

Abstract
The rising prevalence of primary pediatric hypertension and its tracking into adult hypertension point to the importance of determining its pathogenesis to gain insights into its current and emerging management. Considering that the intricate control of BP is governed by a myriad of anatomical, molecular biological, biochemical, and physiological systems, multiple genes are likely to influence an individual’s BP and susceptibility to develop hypertension. The long-term regulation of BP rests on renal and non-renal mechanisms. One renal mechanism relates to sodium transport. The impaired renal sodium handling in primary hypertension and salt sensitivity may be caused by aberrant counter-regulatory natriuretic and anti-natriuretic pathways. The sympathetic nervous and renin-angiotensin-aldosterone systems are examples of antinatriuretic pathways. An important counter-regulatory natriuretic pathway is afforded by the renal autocrine/paracrine dopamine system, aberrations of which are involved in the pathogenesis of hypertension, including that associated with obesity. We present updates on the complex interactions of these two systems with dietary salt intake in relation to obesity, insulin resistance, inflammation, and oxidative stress. We review how insults during pregnancy such as maternal and paternal malnutrition, glucocorticoid exposure, infection, placental insufficiency, and treatments during the neonatal period have long-lasting effects in the regulation of renal function and BP. Moreover, these effects have sex differences. There is a need for early diagnosis, frequent monitoring, and timely management due to increasing evidence of premature target organ damage. Large controlled studies are needed to evaluate the long-term consequences of the treatment of elevated BP during childhood, especially to establish the validity of the current definition and treatment of pediatric hypertension.
Keywords: Pediatric hypertension, Renal dopaminergic system, Salt sensitivity, Fetal programming, Pharmacogenetics, Renin-angiotensin-aldosterone system
Introduction
According to the National Health and Nutrition Examination Survey from 2011 to 2014, 29% of adults have hypertension, with non-Hispanic blacks having the highest prevalence of 41.2% [1]. Twenty-five years ago, the prevalence of pediatric hypertension in the USA was approximately 0.3–1.2%. Current epidemiological data suggest that at least 1 in 10 children is prehypertensive, while 4 in 100 children are hypertensive [2]. This substantial upsurge is attributed to the obesity epidemic and high salt intake, risk factors similar to those for adult primary hypertension [2–5, 6•]. According to the World Health Organization, adult hypertension is the leading risk factor for morbidity in middle-income countries and second only to tobacco smoking in low and high-income countries. The blood pressure (BP) trends in children may not be related to the country’s economic status, although data are not available for low-income countries [4]. In the USA, the prevalence of hypertension in obese children is 11% in 2013 [5]. Comparable prevalence of pediatric hypertension and its risk factors are also seen in Asia [6•]. There has been a 0.19% increase per year in the prevalence of hypertension, adjusted for height, among Chinese children in the past 20 years [7]. The increasing prevalence of hypertension in children may not be related solely to increasing obesity [2–5, 6•, 7].
Elevated BP during childhood is a risk factor for adult hypertension, bearing in mind that not all children with elevated BP have elevated BP as adults and that many adults with hypertension have normal BP during childhood [4]. Indeed, the predictive value of childhood hypertension for adult hypertension has been estimated to vary from 19 to 65% [5, 8]. Nevertheless, there is some predictability of adult BP from childhood values and juvenile target organ damage, which includes left ventricular hypertrophy, carotid intima-media thickening, and decreased neurocognitive performance and brachial flow-mediated dilation [9, 10, 11•]. Higher systolic BP in male children and adolescents with a family history of hypertension increases the risk of developing long-term arterial stiffness, determined by brachial-ankle pulse wave velocity [11•].
Pediatric Hypertension
The consensus-based guidelines of the National High Blood Pressure Education Program and National Heart, Lung, and Blood Institute define pediatric hypertension on the basis of percentiles according to age, height, and sex [12]. Hypertension is defined as systolic blood pressure (SBP) or diastolic BP (DBP) at or above the 95th percentile. Prehypertension is defined as SBP or DBP from 90th to <95th percentile. Stage 1 hypertension is defined as SBP or DBP from 95th to 99th percentile plus 5 mmHg and stage 2 as SBP or DBP >99th percentile, plus 5 mmHg. Normal BP is defined as SBP and DBP that are <90th percentile for sex, age, and height. The accepted BP is the average of three readings of SBP or DBP in a controlled environment after 5 min of rest, with the patient seated and the right arm supported at heart level. The 2016 European Society of Hypertension (ESH) similarly defines hypertension according to the 2004 US Task Force. The 2016 ESH recommends that normotensive children should be reevaluated every 2 years and those with high-normal BP and no organ damage should be reevaluated after 1 year. However, hypertension in older adolescents is defined by the 2016 ESH as BP at the 95th percentile or greater for age, sex, and height. Moreover, patients 16 years or older with hypertension should be graded as for adults.
In addition to normal BP and “usual” hypertension, there are other subclasses of hypertension including: (i) white-coat hypertension, (ii) masked hypertension, (iii) isolated systolic hypertension, (iv) central hypertension, and (v) exercise hypertension. Based on the effect of NaCl intake on BP, the classes of BP are: (i) salt-resistant BP, which includes (a) salt-resistant normotensive and (b) salt-resistant hypertensive, and (ii) salt-sensitive BP, which includes (a) salt-sensitive normotensive and (b) salt-sensitive hypertensive. There is another subclass called inverse salt-sensitive where the BP is increased by a very low salt diet (vide infra).
BP should be measured by the auscultatory method, using the arm with a properly calibrated and validated instrument, usually a mercury sphygmomanometer [13]. If hypertension is detected by an oscillometric method, it must be confirmed by the auscultatory method. Chronic ambulatory BP monitoring is being supported because it can differentiate sustained hypertension from white-coat, masked, non-dipping (decrease in BP <10% while sleeping), and overly dipping hypertension (decrease in BP >20% while sleeping) [14].
The risk for primary hypertension in children is increased several factors, including low birth weight, male sex, African-American ethnicity, sedentary lifestyle, family history of hypertension, and especially by elevated body mass index [2–5, 13]. By contrast, secondary hypertension is linked to non-obese younger children, lower glomerular filtration rate, and higher DBP [15]. Sixty to 90 % of secondary hypertension is accounted for by renal parenchymal, renovascular, and endocrine etiologies [16].
Pathogenesis of Primary Pediatric Hypertension
Fetal Programming
Fetal programming is the association of adverse events in pregnancy with long-lasting effects into adulthood. This concept was introduced by Barker et al. in 1989 when they noted an inverse relationship between birth weight and SBP [17••]. They hypothesized the “thrifty” phenotype where malnutrition of the mother leads to fetal and infant malnutrition, and ultimately to changes in growth, metabolism, and vasculature in the offspring. All of these effects culminate into the metabolic syndrome, which is characterized by hypertension, obesity, dyslipidemia, and insulin resistance. In the Avon Longitudinal study of Parents and Children, Fraser et al. noted that the offspring of hypertensive mothers had elevated BP by 17 years of age; however, there were no differences in insulin, glucose, or lipid values from the normotensive group [18•].
The prenatal manipulations that have been used to study the effect of prenatal environment on the offspring include the following: (i) maternal and paternal nutrition, including the intake of alcohol; (ii) nicotine exposure; (iii) maternal glucocorticoids; (iv) maternal infection; and (v) placental dysfunction [19]. The elevation of BP of the offspring of pregnant mothers with these prenatal manipulations is caused by several mechanisms, including genetics, epigenetics, inflammation, endoplasmic reticulum stress, and oxidative stress [19–22].
Prenatal insults constrain capillary density, endothelial function, and the development of the kidney, which result in adult hypertension [21, 22]. In humans, nephrogenesis continues until 34 to 36 weeks of gestation, after which no new nephrons are formed. Reduced nephron number contributes to fetal programming of adult hypertension [22]. This agrees with Brenner’s hyperfiltration hypothesis which states that the compensatory mechanism of the remaining nephrons results in hastened decline of renal function [23•]. Kidney volume has been used as a surrogate indicator for nephron number. However, kidney volume is positively, rather than negatively, correlated with SBP in children 4–20 years of age [24•]. Furthermore, decreased capillary sprouting increases vascular resistance, leading to increased BP. Preterm infants with excessive exposure to atmospheric oxygen produce free radicals which injure vasculogenesis [25•].
There are sex differences in the fetal programming of hypertension. Experimental models show differences in the effects of developmental insults on males and females through a hormonal milieu (Table 1). Testosterone appears to have a permissive effect, while estrogen has a protective effect, on hypertension in the adult offspring with intrauterine growth retardation (IUGR) [23•, 26••]. Increasing age also leads to sex-specific susceptibility of impaired BP regulation through an age-dependent increase in adiposity, leading to increased plasma leptin that activates the renal sympathetic nerves [23•, 27].
Table 1.
Sex differences in the effects of fetal developmental insults through a hormonal milieu
| Male | Female | |
|---|---|---|
| Hormonal milieu | Testosterone | Estrogen |
| RAAS | ↓a | (−)a |
| Nephron number | ↓a | ↓/(−)a |
| Endothelin production | ↑a | (−)a |
| Reactive oxygen species | ↑a | ↓a |
↓ = decreased, ↑ = increased, (−) = no change
Maternal and Paternal Nutrition
Maternal nutrition plays a role in developmentally programmed hypertension [28, 29]. A high-salt diet during gestation and lactation in Sprague-Daley rats results in increased BP in adult male offspring [30]. Female offspring have lesser increase in BP and even a decrease in BP in some studies [31]. High-salt diet limited to the gestational period or only during weaning [32] may not be associated with hypertension in male or female adult offspring [33]. However, high-salt diet 30 days after birth (dams fed high-salt diet before weaning) increased the BP of adult male offspring [34]. The increase in BP has been related to increased pressor response related to calcium and PKC signaling [35]. Twelve-week-old offspring of Sprague-Dawley dams fed high-salt diet had normal BP but had increased wall thickness of central (aorta, carotid), muscular (mesenteric) and intrapulmonary arteries, regardless of the post-weaning diet [36]. Some offspring of high-salt diet-fed dams had low BP and heart rate, indicative of both left ventricular systolic and diastolic function, and decreased aortic vasodilatory response to nitric oxide [32]. Both high and low maternal salt intakes during pregnancy have been reported to decrease nephron number and increase BP in male offspring [37].
A high fructose diet in pregnant Sprague-Dawley rats also increases BP and aggravates the increase in BP caused by a high-salt diet in male offspring [38]. The mechanisms involved may include the arachidonic acid pathway and the renin-angiotensin system [30]. The increase in BP in the off-spring of dams fed a high-sucrose diet related to increased reactivity to angiotensin II involves calcium signaling, akin to those observed in offspring of pregnant mothers fed a high-salt diet [39•].
High-fat diet in rat dams during pregnancy increases both SBP and DBP; high-fat diet during lactation increases the DBP and other features of the metabolic syndrome in female but not male offspring [40]. A paternal high-fat diet before conception and after birth also leads to hypertension with features of the metabolic syndrome in the offspring [41•]. Statins given to mouse dams during the second half of pregnancy and lactation decreases metabolic risk in both mother and female offspring [42]. The beneficial effect of statins in the female offspring has been related to a decrease in C-reactive protein-induced inflammation [43]. High paternal fat diet may also disturb fetal programming of metabolism through epigenetic changes [41•]. Sirtuins (SIRT1 and SIRT3) are proposed to mediate the fetal programming of obesity, as well as its myriad long-term effects [44•]. Despite the multiple links of maternal obesity to childhood obesity, prenatal weight management did not show any differences in infant growth [45].
High protein diet during pregnancy in Wistar-Kyoto rats does not affect nephron number or BP in adult offspring [46]. However, low protein diet or maternal undernutrition during pregnancy leads to the metabolic syndrome in the offspring [47]. Protein restriction during pregnancy was also associated with decreased nephron number [48] and cardiac dysfunction [49]. In Wistar rats, maternal protein restriction led to increased BP in F1 and F2 but not F3 generation in both adult male and female rats [50•]. F1 but not F2 male offspring of pregnant guinea pigs that had a 30% reduction in food intake during pregnancy also develop hypertension [51]. The high BP in male offspring is glucocorticoid-dependent without modulation of renal angiotensin receptor; however, it is glucocorticoid-independent and associated with decreased renal AT2R expression in female offspring [52]. The cardiac dysfunction may be restricted to the male rat offspring [53]. Maternal protein restriction leads to sympathetic overactivity and oxidative dysfunction at the medulla oblongata of Wistar rat dams [54]. There is an increase in circulating leptin in the adult offspring [55]; leptin can stimulate the sympathetic nervous system in rodents [56•]. Chronic administration of leptinin humans does not increase BP [56•]. Small-for-gestational-age offspring of mothers fed with low-protein diet have higher mu-opioid receptor and dopamine type 1 receptor binding but not with dopamine transporters in mesolimbic brain regions. Changes in these neurotransmitter pathways may affect the development of obesity, attention-deficit/hyperactivity disorder, and addiction [57]. BP was not measured in these studies although mood disorders and cardio-metabolic diseases have genetic overlap [58]. Impaired renal dopamine production or function can result in hypertension [59, 60•, 61–65]. There is increased sodium transport in the renal medullary thick ascending limb due to increased NKCC2 expression in rat dams fed a low-protein diet [66]. The hypertension may be associated with salt sensitivity [67]. Undernutrition in sheep dams leads to increased expression of several extracellular matrix proteins in the carotid arteries, related in part to suppression of miR-29c that may involve glucocorticoids [68•]. Low-protein diet during pregnancy has been reported to lead to enhanced responsiveness to angiotensin II [69], especially in male off-spring [70], and exaggerated proliferative response to vascular injury in the offspring with increased expression of genes related to oxidative stress [71]. By contrast, in the hypothalamus, angiotensin II type 1 receptor is decreased in offspring of Wistar rats fed a low-protein diet [72]; the effect of this apparent difference in central and peripheral angiotensin II expression/function on renal function and BP remains to be determined. miRNAs may play a role in increasing susceptibility to the development of cardio-metabolic disease in off-spring of mothers with abnormal nutrition during gestation or lactation [73]. Some of the programmed effects may be reversible, for example, by inhibition of soluble epoxide hydrolase [74••] and use of antioxidants, such as melatonin and N-acetylcysteine in pregnant rats with nitric oxide deficiency [75••] and seed extract of Euterpe oleracea [76••].
High multivitamin intake during pregnancy in Wistar rats results in high BP, high fasting glucose and insulin in male offspring [77, 78•]. However, both male and female offspring fed an obesogenic diet led to increased body weight, glucose intolerance, and high BP [79]. This was prevented by continued high multivitamin or folic acid intake [80].
Maternal Glucocorticoids
Excessive exposure to endogenous or exogenous synthetic glucocorticoids during pregnancy is associated with low birth weight and hypertension in animal models [37]. Steroid administration to pregnant sheep and rats [81–83] but not mice are associated with increased BP in adult offspring [84••] that is sex-specific. Betamethasone administered to pregnant ewes caused hypertension in the 0.5-month-old male offspring that has been related to decreased expression of angiotensin 1–7 Mas receptor in the dorsal medulla and increased angiotensin-converting enzyme in the cerebrospinal fluid [85, 86]. Steroids given to pregnant mice cause dysregulation of the RAAS in 6- and 12-month-old male but not female offspring [87]. Short-term administration of corticosteroids to pregnant mice decreases nephron number in female and male offspring [87]. At 6 months of age, male but not female offspring had increased plasma aldosterone, renal expression of angiotensin II, and Na+,K+/ATPase alpha1 subunit and sodium ion channels; blood measure was not measured [87]. However, at 12 months of age, the male but not female mice had decreased BP [88]. Other studies have reported an increase in BP in 6–7-month-old female offspring [83]. The increased BP of male offspring of pregnant rats given dexamethasone has also been related to increased levels of asymmetric dimethylarginine (an endogenous inhibitor of nitric oxide synthase) and increased expression of renal NCC and NHE3, effects that were prevented by administration to the mother rat of L-citrulline, which can be converted to arginine, a substrate of nitric oxide synthase [89]. Placental 11β-hydroxysteroid dehydrogenase 2 (11β-HSD2) inactivates the conversion of cortisol to cortisone in humans; thus, a deficiency or dysfunction of the enzyme (e.g., in cases of zinc deficiency) causes excessive fetal exposure to cortisol [90]. Preeclampsia, preterm labor, IUGR, and treatment with dexamethasone or betamethasone to hasten lung maturity all compound the increased exposure to corticosteroids. Long-term consequences are numerous, which include increased the risk for allergies, infection that may be related to decreased immunity, insulin resistance, type I diabetes mellitus, and reduction in nephron number [91], in addition to hypertension [92, 93]. However, maternal glucocorticoids may not directly cause the hypertension in the offspring [94]. It has been suggested that the inconsistency in the increase in BP after prenatal exposure to glucocorticoids in rodents could indicate that hypertension becomes apparent only under stressed conditions and that the use of tail cuff to measure BP contributed to the irregularity [95]. The absence of glucocorticoid-inducible kinase SGK1 in the mother prevents the ability of prenatal protein restriction to increase the BP in adult male and female offspring.
Maternal Infection
There is a positive relationship between infection during pregnancy and adverse outcomes in the off-spring, including cardiovascular diseases [96•, 97•]. Maternal bacterial infection has been mimicked by the systemic administration of the gram-negative bacterial endotoxin lipopolysaccharide (LPS) to dams. Exposure of rat dams to LPS results in increased BP in the offspring that is related to oxidative stress and inflammation [96•, 97•, 98, 99]. The increased BP was found in both male and female offspring [100]. The increase in BP caused by prenatal exposure to LPS has been suggested to be the result of increased sympathetic nerve activity, increased activity of the RAAS, impaired endothelium-dependent and endothelium-independent vascular relaxation, and decreased activity of the renal dopaminergic system [96•, 97•, 98]. The impaired vascular relaxation is caused by decreased vascular expression of connexin 37, endothelial nitric oxide synthase, NO production, and soluble guanylyl cyclase [96•]. As will be discussed below, the renal dopaminergic system is important in the regulation of BP by enabling the kidney to excrete a sodium load during conditions of normal or moderate increase (5–10% acute saline load, or about 200–250 mmol sodium intake/day) in sodium intake. The increase in reactive oxygen species (ROS) in the offspring of dams that received LPS also increased the renal expression and activity of G protein-coupled receptor kinase type 2 (GRK2) and type 4 (GRK4). These kinases are upstream of dopamine receptors (D1R and D3R) and angiotensin type 1 receptor (AT1R). Variants of human GRK4 impair D1R and D3R function but increase AT1R expression and function [101••] which ultimately lead to hypertension in humans and transgenic mice expressing human GRK4γ142V. The impairment of renal dopamine receptor function also impairs the ability of the kidney to excrete an oral sodium load (gastro-renal communication) [102•]. There may be differences in organ response to maternal LPS administration because D2R not D1R expression is decreased in the prefrontral cortex of Sprague-Dawley rat offspring [103]. These apparent discrepancies need to be sorted out because the administration of a proinflammatory cytokine inductor, polyriboinosinic polyribocytidilic acid (poly[I:C]), during gestational day 14–16 in Sprague-Dawley rats increased baseline extracellular dopamine levels in the nucleus accumbens, but not in the prefrontal cortex of their offspring; their ventral tegmental neurons had reduced activity but normal D2R autoreceptor activity [104].
Interestingly, although prenatal exposure to LPS or high-fat diet increased the BP of the offspring, the combination of prenatal exposure to LPS and pre- and post-natal high-fat diet was associated with normalization of BP [98]. This discordant result has been suggested to be the result of adaptive response to inflammation because a high-fat diet can increase plasma LPS levels [98, 105].
Placental Dysfunction
Utero-placental insufficiency is a leading cause of IUGR and low birth weight even with normal maternal nutrition. There are several models of utero-placental insufficiency, including ligation of either the ovarian or uterine arteries [106]. F1 and F2 male but not female offspring of Wistar-Kyoto rat dams with uteroplacental insufficiency caused by bilateral uterine vessel ligation developed hypertension at 6 months of age that could be related, in part, to impaired vasorelaxation and arterial stiffness, especially in the mesenteric artery [107, 108]. Female offspring which were not hypertensive had normal mesenteric, renal, and femoral artery stiffness but had uterine artery endothelial dysfunction and increased wall stiffness [109••]. Utero-placental insufficiency has been shown to decrease nephron number and cyclooxygenase-2 (COX-2) in a rat model [110] and to increase umbilical and carotid artery stiffness in sheep dams due to disrupted extracellular matrix deposition [111]. Increased markers of renal apoptosis and decreased urinary sodium excretion [112] were also noted in rats and newborn piglets. There are sex differences in the development of hypertension in the IUGR offspring; 12-week-old male rats with IUGR have elevated mean arterial BP compared to females [106]. Furthermore, adult IUGR female rats have higher vascular endothelial growth factor (VEGF) levels than IUGR males but have similar lower VEGF levels at birth [113]. Testosterone has a modulating role in the hypertension of adult male offspring of rat dams with placental insufficiency [114•].
Genetics and Pharmacogenetics of Primary Hypertension: Role of GRK4
Hypertension, a complex trait caused by interactions of genetic, epigenetic, environmental, and behavioral factors, is a major public health problem because of its high prevalence and increased risk for cardiovascular and renal diseases [115]. Considering that the intricate control of BP is governed by a gamut of anatomical, molecular biological, biochemical, and physiological systems, multiple genes are likely to influence an individual’s BP and susceptibility to develop hypertension. The long-term regulation of BP rests on renal and non-renal mechanisms [116••]. One renal mechanism relates to sodium transport. The impaired renal sodium handling in primary hypertension and salt sensitivity could be caused by aberrant counter-regulatory natriuretic and anti-natriuretic pathways. The sympathetic nervous and RAAS are examples of antinatriuretic pathways. An important counter-regulatory natriuretic pathway is afforded by the renal autocrine/paracrine dopamine system, aberrations of which are involved in the pathogenesis of hypertension [116••], including that associated with obesity. As indicated earlier, LPS administration to rat dams induces the hypertensive phenotype in the offspring that is related to an increase in the renal expression and activity of GRK2 and GRK4 [97•]. Because GRK4 fulfills all the criteria needed to implicate a gene as a cause of a complex trait, hypertension, in this instance, only GRK4 will be included in this review. The GRK4 gene is one of the few genes that fulfill the criteria for ascribing a gene as causal of a complex disorder. These criteria include gene linkage and gene variant association, in vitro phenotype, with the definitive evidence involving the expression of the variant genes in transgenic animals [64, 117]. Only the variants of genes of AGT that encodes angiotensinogen [118], AGTR1 that encodes the angiotensin II (Ang II) type 1 receptor (AT1R) [119], CYP11B2 that encodes aldosterone synthase [120], and GRK4 have been shown to cause hypertension in transgenic mice [121, 122]. Variants of ATP2B1, STK39 [123], GRK4, and SLC4A5 [124, 125] have been associated with salt sensitivity; GRK4γ486V causes salt-sensitive hypertension in transgenic mice [126].
Genome-wide association studies (GWAS), which have identified only ≈2–5% of the genetic factors believed to influence BP, failed to associate the GRK4 variants with hypertension [127–129]. There are several reasons for this non-association, including stringent correction and requirement for independent replication, resulting in higher type 2 error rates and the absence of specific GRK4 gene variants in the Affymetrix and Illumina chips (vide infra). Nevertheless, using gene-targeted studies, the association of GRK4 variants with hypertension has been replicated in several ethnic groups [59], with some exceptions [130, 131]. Additional reasons for non-association in GWAS include failure to include epistasis, epigenetics, environment, behavioral influences, e.g., sodium and potassium intake, and age in the analyses. Nutrition and gut microbiota can influence epigenetics [132]. Increased dietary salt can increase oxidative stress [133] and oxidative stress can influence epigenetics (e.g., histone deacetylase 1 activity) [134]. Felder et al. reported that miR-124 expression is increased in urinary exosomes of salt-sensitive subjects [135•] and can regulate c-Myc [136]. C-Myc can regulate GRK4 [135•], probably by interacting with the GRK4 promoter. This is of interest because of the following reasons: (1) c-Myc is proto-oncogenic [137]; (2) c-Myc is positively associated with hypertension and cancer, at least in males [138]; and (3) increased dietary salt intake increases the risk of gastric cancer [139]. Furthermore, epigenetics can influence gene transcription. Variants in the promoter region of GRK4 can influence its expression [140], and the salt sensitivity of C57BL/6J mice is related to increased renal expression of GRK4 [141]. Aortic and renal expression of GRK4 is also increased in spontaneously hypertensive rats [142, 143] whose high BP can be increased further by high-salt diet [144].
GRK4142V is not included in the Affymetrix and Illumina chips, except for Illumina Human 1M bead chip [101••]. The only Affymetrix chip that has GRK4486V is Genomewide 6. The Illumina chips, except for Illumina Human 1M-Duov3, do not have GRK4486V. Not all the chips have GRK465L either. It should also be noted that in all the GWAS studies, circulating DNAwas used which may not reflect spontaneous somatic mutations in the kidney that can also cause hypertension. We have reported the association of hypertension and certain DNA and miRNA in urine exosomes and urine renal proximal tubule cells [135•, 145].
GRK4 is upstream of genes that regulate renal function and BP, i.e., those of the RAAS and renal dopaminergic system. GRK4 variants impair D1R and D3R function and increase AT1R function [116••, 121]. As stated above, this could be related to the ability of GRK4 variants, via histone deacetylase 1, to positively regulate renal AT1R expression [121]. The effect of renal dopamine on BP is different from that administered systemically [116••]. The normal circulating concentrations of dopamine (picomolar range) are not sufficiently high to activate endogenous dopamine receptors but high nanomolar to low micromolar concentrations can be attained in dopamine-producing tissues [116••]. The GRK family is normally important in maintaining the responsiveness of certain dopamine receptor subtypes, e.g., D1R and D3R. GRK decreases GPCR responsiveness after continued stimulation by agonists through phosphorylation of the receptors and uncoupling them from their G protein complexes [122]. Growing evidence support the association between the allelic variants of GRK4, salt sensitivity, hypertension, and response to antihypertensive drugs [101••]. This has been reviewed by Rayner and Ramesar and Yang et al. [64,146]. A recent metaanalysis showed that GRK4 and DRD1 gene polymorphisms, rs1024323 GRK4 (OR = 1.826) and rs4532 of DRD1 genes (OR = 1.833), are associated with hypertension in Caucasians and East Asians, respectively [59]. GRK4 polymorphisms are not associated with preeclampsia in northern Han Chinese [147]. However, DRD1 (−48G) and DRD4 (−521T) receptors are associated with preeclampsia in a Polish population [148]. Mice harboring GRK4 gene variants such as GRK4γ486V are hypertensive on a high-salt diet, while mice harboring GRK4γ142V variants are hypertensive on a normal salt diet but not affected by a high-salt diet [149••]. These variants with increased constitutive GRK4 activity have been shown to down-regulate the renal dopaminergic system and upregulate RAAS (AT1R) by decreasing and increasing their expression and activities, respectively, in humans [146]. Furthermore, these variants offer new pharmacogenomic approaches in the treatment of hypertension as evidenced by the African-American Study of Kidney Disease (AASK) where GRK465L and GRK4142V predict a reduced response to α-adrenergic blockers [149••]. By contrast, GRK4142V, by itself, is associated with a more rapid response to a β-adrenergic blockade. In another study in two cohorts with primary hypertension without renal disease, as the number of individual GRK4 single nucleotide polymorphisms (SNPs; 65R>L and 142A>V) increase, BP response to a β-adrenergic blockade in a mixed population of black and white individuals decreases [150••]. GRK4R65 or GRK4A142 predicts a good BP response to a decrease in salt intake, whereas GRK465L or GRK4142V predicts a limited response to reduced salt intake [64]. However, the presence of at least three GRK4 allele variants (65L, 142V, and 486V), relative to those with fewer than three is associated with a better response to diuretic therapy [151]. The expression of GRK4486V, but not GRK4142V, in transgenic mice confers salt sensitivity [152] and predicts a response to diuretics in humans with primary hypertension [153]. Among Japanese, GRK4142V predicts a good response to angiotensin receptor blockers [101••].
Target Organ Damage
Systemic target organ damage (TOD) has been clearly documented with elevated BP in children. However, the lowest level of BP that causes TOD has not been determined. One of the earliest changes seen is left ventricular hypertrophy (LVH) without any correlation to severity of BP [154]. Although hypertension is one of the top causes of chronic kidney disease in adults, this is not the case with children. Hypertensive nephrosclerosis, microalbuminuria, and reduced glomerular filtration rates have been reported in children with hypertension [155]. Hypertensive children have been found to have inferior neurocognitive performance, executive function, and decreased cerebrovascular reactivity (CVR) [156•]. Atherosclerotic changes such as increased carotid intimal-medial thickness (cIMT), arteriolar narrowing, and stiffness have also been associated with pediatric hypertension [157]. These pathologic alterations support Folkow’s hypothesis that elevated BP thickens the medial layer of the vessels which be-gets hypertension [158•]. Furthermore, these linked with BP tracking in children in which BP during childhood predicts adult blood BP [4, 8–10, 11•]. Therefore, there is a need for early monitoring and treatment of pediatric hypertension such as ambulatory BP monitoring and pharmacologic and non-pharmacologic management of elevated BP [159]. Figure 1 summarizes the pathogenesis of pediatric hypertension showing the causes and effects at different stages of human life.
Fig. 1.
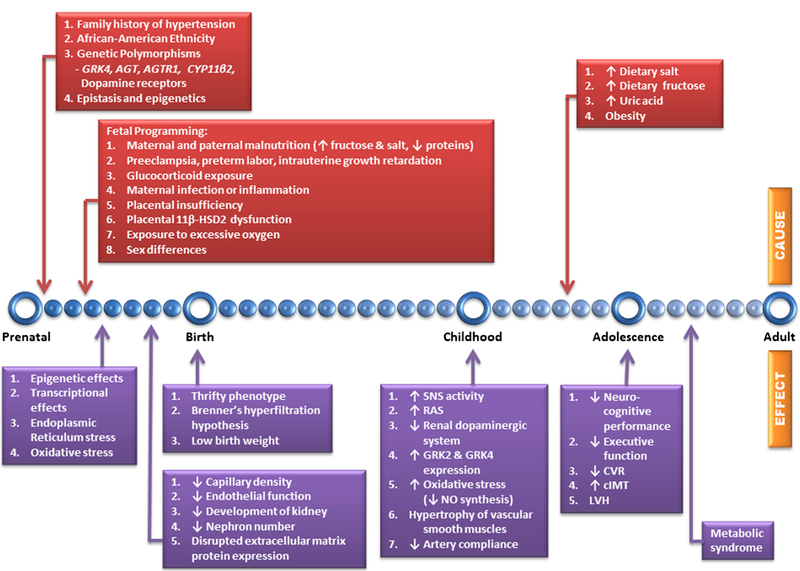
Timeline showing the pathogenesis of pediatric hypertension. Identified underlying causes and short-term and long-term effects are shown above and below the blue line, respectively. Genetic predisposition, early developmental insults, and dietary influences lead to changes in the regulation of BP that are carried into adulthood. GRK4 G protein-coupled receptor kinase type 4, GRK2 G protein-coupled receptor kinase type 2, AGT angiotensinogen gene, AGTR1 angiotensin II (Ang II) type 1 receptor gene, CYP11β2 aldosterone synthase gene, ↑ increase, ↓ decrease, 11β-HSD2 placental 11β-hydroxysteroid dehydrogenase 2, SNS sympathetic nervous system, RAS Renin-angiotensin system, NO nitric oxide, CVR cerebrovascular reactivity, cIMT carotid intimal-medial thickness, LVH left ventricular hypertrophy
Conclusions
Despite the increasing prevalence of hypertensive and prehypertensive children in different parts of the world, pediatric hypertension remains an underdiagnosed condition. Reformed normative data on BP inclusive of ethnicity and not just of age, sex, and height are imperative to define pediatric hypertension across all ethnic backgrounds, especially among African-American and Hispanics. The rise of the obesity epidemic in children shifts the onset of metabolic syndrome to an earlier age. Thus, public health measures to reduce this burden are needed as experimental evidence has documented the interaction of hyperglycemia, hyperinsulinemia, renal dopaminergic system dysfunction, upregulation of the RAAS, and hypertension. Larger controlled studies must be done to evaluate the long-term effect of childhood obesity and dietary salt with adult cardiovascular morbidity and mortality. Studies evaluating the effect of interventions on complicated pregnancies and their offspring must also be reassessed because of the evident theory of fetal programming of hypertension.
Acknowledgments
This work was supported, in part, by grants from the National Institutes of Health (R37 HL023081, R01DK039308, R01HL092196, P01HL068686, and P01HL074940 [PAJose]) and a minigrant from the National Kidney Foundation of Maryland (V.A. Villar).
Footnotes
This article is part of the Topical Collection on Therapeutic Trials
Compliance with Ethical Standards
Conflict of Interest The authors declare no conflicts of interest relevant to this manuscript.
Human and Animal Rights and Informed Consent This article does not contain any studies with human or animal subjects performed by any of the authors.
Papers of particular interest, published recently, have been highlighted as:
Of importance
Of major importance
References
- 1.Yoon SS, Carroll MD, Fryar CD. Hypertension prevalence and control among adults: United States, 2011–2014. NCHS Data Brief 2015;220:1–8. [PubMed] [Google Scholar]
- 2.Falkner B Recent clinical and translational advances in pediatric hypertension. Hypertension 2015;65(5):926–31. doi: 10.1161/HYPERTENSIONAHA.114.03586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bucher BS, Ferrarini A, Weber N, Bullo M, Bianchetti MG, Simonetti GD. Primary hypertension in childhood. Curr Hypertens Rep 2013;15(5):444–52. doi: 10.1007/s11906-013-0378. [DOI] [PubMed] [Google Scholar]
- 4.Roulet C, Bovet P, Brauchli T, Simeoni U, Xi B, Santschi V, et al. Secular trends in blood pressure in children: a systematic review. J Clin Hypertens (Greenwich) 2016; doi: 10.1111/jch.12955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moyer VA. U.S. Preventive Services Task Force. Screening for primary hypertension in children and adolescents: U.S. Preventive Services Task Force recommendation statement. Ann Intern Med 2013;159(9):613–9. [DOI] [PubMed] [Google Scholar]
- 6.•.Lee CG. The emerging epidemic of hypertension in Asian children and adolescents. Curr Hypertens Rep 2014;16:495. doi: 10.1007/s11906-014-0495-z. This study shows that increased prevalence of pediatric hypertension is worldwide and associated with the obesity epidemic. [DOI] [PubMed] [Google Scholar]
- 7.Yan W, Li X, Zhang Y, Niu D, Mu K, Ye Y, et al. Reevaluate secular trends of body size measurements and prevalence of hypertension among Chinese children and adolescents in past two decades. J Hypertens 2016;34(12):2337–43. [DOI] [PubMed] [Google Scholar]
- 8.Thompson M, Dana T, Bougatsos C, Blazina I, Norris SL. Screening for hypertension in children and adolescents to prevent cardiovascular disease. Pediatrics 2013;131(3):490–525. doi: 10.1542/peds.2012-3523. [DOI] [PubMed] [Google Scholar]
- 9.Rao G. Diagnosis, epidemiology, and management of hypertension in children. Pediatrics 2016;138(2) doi: 10.1542/peds.2015-3616. [DOI] [PubMed] [Google Scholar]
- 10.Magnussen CG, Smith KJ. Pediatric blood pressure and adult preclinical markers of cardiovascular disease. Clin Med Insights Blood Disord 2016;4(9):1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.•.Chu C, Dai Y, Mu J, Yang R, Wang M, Yang J, et al. Associations of risk factors in childhood with arterial stiffness 26 years later: the Hanzhong adolescent hypertension cohort. J Hypertens 2017;35(Suppl 1):S10–5. doi: 10.1097/HJH.0000000000001242. Premature target organ damage (long-term arterial stiffness) with pediatric hypertension evaluated by brachial-ankle pulse wave velocity. [DOI] [PubMed] [Google Scholar]
- 12.National High Blood Pressure Education Program Working Group on High Blood Pressure in Children and Adolescents. The fourth report on the diagnosis, evaluation, and treatment of high blood pressure in children and adolescents. Pediatrics 2004;114(2 Suppl 4th Report):555–76. [PubMed] [Google Scholar]
- 13.The Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure National High Blood Pressure Education Program. Bethesda: National Heart, Lung, and Blood Institute (US); 2004. [PubMed] [Google Scholar]
- 14.Viera AJ, Lin FC, Hinderliter AL, Shimbo D, Person SD, Pletcher MJ, et al. Nighttime blood pressure dipping in young adults and coronary artery calcium 10–15 years later: the coronary artery risk development in young adults study. Hypertension 2012;59:1157–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Flynn J, Zhang Y, Solar-Yohay S, Shi V. Clinical and demographic characteristics of children with hypertension. Hypertension 2012;60(4):1047–54. doi: 10.1161/HYPERTENSIONAHA.112.197525. [DOI] [PubMed] [Google Scholar]
- 16.Brady TM, Feld LG. Pediatric approach to hypertension. Semin Nephrol 2009;29(4):379–88. [DOI] [PubMed] [Google Scholar]
- 17.••.Barker DJ. The fetal origins of hypertension. J Hypertens Suppl 1996;14:S117–20. Early studies on “thrifty phenotype” introduced by Barker. [PubMed] [Google Scholar]
- 18.•.Fraser A, Nelson SM, Macdonald-Wallis C, Sattar N, Lawlor DA. Hypertensive disorders of pregnancy and cardiometabolic health in adolescent offspring. Hypertension 2013;62:614–20. The Avon longitudinal study exemplifies fetal programming where insults during pregnancy (gestational hypertension) are associated with increased blood pressure in the offspring. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Von Ehr J, von Versen-Höynck F. Implications of maternal conditions and pregnancy course on offspring’s medical problems in adult life. Arch Gynecol Obstet 2016;294(4):673–9. doi: 10.1007/s00404-016-4178-7. [DOI] [PubMed] [Google Scholar]
- 20.Lopez-Lopez J, Jaramillo PL, Camacho PA, Arbelaez DG, Cohen DD. The link between fetal programming, inflammation, muscular strength, and blood pressure. Mediat Inflamm 2015;710613 doi: 10.1155/2015/710613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lewandowski AJ, Davis EF, Yu G, Digby JE, Boardman H, Whitworth P, et al. Elevated blood pressure in preterm-born off-spring associates with a distinct antiangiogenic state and microvascular abnormalities in adult life. Hypertension 2015;65(3): 607–14. doi: 10.1161/HYPERTENSIONAHA.114.04662. [DOI] [PubMed] [Google Scholar]
- 22.Alexander BT, Dasinger JH, Intapad S. Fetal programming and cardiovascular pathology. Compr Physiol 2015;5(2):997–1025. doi: 10.1002/cphy.c140036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.•.Brenner BM, Lawler EV, Mackenzie HS. The hyperfiltration theory: a paradigm shift in nephrology. Kidney Int 1996;49(6): 1774–7. The Brenner’s hypothesis supports the idea that decreased development of nephrons from prenatal insults leads to compensatory hypertrophy of remaining nephrons, hyperfiltration, and hastened decline of renal function. [DOI] [PubMed] [Google Scholar]
- 24.•.Gurusinghe S, Palvanov A, Bittman ME, Singer P, Frank R, Chorny N, et al. Kidney volume and ambulatory blood pressure in children. J Clin Hypertens (Greenwich) 2016; doi: 10.1111/jch.12954. This study showed that kidney volume is positively correlated with SBP in ages 4 – 20 years. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.•.Shahzad T, Radajewski S, Chao CM, Bellusci S, Ehrhardt H. Pathogenesis of bronchopulmonary dysplasia: when inflammation meets organ development. Mol Cell Pediatr 2016;3(1):23. doi: 10.1186/s40348-016-0051-9. Excessive oxygen exposure in preterm infants can injure vasculogenesis increasing future risk for hypertension. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.••.Grigore D, Ojeda NB, Alexander BT. Sex differences in the fetal programming of hypertension. Gend Med 2008;5(Suppl A): S121–32. doi: 10.1016/j.genm.2008.03.012. Testosterone has permissive effect in the development of hypertension in the offspring as evidenced by decrease in nephron number, increase in endothelin production and increase in reactive oxygen species. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ojeda NB, Intapad S, Alexander BT. Sex differences in the developmental programming of hypertension. Acta Physiol (Oxf) 2014;210(2):307–16. doi: 10.1111/apha.12206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thornburg KL, O’Tierney PF, Louey S. Review: The placenta is a programming agent for cardiovascular disease. Placenta 2010;31(Suppl):S54–9. doi: 10.1016/j.placenta.2010.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Baum M Role of the kidney in the prenatal and early postnatal programming of hypertension. Am J Physiol Ren Physiol 2010;298(2):F235–47. doi: 10.1152/ajprenal.00288.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tain YL, Lee WC, Wu KL, Leu S, Chan JY. Targeting arachidonic acid pathway to prevent programmed hypertension in maternal fructose-fed male adult rat offspring. J Nutr Biochem 2016;38: 86–92. [DOI] [PubMed] [Google Scholar]
- 31.Gray C, Al-Dujaili EA, Sparrow AJ, Gardiner SM, Craigon J, Welham SJ, et al. Excess maternal salt intake produces sex-specific hypertension in offspring: putative roles for kidney and gastrointestinal sodium handling. PLoS One 2013;8(8):e72682. doi: 10.1371/journal.pone.0072682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Maruyama K, Kagota S, Van Vliet BN, Wakuda H, Shinozuka K. A maternal high salt diet disturbs cardiac and vascular function of offspring. Life Sci 2015;136:42–51. doi: 10.1016/j.lfs.2015.06.023. [DOI] [PubMed] [Google Scholar]
- 33.Porter JP, King SH, Honeycutt AD. Prenatal high-salt diet in the Sprague-Dawley rat programs blood pressure and heart rate hyperresponsiveness to stress in adult female offspring. Am J Phys Regul Integr Comp Phys 2007;293(1):R334–42. [DOI] [PubMed] [Google Scholar]
- 34.Contreras RJ, Wong DL, Henderson R, Curtis KS, Smith JC. High dietary NaCl early in development enhances mean arterial pressure of adult rats. Physiol Behav 2000;71(1–2):173–81. [DOI] [PubMed] [Google Scholar]
- 35.Bo L, Jiang L, Zhou A, Wu C, Li J, Gao Q, et al. Maternal high-salt diets affected pressor responses and microvasoconstriction via PKC/BK channel signaling pathways in rat offspring. Mol Nutr Food Res 2015;59(6):1190–9. doi: 10.1002/mnfr.201400841. [DOI] [PubMed] [Google Scholar]
- 36.Piecha G, Koleganova N, Ritz E, Müller A, Fedorova OV, Bagrov AY, et al. High salt intake causes adverse fetal programming—vascular effects beyond blood pressure. Nephrol Dial Transplant 2012;27(9):3464–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Koleganova N, Piecha G, Ritz E, Becker LE, Müller A, Weckbach M, et al. Both high and low maternal salt intake in pregnancy alter kidney development in the offspring. Am J Physiol Ren Physiol 2011;301(2):F344–54. [DOI] [PubMed] [Google Scholar]
- 38.Tain YL, Lee WC, Leu S, Wu K, Chan J. High salt exacerbates programmed hypertension in maternal fructose-fed male offspring. Nutr Metab Cardiovasc Dis 2015;25(12):1146–51. doi: 10.1016/j.numecd.2015.08.002. [DOI] [PubMed] [Google Scholar]
- 39.•.Li S, Fang Q, Zhou A, Wu L, Shi A, Cao L, et al. Intake of high sucrose during pregnancy altered large-conductance Ca2+-activated K+ channels and vessel tone in offspring’s mesenteric arteries. Hypertens Res 2013;36(2):158–65. Not only high maternal salt intake but also high sucrose diet increased the offspring’s sensitivity to angiotensin II similar to a maternal high salt intake. [DOI] [PubMed] [Google Scholar]
- 40.Khan IY, Dekou V, Douglas G, Jensen R, Hanson MA, Poston L, et al. A high-fat diet during rat pregnancy or suckling induces cardiovascular dysfunction in adult offspring. Am J Phys Regul Integr Comp Phys 2005;288(1):R127–33. [DOI] [PubMed] [Google Scholar]
- 41.•.Masuyama H, Mitsui T, Eguchi T, Tamada S, Hiramatsu Y. The effects of paternal high-fat diet exposure on offspring metabolism with epigenetic changes in the mouse adiponectin and leptin gene promoters. Am J Physiol Endocrinol Metab 2016;311(1):E236–45. doi: 10.1152/ajpendo.00095.2016. High paternal fat diet also plays a role in metabolic syndrome in the offspring. [DOI] [PubMed] [Google Scholar]
- 42.Elahi MM, Cagampang FR, Mukhtar D, Anthony FW, Ohri SK, Hanson MA. Long-term maternal high-fat feeding from weaning through pregnancy and lactation predisposes offspring to hypertension, raised plasma lipids and fatty liver in mice. Br J Nutr 2009;102(4):514. [DOI] [PubMed] [Google Scholar]
- 43.Elahi MM, Matata BM. Effects of maternal high-fat diet and statin treatment on bone marrow endothelial progenitor cells and cardiovascular risk factors in female mice offspring fed a similar diet. Nutrition 2017;35:6–13. doi: 10.1016/j.nut.2016.10.01. [DOI] [PubMed] [Google Scholar]
- 44.•.Nguyen LT, Chen H, Pollock CA, Saad S. Sirtuins-mediators of maternal obesity-induced complications in offspring? FASEB J 2016;30(4):1383–90. doi: 10.1096/fj.15-280743. Sirtuins are proposed as one of the mediators of the fetal programming of childhood obesity. [DOI] [PubMed] [Google Scholar]
- 45.Gregory EF, Goldshore MA, Henderson JL, Weatherford RD, Showell NN. Infant growth following maternal participation in a gestational weight management intervention. Child Obes 2016;12(3):219–25. doi: 10.1089/chi.2015.0238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zimanyi MA, Bertram JF, Black MJ. Nephron number and blood pressure in rat offspring with maternal high-protein diet. Pediatr Nephrol 2002;17(12):1000–4. [DOI] [PubMed] [Google Scholar]
- 47.Lee JH, Lee H, Lee SM, Kang PJ, Kim KC, Hong YM. Changes of blood pressure, abdominal visceral fat tissue and gene expressions in fetal programming induced rat model after amlodipinelosartan combination treatment. Clin Hypertens 2016;22:12. doi: 10.1186/s40885-016-0046-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hoppe CC, Evans RG, Bertram JF, Moritz KM. Effects of dietary protein restriction on nephron number in the mouse. Am J Phys Regul Integr Comp Phys 2007;292:R1768–74. [DOI] [PubMed] [Google Scholar]
- 49.de Belchior AC, Freire DD Jr, da Costa CP, Vassallo DV, Padilha AS, Dos Santos L. Maternal protein restriction compromises myocardial contractility in the young adult rat by changing proteins involved in calcium handling. J Appl Physiol 2016;120(3):344–50. doi: 10.1152/japplphysiol.00246.2015. [DOI] [PubMed] [Google Scholar]
- 50.•.Harrison M, Langley-Evans SC. Intergenerational programming of impaired nephrogenesis and hypertension in rats following maternal protein restriction during pregnancy. Br J Nutr 2009;101(7):1020–30. doi: 10.1017/S0007114508057607. This study demonstrates that maternal protein restriction is associated with 2 generations of offsprings with high blood pressure. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bertram C, Khan O, Ohri S, Phillips DI, Matthews SG, Hanson MA. Transgenerational effects of prenatal nutrient restriction on cardiovascular and hypothalamic-pituitary-adrenal function. J Physiol 2008;586(8):2217–29. doi: 10.1113/jphysiol.2007.147967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.McMullen S, Langley-Evans SC. Maternal low-protein diet in rat pregnancy programs blood pressure through sex-specific mechanisms. Am J Phys Regul Integr Comp Phys 2005;288(1):R85–90. [DOI] [PubMed] [Google Scholar]
- 53.Harvey TJ, Murphy RM, Morrison JL, Posterino GS. Maternal nutrient restriction alters Ca2+ handling properties and contractile function of isolated left ventricle bundles in male but not female juvenile rats. PLoS One 2015;10(9):e0138388. doi: 10.1371/journal.pone.0138388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.de Brito Alves JL, de Oliveira JM, Ferreira DJ, Barros MA, Nogueira VO, Alves DS, et al. Maternal protein restriction induced-hypertension is associated to oxidative disruption at transcriptional and functional levels in the medulla oblongata. Clin Exp Pharmacol Physiol 2016;43(12):1177–84. doi: 10.1111/1440-1681.12667. [DOI] [PubMed] [Google Scholar]
- 55.Wattez JS, Delahaye F, Barella LF, Dickes-Coopman A, Montel V, Breton C, et al. Short- and long-term effects of maternal perinatal undernutrition are lowered by cross-fostering during lactation in the male rat. J Dev Orig Health Dis 2014;5(2):109–20. doi: 10.1017/S2040174413000548. [DOI] [PubMed] [Google Scholar]
- 56.•.Simonds SE, Pryor JT, Cowley MA. Does leptin cause an increase in blood pressure in animals and humans? Curr Opin Nephrol Hypertens 2017;26(1):20–5. Chronic administration of leptin does not increase blood pressure. [DOI] [PubMed] [Google Scholar]
- 57.Thanos PK, Zhuo J, Robison L, Kim R, Ananth M, Choai I, et al. Suboptimal maternal diets alter mu opioid receptor and dopamine type 1 receptor binding but exert no effect on dopamine transporters in the offspring brain. Int J Dev Neurosci 2016. September 22 pii: S0736–5748(16)30211–8. doi: 10.1016/j.ijdevneu.2016.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Amare AT, Schubert KO, Klingler-Hoffmann M, Cohen-Woods S, Baune BT. The genetic overlap between mood disorders and cardiometabolic diseases: a systematic review of genome wide and candidate gene studies. Transl Psychiatry 2017;7(1):e1007. doi: 10.1038/tp.2016.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang H, Sun ZQ, Liu SS, Yang LN. Association between GRK4 and DRD1 gene polymorphisms and hypertension: a meta-analysis. Clin Interv Aging 2015;11:17–27. doi: 10.2147/CIA.S94510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.•.Armando I, Konkalmatt P, Felder RA, Jose PA. The renal dopaminergic system: novel diagnostic and therapeutic approaches in hypertension and kidney disease. Transl Res 2015;165(4):505–11. This is an updated review on the renal dopaminergic system and how dysfunction of which would cause decreased sodium excretion and increased blood pressure. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Choi MR, Kouyoumdzian NM, Rukavina Mikusic NL, Kravetz MC, Rosón MI, Rodríguez Fermepin M, et al. Renal dopaminergic system: pathophysiological implications and clinical perspectives. World J Nephrol 2015;4(2):196–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang MZ, Harris RC. Antihypertensive mechanisms of intrarenal dopamine. Curr Opin Nephrol Hypertens 2015;24(2):117–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Asghar M, Tayebati SK, Lokhandwala MF, Hussain T. Potential dopamine-1 receptor stimulation in hypertension management. Curr Hypertens Rep 2011;13(4):294–302. [DOI] [PubMed] [Google Scholar]
- 64.Rayner B, Ramesar R. The importance of G protein-coupled receptor kinase 4 (GRK4) in pathogenesis of salt sensitivity, salt sensitive hypertension and response to antihypertensive treatment. Int J Mol Sci 2015;16(3):5741–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rayner BL, Spence JD. Hypertension in blacks: insights from Africa. J Hypertens 2017;35(2):234–9. doi: 10.1097/HJH.000000000000117. [DOI] [PubMed] [Google Scholar]
- 66.Dagan A, Habib S, Gattineni J, Dwarakanath V, Baum M. Prenatal programming of rat thick ascending limb chloride transport by low-protein diet and dexamethasone. Am J Phys Regul Integr Comp Phys 2009;297(1):R93–9. doi: 10.1152/ajpregu.91006.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wesseling S, Koeners MP, Joles JA. Salt sensitivity of blood pressure: developmental and sex-related effects. Am J Clin Nutr 2011;94(6 Suppl):1928S–32S. [DOI] [PubMed] [Google Scholar]
- 68.•.Khorram O, Chuang TD, Pearce WJ. Long-term effects of maternal undernutrition on offspring carotid artery remodeling: role of miR-29c. J Dev Orig Health Dis 2015. August;6(4):342–9. doi: 10.1017/S2040174415001208. Epub 2015 May 26. Maternal undernutrition (decreased protein intake) increases extracellular matrix proteins in the carotid arteries related to suppression of miR-29C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sathishkumar K, Balakrishnan MP, Yallampalli C. Enhanced mesenteric arterial responsiveness to angiotensin II is androgen receptor-dependent in prenatally protein-restricted adult female rat offspring. Biol Reprod 2015;92(2):55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cooke CL, Zhao L, Gysler S, Arany E, Regnault TR. Sex-specific effects of low protein diet on in utero programming of renal G-protein coupled receptors. J Dev Orig Health Dis 2014;5(1):36–44. doi: 10.1017/S2040174413000524. [DOI] [PubMed] [Google Scholar]
- 71.Chisaka T, Mogi M, Nakaoka H, Kan-No H, Tsukuda K, Wang XL, et al. Low-protein diet-induced fetal growth restriction leads to exaggerated proliferative response to vascular injury in postnatal life. Am J Hypertens 2016;29(1):54–62. [DOI] [PubMed] [Google Scholar]
- 72.de Lima MC, Scabora JE, Lopes A, Mesquita FF, Torres D, Boer PA, et al. Early changes of hypothalamic angiotensin II receptors expression in gestational protein-restricted offspring: effect on water intake, blood pressure and renal sodium handling. J Renin-Angiotensin-Aldosterone Syst 2013;14(3):271–82. doi: 10.1177/1470320312456328. [DOI] [PubMed] [Google Scholar]
- 73.Casas-Agustench P, Iglesias-Gutiérrez E, Dávalos A. Mother’s nutritional miRNA legacy: nutrition during pregnancy and its possible implications to develop cardiometabolic disease in later life. Pharmacol Res 2015;100:322–34. [DOI] [PubMed] [Google Scholar]
- 74.••.Koeners MP, Wesseling S, Ulu A, Sepúlveda RL, Morisseau C, Braam B, et al. Soluble epoxide hydrolase in the generation and maintenance of high blood pressure in spontaneously hypertensive rats. Am J Physiol Endocrinol Metab 2011;300(4):E691–8. doi: 10.1152/ajpendo.00710.2010. Fetal programming may be reversed through inhibition of soluble epoxide hydrolase. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.••.Tain YL, Lee CT, Chan JY, Hsu CN. Maternal melatonin or N-acetylcysteine therapy regulates hydrogen sulfide-generating pathway and renal transcriptome to prevent prenatal NG-Nitro-L-arginine-methyl ester (L-NAME)-induced fetal programming of hypertension in adult male offspring. Am J Obstet Gynecol 2016;215(5):636.e1–636.e72. Maternal antioxidant treatment with melatonin and N-acetylcysteine may reverse fetal programming of hypertension. [DOI] [PubMed] [Google Scholar]
- 76.••.de Bem GF, da Costa CA, de Oliveira PR, Cordeiro VS, Santos IB, de Carvalho LC, et al. Protective effect of Euterpe oleracea Mart (açaí) extract on programmed changes in the adult rat off-spring caused by maternal protein restriction during pregnancy. J Pharm Pharmacol 2014;66(9):1328–38. doi: 10.1111/jphp.12258. This study shows that fetal programming may be reversible in experimental conditions by administration of acai extract. [DOI] [PubMed] [Google Scholar]
- 77.Szeto IM, Aziz A, Das PJ, Taha AY, Okubo N, Reza-Lopez S, et al. High multivitamin intake by Wistar rats during pregnancy results in increased food intake and components of the metabolic syndrome in male offspring. Am J Phys Regul Integr Comp Phys 2008;295(2):R575–82. doi: 10.1152/ajpregu.90354.2008. [DOI] [PubMed] [Google Scholar]
- 78.•.Pannia E, Cho CE, Kubant R, Sánchez-Hernández D, Huot PS, Chatterjee D, et al. A high multivitamin diet fed to Wistar rat dams during pregnancy increases maternal weight gain later in life and alters homeostatic, hedonic and peripheral regulatory systems of energy balance. Behav Brain Res 2015;278:1–11. doi: 10.1016/j.bbr.2014.09.019. Excessive maternal vitamin intake alters the metabolism of offspring. [DOI] [PubMed] [Google Scholar]
- 79.Szeto IM, Das PJ, Aziz A, Anderson GH. Multivitamin supplementation of Wistar rats during pregnancy accelerates the development of obesity in offspring fed an obesogenic diet. Int J Obes 2009;33(3):364–72. doi: 10.1038/ijo.2008.281. [DOI] [PubMed] [Google Scholar]
- 80.Cho CE, Sánchez-Hernández D, Reza-López SA, Huot PS, Kim YI, Anderson GH. Obesogenic phenotype of offspring of dams fed a high multivitamin diet is prevented by a post-weaning high multivitamin or high folate diet. Int J Obes 2013;37(9):1177–82. doi: 10.1038/ijo.2012.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Dodic M, Abouantoun T, O’Connor A, Wintour EM, Moritz KM. Programming effects of short prenatal exposure to dexamethasone in sheep. Hypertension 2002;40:729–34. [DOI] [PubMed] [Google Scholar]
- 82.Dodic M, Samuel C, Moritz KM, Wintour EM, Morgan J, et al. Impaired cardiac functional reserve and left ventricular hypertrophy in adult sheep after prenatal dexamethasone exposure. Circ Res 2002;89:623–9. [DOI] [PubMed] [Google Scholar]
- 83.O’Regan D, Kenyon CJ, Seckl JR, Holmes MC. Glucocorticoid exposure in late gestation in the rat permanently programs gender-specific differences in adult cardiovascular and metabolic physiology. Am J Physiol Endocrinol Metab 2004;287:E863–70. [DOI] [PubMed] [Google Scholar]
- 84.••.Dickinson H, Walker DW, Wintour EM, Moritz KM. Maternal dexamethasone treatment at midgestation reduces nephron number and alters renal gene expression in the fetal spiny mouse. Am J Phys Regul Integr Comp Phys 2007;292:R453–61. Prenatal exposure to glucocorticoids reduces nephron numbers increasing risk of hypertension later in life. [DOI] [PubMed] [Google Scholar]
- 85.Marshall AC, Shaltout HA, Nautiyal M, Rose JC, Chappell MC, Diz D. Fetal betamethasone exposure attenuates angiotensin-(1–7)-Mas receptor expression in the dorsal medulla of adult sheep. Peptides 2013. June;44:25–31.. doi: 10.1016/j.peptides.2013.03.018. Epub 2013 Mar 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Marshall AC, Shaltout HA, Pirro NT, Rose JC, Diz DI, Chappell MC. Antenatal betamethasone exposure is associated with lower ANG-(1–7) and increased ACE in the CSF of adult sheep. Am J Phys Regul Integr Comp Phys 2013;305(7):R679–88. doi: 10.1152/ajpregu.00321.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Cuffe JS, Burgess DJ, O’Sullivan L, Singh RR, Moritz KM. Maternal corticosterone exposure in the mouse programs sex-specific renal adaptations in the renin-angiotensin-aldosterone system in 6-month offspring. Physiol Rep 2016. April;4(8). pii: e12754. doi: 10.14814/phy2.12754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.O’Sullivan L, Cuffe JS, Koning A, Singh RR, Paravicini TM, Moritz KM. Excess prenatal corticosterone exposure results in albuminuria, sex-specific hypotension, and altered heart rate responses to restraint stress in aged adult mice. Am J Physiol Ren Physiol 2015;308(10):F1065–73. doi: 10.1152/ajprenal.00676.2014. [DOI] [PubMed] [Google Scholar]
- 89.Tain YL, Sheen JM, Chen CC, Yu HR, Tiao MM, Kuo HC, et al. Maternal citrulline supplementation prevents prenatal dexamethasone-induced programmed hypertension. Free Radic Res 2014;48(5):580–6. doi: 10.3109/10715762.2014.895341. [DOI] [PubMed] [Google Scholar]
- 90.Benediktsson R, Lindsay RS, Noble J, Seckl JR, Edwards CR. Glucocorticoid exposure in utero: new model for adult hypertension. Lancet 1993;341:339–41. [DOI] [PubMed] [Google Scholar]
- 91.Dickinson H, Walker DW, Wintour EM, Moritz K. Maternal dexamethasone treatment at midgestation reduces nephron number and alters renal gene expression in the fetal spiny mouse. Am J Phys Regul Integr Comp Phys 2007;292(1):R453–61. [DOI] [PubMed] [Google Scholar]
- 92.Sheen JM, Hsieh CS, Tain YL, Li SW, Yu HR, Chen CC, et al. Programming effects of prenatal glucocorticoid exposure with a postnatal high-fat diet in diabetes mellitus. Int J Mol Sci 2016;17(4):533. doi: 10.3390/ijms17040533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Rexhepaj R, Boini KM, Huang DY, Amann K, Artunc F, Wang K, et al. Role of maternal glucocorticoid inducible kinase SGK1 in fetal programming of blood pressure in response to prenatal diet. Am J Phys Regul Integr Comp Phys 2008;294(6):R2008–13. doi: 10.1152/ajpregu.00737.2007. [DOI] [PubMed] [Google Scholar]
- 94.Woods LL. Maternal glucocorticoids and prenatal programming of hypertension. Am J Phys Regul Integr Comp Phys 2006;291(4):R1069–75. [DOI] [PubMed] [Google Scholar]
- 95.O’Sullivan L, Cuffe JS, Paravicini TM, Campbell S, Dickinson H, Singh RR, et al. Prenatal exposure to dexamethasone in the mouse alters cardiac growth patterns and increases pulse pressure in aged male offspring. PLoS One 2013;8(7):e69149. doi: 10.1371/journal.pone.0069149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.•.Wang X, Wang J, Luo H, Chen C, Pei F, Cai Y, et al. Prenatal lipopolysaccharide exposure causes mesenteric vascular dysfunction through the nitric oxide and cyclic guanosine monophosphate pathway in offspring. Free Radic Biol Med 2015;86:322–30. doi: 10.1016/j.freeradbiomed.2015.05.040. Prenatal LPS exposure increases oxidative stress which causes vascular dysfunction in offspring. [DOI] [PubMed] [Google Scholar]
- 97.•.Wang X, Luo H, Chen C, Chen K, Wang J, Cai Y, et al. Prenatal lipopolysaccharide exposure results in dysfunction of the renal dopamine D1 receptor in offspring. Free Radic Biol Med 2014;76:242–50. Prenatal LPS exposure causes dysfunction of renal dopaminergic system decreasing sodium excretion. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Hao XQ, Zhang HG, Yuan ZB, Yang DL, Hao LY, Li XH. Prenatal exposure to lipopolysaccharide alters the intrarenal renin-angiotensin system and renal damage in offspring rats. Hypertens Res 2010;33(1):76–82. doi: 10.1038/hr.2009.185. [DOI] [PubMed] [Google Scholar]
- 99.Gao M, Zhang X, Chen X, Mi C, Tang Y, Zhou J, et al. Prenatal exposure to lipopolysaccharide results in local RAS activation in the adipose tissue of rat offspring. PLoS One 2014. October 31;9(10): e111376. doi: 10.1371/journal.pone.0111376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Wei YL, Li XH, Zhou JZ. Prenatal exposure to lipopolysaccharide results in increases in blood pressure and body weight in rats. Acta Pharmacol Sin 2007;28(5):651–6. [DOI] [PubMed] [Google Scholar]
- 101.••.Sanada H, Yoneda M, Yatabe J, Williams SM, Bartlett J, White MJ, et al. Common variants of the G protein-coupled receptor type 4 are associated with human essential hypertension and predict the blood pressure response to angiotensin receptor blockade. Pharmacogenomics J 2016;16(1):3–9. doi: 10.1038/tpj.2015.6. This study reviews the GRK4 variants and their response to antihypertensives (angiotensin receptor blocker). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.•.Jose PA, Felder RA, Yang Z, Zeng C, Eisner GM. Gastrorenal axis. Hypertension 2016;67(6):1056–63. An oral sodium load causes gastrin release which then stimulates the renal dopaminergic system to excrete the sodium. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Baharnoori M, Bhardwaj SK, Srivastava LK. Effect of maternal lipopolysaccharide administration on the development of dopaminergic receptors and transporter in the rat offspring. PLoS One 2013;8(1):e54439. doi: 10.1371/journal.pone.0054439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Luchicchi A, Lecca S, Melis M, De Felice M, Cadeddu F, Frau R, et al. Maternal immune activation disrupts dopamine system in the offspring. Int J Neuropsychopharmacol 2016. July 5;19(7). pii: pyw007. doi: 10.1093/ijnp/pyw007. Print 2016 Jul. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Trøseid M, Nestvold TK, Rudi K, Thoresen H, Nielsen EW, Lappegård KT. Plasma lipopolysaccharide is closely associated with glycemic control and abdominal obesity: evidence from bariatric surgery. Diabetes Care 2013;36(11):3627–32. doi: 10.2337/dc13-0451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Alexander BT. Placental insufficiency leads to development of hypertension in growth-restricted offspring. Hypertension 41(3): 457–62. [DOI] [PubMed] [Google Scholar]
- 107.Master JS, Zimanyi MA, Yin KV, Moritz KM, Gallo LA, Tran M, et al. Transgenerational left ventricular hypertrophy and hypertension in offspring after uteroplacental insufficiency in male rats. Clin Exp Pharmacol Physiol 2014;41(11):884–9. [DOI] [PubMed] [Google Scholar]
- 108.Tare M, Parkington HC, Bubb KJ, Wlodek ME. Uteroplacental insufficiency and lactational environment separately influence arterial stiffness and vascular function in adult male rats. Hypertension 2012;60(2):378–86. doi: 10.1161/HYPERTENSIONAHA.112.190876. [DOI] [PubMed] [Google Scholar]
- 109.••.Mazzuca MQ, Wlodek ME, Dragomir NM, Parkington HC, Tare M. Uteroplacental insufficiency programs regional vascular dysfunction and alters arterial stiffness in female offspring. J Physiol 2010;588(Pt 11):1997–2010. doi: 10.1113/jphysiol.2010.187849. This study discusses the role of uteroplacental insufficiency in fetal programming of hypertension. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Gallo LA, Tran M, Cullen-McEwen LA, Denton KM, Jefferies AJ, Moritz KM, et al. Transgenerational programming of fetal nephron deficits and sex-specific adult hypertension in rats. Reprod Fertil Dev 2014;26(7):1032–43. doi: 10.1071/RD13133. [DOI] [PubMed] [Google Scholar]
- 111.Dodson RB, Rozance PJ, Fleenor BS, Petrash CC, Shoemaker LG, Hunter KS, et al. Increased arterial stiffness and extracellular matrix reorganization in intrauterine growth-restricted fetal sheep. Pediatr Res 2013;73(2):147–54. doi: 10.1038/pr.2012.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Bauer R, Walter B, Bauer K, Klupsch R, Patt S, Zwiener U. Intrauterine growth restriction reduces nephron number and renal excretory function in newborn piglets. Acta Physiol Scand 2002;176(2):83–90. [DOI] [PubMed] [Google Scholar]
- 113.Baserga M, Bares AL, Hale MA, Callaway CW, McKnight RA. Lane PH, et al. Uteroplacental insufficiency affects kidney VEGF expression in a model of IUGR with compensatory glomerular hypertrophy and hypertension. Early Hum Dev 2009;85(6):361–7. doi: 10.1016/j.earlhumdev.2008.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.•.Ojeda NB, Grigore D, Yanes LL, Iliescu R, Robertson EB, Zhang H, et al. Testosterone contributes to marked elevations in mean arterial pressure in adult male intrauterine growth restricted offspring. Am J Phys Regul Integr Comp Phys 2007;292(2):R758–63. Sex differences is also present in uteroplacental insufficiency modulation of blood pressure. [DOI] [PubMed] [Google Scholar]
- 115.US Burden of Disease Collaborators. The state of US health, 1990–2010: burden of diseases, injuries, and risk factors. JAMA 2013;310:591–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.••.Armando I, Villar VA, Jose PA. Dopamine and renal function and blood pressure regulation. Compr Physiol 2011;1(3):1075–117. doi: 10.1002/cphy.c100032. Comprehensive review of the renal dopaminergic system and GRK4 and role in blood pressure regulation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Glazier AM, Nadeau JH, Aitman TJ. Finding genes that underlie complex traits. Science 2002;298:2345–9. [DOI] [PubMed] [Google Scholar]
- 118.Jain S, Tillinger A, Mopidevi B, Pandey VG, Chauhan CK, Fiering SN, et al. Transgenic mice with −6A haplotype of the human angiotensinogen gene have increased blood pressure compared with −6G haplotype. J Biol Chem 2010;285(52):41172–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Jain S, Prater A, Pandey V, Rana A, Puri N, Kumar A. A haplotype of angiotensin receptor type 1 associated with human hypertension increases blood pressure in transgenic mice. J Biol Chem 2013;288(52):37048–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Mopidevi B, Kaw MK, Puri N, Ponnala M, Jain S, Rana A, et al. Variable transcriptional regulation of the human aldosterone synthase gene causes salt-dependent high blood pressure in transgenic mice. Circ Cardiovasc Genet 2015;8(1):30–9. doi: 10.1161/CIRCGENETICS.114.000694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Wang Z, Zeng C, Villar VA, Chen SY, Konkalmatt P, Wang X, et al. Human GRK4γ142V variant promotes angiotensin II type I receptor-mediated hypertension via renal histone deacetylase type 1 inhibition. Hypertension 2016;67(2):325–34. doi: 10.1161/HYPERTENSIONAHA.115.05962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Felder RA, Sanada H, Xu J, Yu PY, Wang Z, Watanabe H, et al. G protein-coupled receptor kinase 4 gene variants in human essential hypertension. Proc Natl Acad Sci U S A 2002;99(6):3872–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Rhee MY, Yang SJ, Oh SW, Park Y, Kim CI, Park HK, et al. Novel genetic variations associated with salt sensitivity in the Korean population. Hypertens Res 2011;34(5):606–11. doi: 10.1038/hr.2010.278. [DOI] [PubMed] [Google Scholar]
- 124.Carey RM, Schoeffel CD, Gildea JJ, Jones JE, McGrath HE, Gordon LN, et al. Salt sensitivity of blood pressure is associated with polymorphisms in the sodium-bicarbonate cotransporter. Hypertension 2012;60(5):1359–66. doi: 10.1161/HYPERTENSIONAHA.112.196071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Wang Y, O’Connell JR, McArdle PF, Wade JB, Dorff SE, Shah SJ, et al. Whole-genome association study identifies STK39 as a hypertension susceptibility gene. Proc Natl Acad Sci U S A 2009;106:226–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Diao Z, Asico LD, Villar VA, Zheng X, Cuevas S, Armando I, et al. Increased renal oxidative stress in salt-sensitive human GRK4γ486V transgenic mice. Free Radic Biol Med 2017;106: 80–90. doi: 10.1016/j.freeradbiomed.2017.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Cabrera CP, Ng FL, Warren HR, Barnes MR, Munroe PB, Caulfield MJ. Exploring hypertension genome-wide association studies findings and impact on pathophysiology, pathways, and pharmacogenetics. Wiley Interdiscip Rev Syst Biol Med 2015;7(2):73–90. [DOI] [PubMed] [Google Scholar]
- 128.Padmanabhan S, Caulfield M, Dominiczak AF. Genetic and molecular aspects of hypertension. Circ Res 2015;116(6):937–59. doi: 10.1161/CIRCRESAHA.116.303647. [DOI] [PubMed] [Google Scholar]
- 129.Huan T, Esko T, Peters MJ, Pilling LC, Schramm K, Schurmann C, et al. International consortium for blood pressure GWAS (ICBP). A meta-analysis of gene expression signatures of blood pressure and hypertension. PLoS Genet 2015;11(3):e1005035. doi: 10.1371/journal.pgen.1005035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Staessen JA, Kuznetsova T, Zhang H, Maillard M, Bochud M, Hasenkaml S, et al. Blood pressure and renal sodium handling in relation to genetic variation in the DRD1 promoter and GRK4. Hypertension 2008;51:1643–50. [DOI] [PubMed] [Google Scholar]
- 131.Martinez Cantarin MP, Ertel A, Deloach S, Fortina P, Scott K, Burns TL, et al. Variants in genes involved in functional pathways associated with hypertension in African Americans. Clin Transl Sci 2010;3(6):279–86. doi: 10.1111/j.1752-8062.2010.00242.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Stilling RM, Dinan TG, Cryan JF. Microbial genes, brain & behavior - epigenetic regulation of the gut-brain axis. Genes Brain Behav 2014;13:69–86. [DOI] [PubMed] [Google Scholar]
- 133.Banday AA, Lau YS, Lokhandwala MF. Oxidative stress causes renal dopamine D1 receptor dysfunction and salt-sensitive hypertension in Sprague-Dawley rats. Hypertension 2008;51:367–75. [DOI] [PubMed] [Google Scholar]
- 134.Doyle K, Fitzpatrick FA. Redox signaling, alkylation (carbonylation) of conserved cysteines inactivates class I histone deacetylases 1, 2, and 3 and antagonizes their transcriptional repressor function. J Biol Chem 2010;285:17417–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.•.Gildea JJ, Carlson JM, Schoeffel CD, Carey RM, Felder RA. Urinary exosome miRNome analysis and its applications to salt sensitivity of blood pressure. Clin Biochem 2013;46:1131–4. This study discusses new methods of diagnosing salt sensitivity through increased miR-124 expression in urinary exosomes. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Walesky C, Edwards G, Borude P, Gunewardena S, O’Neil M, Yoo B, et al. Hepatocyte nuclear factor 4 alpha deletion promotes diethylnitrosamine-induced hepatocellular carcinoma in rodents. Hepatology 2013;57:2480–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Heideman MR, Wilting RH. Yanover, Velds a, de Jong J, Kerkhoven RM, et al. dosage-dependent tumor suppression by histone deacetylases 1 and 2 through regulation of c-Myc collaborating genes and p53 function. Blood 2013;121:2038–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Stocks T, Van Hemelrijck M, Manjer J, Bjørge T, Ulmer H, Hallmans G, et al. Blood pressure and risk of cancer incidence and mortality in the metabolic syndrome and cancer project. Hypertension 2012;59:802–10. [DOI] [PubMed] [Google Scholar]
- 139.D’Elia L, Galletti F, Strazzullo P. Dietary salt intake and risk of gastric cancer. Cancer Treat Res 2014;159:83–95. [DOI] [PubMed] [Google Scholar]
- 140.Hasenkamp S, Telgmann R, Staessen JA, Hagedorn C, Dördelmann C, Bek M, et al. Characterization and functional analyses of the human G protein-coupled receptor kinase 4 gene promoter. Hypertension 2008;52:737–46. [DOI] [PubMed] [Google Scholar]
- 141.Escano CS, Armando I, Wang X, Asico LD, Pascua A, Yang Y, et al. Renal dopaminergic defect in C57Bl/6J mice. Am J Phys Regul Integr Comp Phys 2009;297:R1660–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Zhao Y, Vanhoutte PM, Leung SW. α1-Adrenoceptor activation of PKC-ε causes heterologous desensitization of thromboxane receptors in the aorta of spontaneously hypertensive rats. Br J Pharmacol 2015;172(14):3687–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Sanada H, Yatabe J, Midorikawa S, Katoh T, Hashimoto S, Watanabe T, et al. Amelioration of genetic hypertension by suppression of renal G protein-coupled receptor kinase type 4 expression. Hypertension 2006;47(6):1131–9. [DOI] [PubMed] [Google Scholar]
- 144.Rassler B The renin-angiotensin system in the development of salt-sensitive hypertension in animal models and humans. Pharmaceuticals (Basel) 2010;3(4):940–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Gildea JJ, Lahiff DT, Van Sciver RE, Weiss RS, Shah N, McGrath HE, et al. A linear relationship between the ex-vivo sodium mediated expression of two sodium regulatory pathways as a surrogate marker of salt sensitivity of blood pressure in exfoliated human renal proximal tubule cells: the virtual renal biopsy. Clin Chim Acta 2013;421:236–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Yang J, Villar VA, Armando I, Jose PA, Zeng C. G protein-coupled receptor kinases: crucial regulators of blood pressure. J Am Heart Assoc 2016. July 7;5(7). pii: e003519. doi: 10.1161/JAHA.116.003519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Sun CJ, Zhang WY. G protein-coupled receptor kinase 4 gene variants are not associated with preeclampsia in Northern Han Chinese. Hypertens Res 2010;33(7):683–7. doi: 10.1038/hr.2010.57. [DOI] [PubMed] [Google Scholar]
- 148.Wolski H, Marek P, Drews K, Barlik M, Kurzawińska G, Oarowski M, et al. DRD1 and DRD4 dopamine receptors in the etiology of preeclampsia. Ginekol Pol 2015;86(9):672–7. [DOI] [PubMed] [Google Scholar]
- 149.••.Bhatnagar V, O’Connor DT, Brophy VH, Schork NJ, Richard E, Salem RM, et al. AASK Study Investigators. G-protein-coupled receptor kinase 4 polymorphisms and blood pressure response to metoprolol among African Americans: sex-specificity and interactions. Am J Hypertens 2009;22(3):332–8. doi: 10.1038/ajh.2008.341. AASK study discusses the GRK4 polymorphisms among African Americans and response to beta blockers. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.••.Vandell AG, Lobmeyer MT, Gawronski BE, Langaee TY, Gong Y, Gums JG, et al. G protein receptor kinase 4 polymorphisms: β-blocker pharmacogenetics and treatment-related outcomes in hypertension. Hypertension 2012;60(4):957–64. doi: 10.1161/HYPERTENSIONAHA.112.198721. This study discusses the different GRK4 polymorphisms and response to beta blockers and risk for adverse outcomes. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Wagner F, Malice MP, Wiegert E, McGrath HE, Gildea J, Mitta S, et al. A comparison of the natriuretic and kaliuretic effects of cicletanine and hydrochlorothiazide in prehypertensive and hypertensive humans. J Hypertens 2012;30(4):819–27. doi: 10.1097/HJH.0b013e32835022a8. [DOI] [PubMed] [Google Scholar]
- 152.Wang Z, Asico LD, Escano CS, Felder RA, Jose PA. Human G protein-coupled receptor kinase type 4 γ (hGRK4γ) wild-type prevents salt sensitivity while its variant, hGatatRK4γ486V, promotes salt sensitivity in transgenic mice: role of genetic background. Hypertension 2006;48:e27. [Google Scholar]
- 153.Sanada H, Yatabe J, Yatabe MS, Yokokawa H, Williams S, Bartlett J, et al. G protein coupled receptor type 4 gene variants and response to antihypertensive medication. Circulation 2009;120:S1087. [Google Scholar]
- 154.Litwin M, Niemirska A, Sladowska J, Antoniewicz J, Daszkowska J, Wierzbicka A, et al. Left ventricular hypertrophy and arterial wall thickening in children with essential hypertension. Pediatr Nephrol 2006;21:811–9. [DOI] [PubMed] [Google Scholar]
- 155.Flynn JT. Pediatric hypertension update. Curr Opin Nephrol Hypertens 2010;19:292–7. [DOI] [PubMed] [Google Scholar]
- 156.•.Kupferman JC, Lande MB, Adams HR, Pavlakis SG. Primary hypertension and neurocognitive and executive functioning in school-age children. Pediatr Nephrol 2013;28:401–8. doi: 10.1007/s00467-012-2215-8. Premature target organ damage (brain) of pediatric hypertension. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Litwin M, Niemirska A. Intima-media thickness measurements in children with cardiovascular risk factors. Pediatr Nephrol 2009;24:707–19. [DOI] [PubMed] [Google Scholar]
- 158.•.Folkow B. Structure and function of the arteries in hypertension. Am Heart J 1987;114:938–48. Folkow’s hypothesis that hypertension thickens medial layer of blood vessels which begets hypertension. [DOI] [PubMed] [Google Scholar]
- 159.Srinivasan SR, Myers L, Berenson GS. Changes in metabolic syndrome variables since childhood in prehypertensive and hypertensive subjects: the Bogalusa heart study. Hypertension 2006;48: 33–9. [DOI] [PubMed] [Google Scholar]