

Abstract
Background:
Peripapillary sparing is a characteristic that is traditionally described as pathognomonic for Stargardt disease.
Materials and methods:
We present a multi-modal assessment of four f Leber Congenital Amaurosis (LCA) cases with congenital macular atrophy and severely attenuated electroretinogram findings caused by bilallelic mutations in RDH12.
Results:
Fundus autofluorescence imaging revealed a general loss of retinal pigment epithelium across the macula except for the peripapillary region in both eyes of all patients. Spectral domain-optical coherence tomography confirmed relative preservation in this area along with retinal thinning and excavation throughout the rest of the macula. LCA was diagnosed based on clinical exam and retinal imaging, and subsequently confirmed with genetic testing.
Conclusions:
Peripapillary sparing is a novel phenotypic feature of RDH12-associated LCA.
Keywords: Autofluorescence, Leber congenital amaurosis, peripapillary sparing, RDH12, spectral domain optical coherence tomography
INTRODUCTION
Leber congenital amaurosis (LCA) refers to a clinically and genetically heterogeneous group of inherited retinal dystrophies characterized by a progressive, early-onset visual impairment, nystagmus and amaurotic pupils.1,2 The cause of LCA is attributed to mutations in several genes that include GUCY2D, RPE65, SPATA7, AIPL1, CEP290, RPGRIP1, NMNAT1, and RDH12.3,4 The clinical spectrum of LCA comprises clinical features of other inherited retinal disease such as cone-rod dystrophy and retinitis pigmentosa. Peripapillary sparing is a finding considered pathognomonic of Stargardt disease (STGD1), a recessive, juvenile-onset maculopathy caused by mutations in the ATP Binding Cassette Subfamily A Member 4 (ABCA4). Visual symptoms in STGD1 begin in the second or third decade of life (Cideciyan et al., 2005) and can often progress to retina-wide degeneration due to the accumulation of toxic bisretinoid-containing lipofuscin in the retinal pigment epithelium (RPE). Although these are both devastating retinal dystrophies, they have distinct genetic etiologies. This report describes cases of four children with clinical and electrophysiologic manifestations of LCA caused by mutations in RDH12, whose fundus imaging and spectral domain-optical coherence tomography (SD-OCT) demonstrate a unique incidence of peripapillary sparing.
METHODS
Genetic analyses.
DNA isolated from whole blood lymphocytes was sequenced for candidate genes by the Casey Eye Institute Diagnostic Laboratory (Portland, Oregon). One patient (P3) underwent whole exome sequencing performed by the Columbia University Medical Center Pathology Department (New York, NY).
Electroretinography.
Full-field electroretinogram (ffERG) (Diagnosys LLC, Lowell, Massachusetts, USA) recordings were acquired in all patients from both eyes per International Society for Clinical Electrophysiology of Vision (ISCEV) standards in scotopic and photopic states. DTL recording electrodes were utilized for all patients.5
Imaging.
All imaging was performed as previously described by Sujirakul et al.6 After dilation, each patient underwent digital fundus photography and both fundus short-wavelength autofluorescence (SW-AF) and SD-OCT imaging using a Spectralis HRA+OCT device (Heidelberg Engineering, Heidelberg, Germany). For SW-AF imaging, 30-degree views were acquired at 1536 × 1536 pixel resolution using a 486nm wavelength stimulus and 521nm barrier filter. SD-OCT imaging was performed with an 870nm light source and automated real-time infrared reflectance image registration. Horizontal scans were positioned through the fovea (high resolution mode, 9mm window, ART, minimum of 50 averaged images). Microperimetry was performed using the Nidek MP-1 (Nidek Technologies; Padova, Italy).
FINDINGS
Cases 1 and 2
A 16-year-old boy (P1) was diagnosed with LCA13 presented with best-corrected visual acuity (BCVA) of 20/50 in the right eye and 20/100 in the left eye, equally reactive pupils, no relative afferent pupillary defect, and intraocular pressure of 14 mmHg in both eyes. Ophthalmoscopy revealed extensive macular atrophy, peripheral pigment migration, as well as peripapillary and para-arterial sparing.
SD-OCT demonstrated marked retinal thinning and excavation at the macula, with sparing of RPEatrophy in the peripapillary region of each eye (Fig. 1A, inset). SW-AF imaging (Heidelberg Spectralis HRA+ OCT version 1.7.0.0; Heidelberg Engineering, Heidelberg, Germany) revealed a pattern of hypoautofluorescence consistent with the loss of RPE throughout the macula with the exception of the peripapillary region of each eye (Fig. 1B).
Figure 1.
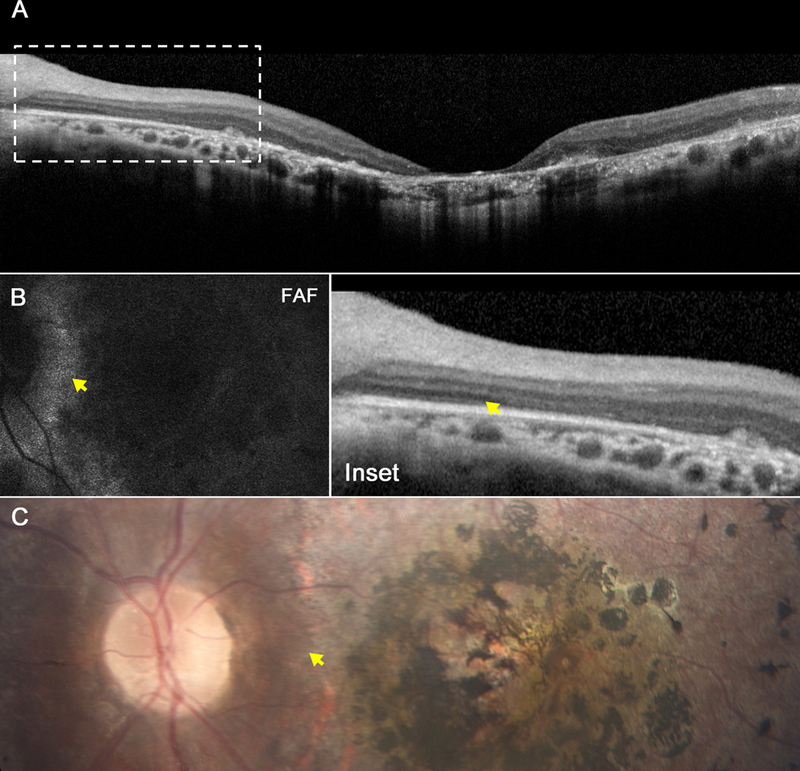
Retinal images from the 16-year-old brother (P1). Spectral domain-optical coherence tomography (SD-OCT) reveals loss of photoreceptors and ellipsoid nuclei throughout the macula with exception of the peripapillary region (A), and magnification demonstrates preservation of the retinal pigment epithelium and inner segment ellipsoid band (inset). These findings correlate with fundus autofluorescence images (B), which reveal hypofluorescence throughout the macula except for in the peripapillary region, as well as color fundus photography (C), which demonstrates atrophy of the retinal pigment epithelium with a bone spicule-like pattern, with sparing of the peripapillary region. Yellow arrows in each image mark the edge of sparing and in SD-OCT, the loss of retinal architecture.
ffERG revealed extinguished photopic and scotopic responses consistent with a generalized loss of cone and rod function, respectively. Microperimetry exhibited marked decreases in visual sensitivities over the fovea and a complete loss of visual function in the parafoveal region. Fixation was foveal in an area of low-decibel cone sensitivity (Supplemental Fig. 1). The peripapillary region was not sampled.
The patient’s 21-year-old sister (P2) had an identical macular phenotype, with the additional finding of horizontal nystagmus (Fig 2, Fig 3). Direct sequencing of several candidate genes identified homozygous missense mutations p.L99I in exon 3, predicted to be deleterious by SIFT in both siblings. Microperimetry in this case showed foveal fixation with complete loss of visual function in the sampled area, which did not include the parapapillary area (Supplemental Fig. 1).
Figure 2.
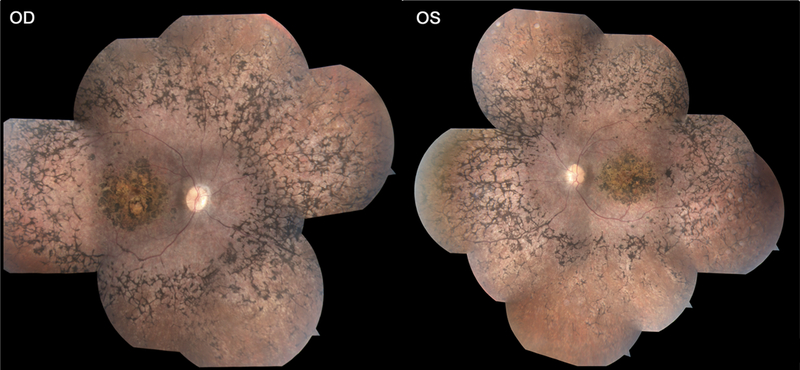
Color fundus photography of P2 reveals localized macular atrophy of the retinal pigment epithelium circumscribed by a confluent bone spicule pigment pattern in the beyond the vascular arcades in the periphery in both eyes.
Figure 3.
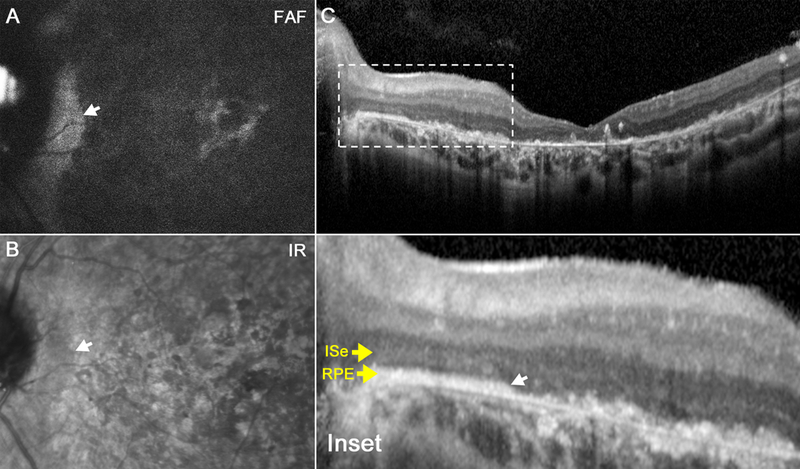
Scanning laser ophthalmoscopy images from the 21-year-old sister (P2). Fundus autofluorescence and infrared images reveal macula-wide hypoautofluorescence (a) and hyporeflectance (b), respectively, with peripapillary sparing in each eye. Spectral domain-optical coherence tomography reveals loss of photoreceptors and ellipsoid nuclei in a similar pattern (C). Magnification reveals intact retinal architecture limited to the peripapillary region (inset).
Case 3
An independent case of an 8-year-old girl (P3) has striking similarities with the siblings described above. The patient had BCVA of 20/60 in the right eye and 20/80 in left, with eccentric fixation, equally reactive pupils, no relative afferent pupillary defect, and normal intraocular pressures in both eyes. Ophthalmoscopy revealed dense intraretinal pigmentation in the macular and peripherally with peripapillary sparing. SD-OCT, FAF, and CFP findings were similar to the previously described cases. ffERG on this patient revealed extinguished photopic and scotopic responses indicating generalized retinal dysfunction affecting both rods and cones. Whole exome sequencing identified homozygous missense mutations (p.V233D) in the RDH12 gene.
Case 4
A 10-year-old boy (P4) presented for evaluation after previously being diagnosed with retinitis pigmentosa at the age of 3-years-old. He complained of night blindness but had no history of nystagmus. Past medical history was unremarkable and no other family members were similarly affected. The patient had a BCVA of 20/30 in the right eye and 20/60 in the left. Dilated fundus exam revealed depigmentation atrophy of both maculae with bilateral peripapillary sparing (Fig. 4D and 4E). The fundi showed extensive mottling but very little pigment migration. The far peripheral retina had a salt-and-pepper appearance. SD-OCT showed marked atrophy of the RPE (Fig. 4A), however, with intact retinal cell layers in the peripapillary regions (Fig. 4C), correlating to AF findings bilaterally (Fig. 4B). ffERG showed extinguished rod responses and diminished 30 Hz-flicker responses of approximately 10μV in both eyes confirming a rod-cone dystrophy. Genetic testing was performed at Casey Eye Institute (Portland, Oregon), identifying a c.806_820delCCCTG and p.Arg295Stop:c.C>T mutation in the RDH12 gene, for which each parent was a carrier of one allele, confirming the diagnosis of early onset retinal dystrophy.
Figure 4.
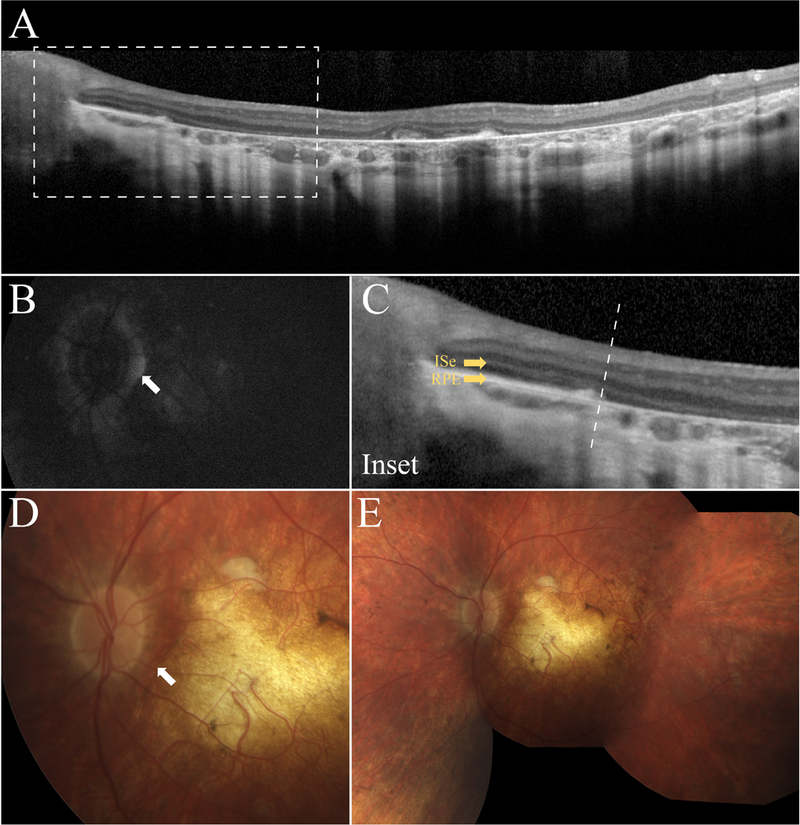
Retinal imaging of the 10-year old boy (P4). SD-OCT shows extensive RPE and photoreceptor atrophy in the macula (A) and relative preservation of the RPE and retinal structure in the peripapillary region (C). SW-AF (B) imaging and color fundus photography (D and E, respectively) corroborate SD-OCT findings and show ubiquitous hypoautoflourescence and hyporeflectance that is spared immediately surrounding the optic nerve.
DISCUSSION
These patients are, to our knowledge, the first reported case series of LCA with lack of RPE atrophy in the peripapillary region, a finding that is traditionally reserved for Stargardt disease (STGD1) in the context of retinal degenerations.7,8
RDH12, an LCA candidate gene, maps to chromosome 14q23 (8Mb from the LCA3 locus), and has been localized to photoreceptor inner segments. 9 A missense or frameshift mutation in RDH12 is associated with an autosomal recessive progressive retinal degeneration characterized by severe and progressive rod-cone dystrophy with severe macular atrophy that leads to visual loss before the second decade of life.1 Structurally, an OCT in this condition typically reveals a distorted retinal architecture that can vary in thickness and prohibits identification of normal laminae.9 The L99I variant of RDH12 found in P1 has been described to include reduced visual acuity and hyperpigmentation in the mid-periphery of the fundus.10 Our four RDH12 cases demonstrate macular atrophy and hyperpigmentation similar to those reported in the literature, but also uniquely exhibit peripapillary and para-arterial sparing.
Peripapillary sparing in STGD1 has been attributed to this region’s favorable photoreceptor-to-RPE ratio and thicker overlying retinal nerve fiber layer, creating a reduction in load of light on the photoreceptor-RPE complex, and, thus, less photo-oxidative damage.7 The role of RDH12 in the visual cycle is well-characterized and dysfunction of this enzyme leads to decreased synthesis of 11-cis-retinal.11 Reduced light stimulus near the peripapillary region may also theoretically temper the degenerative process as RDH12 plays a key role in phototransduction. However, while these reasons may contribute to the observed phenotype, gene-specific features are likely responsible for our findings. Aside from its presence in STGD1, peripapillary sparing has been described in RDS/Peripherin 2 Pattern Dystrophy12 and in one case report of pigmentary retinal dystrophy in which RPE mottling and pigment clumping spared the peripapillary and foveal regions.13 Interestingly, Cideciyan et al. provided a functional assessment of the peripapillary region in Stargardt disease.7 In patients with an eccentric focus, 30% had a preferred locus in the peripapillary retina, suggesting superior retinal function in this region. For P1 and P2 who underwent microperimetry testing, fixation was foveal and sensitivity was not measured in the peripapillary region. Future studies are needed to discern the retinal function in the peripapillary region of RDH12-associated LCA patients, and assess if these findings correlate to the structural observations we describe on multimodal imaging.
Concurrent incidence of STGD1 and LCA in our patients was considered clinically unlikely, as the patients’ phenotype was typical of LCA. The ophthalmic exam revealed reduced perception of light, nystagmus, and pigmentary changes, which is consistent with previous descriptions of LCA.14,15 ERG findings corroborated clinical exams, demonstrating undetectable waveforms, which are consistent with the LCA phenotype.14 Such a finding would be considered highly unlikely in STGD1, in which only a fraction of the diseased population demonstrates any ERG changes. STGD1 ERG group I, which is characterized by a normal scotopic and photopic ERG, displays a trend toward relative functional and structural sparing in the peripapillary region. Those in STGD1 ERG groups II or III display altered ERG limited to macular involvement as well as peripapillary atrophy.8
The p.D2177N variant of ABCA4 found in the siblings (P1 and P2) has been reported to occur in 8% of patients with age-related macular degeneration (AMD)16,17; however, a subsequent study showed that the frequency of this variant was not statistically associated with an increased risk of developing AMD.18 This variant is predicted benign and unlikely to be a disease-causing allele for STGD1 ruling out any trans modifier effects. Although variations in ABCA4 have been extensively associated with STGD1, no such phenotypic correlations have been ascribed to D2177N. In addition, mutations in RDH12 have not been reported in the setting of STGD1.
Whole exome sequencing may allow us to detect specific variants responsible for the patients’ phenotype; however, our findings prompt clinical awareness of shared manifestations in conditions with distinct genetic origins. This case demonstrates a novel presentation of LCA as a subphenotypic overlap of STGD1, and may be representative of a dual effect of RDH12 and ABCA4.
Supplementary Material
Funding
Jonas Children’s Vision Care, and Bernard & Shirlee Brown Glaucoma Laboratory are supported by the National Institutes of Health [5P30EY019007, R01EY018213, R01EY024698, R01EY026682, R21AG050437], National Cancer Institute Core [5P30CA013696], the Research to Prevent Blindness (RPB) Physician-Scientist Award, unrestricted funds from RPB, New York, NY, USA. J.D.S is supported by the RPB Medical Student Research Fellowship. S.H.T. is a member of the RD-CURE Consortium and is supported by the Tistou and Charlotte Kerstan Foundation, the Schneeweiss Stem Cell Fund, New York State [C029572], the Foundation Fighting Blindness New York Regional Research Center Grant [C-NY05–0705-0312], the Crowley Family Fund, and the Gebroe Family Foundation.
Footnotes
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article.
REFERENCES
- 1.Perrault I, Hanein S, Gerber S, et al. Retinal dehydrogenase 12 (RDH12) mutations in leber congenital amaurosis. Am J Hum Genet 2004;75:639–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang X, Wang H, Sun V, et al. Comprehensive molecular diagnosis of 179 Leber congenital amaurosis and juvenile retinitis pigmentosa patients by targeted next generation sequencing. J Med Genet 2013;50:674–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang S, Zhang Q, Zhang X, Wang Z, Zhao P. Clinical and genetic characteristics of Leber congenital amaurosis with novel mutations in known genes based on a Chinese eastern coast Han population. Graefes Arch Clin Exp Ophthalmol 2016. [DOI] [PubMed] [Google Scholar]
- 4.Srilekha S, Arokiasamy T, Srikrupa NN, et al. Homozygosity Mapping in Leber Congenital Amaurosis and Autosomal Recessive Retinitis Pigmentosa in South Indian Families. PLoS One 2015;10:e0131679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McCulloch DL, Marmor MF, Brigell MG, et al. ISCEV Standard for full-field clinical electroretinography (2015 update). Doc Ophthalmol 2015;130:1–12. [DOI] [PubMed] [Google Scholar]
- 6.Sujirakul T, Davis R, Erol D, et al. Bilateral Concordance of the Fundus Hyperautofluorescent Ring in Typical Retinitis Pigmentosa Patients. Ophthalmic Genet 2015;36:113–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cideciyan AV, Swider M, Aleman TS, et al. ABCA4-associated retinal degenerations spare structure and function of the human parapapillary retina. Invest Ophthalmol Vis Sci 2005;46:4739–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Burke TR, Rhee DW, Smith RT, et al. Quantification of peripapillary sparing and macular involvement in Stargardt disease (STGD1). Investigative ophthalmology & visual science 2011;52:8006–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jacobson SG, Cideciyan AV, Aleman TS, et al. RDH12 and RPE65, visual cycle genes causing leber congenital amaurosis, differ in disease expression. Investigative ophthalmology & visual science 2007;48:332–8. [DOI] [PubMed] [Google Scholar]
- 10.Valverde D, Pereiro I, Vallespin E, Ayuso C, Borrego S, Baiget M. Complexity of phenotype-genotype correlations in Spanish patients with RDH12 mutations. Investigative ophthalmology & visual science 2009;50:1065–8. [DOI] [PubMed] [Google Scholar]
- 11.Thompson DA, Janecke AR, Lange J, et al. Retinal degeneration associated with RDH12 mutations results from decreased 11-cis retinal synthesis due to disruption of the visual cycle. Hum Mol Genet 2005;14:3865–75. [DOI] [PubMed] [Google Scholar]
- 12.Duncker T, Tsang SH, Woods RL, et al. Quantitative Fundus Autofluorescence and Optical Coherence Tomography in PRPH2/RDS- and ABCA4-Associated Disease Exhibiting Phenotypic Overlap. Invest Ophthalmol Vis Sci 2015;56:3159–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Piccolino FC, Calabria G, Polizzi A, Fioretto M. Pigmentary retinal dystrophy associated with pigmentary glaucoma. Graefe’s archive for clinical and experimental ophthalmology = Albrecht von Graefes Archiv fur klinische und experimentelle Ophthalmologie 1989;227:335–9. [DOI] [PubMed] [Google Scholar]
- 14.Scholl HP, Chong NH, Robson AG, Holder GE, Moore AT, Bird AC. Fundus autofluorescence in patients with leber congenital amaurosis. Investigative ophthalmology & visual science 2004;45:2747–52. [DOI] [PubMed] [Google Scholar]
- 15.Mackay DS, Dev Borman A, Moradi P, et al. RDH12 retinopathy: novel mutations and phenotypic description. Molecular vision 2011;17:2706–16. [PMC free article] [PubMed] [Google Scholar]
- 16.Allikmets R Further evidence for an association of ABCR alleles with age-related macular degeneration. The International ABCR Screening Consortium. American journal of human genetics 2000;67:487–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang R, Wang LY, Wang YF, et al. Associations of the G1961E and D2177N variants in ABCA4 and the risk of age-related macular degeneration. Gene 2015;567:51–7. [DOI] [PubMed] [Google Scholar]
- 18.Guymer RH, Heon E, Lotery AJ, et al. Variation of codons 1961 and 2177 of the Stargardt disease gene is not associated with age-related macular degeneration. Archives of ophthalmology 2001;119:745–51. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.