bound in a fishhook-like conformation in the central cavity (Fig. 6, C and D). This substrate conformation is apparently stabilized by two symmetric D(E)N pairs from each domain: Asp-30TM2 and Asn-31TM2 from the N-domain, and Glu-351TM11 and Asn-352TM11 from the C-domain. Asp-30 directly interacts with the amine group of LPE or the hydroxyl groups of LPG, whereas the side chains of Glu-351 and Asn-352 are positioned favorably to interact with the carbonyl oxygen atom of the fatty acid chain from another side of the cavity. In the bottom of the cavity, Lys-120TM4 stabilizes the phosphoryl group of the LPLs via a salt-bridge interaction.
The importance of these head group interactions for LPL transport activity was demonstrated using in vivo LPE acylation assays (Fig. 7, A and B). In the assay, LplTKp WT assimilated exogenous [32P]LPE into OM-depleted spheroplasts and 40% of added LPE was converted to diacyl-PE by endogenous Aas in 30 min. Remarkably, mutation of Asp-30 to asparagine completely abrogated the LPE acylation activity, whereas substitution of Asn-352 with a cysteine residue drastically reduced the activity by >80%. Substantial reductions (30–40%) were also observed in the mutants of E351Q and K120C. The loss of LPE acylation activity is clearly attributed to the changes in LplTKp-mediated LPL uptake given the facts that (i) disrupting the mutant spheroplasts using Triton X-100 detergent retrieved the full Aas activities (Fig. 7, A and B); (ii) the mutant proteins were present in spheroplasts in similar levels compared with WT as examined using immunoblotting (Fig. 7C).
Figure 7.

LPE acylation assays in LplTKp spheroplasts. A, TLC images showing conversion of LPE to PE ± 1% Triton X-100 mediated by LplTKp WT and mutants. E. coli ΔlplT harboring empty vector served as (−) control. B, the relative LPE acylation activity (% of WT) of mutants. C, Western blotting of LplTKp WT and mutant proteins extracted from spheroplasts and developed using anti-His antibody. The same amount of total protein was loaded in each lane.
To help understand the role of these head group interacting residues, we also tested substrate docking to in silico LplTKp mutants. D30N and K120C mutants were generated in silico using University of California San Francisco, Chimera rotamers module (22, 23). Both mutant models exhibited significantly reduced ability to bind to 18:1 LPE and LPG based on their docking scores (Fig. 2B). Taken together, these data suggest that LplTKp utilizes the central cavity for LPL substrate binding.
Molecular determinant of head group discrimination
In the docked models, the carboxylate group of Asp-30 appears to be in the critical position to interact with the head group moiety of LPE or LPG and may contribute to the LPL head group selectivity of LplT. To test this hypothesis, we compared the transport activities of LplTKp WT and the D30N mutant using [32P]LPE and LPG. The results showed that LplTKp WT transports LPE and LPG with similar kinetic parameters: 2.3 ± 0.1 μm (Km) and 98.4 ± 7.3 nmol/g dry weight/h (Vmax) for LPE; 2.2 ± 0.2 μm (Km) and 80.2 ± 6.4 nmol/g dry weight/h (Vmax) for LPG. Consistent to the acylation assay, the mutation of D30N significantly reduced the transport activities for both LPE and LPG. Kinetic analysis showed reductions of both Km and Vmax for LPE by 4- or 2-fold to 9.5 ± 1.0 μm (Km) and 64.4 ± 5.6 nmol/g dry weight/h (Vmax), suggesting that the carboxylate group of Asp-30 is involved in both substrate-binding and transport. Similar changes were also observed in transport of LPG (Vmax = 56.4 ± 4.3 nmol/g dry weight/h, Km = 6.9 ± 0.7 μm). These results support our model prediction that both substrates share similar binding conformations in the central cavity (Fig. 6, C and D). In fact, the importance of head group interaction with Asp-30 was also demonstrated in LPA docking; i.e. C18:1 LPA was able to dock in the binding site in a fishing-hook pose similar to that of LPE and LPG (Fig. 5C). But the lack of a head group moiety led to increased free energy based on the docking score (Fig. 2A).
LplT cannot transport LPC (8). The head group of LPC also carries a cationic amine group, which could potentially interact with Asp-30. However, the most energetically favorable docked pose for 18:1 LPC precludes such an interaction. LPC is positioned in the opposite orientation compared with LPE and LPG in the central cavity, i.e. a head-to-tail orientation along the intracellular-to-extracellular direction (Fig. 5D). For C18:1 LPC, at a total volume of ∼763 Å3, is bulkier than C18:1 LPE (∼677 Å3) and C18:1 LPG (∼704 Å3), which could prevent access to the head group–binding site in the middle of the membrane. In fact, the reason for the exclusion of the LPC head group from the polar region of the central cavity becomes apparent when an LPC molecule was superposed onto the docked LPE molecule in the LplTKp model (Fig. 6D). The bulky trimethyl moiety of LPC experiences severe van der Waals clashes with Asp-30, preventing its direct interaction with the head group of LPC.
LPL acyl tail interaction
The central cavity toward the extracellular side is primarily lined by aromatic and aliphatic residues. Three residues from TM2 including Leu-34TM2, Phe-35TM2, and Leu-38TM2 occur within 4 Å of the acyl chain of the docked 18:1 LPE/LPG (Fig. 8A) and could represent important nonpolar interactions for LPL recognition and transport. We found that mutations of L34F, F35N, and L38F reduced LPE acylation activity modestly by 20–40% (Fig. 7, A and B). We hypothesized that these aliphatic residues provide a favorable “greasy patch” to accommodate the acyl chain of LPL during the transport cycle. In fact, a similar structural feature was found to be crucial for the transport activity of MFSD2A (24). Hydrophobic interactions with the monoacyl chain may facilitate LPL flipping in general.
Figure 8.

A, the conformations of the acyl chain interaction region. The acyl chain of 18:1 LPE is sandwiched between hydrophobic residues on TM2 and Arg-236 from TM7. The residues are depicted as pink sticks. B, V-shaped grooves between TM2 and -11 (orange dashed lines) and between TM5 and -8 (yellow dashed lines), the potential membrane entry. Residue Ile-148 is displayed as a sphere in magenta. C, TLC images of the total phospholipids extracted from spheroplasts generated from E. coli BL21(DE3) ΔlplT expressing LplTKp mutants.
An interesting question is whether the large monoacyl fatty chain also plays any role in maintaining the substrate selectivity. To gain any molecular insight into this question, we performed additional docking studies using shorter chain substrates including 16:1 and 14:1 LPE, LPG, and LPC molecules. The docking scores for the best scoring poses are presented in Fig. 2C. From the docking scores it is evident that, with a decrease in acyl chain length, the docking scores (kcal/mol) improve for docked ligands (especially for LPC), whereas the discriminatory capability of the LplTKp model seems to diminish when the acyl chain length is reduced to 14. Examination of the docking poses show that all six shorter acyl chain LPLs are snuggly buried within the central pocket with their acyl chains pointing toward the extracellular side (Fig. S2). The docking conformations of 16:1 and 14:1 LPC exhibit an orientation that is reversed compared with that of the 18:1 LPC. Despite the reversed orientation, analysis of intramolecular contacts indicates that the trimethyl moiety of 16:1 LPC still encounters severe van der Waals clashes with the side chain atoms of polar residues within the pocket. Such clashes are much less with 14:1 LPC. These observations suggest that the nature of both the head group and acyl chain could potentially contribute to the substrate selectivity of LplT.
A positively charged residue Arg-236TM7 was found in the largely apolar region of the central cavity (Fig. 8A). Mutation of R236M led to nearly 40% reduction of the LplTKp activity based on the LPL acylation assay (Fig. 7). Given its strategic location, we anticipate that the basic side chain of Arg-236 is involved in the early phases of LplTKp's interaction with the head group of LPLs. Also, its potential role in forming an extracellular “gate” with Asp-30/Asn-31, similar to the one observed in E. coli GlpT (25), cannot be ruled out.
Substrate accessing pathway for LPL flipping
The central cavity is the only substrate translocation pathway found in the LplTKp model, thus it may also be used for intramembranous LPL flipping. The five residues in the head group–binding site appeared to be essential for the flipping activity; i.e. spheroplasts of D30N, K120C, N31C, E351Q, or N352C accumulated both LPE and LPG to the ΔlplT level (Fig. 8C, Table 1). These results suggest that LplTKp utilizes the same substrate-binding site for both intramembranous LPL flipping and extracellular LPL uptake.
One important question is how LPL substrates gain access to the central translocation pathway from the membrane bilayer. In the LplTKp model, the outward-open conformation creates two V-shaped grooves between TMs 5 and 8 and between TMs 2 and 11 (Fig. 8B). These two grooves open toward the outer leaflet of the membrane and may serve as membrane entry points for LPL substrates. In support of this idea, we have found that the hydrophobic residues predicted for the acyl chain interaction on TM2 become more prominent for the LPL flipping activity compared with their modest roles in LPL uptake. Mutations of L34F, F35N, and L38F led to accumulation of ∼15% PE and ∼8% PG, similar to the levels observed in ΔlplT (Fig. 8C, Table 1). Furthermore, we found that mutation of Ile-148TM5 located in the middle of the groove between TMs 5 and 8 to a bulky phenylalanine caused a significant increase in LPL levels by >40% compared with WT (Fig. 8C, Table 1), whereas its LPL uptake activity remains fully active (Fig. 7). These results suggest hydrophobic interaction plays a critical role in mediating LPL intramembranous flipping.
Intracellular exit
In the outward-open model of LplTKp, LPE/LPG are bound in the extracellular half of the translocation pathway and the pathway is apparently closed at the intracellular side of the molecule. Several polar residues including Gln-23TM1, Asn-137TM5, Asn-327TM10, Gln-331TM10, Gln-347TM11, and Asn-348TM11 were found toward the intracellular side of the central pathway (Fig. 9A). Although these residues have no direct interactions with LPE/LPG, we asked whether these residues facilitate translocation of the substrate through the pathway. However, mutation of these residues to cysteine, respectively, yielded no effect on LPL transport activity (Fig. 9B) and no LPL accumulation was detected in N137C spheroplasts (Fig. 7). Although any large structural rearrangements in the pathway cannot be ruled out, our results suggest that LplTKp utilizes a different exit than the central port to deliver LPLs to the inner leaflet of the membrane.
Figure 9.

A, polar residues lining the translocation pathway toward the intracellular side of the membrane. 18:1 LPE is depicted in yellow. B, transport activities of LplTKp WT and mutants. 4 μm [32P]LPE or -LPG was added into spheroplasts for 30 min at 37 °C. Error bars represent S.D. of three replicate experiments.
Discussion
Maintaining phospholipid homeostasis is important for the structure of cell membranes and the activity of membrane proteins (26, 27). This process is facilitated by multiple mechanisms in which lipid flippases and scramblases play an essential role (28, 29). LplT is unique among the known flippases due to its substrate specificity for LPLs (8). In this study, we proposed a new dual LPL-flipping mechanism of MFS transporters. We took advantage of conserved MFS protein structures to create a structural model of LplTKp. The model, supported by biochemical characterizations, suggests that LPL transport requires recognition of both the head group and fatty acyl chain. Direct interaction between the carboxylate group of Asp-30 and the head group moiety is important for substrate binding and selectivity (Fig. 7, A and B). In the model, LPE/LPG are stabilized in a fishhook-like conformation primarily by the two D(E)N motifs in the binding site (Fig. 6, C and D). This substrate-binding conformation lays the glycerol 3-phosphate backbone in the bottom of the binding cavity and inevitably places its sn-2 -OH group at an inaccessible position, precluding the sn-2 acyl chain of any diacyl phospholipids from penetrating the narrow central cavity.
The docking with different acyl chain substrates also suggest a role for the long acyl chain in maintaining the selectivity of LPE/LPG versus LPC of LplT. We found, in silico, that the discrimination of LPC is strictly maintained with C16–C18 LPLs, but is quickly diminished when the chain length is reduced to 14 (Fig. 2C). It seems that the longer acyl chain enhances structural hindrance of the PC head group despite the nature of its flexibility. LPC is not synthesized in E. coli cells, but is present abundantly in body fluids (30). C16–20 LPC are the most prominent LPC species generated by host endothelial lipase (30). Therefore, establishing the distinguishability among the LPLs at the level of C16–18 could be sufficient to prevent assimilation of exogenous LPC into the bacterial cells. Further testing of this hypothesis may help gain novel insights into the mechanism of substrate binding and selectivity of LplT.
LplTKp catalyzes both exogenous LPL uptake and intramembranous flipping of LPLs across the bacterial IM. Our results strongly suggest that both events take place in the same translocation pathway between the N- and C-domains (Figs. 7, A and B, and 8C). The ability of LplTKp to assimilate LPLs from the extracellular space is remarkably efficient due to its high substrate binding affinity (micromolar range). LPLs exist as a mixture of micelles and monomeric forms in solution due to their high critical micelle concentrations (31). Based on the model, monomeric LPE/LPG molecules may access the central cavity directly in a diving-like pose. The residue Arg-236 that is located near the periplasmic entry region (Fig. 8A) could mediate the passage of the polar group through the apolar region toward the head group–binding site.
However, this extracellular entry is apparently inaccessible to LPLs in the membrane because LPLs generated in the outer leaflet of the IM stay in the bilayer as demonstrated in ΔlplT spheroplasts (Fig. 1B) and the fact that no spontaneous incorporation of LPLs was observed in both spheroplasts and ISO control vesicles (Fig. 1D). The cavity-lining residues, responsible for the binding of LPL head group, are also essential for LPL-flipping activity (Fig. 8C), suggesting that membrane LPLs are positioned in the central cavity in a similar asymmetric orientation. According to the MFS rocker-switch mechanism, substrate binding in the cavity may induce a rigid body rotation between the N- and C-domains along TMs 5 and 11, resulting in two V-shaped openings on both sides of the translocation pathway (32). Mutation of Ile-148 in the groove between TMs 5 and 7–8 only impairs the LPL flipping activity without any interruption of LPL uptake (Fig. 7, A and B, and 8C), arguing for its specific role in mediating the membrane LPL entry rather than for general transporter operation. We envision that the V-shaped grooves between the N- and C-domains serve as a membrane-embedded entry for slender LPL accessing into the central-binding cavity from the outer leaflet (Fig. 8B). This hypothesis is further supported by mutation of hydrophobic residues on TM2. Mutation of L34F, F35N, or L38F completely abolished the LPL-flipping activity based on the levels of LPLs accumulated in spheroplasts (Fig. 8C). These residues are located on the periplasmic half of the pathway and adjacent to the V-shaped grooves (Fig. 8A). It is possible that they help orientate LPL substrates from the membrane toward the head group–binding site for translocation.
It has long been enigmatic how LPL substrates pass through their translocation pathway across the membrane. How LplT achieves alternating-access operation is still unclear because mutations near the cytoplasmic exit gave no effect on the transport activity (Fig. 9). Unlike other MFS transporters that move substrates through the translocation pathway, the major function of LplT is to orientate the LPL to the right position for acylation by Aas in the membrane. In the LplTKp model, substrate bound in the elongated cavity arranges its head group to crossover the mid-line of the bilayer. It is possible that an exit is then created on the membrane interface to release substrates laterally into the inner leaflet of the membrane. Despite the fact that LplT and Aas may work independently (9), whether Aas is involved in LPL exiting from LplT is still unknown. Flipped LPLs may need to be acylated immediately by Aas to eliminate any membrane disruptive effect in the inner leaflet. In many bacteria including E. coli and K. pneumoniae, the genes of LplT and Aas are adjacently encoded in the same operon (7). However, in other Gram-negative microorganisms, LplT and Aas are predicted to form a fusion protein. Future investigation of any protein interaction between LplT and Aas may help to understand LPL substrate exiting from LplT.
LplT is an energy-independent transporter (7) and its transport activity should be driven by the concentration gradient of substrates across the bilayer. However, the extremely asymmetric polarity in the central translocation pathway may only support LPL influx. No significant role of those polar residues toward the cytoplasm in the pathway (Fig. 9) perhaps also prohibits LPL substrate binding from the cytoplasmic surface. Considering the facts that phospholipid degradation occurs mostly in the OM and the outer leaflet of the IM and no acyltransferase has been found in the periplasm (33), it may be unnecessary to maintain an LPL efflux activity in the bacterial membrane.
The functional role of this dual-substrate accessing mechanism in LplT-mediated bacterial membrane homeostasis is still unclear. Our recent study has showed that LplT/Aas maintain the structure and stability of both the IM and OM (9). The dual-substrate accessing mechanism may facilitate the functional versatility of LplT in the bacterial membrane envelope. In addition to LPLs generated from Lpp biosynthesis, LPLs can be produced from OM hydrolysis by secretory phospholipase A2 serving as a potent host anti-bacterial mechanism (33). We found that inactivation of LplT in E. coli greatly facilitates mammalian phospholipase A2-mediated attack (9). The dual-access mechanism may enable LplT to scavenge LPLs generated from multiple phospholipid degradation pathways (33). Whether this LPL transport mechanism is applicable to other LplT members requires further characterization. What is clear is that LplT has orthologues across multiple species. On the basis of the conservation of characteristic residues that interact with LPL substrates in the modeled structures, respectively, 150 other sequence homologs (pairwise sequence identity of 35.2–85.6% with LplTKp) from different prokaryotic species may now be classified as lysophospholipid transporters with a similar transporting mechanism (Fig. 6, A and B). In all of these sequences, the cavity-lining residues (Asp-30, Asn-31, Lys-120, Asn-137, Arg-236, Glu-351, Asn-352, Leu-34, Phe-35, and Leu-38, LplTKp numbering) are highly conserved, as is the residue located in the middle of the groove between TMs 5 and 8 (Ile-148). Therefore, the transport mechanism demonstrated in LplTKp may serve as a model to understand the role of LplT in lipid homeostasis.
Experimental procedures
Preparation of E. coli spheroplasts
Spheroplasts were generated from E. coli cells using a lysozyme-LiCl approach as we previously used (8). Prior to preparation, LplTKp WT or mutant proteins with a His tag at the N terminus was expressed in E. coli BL21(DE3) strains using a pET vector. The mutations were introduced using a standard site-directed mutagenesis method. E. coli cells were grown in LB broth at 37 °C until an A600 of 0.5. Protein expression was induced by adding 0.2 mm isopropyl β-d-1-thiogalactopyranoside for 2 h at 25 °C. Cells were washed with a buffer containing 10 mm HEPES (pH 7.5), 0.75 m sucrose, 10 mm MgSO4, and 460 mm LiCl. After addition of 1 mg/ml of lysozyme, cell suspensions were ice-chilled, warmed to room temperature, and then incubated with gentle shaking at 30 °C for 30 min. Intact spheroplasts were collected by centrifugation (3,000 × g for 10 min) at room temperature and resuspended at 10 mg/ml of total protein concentration in the above buffer without LiCl for immediate use. Spheroplast formation and stability were thoroughly monitored nephelometrically by following the A600 of 100 μl of a spheroplast suspension in 2 ml of either water or a solution supplemented with 10 mm MgCl2, 0.75 m sucrose, respectively.
LPE acylation assay
LPE acylation assays were performed in spheroplasts generated from E. coli BL21(DE3) ΔlplT mutant strains as previously described (8). Prior to assays, [32P]LPE and -LPG were prepared by digestion of metabolically 32P-labeled PE or PG using venom PLA2 and then purified by thin-layer chromatography (TLC). [32P]LPE (105 cpm for each assay) mixed with 10 μm (final concentration) 18:1 LPE (Avanti lipids) was added into spheroplast suspensions for 30 min. The reactions were terminated by adding chloroform/methanol (1:2) to extract total lipids using the Bligh-Dyer method. Lipids were separated on Silica Gel G thin-layer plates and developed with chloroform/methanol/ammonia/H2O (60:37.5:1:3, v/v). The dried plate was exposed to a Storage Phosphor Screen overnight. PE and LPE were visualized and quantified using Molecular Imager FX. Stored images were processed and quantified using Quantity One software. LPE acylation activity is expressed as mole % of PE/(PE + LPE).
Analysis of LPL contents in IM
LPL contents in the E. coli inner membrane was measured by TLC analysis of LPLs in spheroplasts. Cells were grown in the presence of 5 μCi/ml of [32P]orthophosphate. Spheroplasts were generated using the same approach as described above. Spheroplasts were washed carefully three times with the buffer prior to lipid extraction and TLC analysis. The content of each individual lipid species is expressed as mole % of the total phospholipid composition.
Immunoblotting
The Lpp protein in E. coli was assayed by following a published protocol (13). Crude extracts from PAP9502 (BW25113 ybeX-(kan-rpoCter-paraB)-lnt), W3110 WT, ΔlplT, and Δaas strains were prepared from cells grown in LB broth containing 0.2% arabinose or glucose for 8 generations at 37 °C. The proteins were separated on urea SDS gel and developed with an anti-Lpp antibody. The expression of LplTKp WT and variants were analyzed using an anti-His antibody.
LPL incorporation in inside-out vesicles
Inside-out membrane vesicles were prepared using a low-pressure homogenization approach described earlier (34). Briefly, cells suspended in cold Tris-HCl (50 mm, pH 8.0) were ruptured by single passage through a C3 homogenizer (Avestin) at 4,000 p.s.i. After removing cell debris, the supernatant was pelleted by ultracentrifugation at 40,000 rpm using a Ti45 rotor for 1 h. Membrane fractions were rinsed and homogenized in the same Tris-HCl buffer. Prior to assays, vesicles were diluted into a buffer containing 0.1 m HEPES (pH 7.5), 100 mm KCl, and 2 mm NADH to 0.13 mg/ml of total protein concentration. The reactions were triggered by adding 10 μm [32P]LPE (105 cpm) for the indicated times at 37 °C. The reactions were terminated by filtration through a 0.22-μm membrane on a Millipore filtration manifold and washed with the reaction buffer. The radioactivity in the membrane was counted using a liquid scintillation counter to calculate transport activity.
LPL transport assay in spheroplasts
Transport assays were carried out in spheroplasts generated from E. coli BL21(DE3) Δaas-lplT strains using a silicon oil-based approach as we described previously (8). Briefly, [32P]LPE or LPG mixed with synthetic LPE (18:1) or LPG (18:1) were added to the spheroplasts at different concentrations for 30 min at 37 °C. After incubation, 0.5-ml reactions were layered onto 0.15 ml of 22% perchloric acid and 0.50 ml of silicone oil in Eppendorf tubes and centrifuged at 14,000 × g for 5 min at room temperature to separate spheroplasts from free substrates. The radioactivity in the perchloric acid phase was quantified by a liquid scintillation counter to calculate transport activity. Michaelis-Menten kinetic constants were analyzed using the GraphPad Prism software.
Three-dimensional structure prediction
The structural model of LplTKp was constructed using a similar homology modeling approach previously used for the human sodium-dependent LPC transporter MFSD2A with modifications (24). The three steps in the modeling approach are as follows. 1) The initial templates were identified using four automated protein structure prediction programs HHpred (15), Phyre2 (16), RaptorX (17), and I-TASSER (18). The most commonly identified templates that were used by the structure prediction programs include PDB 3O7Q, 5AYN, 3WDO (outward-open conformation), and 4ZP0 and 1PW4 (inward-open conformation) (Table S2). We also included PDB 5AYO as one of the templates whose primary sequence is identical to that of PDB 5AYN but the solved structure is in the inward-open conformation. 2) In addition to the template-query alignments generated by the structure prediction programs, to further improve sampling, pairwise alignments between each of the template sequences and LplTKp were generated using five different alignment approaches, namely pairwise, and profile-profile alignment using AlignMe (35) and MUSCLE (36), and pairwise alignment using PRALINE (37). Depending on the chosen template and alignment approach used, 30 different combinations were considered and 500 homology models were constructed for each template-alignment combination using the automodel class in MODELLER v9.14 (38). The modeling approach comprises structure optimization by conjugate gradients and refinement by molecular dynamics and simulated annealing. No additional post-modeling structural refinement of the models was performed. The models generated from each combination were evaluated by the discrete optimized protein energy score (39), and the 20 best ranking models were selected (total 600 models). R.m.s. deviation values between each template and their corresponding models (top 20) were calculated and average values corresponding to each alignment method used are summarized in Table S1. We also included models predicted by HHpred (10 models), Phyre2 (20 models), RaptorX (5 models), and I-TASSER (4 models), based on single templates. HHpred, Phyre2, and RaptorX also created one model each based on multiple templates that were also included for validation. In total, the 42 models generated by the four protein structure prediction programs along with the 600 models independently generated using MODELLER were considered for further validation.
Structural validation
Because of the very low sequence homology shared by the chosen templates and query, selection of the best structural model from among the 642 models for further interpretation solely based on parameters such as template query sequence identity, similarity, r.m.s. deviation, and model discrete optimized protein energy scores would be flawed and incomplete. To improve our confidence in the accuracy of a given model, we docked known substrates and nonsubstrates to the models and analyzed the docked scores and conformations. We reasoned that the more accurate a given model is the better it could be at distinguishing substrates from nonsubstrates. Thus, each of the 642 models was docked with a chemical library containing LPE and LPG, LPC, and LPA using DOCK v3.6 (19). The docked poses for each of the four compounds were scored and ranked on the basis of the docking energy function. The final model that was generated by MODELLER based on the template structure of a putative MFS transporter YajREc protein using the alignment created with PRALINE was chosen based on the best docking score qualitatively matching the substrate selectivity preferences (LPE ≈ LPG > LPA ≫ LPC) previously reported for LplTKp (8).
Author contributions
Y. L., R. K. D., J. Z. Z., H. F., and L. Z. data curation; Y. L., H. F., and L. Z. formal analysis; Y. L., R. K. D., J. Z. Z., H. F., and L. Z. investigation; Y. L., R. K. D., J. Z. Z., H. F., and L. Z. visualization; Y. L., R. K. D., H. F., and L. Z. methodology; Y. L., H. F., and L. Z. writing-review and editing; R. K. D. and H. F. software; H. F. and L. Z. conceptualization; H. F. and L. Z. resources; H. F. and L. Z. supervision; H. F. and L. Z. funding acquisition; H. F. and L. Z. validation; H. F. and L. Z. writing-original draft; H. F. and L. Z. project administration.
Supplementary Material
Acknowledgments
We thank William Dowhan for use of a phosphoimager, Shin-ichiro Narita for the anti-Lpp antibody, and Nienke Buddelmeijer for the E. coli PAP9502 strain. The computational work for this article was partially performed on resources of the National Supercomputing Centre, Singapore.
This work was supported by National Institutes of Health Grants R01GM097290 and R01GM098572 and American Heart Association Grant 18TPA34230046 (to L. Z.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1 and S2 and Tables S1 and S2.
- MFS
- major facilitator superfamily
- LplT
- lysophospholipid transporter
- LPL
- lysophospholipid
- Aas
- 2-acyl lyso-PE acyltransferase/acyl-acyl carrier protein synthetase
- LPE
- lysophosphatidylethanolamine
- LPC
- lysophosphatidylcholine
- LPA
- lysophosphatidic acid
- LPG
- lysophosphatidylglycerol
- PE
- phosphatidylethanolamine
- PG
- phosphatidylglycerol
- Lnt
- lipoprotein acyltransferase
- LplTKp
- LplT protein from Klebsiella pneumoniae
- r.m.s. deviation
- root mean square deviation
- IM
- inner membrane
- OM
- outer membrane
- TM
- transmembrane
- ISO
- inside-out vesicles
- Lpp
- lipoprotein
- PDB
- Protein Data Bank.
References
- 1. Pao S. S., Paulsen I. T., and Saier M. H. (1998) Major facilitator superfamily. Microbiol. Mol. Biol. Rev. 62, 1–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Yan N. (2015) Structural biology of the major facilitator superfamily transporters. Annu. Rev. Biophys. 44, 257–283 10.1146/annurev-biophys-060414-033901 [DOI] [PubMed] [Google Scholar]
- 3. Abramson J., Smirnova I., Kasho V., Verner G., Kaback H. R., and Iwata S. (2003) Structure and mechanism of the lactose permease of Escherichia coli. Science 301, 610–615 10.1126/science.1088196 [DOI] [PubMed] [Google Scholar]
- 4. Deng D., Xu C., Sun P., Wu J., Yan C., Hu M., and Yan N. (2014) Crystal structure of the human glucose transporter GLUT1. Nature 510, 121–125 10.1038/nature13306 [DOI] [PubMed] [Google Scholar]
- 5. Jackowski S., and Rock C. O. (1986) Transfer of fatty acids from the 1-position of phosphatidylethanolamine to the major outer membrane lipoprotein of Escherichia coli. J. Biol. Chem. 261, 11328–11333 [PubMed] [Google Scholar]
- 6. Fuller N., and Rand R. P. (2001) The influence of lysolipids on the spontaneous curvature and bending elasticity of phospholipid membranes. Biophys. J. 81, 243–254 10.1016/S0006-3495(01)75695-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Harvat E. M., Zhang Y. M., Tran C. V., Zhang Z., Frank M. W., Rock C. O., and Saier M. H. Jr. (2005) Lysophospholipid flipping across the Escherichia coli inner membrane catalyzed by a transporter (LplT) belonging to the major facilitator superfamily. J. Biol. Chem. 280, 12028–12034 10.1074/jbc.M414368200 [DOI] [PubMed] [Google Scholar]
- 8. Lin Y., Bogdanov M., Tong S., Guan Z., and Zheng L. (2016) Substrate selectivity of lysophospholipid transporter LplT involved in membrane phospholipid remodeling in Escherichia coli. J. Biol. Chem. 291, 2136–2149 10.1074/jbc.M115.700419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lin Y., Bogdanov M., Lu S., Guan Z., Margolin W., Weiss J., and Zheng L. (2018) The phospholipid-repair system LplT/Aas in Gram-negative bacteria protects the bacterial membrane envelope from host phospholipase A2 attack. J. Biol. Chem. 293, 3386–3398 10.1074/jbc.RA117.001231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Nguyen L. N., Ma D., Shui G., Wong P., Cazenave-Gassiot A., Zhang X., Wenk M. R., Goh E. L., and Silver D. L. (2014) Mfsd2a is a transporter for the essential ω-3 fatty acid docosahexaenoic acid. Nature 509, 503–506 10.1038/nature13241 [DOI] [PubMed] [Google Scholar]
- 11. Ben-Zvi A., Lacoste B., Kur E., Andreone B. J., Mayshar Y., Yan H., and Gu C. (2014) MSFD2A is critical for the formation and function of the blood brain barrier. Nature 509, 507–511 10.1038/nature13324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhao Z., and Zlokovic B. V. (2014) Blood-brain barrier: a dual life of MFSD2A? Neuron 82, 728–730 10.1016/j.neuron.2014.05.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gélis-Jeanvoine S., Lory S., Oberto J., and Buddelmeijer N. (2015) Residues located on membrane-embedded flexible loops are essential for the second step of the apolipoprotein N-acyltransferase reaction. Mol. Microbiol. 95, 692–705 10.1111/mmi.12897 [DOI] [PubMed] [Google Scholar]
- 14. Altschul S. F., Madden T. L., Schäffer A. A., Zhang J., Zhang Z., Miller W., and Lipman D. J. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402 10.1093/nar/25.17.3389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Söding J., Biegert A., and Lupas A. N. (2005) The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 33, W244–248 10.1093/nar/gki408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kelley L. A., Mezulis S., Yates C. M., Wass M. N., and Sternberg M. J. (2015) The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 10, 845–858 10.1038/nprot.2015.053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Källberg M., Wang H., Wang S., Peng J., Wang Z., Lu H., and Xu J. (2012) Template-based protein structure modeling using the RaptorX web server. Nat. Protoc. 7, 1511–1522 10.1038/nprot.2012.085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhang Y. (2008) I-TASSER server for protein 3D structure prediction. BMC Bioinformatics 9, 40 10.1186/1471-2105-9-40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mysinger M. M., and Shoichet B. K. (2010) Rapid context-dependent ligand desolvation in molecular docking. J. Chem. Inf. Model 50, 1561–1573 10.1021/ci100214a [DOI] [PubMed] [Google Scholar]
- 20. Morein S., Andersson A., Rilfors L., and Lindblom G. (1996) Wild-type Escherichia coli cells regulate the membrane lipid composition in a “window” between gel and non-lamellar structures. J. Biol. Chem. 271, 6801–6809 10.1074/jbc.271.12.6801 [DOI] [PubMed] [Google Scholar]
- 21. Jiang D., Zhao Y., Wang X., Fan J., Heng J., Liu X., Feng W., Kang X., Huang B., Liu J., and Zhang X. C. (2013) Structure of the YajR transporter suggests a transport mechanism based on the conserved motif A. Proc. Natl. Acad. Sci. U.S.A. 110, 14664–14669 10.1073/pnas.1308127110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., and Ferrin T. E. (2004) UCSF Chimera: a visualization system for exploratory research and analysis. J. Comp. Chem. 25, 1605–1612 10.1002/jcc.20084 [DOI] [PubMed] [Google Scholar]
- 23. Shapovalov M. V., and Dunbrack R. L. Jr. (2011) A smoothed backbone-dependent rotamer library for proteins derived from adaptive kernel density estimates and regressions. Structure 19, 844–858 10.1016/j.str.2011.03.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Quek D. Q., Nguyen L. N., Fan H., and Silver D. L. (2016) Structural insights into the transport mechanism of the human sodium-dependent lysophosphatidylcholine transporter Mfsd2a. J. Biol. Chem. 291, 9383–9394 10.1074/jbc.M116.721035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Huang Y., Lemieux M. J., Song J., Auer M., and Wang D. N. (2003) Structure and mechanism of the glycerol-3-phosphate transporter from Escherichia coli. Science 301, 616–620 10.1126/science.1087619 [DOI] [PubMed] [Google Scholar]
- 26. Zhang Y. M., and Rock C. O. (2008) Membrane lipid homeostasis in bacteria. Nat. Rev. Microbiol. 6, 222–233 10.1038/nrmicro1839 [DOI] [PubMed] [Google Scholar]
- 27. Holthuis J. C., and Menon A. K. (2014) Lipid landscapes and pipelines in membrane homeostasis. Nature 510, 48 10.1038/nature13474 [DOI] [PubMed] [Google Scholar]
- 28. Pomorski T., and Menon A. K. (2006) Lipid flippases and their biological functions. Cell. Mol. Life Sci. 63, 2908–2921 10.1007/s00018-006-6167-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sanyal S., and Menon A. K. (2009) Flipping lipids: why an' what's the reason for? ACS Chem. Biol. 4, 895–909 10.1021/cb900163d [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Riederer M., Ojala P. J., Hrzenjak A., Graier W. F., Malli R., Tritscher M., Hermansson M., Watzer B., Schweer H., Desoye G., Heinemann A., and Frank S. (2010) Acyl chain-dependent effect of lysophosphatidylcholine on endothelial prostacyclin production. J. Lipid Res. 51, 2957–2966 10.1194/jlr.M006536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Stafford R. E., Fanni T., and Dennis E. A. (1989) Interfacial properties and critical micelle concentration of lysophospholipids. Biochemistry 28, 5113–5120 10.1021/bi00438a031 [DOI] [PubMed] [Google Scholar]
- 32. Quistgaard E. M., Löw C., Guettou F., and Nordlund P. (2016) Understanding transport by the major facilitator superfamily (MFS): structures pave the way. Nat. Rev. Mol. Cell Biol. 17, 123–132 10.1038/nrm.2015.25 [DOI] [PubMed] [Google Scholar]
- 33. Zheng L., Lin Y., Lu S., Zhang J., and Bogdanov M. (2017) Biogenesis, transport and remodeling of lysophospholipids in Gram-negative bacteria. Biochim. Biophys. Acta 1862, 1404–1413 10.1016/j.bbalip.2016.11.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wu M., Tong S., Waltersperger S., Diederichs K., Wang M., and Zheng L. (2013) Crystal structure of Ca(2+)/H(+) antiporter protein YfkE reveals the mechanisms of Ca2+ efflux and its pH regulation. Proc. Natl. Acad. Sci. U.S.A. 110, 11367–11372 10.1073/pnas.1302515110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Stamm M., Staritzbichler R., Khafizov K., and Forrest L. R. (2014) AlignMe: a membrane protein sequence alignment web server. Nucleic Acids Res. 42, W246–251 10.1093/nar/gku291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Edgar R. C. (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 10.1093/nar/gkh340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Simossis V. A., and Heringa J. (2005) PRALINE: a multiple sequence alignment toolbox that integrates homology-extended and secondary structure information. Nucleic Acids Res. 33, W289–294 10.1093/nar/gki390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sali A., and Blundell T. L. (1993) Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 234, 779–815 10.1006/jmbi.1993.1626 [DOI] [PubMed] [Google Scholar]
- 39. Shen M. Y., and Sali A. (2006) Statistical potential for assessment and prediction of protein structures. Protein Sci. 15, 2507–2524 10.1110/ps.062416606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Jurrus E., Engel D., Star K., Monson K., Brandi J., Felberg L., E., Brookes D. H., Wilson L., Chen J., Liles K., Chun M., Li P., Gohara D., W., Dolinsky T., Konecny R., et al. (2018) Improvements to the APBS biomolecular solvation software suite. Protein Sci. 27, 112–128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gershon C., Guy N., Haim A., Fabian G., Eric M., Itay M., Tal P., and Nir B. T. (2013) ConSurf: using evolutionary data to raise testable hypotheses about protein function. Isr. J. Chem. 53, 199–206 10.1002/ijch.201200096. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.