

Abstract
Accurate gene expression requires the targeting of transcription factors (TFs) to regulatory sequences often occluded within nucleosomes. The ability to target a TF binding site (TFBS) within a nucleosome has been the defining characteristic for a special class of TFs known as pioneer factors. Recent studies suggest TP53 functions as a pioneer factor that can target its TFBS within nucleosomes, but it remains unclear how TP53 binds to nucleosomal DNA. To comprehensively examine TP53 nucleosome binding, we competitively bound TP53 to multiple in vitro–formed nucleosomes containing a high- or low-affinity TP53 TFBS located at differing translational and rotational positions within the nucleosome. Stable TP53–nucleosome complexes were isolated and quantified using next-generation sequencing. Our results demonstrate TP53 binding is limited to nucleosome edges with significant binding inhibition occurring within 50 bp of the nucleosome dyad. Binding site affinity only affects TP53 binding for TFBSs located at the same nucleosomal positions; otherwise, nucleosome position takes precedence. Furthermore, TP53 has strong nonspecific nucleosome binding facilitating its interaction with chromatin. Our in vitro findings were confirmed by examining TP53-induced binding in a cell line model, showing induced binding at nucleosome edges flanked by a nucleosome-free region. Overall, our results suggest that the pioneering capabilities of TP53 are driven by nonspecific nucleosome binding with specific binding at nucleosome edges.
Appropriate gene expression is vital for the success of countless cellular processes, including growth, development, and metabolism. Among the factors known to regulate gene expression is a highly characterized subset of proteins known as transcription factors (TFs). TFs regulate genes by binding to specific DNA sequences in the genomic vicinity of each regulated gene and inducing a change in expression. TFs recognize degenerate sites as TF binding sites (TFBSs) that appear thousands of times across a given eukaryotic genome. In vivo, most TFBSs are never targeted, likely due to the fact that they are inaccessible due to binding by nucleosomes.
Nucleosomes are the primary unit of chromatin structure composed of a 147-bp section of DNA wrapped around a histone protein core with neighboring nucleosomes separated by accessible linker DNA (Kornberg and Lorch 1999; Jiang and Pugh 2009). In theory, the steric hindrance of nucleosome–DNA interactions could inhibit TFs binding to all nucleosome-bound DNA. In reality, however, nucleosome inhibition of TFs binding is variable both across a genome and even within nucleosomes (Buck and Lieb 2006). As DNA duplex molecule bends and twists around the histone octamer, one side of it directly contacts the histone surface and gets buried inside, while the other side is exposed to the solvent and is accessible, with a concealed/exposed periodicity of 5 to 6 bp. This well-organized architecture maintains nucleosome stability via electrostatic interactions and hydrogen bonds between histone protein side chains and phosphate groups in the DNA backbone. As a result, this highly distorted and partially overlaid nucleosomal DNA cannot be accessed readily by TFs (Vermaak et al. 2003; Luger et al. 2012).
The location of TFBSs within a nucleosome can significantly affect TF–nucleosome binding. TFBSs can have various positions within a nucleosome core (known as translational setting), from near the edge to the center of the nucleosome. For the glucocorticoid receptor, a TFBS located near the edge is bound fourfold better compared with an identical TFBS positioned 20 bp from the dyad (Li and Wrange 1993). Other TFs are inhibited by the translational settings by differing amounts from two- to 100-fold (Vettese-Dadey et al. 1994; Blomquist et al. 1996; Angelov et al. 2004). These divergences in nucleosome inhibition of TF binding are likely driven by differences in how TFs recognize their binding sites on nucleosomes. The orientation of a TFBS on a nucleosome (known as “rotational setting”) also influences how nucleosomes inhibit TF binding. TFBSs located along a nucleosome surface can face either inward or outward due to the twisting of DNA's helical structure. For FOXA, a TFBS located near the nucleosome dyad at a specific rotational setting is significantly bound in in vitro binding assays, while a rotational shifted TFBS located 5 bp away is not bound (Sekiya et al. 2009).
FOXA represents a special class of TFs known as pioneer factors. Pioneer factors were first described in 2002, as regulatory proteins capable of targeting DNA sequences even within compacted, closed chromatin, while other TFs cannot (Cirillo et al. 2002). Among all known TFs, only a few have been characterized as pioneer TFs (Iwafuchi-Doi and Zaret 2014). Recent studies suggest that TP53 functions as a pioneer factor at some of its binding sites (Sammons et al. 2015). Sammons et al. (2015) showed that activated TP53 bound to 4416 new binding sites, 44% of which reside within inactive (H3K4me1- and H3K4me3-) and inaccessible chromatin (Sammons et al. 2015). At the CDKN1A gene, TP53 binds at a higher affinity to its binding site within chromatin compared with naked DNA (Espinosa and Emerson 2001). Furthermore, throughout the human genome TP53 TFBSs (p53BS) occur in regions with strong nucleosome positioning sequences (Lidor Nili et al. 2010). These findings suggest that TP53 functions as a pioneer factor that can target its binding sites within nucleosomes.
TP53 is a DNA binding TF that acts as a tumor suppressor and has its DNA-binding domain being frequently mutated during cancer (Rivlin et al. 2011). TP53 integrates multiple stress-induced signals and acts as a transcriptional regulator for a wide variety of genes involved in DNA repair, cell-cycle arrest, and apoptosis. TP53's ability to regulate these transcriptional responses requires it to specifically target and bind its binding sites throughout the genome. TP53's recognition of naked DNA has been extensively studied and TP53 binds to DNA as a homo-tetramer recognizing two decamers of RRRCWWGYYY (el-Deiry et al. 1992), each decamer is called a “half site” with underlined 4 nucleotides (nt) CWWG as the core, usually one full site consists of two half sites. TP53 binding to nucleosomes has been previously tested with conflicting findings. Laptenko et al. (2011) showed that a p53BS located near the dyad had undetectable binding, while sites near the nucleosome edge are bound. Sahu et al. (2010) showed that TP53 can bind to sites near the nucleosome dyad when in the appropriate rotational position. Therefore, to address these conflicting results, we developed a quantitative and competitive binding assay allowing the direct comparison of multiple nucleosome sequences in a single binding assay.
Results
Nucleosome design and formation
To determine the specificity of TP53 to nucleosomal DNA, we developed a competitive nucleosome binding assay (Fig. 1A). Starting with the Widom 601 nucleosome positioning sequence, 14 templates were designed and compared to nonspecific binding to two control sequences (Fig. 1B). With increasing distance to the dyad axis, three translational settings were tested: dyad (superhelix location [SHL] 0, 0.5), intermediate (SHL 4, 4.5), and edge (SHL 6.5, 7) (Fig. 1B). At each translational position, two rotational settings were tested by shifting the p53BS 5 bp to the right. p53BS accessibility was determined by modeling the TFBS position onto the nucleosome crystal structure formed from the Widom 601 sequence (Makde et al. 2010). In addition, a site in the linker region (SHL 8), which is outside the nucleosome core, was examined as well. We selected two p53BSs: The first is from its in vivo target CDKN1A promoter with relative low affinity; the other one is a modified sequence with high TP53 binding affinity. The two p53BSs were then separately added to Widom 601 DNA by replacing the base pairs at the selected locations. Consequently, we obtained 14 different nucleosomal templates having p53BS with increasing distance to the nucleosome dyad and being placed in either an exposed or concealed orientation (Fig. 1C). Nucleosomes were then assembled using salt gradient dialysis on all nucleosome sequences simultaneously (Supplemental Fig. S1).
Figure 1.

Design of nucleosome templates with p53BS. (A) A 217-bp dsDNA library was designed containing p53BS in various nucleosomal positions. Nucleosomes were formed and purified, generating a nucleosome library that was incubated with increasing amounts of TP53 protein. TP53–nucleosome complexes were separated by EMSA, and the bound and unbound DNA was recovered, quantified by qPCR, and sequenced. (B) A 20-bp-long p53BS (low and high affinity) was placed at different positions 0, 5, 41, 46, 66, 71, and 81 bp away from the dyad. The superhelix location (SHL) is designated for each nucleosome sequence. (C) Within the nucleosome, three different positions (dyad, intermediate, and edge) were chosen with increasing distance to nucleosome dyad.
Since we are modifying the Widom 601 nucleosome positioning sequence by inserting a p53BS, we validated nucleosome formation efficiency for each sequence compared with the unmodified Widom 601 sequence. After nucleosome assembly and gel shift assay, the nucleosomal DNA and free DNA were gel-extracted and sequenced. By comparing the number of reads for each template sequence in the nucleosome band with the number of reads for the 601 control sequence, we can determine the relative nucleosome formation efficiency. Nucleosome formation efficiency for each template was extremely consistent across the replicates with only small differences among nucleosome templates, <20% (Supplemental Fig. S1B).
TP53 is occluded from binding nucleosome core region
To get a comprehensive idea of how TP53 binds to nucleosomes, we combined the traditional TF–nucleosome binding assay with high-throughput sequencing, so that we can analyze multiple positions on the nucleosome simultaneously with one binding reaction. We added TP53 protein to 0.16 pmol purified nucleosome with increasing amount of TP53 (0–2 pmol, 0–286 nM). After a short incubation, the binding reactions were separated on a native polyacrylamide gel to detect the TP53–nucleosome complex (Fig. 2A). The first lane contained only nucleosomes and was used to measure background and input levels for each experimental replicate. As the concentration of TP53 increased, the first supershift band intensity increased and higher-order bands appeared. It is also noteworthy that as the TP53 amount increased, the intensity of nucleosome-only band significantly decreased. To determine the makeup of the shifted complex, we performed a modified western blot from the EMSA gel (see Supplemental Methods). This result confirmed a stable ternary complex composed of TP53 and nucleosome (Supplemental Fig. S2).
Figure 2.

TP53 is occluded from binding the nucleosome core region. (A) Nucleosomes containing 16 different sequences were bound to increasing amounts of TP53 protein and separated by native PAGE. Lanes contain the following: (1) 0.16 pmol nucleosomes; (2–5) 0.16 pmol of nucleosomes with 36, 71, 142, or 286 nM of TP53 (0.25, 0.5, 1, or 2 pmol). Nucleosome and the major supershift bands are indicated (Ss-1, Ss-2, Ss-3). (B–D) Relative supershift for each nucleosome is determined by counting the frequency of each sequence within the supershift and comparing it to nonspecific binding to the 601 sequence. This value is then normalized to the input ratios of nucleosomes (see Equation 1). Error bars, SEM; P-values are shown comparing each nucleosome sequence at a specific TP53 concentration to background levels in the input lane. (B) First supershift band (Ss-1). (C) Second supershift band (Ss-2). (D) Third supershift band (Ss-3). (E) Nucleosome sequences that are lost from the nucleosome band were quantified relative to the input nucleosomes. Strongly bound nucleosomes will generate negative relative shifts.
To determine which nucleosome TP53 preferentially binds, all shifted bands were gel extracted, DNA purified, quantified by qPCR, and sequenced. The sequencing results were then analyzed and mapped back to the original 16 nucleosome sequences. The resulting data set was then analyzed by multiple methods, all producing the same conclusions. In the first method, raw sequence counts were analyzed by comparing each template sequence to the control nonspecific sequence in the same binding experiment (see Equation 1). By comparing each nucleosome sequence to the nonspecific control sequences from the same lane, loading, PCR amplification, and next-generation sequencing (NGS) are all internally controlled. This approach is performed on each shifted band independently (Fig. 2A–D). Statistical significance for each template sequence was then determined by comparison to background measurements from the TP53-null lane. This analysis demonstrates that nucleosome sequences containing a p53BS located outside of the nucleosome are bound first at the lowest concentrations. Sites located near the nucleosome edge are also bound at low concentrations, while the nucleosomes containing a p53BS located near the dyad (±50 bp) are not specifically bound compared with the control nucleosomes. At higher concentrations of TP53, 142 and 286 nM, binding is no longer significantly different than nonspecific binding to the control nucleosomes.
To confirm these results from the shifted bands, we also examined the nonshifted nucleosome fragments (Fig. 2E). In this analysis, we examined the loss of DNA fragments relative to the starting amount (TP53-null nucleosome band). Nucleosomes that are bound strongly by TP53 will be shifted out of the nucleosome band and generate a negative relative shift. The advantage of this approach is that it allows the determination of binding regardless of TP53/nucleosome complex structure and location of the supershift. Previous studies have shown that TP53 oligomerizes into larger complexes (Stenger et al. 1992; Lee et al. 1994; Chène 2001; Kearns et al. 2016). These results confirm specific binding at nucleosome edges and linker. In this analysis, we do not see a drop-off in binding at higher TP53 concentrations relative to the control because once a specific nucleosome is shifted away from the nucleosome, it will still be accurately counted as missing regardless of where it has been shifted to. This differs from our examination of the supershift fragments because at higher concentrations of TP53, higher-order complexes are created, further shifting the nucleosome sequence and removing them from the supershift count.
To confirm the differences seen in our nucleosome binding experiments are not due to the placement of the p53BS at various locations within 601, we performed a control binding experiment to the pool of 16 DNA sequences (Supplemental Fig. S3). In this assay, all 16 DNA templates were mixed at equal amounts and then added to TP53 protein. The TP53–DNA supershift band was then excised, quantified, and sequenced. The results indicate that there are relatively small differences in binding to templates containing the same p53BS. The high-affinity p53BS is bound stronger than the low-affinity p53BS across all templates as expected.
The two approaches above are unable to estimate the nonspecific binding of TP53 to nucleosomes; therefore to determine both specific and nonspecific binding directly, we performed a more in-depth analysis using the DNA amounts determined by qPCR. Briefly, after isolating the DNA from the native PAGE, the amount of DNA was determined by qPCR using a standard curve generated from a control DNA fragment. This allows us to determine the absolute number of DNA molecules in a shifted or nucleosome band before amplification for NGS library generation. After sequencing, we then convert the relative counts from the sequencing library to an absolute number. These absolute counts were then used to determine the percentage of nucleosomes bound for each template across all TP53 concentrations (Equation 2). The KD was then determined by nonlinear regression (Heffler et al. 2012). As seen with the previous methods, significant supershift occurs for templates with p53BS at the edge or outside of the nucleosome (Fig. 3A,B). In both the high-affinity p53BS and low-affinity p53BS groups, it is clear that p53BS located near the nucleosome edges have the smallest KD values. As the p53BS moves closer into the nucleosome dyad, KD value gradually increases (Fig. 3C) and eventually shows no significant difference from the control non-BS-containing nucleosomes. Interestingly, p53BS affinity only matters when it is placed at nucleosome edges or in the linker; there is no significant difference between high-affinity p53BS and low-affinity p53BS when they are located within 50 bp of the nucleosome dyad (Fig. 3C). All analysis approaches consistently demonstrate that only p53BSs located within the nucleosome edge are bound. Furthermore, binding site affinity only affects TP53 binding for TFBSs located at the same nucleosomal positions. When we look at the same site in the linker region, the high-affinity p53BS has a lower KD than low-affinity p53BS, but when we compare the high-affinity p53BS in the edges (SHL 7) with the low-affinity BS in the linker (SHL 8), the low-affinity group displays smaller KD values, indicating higher binding affinity. This result shows that nucleosome translational position takes precedent over binding site affinity.
Figure 3.
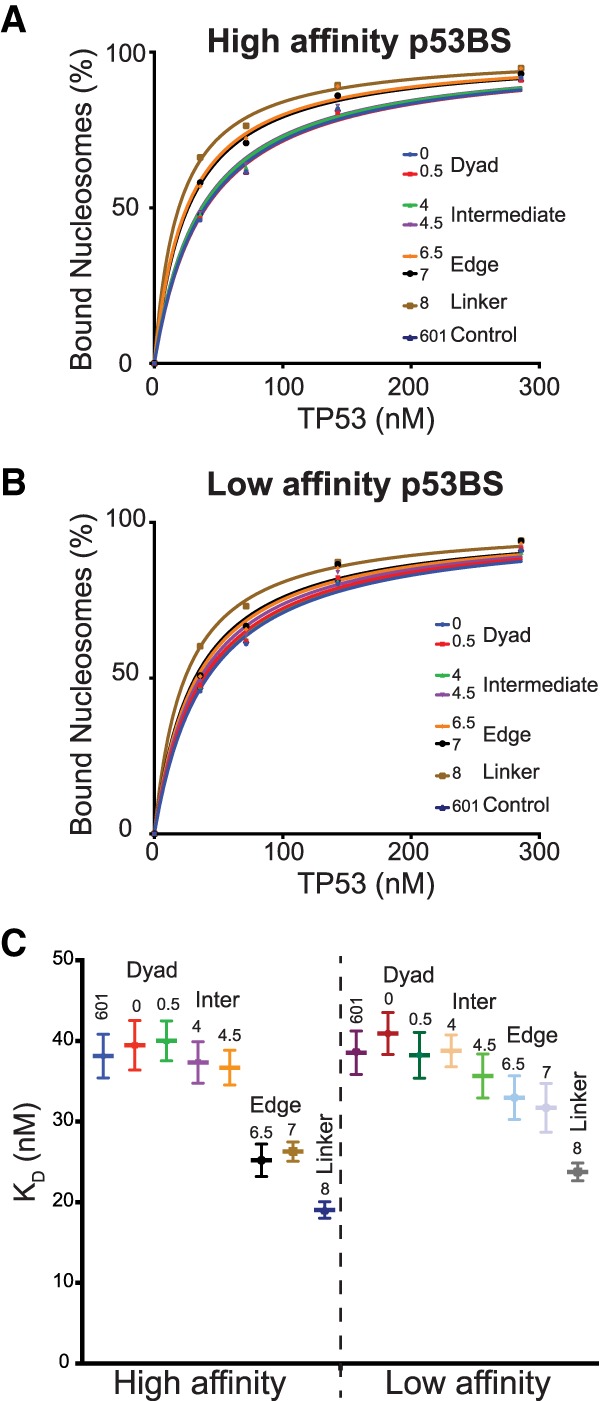
TP53 has higher affinity to p53BS near nucleosome boundaries. (A–C) The sequence results for the nucleosome bands were used to determine the binding affinities, KD, with nonlinear regression. (A) The percentage of nucleosomes bound by TP53 as TP53 is titrated. The location of the high-affinity p53BS is indicated. (B) The percentage of nucleosomes bound by TP53 as TP53 is titrated. The location of the low-affinity p53BS is indicated. (C) KD values with standard errors were plotted for each nucleosome.
TP53 has two DNA-binding domains: a core domain (DBD), which shows sequence-specific binding ability; and a highly basic C-terminal domain (CTD), which binds without specificity (Lee et al. 1995; Reed et al. 1995; Selivanova et al. 1996). To evaluate the role of the DBD and the CTD, we repeated our nucleosome binding assay with mutant or truncated proteins. The DBD mutant TP53C135S and the truncated TP53 (TP53ΔCTD: Gly108-Lys370) were compared to the full-length normal TP53 with 0.25 pmol of nucleosomes (Supplemental Figs. S4, S5). For TP53C135S there is a smear on the EMSA gel suggestive of nonspecific binding. For TP53ΔCTD there is little to no smearing, suggesting limited binding. TP53C135S appears to bind nonspecifically as evident by the low relative supershift for all nucleosome fragments. These results are consistent with previous finding showing that both DNA-binding domains are required for TP53 binding (Sullivan et al. 2018). The purified TP53 variant proteins were obtained from different sources, which could affect the presented interpretation.
TP53 binds nucleosome edges in human lung fibroblasts
To validate the relevance of our in vitro binding results, we examined TP53-induced binding in IMR-90 human lung fibroblasts combined with steady-state nucleosome occupancy as measured by MNase-seq (Kelly et al. 2012; Sammons et al. 2015). To perform this analysis, we reanalyzed the published MNase-seq and ChIP-seq data sets. The TP53 ChIP-seq results are after TP53 is induced and define the specific sites bound by TP53. The MNase-seq data were from the same cell line in a steady state without TP53 activation and define the nucleosome position/occupancy before TP53 binds. TP53 ChIP-seq experiments were performed on IMR-90 cells treated with a DMSO control or after TP53 activation with nutlin. For each bound site, the p53BS was determined from a predefined list of TP53 motif locations. This step ensures that we are examining direct binding by TP53 and have accurate binding locations but will miss ill-defined or nonconsensus binding sequences. The raw MNase-seq data were then extracted, standardized, and visualized centered at the p53BS located within the TP53 ChIP-seq peaks. As shown previously, average nucleosome occupancy peaks at TP53-induced binding sites (Fig. 4A). Closer examination of individual sites after clustering the MNase-seq results shows that p53BSs are located within the nucleosome edge flanked by a region of lower nucleosome occupancy (Fig. 4B). These results suggest that TP53-induced binding in vivo occurs at the edges of nucleosomes, in a manner consistent with our in vitro findings.
Figure 4.
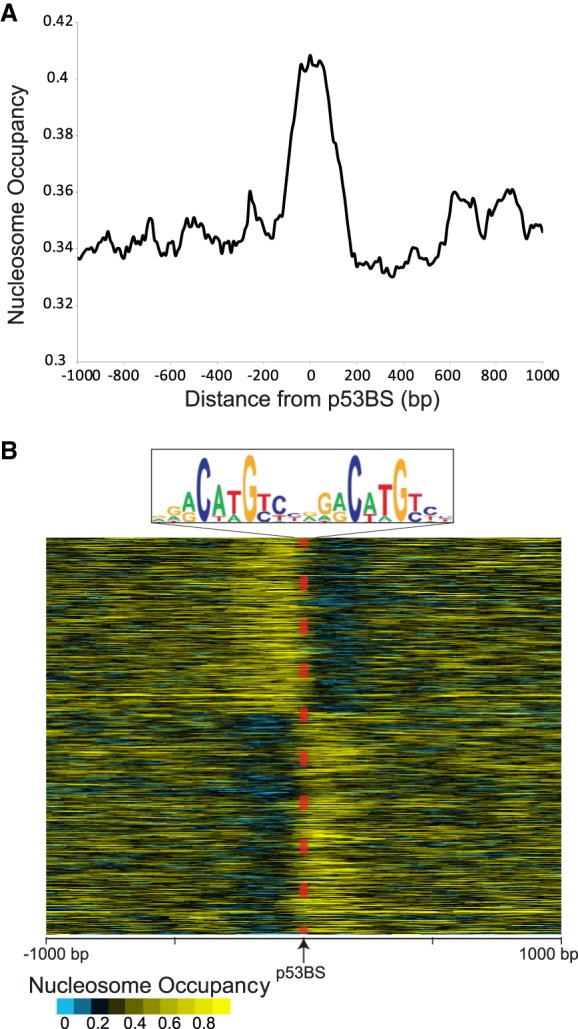
Nucleosome occupancy at TP53 nutlin-induced binding sites. (A) Average nucleosome occupancy at induced TP53 binding sites determined from MNase-seq. TP53 ChIP-seq results from Sammons et al. (2015) with MNase-seq data from Kelly et al. (2012; Sammons et al. 2015). The MNase-seq reads were extracted, standardized (1 billion reads), and extended with ArchTEx (Lai et al. 2012). (B) Nucleosome occupancy at each p53BS (±1000-bp) after nutlin-induced TP53 binding. Standardized and extended MNase-seq reads were clustered by symmetry (Lai and Buck 2010).
Discussion
To identify the rules defining TF targeting in chromatin, we developed a new approach to examine the binding characteristics of TFs to nucleosome DNA. Our approach combines traditional proven nucleosome binding assays with NGS. This approach allowed the direct comparison of multiple nucleosomes in a single experiment. Each nucleosome template sequence was designed allowing the positioning of high or low-affinity p53BS in various translational and rotational settings. Nucleosomes were then generated from the pool of nucleosome sequences, purified, and competitively bound to TP53. Each experiment is controlled at multiple steps. The starting nucleosomes or input to each binding reaction is quantified and sequenced after PAGE (TP53-null lane). This input measurement ensures correction for any variation in nucleosome formation efficiency or starting quantities. Within this input lane, blank bands corresponding to the supershifted TP53–nucleosome complex are also quantified and sequenced. This measurement represents the background for each particular supershift. As can be seen in Figure 2, for the 0 nM sample (gray bar), there is only slight differences between different nucleosome sequences. In addition to examining the supershifted fragments, we also analyzed the nucleosome bands and compared the quantities of each template to the input nucleosomes. In this case we are determining the nucleosome sequences that have shifted out of the nucleosome band after being bound. This analysis does not require us to determine the appropriate supershift band and is resilient to the formation of higher-order TP53–nucleosome complexes seen at higher TP53 concentrations. To ensure the accurate absolute quantification of our results, we measure the DNA concentration of each gel-excised sample by qPCR. By using this amount with the NGS results, we can calculate the percentage of bound nucleosomes for each nucleosome type across all TP53 concentrations. The resulting data were then fitted using nonlinear regression to obtain KD values, allowing the examination of both specific and nonspecific binding. The methodology we have proposed is not limited to TP53–nucleosome binding but can be applied to understand how other pioneer factors bind nucleosomal DNA.
Regardless of the analysis methods we used, all approaches consistently showed that TFBS positioning within a nucleosome affects TP53-binding capability. TP53 displays a strong preference to sites outside the 100 bp surrounding the nucleosome dyad. These results are consistent with a dynamic partial unwrapping near nucleosome edges (Polach and Widom 1995; Li and Widom 2004). In this model, DNA near the entry–exit region is unwrapped from the histone proteins exposing the DNA to TF binding. Once bound, the nucleosome can then rewrap, thus kicking off the TF (Luo et al. 2014). Our results for TP53 suggest that TP53 can access the partially unwrapped nucleosome and remain stably bound. Binding by TP53 to nucleosomes differs when compared to FOXA, which can target its binding sites near nucleosome dyads (McPherson et al. 1993; Chaya et al. 2001). FOXA binds nucleosomal DNA on one side as a monomer (Cirillo and Zaret 2007), whereas TP53 binds as a tetramer complex that partially wraps around the DNA (Malecka et al. 2009; Emamzadah et al. 2011). These differences in how a TF contacts its binding site may explain the ability of some TFs to bind within the nucleosome core.
TP53 displays a strong preference to the nucleosome structure itself and binds nucleosomes in a consensus sequence-independent manner with relative high affinity. Our experiments show that control nucleosomes are bound with a KD of 38.12–38.54 nM compared with 19.04 nM for the high-affinity p53BS located in the linker. Fluorescence recovery after photobleaching (FRAP) supports the model that TP53 binds nonspecifically to chromatin in the nucleus. Hinow et al. (2006) compared wild-type TP53 with a DNA binding mutant and showed indistinguishable nucleus diffusion properties, suggesting that TP53 had significant sequence-independent binding to chromatin (Hinow et al. 2006). Our experiments with mutated TP53C135S and truncated TP53ΔCTD suggest that the sequence-independent nucleosome binding is due to TP53's CTD. Our findings are in agreement with recent studies that show the CTD to be a crucial part for maintaining TP53 full function and ensuring binding stability in vivo (Rodriguez et al. 2000; Espinosa and Emerson 2001; McKinney et al. 2004; Tafvizi et al. 2011; Kim et al. 2012; Laptenko et al. 2015).
Our in vitro results combined with the in vivo binding patterns suggest that TP53 is not a FOXA-like canonical pioneer factor; its specific binding capabilities are limited to nucleosome edges flanked by a nucleosome-free region. This presents a model for TP53 nucleosome binding where TP53 encroaches on nucleosomes from an exposed linker region where TP53's CTD binds nonspecifically and slides along the DNA (Tafvizi et al. 2008, 2011; Khazanov and Levy 2011; Murata et al. 2015). It then gains access to its binding site within the nucleosome edge by the nucleosome partially unwrapping (Fig. 5). Once TP53 binds, it can recruit histone acetyltransferases, further activating the bound enhancer or promoter region (Gu and Roeder 1997; Gu et al. 1997). Our model does not take into consideration the consequences of the role of histone variants like H2A.Z, which has been shown to associate with TP53 binding in vivo (Gevry et al. 2007). All members of the TP53/TP63/TP73 family share a similar DNA-binding domain with high sequence and structural homology (Levrero et al. 2000). Structural studies of DNA-binding domains for TP53, TP63, and TP73 cocrystallized with DNA target sequences reveal an overall conserved conformation and DNA–protein contact sites for the three proteins (Chen et al. 2011; Ethayathulla et al. 2012; Ethayathulla et al. 2013). These structural similarities between family members suggest that TP63 and TP73 will also bind nucleosomal DNA in a similar manner. Genomic studies on TP63 have suggested that TP63 also acts as pioneer TF during epidermal development by binding at regions of the genome with high encoded nucleosome occupancy (Sethi et al. 2014).
Figure 5.
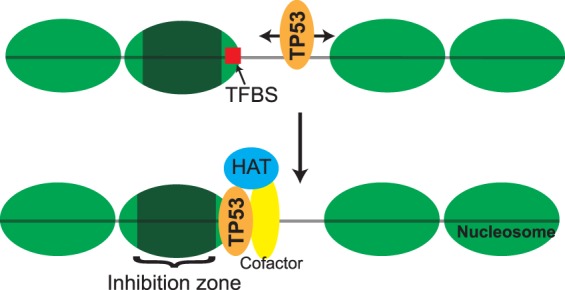
Model of TP53 binding to targets within nucleosomes. TP53 scans DNA in a sequence-independent manner and targets p53BS located at the nucleosome edge. Once bound, TP53 can recruit cofactors and histone acetyltransferases (HATs).
Chromatin accessibility has been recognized as a prerequirement for functional activity at regulatory elements (Tsompana and Buck 2014). Our results demonstrate a more meticulous understanding of how positioning within a nucleosome can affect TF binding. In particular, our results demonstrate that positioning within a nucleosome is more important than the affinity of the underlying binding site and that small differences in positioning, 50 bp, can dramatically affect TF binding. Therefore, to accurately identify the earliest events during gene activation, high-resolution nucleosome maps will be needed with an understanding of which TFs can target their binding sites within the nucleosome. The methodology we have presented here provides a comprehensive approach to examine the rules dictating nucleosome binding by pioneer factors; further studies on all pioneer factors will allow the identification of general themes driving gene regulation by pioneer factors.
Methods
Design of the nucleosome positioning templates
Nucleosomal templates were derived from the Widom 601 strong nucleosome-positioning sequence (Lowary and Widom 1998; Anderson and Widom 2000). The 601 sequence was first scanned for the presence of sequences similar to the p53BS. The original 601 sequence contains a TP53 half-site core (CATG) located just outside the nucleosome edge. To ensure that this sequence does not affect the binding assays, we modified this sequence to AGGT. We called it “601-modified,” which was regarded as an additional control sequence. The original Widom 601 DNA (601) was still used in the study and had indistinguishable results compared with 601-modified. Two p53BS were used: an adapted high-affinity ideal sequence, 5′-GGGCATGTCCGGGCATGTCC-3′ (Veprintsev and Fersht 2008; Noureddine et al. 2009); and a natural lower-affinity sequence from the CDKN1A promoter, 5′-AGACTGGGCATGTCTGGGCA-3′ (Westfall et al. 2003). Within each p53BS, there are two cores (underlined above), which are directly bound by TP53. We design different fragments to ensure that the cores are in either an exposed or concealed orientation as determined by the crystal structure of a nucleosome with the Widom 601 sequence (Makde et al. 2010). Therefore, 14 templates were designed starting from the 217-bp Widom 601 sequence and compared with nonspecific binding to two control sequences (Supplemental Table S1).
In vitro nucleosome reconstitution and purification
The 217-bp nucleosome sequences for these experiments were obtained as double-stranded DNA fragments from Integrated DNA Technologies. All 16 synthesized DNA templates were amplified via PCR and column-purified (Qiagen) and quantified. In vitro nucleosomes were generated from H2A/H2B dimer and H3.1/H4 tetramer (NEB). All 16 nucleosome sequences were mixed at equal molar amounts. Mixed DNAs were then added to histones at octamer/DNA molar ratios of 1.5:1 in 2 M NaCl. Nucleosomes were reconstituted through salt gradient dialysis as previously described (Hayes and Lee 1997), which were further purified by 7%–20% sucrose gradient centrifuge (Fang et al. 2016) and concentrated by 50,000 centrifugal filter units (Millipore, Amicon ultra).
DNA binding assay followed by EMSA
The protein-nucleosome binding assays were carried out four times with the purified nucleosomes mentioned above and human full-length recombinant TP53 protein (Abcam catalog no. ab84768) in 7 µL DNA binding buffer (10 mM Tris-Cl at pH 7.5, 50 mM NaCl, 1 mM DTT, 0.25 mg/mL BSA, 2 mM MgCl2, 0.025% Nonidet P-40 substitute, and 5% glycerol) and then incubated for 10 min on ice and then for 30 min at room temperature. Increasing concentrations of TP53 (0–286 nM; 0–2 pmol) were added to 0.16 pmol purified nucleosomes. Protein binding was detected by mobility shift assay on 4% (w/v) native polyacrylamide gels (acrylamide/bisacrylamide, 29:1, w/w, 7 × 10 cm) in 0.5× Tris borate-EDTA buffers at 100 V at 4°C. After electrophoresis, DNA was imaged by staining with SYBR green (Lonza).
Library construction and sequencing
All visual bands were excised from the gel, as well as the bands at the same locations in the other lanes. Each gel slice was processed separately for a total of 104 samples from six TP53 full-length and two TP53C135S experiments. The DNA concentration for each sample was determined by qPCR using a standard curve generated from the control 601 sequence. Illumina sequencing libraries were generated using standard two-step PCR amplicon methodology with indexing (see Supplemental Methods). The number of cycles for PCR step 1 was determined by the sample concentration determined by qPCR, and ranged from eight to 12 cycles. All samples were multiplexed and sequenced on a MiSeq using 2 × 150-bp paired-end sequencing.
Data analysis
Quality sequence reads were mapped to each specific starting sequence using BLAT (Kent 2002). Processed results after mapping are in Supplemental Table S2. The results were then analyzed relative to control/nonspecific binding (relative supershift) or by determining the binding affinity by fitting a binding curve. Relative supershift is determined from the supershift bands and controls technical variability introduced by gel-excision, PCR, NGS library construction, or NGS sequencing. In this method, each specific nucleosome sequence is measured relative to nonspecific binding (control 601 fragment):
| (1) |
where N is one of the 16 nucleosome sequences, 601 is the control nucleosome sequence, reads supershift is the supershift band, and reads nucleosome band is the nucleosome band in TP53-null lane.
By using the DNA concentrations after gel-excision with the number of each nucleosome sequence in a sample, the absolute number of a particular nucleosome can be determined for each sample. These absolute nucleosome counts when applied to the nucleosome only bands at each TP53 concentration are used to determine the percentage of bound nucleosomes for each nucleosome fragment with the following:
| (2) |
The KD values are then estimated with nonlinear regression in Prism (Heffler et al. 2012).
Analysis of TP53-induced binding in IMR-90 cells
TP53 ChIP-seq binding sites identified after nutlin-induced binding in IMR-90 cells were obtained from GEO accession GSE58740. MNase-seq data from proliferating IMR-90 were obtained from GEO accession GSE21823 and aligned to hg19 with Bowtie 2 (Langmead and Salzberg 2012). Aligning to GRCh38 (hg38) does not change the presented results. p53BS from HOMER (Heinz et al. 2010) were intersected to the TP53-induced binding sites to define the exact location of binding. TP53 ChIP-seq binding sites without a previously defined p53BS were excluded from the analysis. The MNase-seq reads were extracted, standardized (1 billion reads), and extended (120 bp) as done previously with ArchTEx (Lai et al. 2012; Rizzo et al. 2012). Symmetry of resulting MNase-seq data set at p53BS was then determined with ArchAlign with 0-bp shifts and region reversal enabled (Lai and Buck 2010; Givens et al. 2012).
Data access
The unprocessed sequencing data from this study have been submitted to the NCBI BioProject database (BioProject; https://www.ncbi.nlm.nih.gov/bioproject/) under accession number PRJNA498696. Processed results are available in Supplemental Table S2.
Supplementary Material
Acknowledgments
We thank Dr. Jeffrey Hayes and Dr. Laxmi Mishra at University of Rochester Medical Center for suggestions of sucrose gradient centrifuge to purify nucleosomes, and UB Genomics and Bioinformatics Core for next-generation sequencing services. This work was partially supported by Mark Diamond Research Fund FA-16-22 to X.Y. and by the New York State Department of Health C026714.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.234104.117.
References
- Anderson JD, Widom J. 2000. Sequence and position-dependence of the equilibrium accessibility of nucleosomal DNA target sites. J Mol Biol 296: 979–987. 10.1006/jmbi.2000.3531 [DOI] [PubMed] [Google Scholar]
- Angelov D, Lenouvel F, Hans F, Müller CW, Bouvet P, Bednar J, Moudrianakis EN, Cadet J, Dimitrov S. 2004. The histone octamer is invisible when NF-κB binds to the nucleosome. J Biol Chem 279: 42374–42382. 10.1074/jbc.M407235200 [DOI] [PubMed] [Google Scholar]
- Blomquist P, Li Q, Wrange O. 1996. The affinity of nuclear factor 1 for its DNA site is drastically reduced by nucleosome organization irrespective of its rotational or translational position. J Biol Chem 271: 153–159. 10.1074/jbc.271.1.153 [DOI] [PubMed] [Google Scholar]
- Buck MJ, Lieb JD. 2006. A chromatin-mediated mechanism for specification of conditional transcription factor targets. Nat Genet 38: 1446–1451. 10.1038/ng1917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaya D, Hayamizu T, Bustin M, Zaret KS. 2001. Transcription factor FoxA (HNF3) on a nucleosome at an enhancer complex in liver chromatin. J Biol Chem 276: 44385–44389. 10.1074/jbc.M108214200 [DOI] [PubMed] [Google Scholar]
- Chen C, Gorlatova N, Kelman Z, Herzberg O. 2011. Structures of p63 DNA binding domain in complexes with half-site and with spacer-containing full response elements. Proc Natl Acad Sci 108: 6456–6461. 10.1073/pnas.1013657108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chène P. 2001. The role of tetramerization in p53 function. Oncogene 20: 2611–2617. 10.1038/sj.onc.1204373 [DOI] [PubMed] [Google Scholar]
- Cirillo LA, Zaret KS. 2007. Specific interactions of the wing domains of FOXA1 transcription factor with DNA. J Mol Biol 366: 720–724. 10.1016/j.jmb.2006.11.087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirillo LA, Lin FR, Cuesta I, Friedman D, Jarnik M, Zaret KS. 2002. Opening of compacted chromatin by early developmental transcription factors HNF3 (FoxA) and GATA-4. Mol Cell 9: 279–289. 10.1016/S1097-2765(02)00459-8 [DOI] [PubMed] [Google Scholar]
- el-Deiry WS, Kern SE, Pietenpol JA, Kinzler KW, Vogelstein B. 1992. Definition of a consensus binding site for p53. Nat Genet 1: 45–49. 10.1038/ng0492-45 [DOI] [PubMed] [Google Scholar]
- Emamzadah S, Tropia L, Halazonetis TD. 2011. Crystal structure of a multidomain human p53 tetramer bound to the natural CDKN1A (p21) p53-response element. Mol Cancer Res 9: 1493–1499. 10.1158/1541-7786.MCR-11-0351 [DOI] [PubMed] [Google Scholar]
- Espinosa JM, Emerson BM. 2001. Transcriptional regulation by p53 through intrinsic DNA/chromatin binding and site-directed cofactor recruitment. Mol Cell 8: 57–69. 10.1016/S1097-2765(01)00283-0 [DOI] [PubMed] [Google Scholar]
- Ethayathulla AS, Tse PW, Monti P, Nguyen S, Inga A, Fronza G, Viadiu H. 2012. Structure of p73 DNA-binding domain tetramer modulates p73 transactivation. Proc Natl Acad Sci 109: 6066–6071. 10.1073/pnas.1115463109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ethayathulla AS, Nguyen HT, Viadiu H. 2013. Crystal structures of the DNA-binding domain tetramer of the p53 tumor suppressor family member p73 bound to different full-site response elements. J Biol Chem 288: 4744–4754. 10.1074/jbc.M112.408039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang H, Wei S, Lee TH, Hayes JJ. 2016. Chromatin structure-dependent conformations of the H1 CTD. Nucleic Acids Res 44: 9131–9141. 10.1093/nar/gkw586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gevry N, Chan HM, Laflamme L, Livingston DM, Gaudreau L. 2007. p21 transcription is regulated by differential localization of histone H2A.Z. Genes Dev 21: 1869–1881. 10.1101/gad.1545707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Givens RM, Lai WK, Rizzo JM, Bard JE, Mieczkowski PA, Leatherwood J, Huberman JA, Buck MJ. 2012. Chromatin architectures at fission yeast transcriptional promoters and replication origins. Nucleic Acids Res 40: 7176–7189. 10.1093/nar/gks351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu W, Roeder RG. 1997. Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell 90: 595–606. 10.1016/S0092-8674(00)80521-8 [DOI] [PubMed] [Google Scholar]
- Gu W, Shi XL, Roeder RG. 1997. Synergistic activation of transcription by CBP and p53. Nature 387: 819–823. 10.1038/42972 [DOI] [PubMed] [Google Scholar]
- Hayes JJ, Lee KM. 1997. In vitro reconstitution and analysis of mononucleosomes containing defined DNAs and proteins. Methods 12: 2–9. 10.1006/meth.1997.0441 [DOI] [PubMed] [Google Scholar]
- Heffler MA, Walters RD, Kugel JF. 2012. Using electrophoretic mobility shift assays to measure equilibrium dissociation constants: GAL4-p53 binding DNA as a model system. Biochem Mol Biol Educ 40: 383–387. 10.1002/bmb.20649 [DOI] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK. 2010. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 38: 576–589. 10.1016/j.molcel.2010.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinow P, Rogers CE, Barbieri CE, Pietenpol JA, Kenworthy AK, DiBenedetto E. 2006. The DNA binding activity of p53 displays reaction-diffusion kinetics. Biophys J 91: 330–342. 10.1529/biophysj.105.078303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwafuchi-Doi M, Zaret KS. 2014. Pioneer transcription factors in cell reprogramming. Genes Dev 28: 2679–2692. 10.1101/gad.253443.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang C, Pugh BF. 2009. Nucleosome positioning and gene regulation: advances through genomics. Nat Rev Genet 10: 161–172. 10.1038/nrg2522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearns S, Lurz R, Orlova EV, Okorokov AL. 2016. Two p53 tetramers bind one consensus DNA response element. Nucleic Acids Res 44: 6185–6199. 10.1093/nar/gkw215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly TK, Liu Y, Lay FD, Liang G, Berman BP, Jones PA. 2012. Genome-wide mapping of nucleosome positioning and DNA methylation within individual DNA molecules. Genome Res 22: 2497–2506. 10.1101/gr.143008.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WJ. 2002. BLAT: the BLAST-like alignment tool. Genome Res 12: 656–664. 10.1101/gr.229202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khazanov N, Levy Y. 2011. Sliding of p53 along DNA can be modulated by its oligomeric state and by cross-talks between its constituent domains. J Mol Biol 408: 335–355. 10.1016/j.jmb.2011.01.059 [DOI] [PubMed] [Google Scholar]
- Kim H, Kim K, Choi J, Heo K, Baek HJ, Roeder RG, An W. 2012. p53 requires an intact C-terminal domain for DNA binding and transactivation. J Mol Biol 415: 843–854. 10.1016/j.jmb.2011.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornberg RD, Lorch Y. 1999. Twenty-five years of the nucleosome, fundamental particle of the eukaryote chromosome. Cell 98: 285–294. 10.1016/S0092-8674(00)81958-3 [DOI] [PubMed] [Google Scholar]
- Lai WK, Buck MJ. 2010. ArchAlign: coordinate-free chromatin alignment reveals novel architectures. Genome Biol 11: R126 10.1186/gb-2010-11-7-126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai WK, Bard JE, Buck MJ. 2012. ArchTEx: accurate extraction and visualization of next-generation sequence data. Bioinformatics 28: 1021–1023. 10.1093/bioinformatics/bts063 [DOI] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9: 357–359. 10.1038/nmeth.1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laptenko O, Beckerman R, Freulich E, Prives C. 2011. p53 binding to nucleosomes within the p21 promoter in vivo leads to nucleosome loss and transcriptional activation. Proc Natl Acad Sci 108: 10385–10390. 10.1073/pnas.1105680108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laptenko O, Shiff I, Freed-Pastor W, Zupnick A, Mattia M, Freulich E, Shamir I, Kadouri N, Kahan T, Manfredi J, et al. 2015. The p53 C terminus controls site-specific DNA binding and promotes structural changes within the central DNA binding domain. Mol Cell 57: 1034–1046. 10.1016/j.molcel.2015.02.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee W, Harvey TS, Yin Y, Yau P, Litchfield D, Arrowsmith CH. 1994. Solution structure of the tetrameric minimum transforming domain of p53. Nat Struct Biol 1: 877–890. 10.1038/nsb1294-877 [DOI] [PubMed] [Google Scholar]
- Lee S, Elenbaas B, Levine A, Griffith J. 1995. p53 and its 14 kDa C-terminal domain recognize primary DNA damage in the form of insertion/deletion mismatches. Cell 81: 1013–1020. 10.1016/S0092-8674(05)80006-6 [DOI] [PubMed] [Google Scholar]
- Levrero M, De Laurenzi V, Costanzo A, Gong J, Wang JY, Melino G. 2000. The p53/p63/p73 family of transcription factors: overlapping and distinct functions. J Cell Sci 113(Pt.10): 1661–1670. [DOI] [PubMed] [Google Scholar]
- Li G, Widom J. 2004. Nucleosomes facilitate their own invasion. Nat Struct Mol Biol 11: 763–769. 10.1038/nsmb801 [DOI] [PubMed] [Google Scholar]
- Li Q, Wrange O. 1993. Translational positioning of a nucleosomal glucocorticoid response element modulates glucocorticoid receptor affinity. Genes Dev 7: 2471–2482. 10.1101/gad.7.12a.2471 [DOI] [PubMed] [Google Scholar]
- Lidor Nili E, Field Y, Lubling Y, Widom J, Oren M, Segal E. 2010. p53 binds preferentially to genomic regions with high DNA-encoded nucleosome occupancy. Genome Res 20: 1361–1368. 10.1101/gr.103945.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowary PT, Widom J. 1998. New DNA sequence rules for high affinity binding to histone octamer and sequence-directed nucleosome positioning. J Mol Biol 276: 19–42. 10.1006/jmbi.1997.1494 [DOI] [PubMed] [Google Scholar]
- Luger K, Dechassa ML, Tremethick DJ. 2012. New insights into nucleosome and chromatin structure: an ordered state or a disordered affair? Nat Rev Mol Cell Biol 13: 436–447. 10.1038/nrm3382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Y, North JA, Rose SD, Poirier MG. 2014. Nucleosomes accelerate transcription factor dissociation. Nucleic Acids Res 42: 3017–3027. 10.1093/nar/gkt1319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makde RD, England JR, Yennawar HP, Tan S. 2010. Structure of RCC1 chromatin factor bound to the nucleosome core particle. Nature 467: 562–566. 10.1038/nature09321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malecka KA, Ho WC, Marmorstein R. 2009. Crystal structure of a p53 core tetramer bound to DNA. Oncogene 28: 325–333. 10.1038/onc.2008.400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinney K, Mattia M, Gottifredi V, Prives C. 2004. p53 linear diffusion along DNA requires its C terminus. Mol Cell 16: 413–424. 10.1016/j.molcel.2004.09.032 [DOI] [PubMed] [Google Scholar]
- McPherson CE, Shim EY, Friedman DS, Zaret KS. 1993. An active tissue-specific enhancer and bound transcription factors existing in a precisely positioned nucleosomal array. Cell 75: 387–398. 10.1016/0092-8674(93)80079-T [DOI] [PubMed] [Google Scholar]
- Murata A, Ito Y, Kashima R, Kanbayashi S, Nanatani K, Igarashi C, Okumura M, Inaba K, Tokino T, Takahashi S, et al. 2015. One-dimensional sliding of p53 along DNA is accelerated in the presence of Ca2+ or Mg2+ at millimolar concentrations. J Mol Biol 427: 2663–2678. 10.1016/j.jmb.2015.06.016 [DOI] [PubMed] [Google Scholar]
- Noureddine MA, Menendez D, Campbell MR, Bandele OJ, Horvath MM, Wang X, Pittman GS, Chorley BN, Resnick MA, Bell DA. 2009. Probing the functional impact of sequence variation on p53-DNA interactions using a novel microsphere assay for protein-DNA binding with human cell extracts. PLoS Genet 5: e1000462 10.1371/journal.pgen.1000462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polach KJ, Widom J. 1995. Mechanism of protein access to specific DNA sequences in chromatin: a dynamic equilibrium model for gene regulation. J Mol Biol 254: 130–149. 10.1006/jmbi.1995.0606 [DOI] [PubMed] [Google Scholar]
- Reed M, Woelker B, Wang P, Wang Y, Anderson ME, Tegtmeyer P. 1995. The C-terminal domain of p53 recognizes DNA damaged by ionizing radiation. Proc Natl Acad Sci 92: 9455–9459. 10.1073/pnas.92.21.9455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivlin N, Brosh R, Oren M, Rotter V. 2011. Mutations in the p53 tumor suppressor gene: important milestones at the various steps of tumorigenesis. Genes Cancer 2: 466–474. 10.1177/1947601911408889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzo JM, Bard JE, Buck MJ. 2012. Standardized collection of MNase-seq experiments enables unbiased dataset comparisons. BMC Mol Biol 13: 15 10.1186/1471-2199-13-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez MS, Desterro JM, Lain S, Lane DP, Hay RT. 2000. Multiple C-terminal lysine residues target p53 for ubiquitin-proteasome-mediated degradation. Mol Cell Biol 20: 8458–8467. 10.1128/MCB.20.22.8458-8467.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahu G, Wang D, Chen CB, Zhurkin VB, Harrington RE, Appella E, Hager GL, Nagaich AK. 2010. p53 binding to nucleosomal DNA depends on the rotational positioning of DNA response element. J Biol Chem 285: 1321–1332. 10.1074/jbc.M109.081182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sammons MA, Zhu J, Drake AM, Berger SL. 2015. TP53 engagement with the genome occurs in distinct local chromatin environments via pioneer factor activity. Genome Res 25: 179–188. 10.1101/gr.181883.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekiya T, Muthurajan UM, Luger K, Tulin AV, Zaret KS. 2009. Nucleosome-binding affinity as a primary determinant of the nuclear mobility of the pioneer transcription factor FoxA. Genes Dev 23: 804–809. 10.1101/gad.1775509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selivanova G, Iotsova V, Kiseleva E, Strom M, Bakalkin G, Grafstrom RC, Wiman KG. 1996. The single-stranded DNA end binding site of p53 coincides with the C-terminal regulatory region. Nucleic Acids Res 24: 3560–3567. 10.1093/nar/24.18.3560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sethi I, Sinha S, Buck MJ. 2014. Role of chromatin and transcriptional co-regulators in mediating p63-genome interactions in keratinocytes. BMC Genomics 15: 1042 10.1186/1471-2164-15-1042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenger JE, Mayr GA, Mann K, Tegtmeyer P. 1992. Formation of stable p53 homotetramers and multiples of tetramers. Mol Carcinog 5: 102–106. 10.1002/mc.2940050204 [DOI] [PubMed] [Google Scholar]
- Sullivan KD, Galbraith MD, Andrysik Z, Espinosa JM. 2018. Mechanisms of transcriptional regulation by p53. Cell Death Differ 25: 133–143. 10.1038/cdd.2017.174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tafvizi A, Huang F, Leith JS, Fersht AR, Mirny LA, van Oijen AM. 2008. Tumor suppressor p53 slides on DNA with low friction and high stability. Biophys J 95: L01–L03. 10.1529/biophysj.108.134122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tafvizi A, Huang F, Fersht AR, Mirny LA, van Oijen AM. 2011. A single-molecule characterization of p53 search on DNA. Proc Natl Acad Sci 108: 563–568. 10.1073/pnas.1016020107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsompana M, Buck MJ. 2014. Chromatin accessibility: a window into the genome. Epigenetics Chromatin 7: 33 10.1186/1756-8935-7-33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veprintsev DB, Fersht AR. 2008. Algorithm for prediction of tumour suppressor p53 affinity for binding sites in DNA. Nucleic Acids Res 36: 1589–1598. 10.1093/nar/gkm1040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermaak D, Ahmad K, Henikoff S. 2003. Maintenance of chromatin states: an open-and-shut case. Curr Opin Cell Biol 15: 266–274. 10.1016/S0955-0674(03)00043-7 [DOI] [PubMed] [Google Scholar]
- Vettese-Dadey M, Walter P, Chen H, Juan LJ, Workman JL. 1994. Role of the histone amino termini in facilitated binding of a transcription factor, GAL4-AH, to nucleosome cores. Mol Cell Biol 14: 970–981. 10.1128/MCB.14.2.970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westfall MD, Mays DJ, Sniezek JC, Pietenpol JA. 2003. The ΔNp63α phosphoprotein binds the p21 and 14-3-3σ promoters in vivo and has transcriptional repressor activity that is reduced by Hay-Wells syndrome-derived mutations. Mol Cell Biol 23: 2264–2276. 10.1128/MCB.23.7.2264-2276.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.