

ABSTRACT
Guillain-Barré syndrome (GBS), the most common cause of acute neuromuscular weakness and paralysis worldwide, encompasses a group of acute immune-mediated disorders restricted to peripheral nerves and roots. Immune-mediated attack of peripheral nervous system myelin, axons or both is presumed to be triggered by molecular mimicry, with both cell- and humoral-dependent mechanisms implicated in disease pathogenesis. Good circumstantial evidence exists for a pathogenic role for molecular mimicry in GBS pathogenesis, especially with its axonal forms, providing insights that could guide future immunotherapy. Intravenous immunoglobulin (IVIg) and plasma exchange (PE) are the most commonly prescribed immunotherapies for GBS with variable efficacy dependent on GBS subtype, severity at initial presentation and other clinical and electrophysiologic prognostic factors. The mechanisms of action of IVIg and PE are not known definitely. Despite recent significant advances in molecular biology that provide insights into GBS pathogenesis, no advances in therapeutics or significant improvements in patient outcomes have occurred over the past three decades. We summarize the clinical aspects of GBS, its current pathogenesis and immunotherapy, and highlight the potential of leukocyte trafficking inhibitors as novel disease-specific immunotherapeutic drugs.
KEYWORDS: Blood-Nerve Barrier, Guillain-Barré syndrome, Leukocyte Trafficking, Immunopathology, Immunotherapy, Peripheral Nerve Inflammation
Introduction
Guillain-Barré syndrome (GBS) is an acute onset, monophasic, immune-mediated peripheral nerve and nerve root disorder (termed polyradiculoneuropathy), which was first recognized as a distinct medical condition in 1916.1 GBS has become the most common cause of acute flaccid paralysis worldwide, following the near-eradication of poliomyelitis, and is a neurological emergency.2,3 The reported incidence of GBS in Europe and North America ranges from 1 to 2 cases per 100,000 adults, and 0.4 to 1.4 cases per 100,000 children per year.4-7 There also appears to be a linear increase in incidence with age and a slightly higher frequency in males compared to females.8 Despite significant advances in understanding the pathogenesis of immune-mediated disorders and the development of targeted molecular based therapies, current immunotherapies for GBS are non-specific and only partly efficacious. Improved collaboration is needed between neurologists, neuroscientists, immunologists, medical chemists and pharmacologists to deduce and design more effective immunotherapies for GBS.
The typical GBS clinical presentation is a sudden onset of rapidly progressive and symmetrical weakness of the limbs, with or without peripheral sensory disturbance, reduction in or loss of tendon reflexes (termed hyporeflexia and areflexia respectively), and cerebrospinal fluid (CSF) analyses showing elevated protein concentrations with a normal white cell count, termed albuminocytologic dissociation, to distinguish it from infections that typically demonstrate elevated protein and white cell counts.1,9 GBS often follows an antecedent infection, such as certain bacterial or viral infections or minor trauma. The symptoms typically reach maximal severity within four weeks from symptom onset. Most patients generally require hospitalization for treatment, with close cardiopulmonary monitoring performed. About 20–30% patients of patients require mechanical ventilation during the course of the illness.5,10 Many patients also develop symptoms or signs of autonomic nervous system dysfunction, termed dysautonomia.11 These commonly consist of sinus tachycardia, cardiac arrhythmias, labile blood pressure, orthostatic hypotension, increased sweating, as well as bladder and gastrointestinal dysfunction.
Varying degrees of sensory loss and neuropathic or radicular pain occur in GBS, and this can be debilitating. Persistent neuropathic pain may arise 2 weeks preceding weakness in 36% of cases, while 66% of patients in the acute phase and 38% a year after disease onset are affected.12 In some cases, chronic sensory loss and neuropathic pain persist indefinitely in addition to residual motor deficits. Respiratory failure and death may occur in an average of 5% of affected patients despite current therapies and advancement in intensive care.2,7,13–15 The estimated total average annual cost of GBS to the United States was ∼$1.7 billion in 2004 (equivalent to ∼$2.25 billion in 2018), predominantly due to premature death and lost productivity due to permanent disability.16
GBS is diagnosed based on clinical features, electrophysiological studies and cerebrospinal fluid analysis. GBS is a heterogeneous disorder, and has several variants which share key clinical features, and are classified by their distinct clinical phenomenological, electrophysiological and pathological features. Common GBS variants include the ‘classic’ acute inflammatory demyelinating polyradiculoneuropathy (AIDP),17,18 with electrophysiological studies primarily showing changes in peripheral nerve myelin status, termed demyelination, and variants showing primary dysfunction or loss of peripheral nervous system axons that include acute motor axonal neuropathy (AMAN), initially reported in Northern China as Chinese paralytic illness, with only motor axons involved,19,20 and acute motor and sensory axonal neuropathy (AMSAN), when motor and sensory axons are affected.21 In Europe and North America, AIDP is the most common variant, accounting for up to 90 % of GBS cases,22 although in a recent Italian study, AIDP was seen in 58% of GBS cases.23 The axonal variants are more common in Asia and South America, accounting for 30% to 70% of GBS cases.24-28
Another well-known GBS variant is the Miller Fisher syndrome (MFS), recognized as the classic triad of areflexia, ataxia and ophthalmoplegia.29 MFS is estimated to represent ∼1-5% of GBS cases from Western countries, although it may account for 19–25% of cases from Asian countries.8,13,30 There are less common GBS variants, such as pharyngeal-cervical-brachial weakness, paraparetic GBS, bifacial weakness with distal paresthesia, polyneuritis cranialis, pure sensory neuropathy, acute pandysautonomia and oropharyngeal variants.31-34 Several diagnostic criteria for clinical and research purposes have been developed recently that also guide institution of therapy.35,36
Pathogenesis
Antecedent infections, typically within 4 weeks of neurological symptom onset, commonly occur in GBS patients, resulting in the commonly cited molecular mimicry hypothesis, in which the immune system becomes activated in response to infectious antigen with structural similarity to peripheral nerve myelin or axonal components, with resultant tissue-specific peripheral nerve and nerve root injury in susceptible individuals.37-39 Epidemiological data implies that about two-thirds of GBS adult patients had a prior respiratory or gastrointestinal infection.40,41 Children exhibited a greater percentage of these infections than adults (67–85%), with respiratory infections (22–70%) being more common than gastrointestinal infections (6–26%).42 Multiple antecedent infections have been reported in GBS patients, but only a few micro-organisms have been shown to have a clear association with pathogenesis based on case-control studies. The most commonly associated infectious pathogen is Campylobacter jejuni (C. jejuni), found in 25–50% of adult GBS patients, with a higher frequency observed in Asian countries and in patients with more severe forms of GBS with axonal degeneration.43,44 Several other infections implicated in GBS pathogenesis include Mycoplasma pneumonia,45 cytomegalovirus (CMV),46 Epstein-Barr virus,47 Haemophilus influenza,48 influenza A virus49 and human immunodeficiency virus.50 The increased incidence of GBS associated with epidemics of arthropod-borne virus infectious in previously unexposed populations has been observed with West Nile virus,51 and more recently with Zika virus.52,53 However, it is important to recognize that most individuals infected by these pathogens do not develop GBS, emphasizing the importance of host factors, such as genetic susceptibility, in developing the peripheral nerve/ nerve root-specific disease.
Pathophysiology and immunopathology
As stated previously, the preceding infections are pathogenically associated with GBS, and may play an essential role in triggering the initial peripheral nerve/ nerve root-specific systemic immune system activation that causes cross-reactive humoral and cellular immune responses with resultant demyelination, axonal injury or both involving peripheral nerves and nerve roots in susceptible individuals. Although GBS variants share some key clinical features, they have distinctive pathophysiologic characteristics with overlap that could guide immunotherapeutic strategies. Thus, it is important to understand GBS pathogenesis to decipher potential efficacious treatment targets with minimal systemic adverse effects.
Acute Inflammatory Demyelinating Polyradiculoneuropathy (AIDP)
AIDP, the most common GBS variant, is characterized by demyelination detected by electrophysiologic studies and histopathological assessment of peripheral nerves. Myelin is formed by concentric rings of Schwann cell cytoplasm that surrounds a segment of peripheral axons. The immunological cascade that may trigger and induce demyelination in peripheral nerves and nerve roots in AIDP patients is complex and incompletely understood. There is a wider range of pathogenic mechanisms postulated in AIDP compared to AMAN. In addition, no definitive antigen targets, including peripheral nerve myelin proteins or peptide derivatives, have been identified in patient's sera or cerebrospinal fluid despite extensive studies, including insights derived from a representative animal model, experimental autoimmune neuritis (EAN). The classic histopathological profile of AIDP is diffuse multifocal mononuclear leukocyte infiltration with associated demyelination involving peripheral nerves and nerve roots (Figure 1). The distribution of inflammation corresponds to the degree clinical involvement.17 Subsequent observational studies have implicated both cell- and antibody-mediated effector mechanisms in AIDP pathogenesis.
Figure 1.
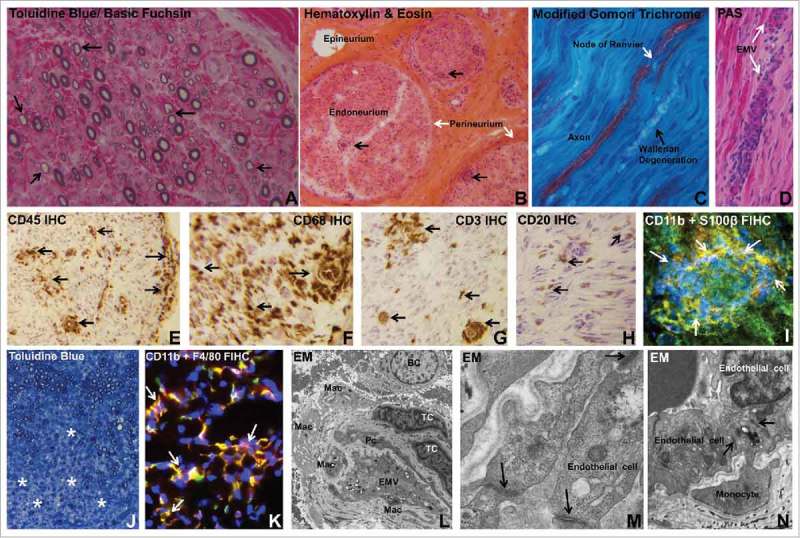
Histopathologic features of Guillain-Barré syndrome. Digital photomicrographs depict essential histopathological features of AIDP, the most common GBS variant in Western countries. Semi-thin plastic embedded axial section of the sural nerve biopsy of an AIDP patient shows thinly myelinated large diameter axons (black arrows), indicative of demyelination (A). Frozen thick axial section of the sural nerve biopsy shows mononuclear cells within the endoneurium with some perivascular foci (black arrows) in an AIDP patient (B). Longitudinal frozen thick section from the same AIDP patient's sural nerve biopsy shows reduction in large diameter axon density with evidence of Wallerian degeneration (black arrow). A node of Ranvier (white arrow) in an intact axon is shown (C). Leukocyte trafficking from endoneurial microvessels (EMV, white arrows) is another characteristic pathological finding in AIDP, as shown in the frozen longitudinal section of the sural nerve biopsy of an untreated AIDP patient stained with Periodic Acid Schiff (PAS) with high affinity for blood vessel walls that are rich in glycogen (D). Multifocal leukocyte infiltration (CD45+, black arrows) is commonly seen in AIDP patient nerve biopsies, as shown by immunohistochemistry (IHC) of a frozen thick axial section of a sural nerve biopsy (E). Macrophages (CD68+, black arrows) are the most prevalent endoneurial leukocyte subpopulation in AIDP (F), followed by CD3+ T-cells (black arrows, G) and CD20+ B-cells (black arrows, H). CD11b+ mononuclear leukocytes (white arrows) infiltrating into peripheral nerve endoneurium associated with demyelination of S100β Schwann cells is shown in a frozen thick section of an untreated AIDP patient sural nerve biopsy using indirect fluorescent immunohistochemistry (FIHC; I). An animal model of GBS, severe murine experimental autoimmune neuritis, recapitulates essential pathological features of AIDP, with diffuse areas of demyelination associated with mononuclear leukocyte infiltration (*) seen in the sciatic nerve endoneurium on a semi-thin, plastic-embedded axial section of an affected mouse at peak severity (J). As seen in AIDP, the most prevalent inflammatory leukocyte subpopulation observed in this murine GBS model is the monocyte/macrophage (F4/80+) and these cells are predominantly CD11b+ (white arrows, K) as shown in a frozen axial FIHC section. Electron microscopy (EM) demonstrates macrophages (Mac), T-cells (TC) and B-cells (BC) in the endoneurium of an AIDP patient sural nerve biopsy close to an endoneurial microvessel (EMV) that forms the blood-nerve barrier, sharing its basement membrane with a pericyte (Pc), as shown in L. In this region of leukocyte infiltration, higher magnification demonstrates intact electron dense interendothelial cell tight junctions (black arrows, M). In another region showing active monocyte transmigration, intact tight junctions persist between endoneurial endothelial cells and the migrating monocyte during paracellular diapedesis (black arrows, N). Unless indicated, the stains or histological technique performed on each section are indicated at the upper left corner of the photomicrograph.
Monocytes/ macrophages are the most commonly detected leukocyte subpopulation in AIDP and EAN peripheral nerves, with ultrastructural examination demonstrating invasion of myelin sheaths of structurally intact axons, hypothesized to be a response to specific antibody-antigen complexes on the myelin surface, on Schwann cells (the supportive glial cells of the peripheral nervous system) or at the node of Ranvier between myelin segments.18,54–56 Activated monocytes and tissue-resident macrophages may further enhance the pathogenic immune response, worsen local inflammation and induce axonal injury via cytokine and other pro-inflammatory molecule release. T-cells are the next most prevalent leukocyte subpopulation in peripheral nerve biopsies in AIDP and EAN, with CD4+ T-helper cells (polarized to Th1 and Th17 cytokine production) implicated in pathogenesis via endogenous chemokine and other pro-inflammatory cytokine production in peripheral nerves and nerve roots that enhance inflammation and activate macrophages. Although, definitive evidence of antigen-specific T-cell proliferation and cytokine secretion to myelin proteins/peptides in vitro is lacking,57 there is more recent evidence demonstrating increased pro-inflammatory cytokine interferon-γ expression by T-cells in response to stimulation with ganglioside GM1 in vitro.58 It is important to recognize that GM1 antibodies are not elevated in AIDP patients. In addition, alterations in the number, but not the function, of circulating CD4+ CD25+ FoxP3+ regulatory T cells have been shown during the acute phase of GBS.59,60
B-cells are also detected within peripheral nerve endoneurium in AIDP and EAN,61,62 implying endogenous secretion of pathogenic polyclonal antibodies or other pro-inflammatory molecules that may directly contribute to demyelination. Most commentaries suggest circulating pathogenic autoantibodies generated by activated B-cells gain access into peripheral nerves and nerve roots and cause demyelination or Schwann cell injury by antibody-dependent cellular cytotoxicity mediated by macrophages or complement fixation and activation resulting in membrane attack complex (C5b-9).63,64 In support of this, membrane bound complement components (e.g. complement activation marker, C3d and C5b-9) and immunoglobulin deposition have been described in peripheral nerve biopsies of AIDP patients.65-67 Since demyelinaton occurs in a multifocal pattern throughout the length of multiple peripheral nerves and nerve roots (based on electrophysiological and histopathological observations, see Figure 1), the assumption is that these pathogenic autoantibodies readily cross the blood-nerve barrier in affected AIDP patients.
Despite the known efficacy of antibody-modulating therapies, circulating antibodies against myelin glycoproteins, such as myelin protein zero (the most prevalent peripheral nerve myelin protein) and peripheral nerve myelin 22 or the basic peripheral nerve protein P2 are absent or uncommonly detected in AIDP.68-70 Taking into account the variety of different infections associated with GBS and lack of definite evidence of specific autoantibodies against myelin components, a recent AIDP study hypothesized that primary peripheral nerve/ nerve root damage occurs as a consequence of innate immunity-associated local inflammation following neurotropic virus egress, with polyclonal autoantibody production occurring as optional complementary secondary process.71 The reader is also referred a recent comprehensive review article summarizing current knowledge on AIDP pathogenesis.61
Axonal variants of GBS (AMAN and AMSAN)
Guided by histopathological observations, the axonal variants of GBS, particularly AMAN, have a more defined pathogenesis based on demonstrated antibody- and complement-mediated destruction of the membrane surface of axons (known as axolemma) in autopsy cases and evidence of molecular mimicry between microbial epitopes and axolemmal molecules, particularly gangliosides or ganglioside complexes which are abundant in peripheral nerves. Gangliosides consist of signature sugar residues bearing one or more sialic acid molecules.25,63,72–75 Preceding C. jejuni infection commonly occurs in AMAN. Bacterial lipo-oligosaccharides are structurally similar to carbohydrate moieties of sugar residues of gangliosides, including GM1, GD1a, GQ1b and GalNAc-GD1a that are present on motor axolemma.75-79 As part the humoral immune response to the bacterial infection, antibodies are systemically generated that may cross-react with specific gangliosides or ganglioside complexes in peripheral nerves. The pathogenic role of anti-ganglioside antibodies in axonal GBS variants is supported by correlative human studies demonstrating a relationship between specific antibodies and clinical phenotype and outcomes, as well as animal studies using passive transfer or gene knockout.14,23,25,26,28,34,38,47,76–78,80–82
Gangliosides located at or near the node of Ranvier (the part of a myelinated axon that lacks myelin and is rich in Na+ channels needed for rapid impulse conduction, see Figure 1) are preferentially targeted by cross-reactive autoantibodies in axonal variants of GBS.21,25,72–74 Complement-fixing immunoglobulins, such as IgG1 and IgG3, bind to specific gangliosides, generate C5b-9 and cause axonal injury with subsequent degeneration or alteration of Na+ channel function, causing transient conduction block, which may explain the rapid recovery of some AMAN cases following therapy.25,83,84 Furthermore, endogenous macrophages may invade nodes of Ranvier or internodal periaxonal space of myelinated axons, and scavenge axons beneath its Schwann cell membrane, with resulting axonal degeneration.21,34,72,73 The extent of macrophage-mediated axonal degeneration could predict the clinical outcomes, with more widespread axonal degeneration associated with poor recovery.19,21,83,85 Poorer outcomes are more commonly seen in AMSAN compared to AMAN.21,25,38,83 It is not fully understood why circulating anti-ganglioside antibodies generated as a consequence of bacterial infections only cause axonal GBS in a small percentage of individuals, and do not cause tissue injury in the brain, particularly at the circumventricular organs that lack the restrictive tight junction-forming blood-brain barrier, or other tissues such as the kidney that are also rich in gangliosides and lack restrictive endothelial microvessels.86
Miller Fisher syndrome (MFS)
MFS patients frequently have circulating antibodies against GD1b, GD3, GT1a and GQ1b gangliosides as a consequence of molecular mimicry of bacterial lipo-oligosaccharides.37,39 Oculomotor and bulbar nerves, as well as extraocular muscle motor end plate (terminal axon) have been shown to have high ganglioside densities, particularly GQ1b and GT1a.87,88 Over 95% of MFS patients have anti-GQ1b IgG antibodies (IgG/ IgM/ IgA anti-GQ1b antibodies),30,89,90implying a potential role in disease pathogenesis or reliable diagnostic biomarker. Anti-GQ1b antibodies have been shown to activate complement at neuromuscular junctions in vitro and in mice ex vivo.91 As a consequence, complement activation at the presynaptic nerve terminal and perisynaptic Schwann cells has been proposed as the primary pathogenic mechanism in MFS, with generally good patient outcomes without residual deficits, even without immune modulatory therapy.30,75,90,92,93
Immunotherapies
GBS is an acute immune-mediated disorder of peripheral nerves and nerve roots, thus, immune-modulating therapies are generally administered to improve outcomes and prevent disability. Several randomized controlled trials (RCTs) have established that Plasma Exchange (PE) and intravenous immunoglobulin (IVIg) are effective GBS immunotherapies. Early utilization of PE or IVIg, prior to irreversible axonal damage, is equally effective to the other in improving neurological outcomes.94-98 However, subsets of patients respond slowly, partially respond or worsen on either therapy. Other immunotherapies, such as corticosteroids, have been shown to lack efficacy based on clinical trials and meta-analyses. Several immune-modulatory therapies had been proposed based on in vitro studies, in situ observations or animal model data that have failed to translate99 or have been tried in smaller cohorts or individual GBS cases without conclusive efficacy. There is a significant need to better understand GBS pathogenesis, ascertain pathways that are realistically amenable to pharmacologic blockade and develop targeted molecular therapies for different GBS variants or subsets of patients refractory to either PE or IVIg, or predicted to have poor outcomes based on clinical and electrophysiological criteria.
Plasma exchange (PE)
In 1959, a patient with thrombotic thrombocytopenic purpura recovered after treatment with fresh frozen plasma exchange, implying that PE may be beneficial for autoimmune disorders.100 In 1978, PE was first used to treat a patient with acute polyneuropathy who rapidly recovered, advocating potential efficacy in GBS.101 In a RCT with 245 GBS patients, PE was established as an effective treatment95 with further confirmation of efficacy by a subsequent larger clinical trial.102 PE became the first validated therapy for GBS and was considered as “gold standard” due to its status as an evidence-based efficacious immunotherapy.
The Quality Standards Subcommittee of the American Academy of Neurology (AAN) subsequently provided evidence-based guidelines for physician practice as follows: PE is beneficial for non-ambulatory GBS patients within 4 weeks of symptoms onset (Level A, Class II evidence) and for ambulatory patients, PE is recommended within 2 weeks of onset (Level B, limited Class II evidence).103,104 The therapeutic response may be better within 2 weeks of disease onset, especially in non-ambulatory patients. The treatment typically consists of five exchanges, one plasma volume each time (about 50 mL/kg body weight), administered over 1–2 weeks.103,105 For mild GBS patients, two exchanges resulted in quicker recovery compared to untreated patients. For moderate or severe GBS patients, at least four exchanges are needed to improve outcomes.106 PE has been shown to reduce the likelihood and duration of mechanical ventilation, reduce the time required to walk with assistance and increase the likelihood of fully recovering muscle strength after one year.97,106,107 Continuous flow plasma exchange machines may be superior to intermittent flow machines and albumin is deemed superior to fresh frozen plasma as the exchange fluid.102,107
The mechanism of action of PE is not clearly known; however, it may nonspecifically remove circulating autoantibodies, immune complexes, complement factors, cytokines and other pro-inflammatory humoral mediators that contribute to GBS immunopathogenesis.15,107 Based on clinical observations, PE may reduce the extent of demyelination or axonal injury with hastening of clinical recovery compared with supportive care alone.103 PE is associated with significant adverse effects which include hemodynamic instability, dilutional coagulopathy, hypocalcemia, septicemia, thrombosis, pneumonia, complications from central venous access and allergic reactions.108 Citrate infused as part of the exchange fluid may induce metabolic acidosis or hypocalcemia. Hemostatic disorders, unstable cardiovascular status, active infection, and pregnancy are considered relative contraindications to PE.102 Lack of widespread access to plasma exchange (typically restricted to tertiary care hospitals due to need for specialized equipment and clinical expertise), need for close monitoring and its potentially serious adverse effects have limited the general use of PE for GBS. In addition, prolonged hospitalization and its associated increased direct and indirect medical costs,109,110 have resulted in PE being a relative restricted immunotherapy for GBS.
Intravenous immunoglobulin (IVIg)
Intravenous immunoglobulin (IVIg), derived from pooled purified immunoglobulins from thousands of blood donors, is the other proven immunotherapy for GBS. In 1988, a study reported the use of IVIg in eight severe GBS patients, with beneficial effect in some94 at a time when PE was considered standard of care.103 The first RCT of IVIg in GBS confirmed efficacy similar to PE in 1992.96 The Quality Standards Subcommittee of the AAN recommended IVIg in hastening recovery for GBS patients who require aid to walk within 2 weeks (Level A, Class I evidence), or 4 weeks (Level B recommendation) of symptom onset.103 IVIg therapy has been shown to hasten recovery in children compared with supportive therapy alone based on limited evidence from three open labeled trials.111 The typical IVIg dosage for GBS is 0.4 g/kg body weight daily for five consecutive days, with a total of 2 g/kg. Administering the total dose over 2 days is equally efficacious when compared to 5 days.112 A study evaluating IVIg pharmacokinetics in GBS patients demonstrated significantly better outcomes, such as likelihood of independent ambulation after 6 months, in patients with higher increases in serum IgG levels after IVIg administration, especially at two weeks after treatment. The study authors advocated administration of a higher dose or a second course of IVIg in patients with low serum IgG increases.113
IVIg is hypothesized to modulate the immune system in GBS in several ways; however, the definitive mechanism(s) has/have not been established. Possibilities include: restraint of autoantibody production and autoantibody neutralization via anti-idiotypic antibodies114; inhibition of complement activation and membrane attack complex formation115; modulating the expression and function of Fc receptors on macrophages and other effector cells, suppression of cytokine, chemokine and adhesion molecules, modulation of T-cell functions and interference with pathogenic antigen recognition.116 The net effect, as postulated based on clinical observations, is a reduction in demyelination and axonal injury with resultant hastening of clinical recovery and better outcomes. It is important to recognize that IVIg is not a single drug, and the constituent immunoglobulins may vary depending on manufacturer and donor source.
Broadly speaking, adverse events associated with IVIg administration are usually minor and rare, occurring in less than 10% of GBS patients. The most significant adverse reactions of IVIg reported in GBS clinical trials include myocardial infarction, renal failure, vomiting and headache due to meningeal irritation (known as meningismus).96,98,103 Elevated serum viscosity, high triglycerides, or hypergammaglobulinemia can be considered as relative contraindications to IVIg, due to increasing risk of thromboembolic events.116,117 IVIg is not contraindicated in pregnancy. IVIg should be used with caution in GBS patients with coronary artery disease, congestive heart failure, recent deep vein thrombosis, preexisting kidney disease, and should be avoided in patients with selective IgA deficiency due to the risk of anaphylaxis. Slowing the rate of infusion, administering intravenous fluids following transfusion, using low osmolality brands, and screening for IgA deficiency should aid reduce the risk of adverse events.116,117
Limitations of PE and IVIg in GBS
RCTs have shown that PE and IVIg are equal effective in treating GBS.112 However, despite similar efficacy, IVIg is more widely used for GBS, due to its higher availability, non-requirement of specialized equipment to administer and its relatively reduced risk for adverse effects. However, the decision to perform PE or administer IVIg may depend on the patient's clinical circumstance (with respect to relative contraindications to either immunotherapy) and local factors such as cost and availability of specialized staff and equipment for PE. Despite known efficacy in GBS, patients treated with either PE or IVIg may not respond, show partial response, fluctuate or worsen clinically during treatment. Although it may seem logical to administer IVIg after performing PE to maximize the immune modulatory effect of IVIg following removal of pathogenic humoral factors, there is no evidence that combination therapy or repeated therapy with the same immunotherapy is more effective or associated with better outcomes in the short- or long-term compared to that standard recommended therapy in GBS patients.14,106,112,118
Both PE and IVIg are nonspecific and could also modulate anti-inflammatory molecules or signaling mechanisms that are protective or reparative in GBS patients in addition to the pro-inflammatory cascade. Taking into account slow treatment effects with common residual deficits in patients with motor function recovery, partial efficacy, treatment-related fluctuations, treatment failures and the potential complications of current therapies, there is a need to develop more effective, disease-specific immunotherapeutic agents for GBS.
Corticosteroids
Due to the fact that GBS is an immune-mediated polyradiculoneuropathy, corticosteroids could intuitively treat this group of disorders. In 1952, corticosteroids were first successfully reported in treating a GBS patient, and were anecdotally used as a treatment option for many years. However, subsequent studies performed the 1970s indicated that corticosteroids were ineffective in GBS.119 A Cochrane systematic review of six clinical trials with 587 GBS patients treated with different forms and dosage of corticosteroids showed no significant difference in mortality or disability grade compared with patients taking placebo. Steroids are not recommended for the treatment of GBS patients based on current evidence (Level A, Class I evidence).103,120 In four additional trials with a total of 120 patients, patients taking oral corticosteroids had less clinical improvement after four weeks compared to patients not receiving corticosteroids, suggesting that oral corticosteroids may delay long-term recovery in GBS patients.120
While intravenous methylprednisolone alone does not produce significant benefit or harm in GBS patients, the combination of IVIg and intravenous methylprednisolone demonstrated no significant effect on long-term outcomes compared with IVIg alone.111,121 Despite the general efficacy of corticosteroids in immune-mediated disorders and in the EAN animal model of GBS, it has been clearly established to be non-efficacious in GBS, including the AIDP variant characterized by inflammatory cell infiltration into peripheral nerves and nerve roots. It is hypothesized that anti-reparative effects of corticosteroids following axonal injury may counteract its initial anti-inflammatory effects in GBS, resulting in no appreciable benefit compared to placebo.120
Other Immunotherapies for GBS
A recent Cochrane systematic review for other pharmacological agents for GBS apart from IVIg, PE and corticosteroids122demonstrated very low quality evidence for four different treatments published in four studies as potential immunotherapies for GBS. These include CSF filtration in 37 patients compared to PE,123 interferon β-1a (a cellular immunomodulatory drug effective in multiple sclerosis) compared to placebo in 19 patients,124-126 brain-derived neurotrophic factor (a growth factor known to protect against degeneration or induce regeneration in motor axons) in 10 patients compared to placebo127 and Chinese herbal medicine, Tripterygium Wilfordii polyglycoside (an extract of Thunder God Vine with presumed anti-inflammatory, anti-proliferative and immunosuppressive properties) compared to intravenous corticosteroids in 43 patients.128 It was only the latter therapy that showed beneficial outcomes with improvement in disability grade 8 weeks after treatment associated with reduced serum interleukin-6 levels128; however, it is important to note that some GBS patients in that cohort also responded to corticosteroid therapy, which in general terms, is not efficacious in GBS. Further studies may be required to elucidate the biologically active ingredients in Tripterygium Wilfordii polyglycoside and potential mechanism(s) of action in immune-mediated peripheral nerve disorders.
Future proposed immunotherapies
With advancements in the development of efficacious molecular-based targeted biologic therapies for immune-mediated disorders and cancers over the last 20–30 years, there is a drive to identify drugs that safely treat GBS with superior outcomes to PE and IVIg, especially in patients with suboptimal responses to these immunotherapies. Eculizumab, a humanized monoclonal antibody that specifically binds to complement component 5 (C5) and potently inhibits generation of pro-inflammatory C5a and C5b-9 (implicated in axonal injury in GBS) is currently undergoing clinical trials in the United Kingdom and Japan to determine efficacy in GBS guided by prior animal model studies that demonstrated beneficial effects.129,130 Other biologic agents are being proposed as potential future therapies based on animal model data or small case reports.131 Due to the proposed pathogenic differences between demyelinating and axonal variants of GBS, future immunotherapy RCTs may require randomization based on electrophysiologic characterization or perform subgroup analyses dependent on relative numbers of these variants in addition to other demographic factors and measures of disease severity. Table 1 summarizes current GBS immunotherapies, including known drugs in clinical development, their molecular targets/ mechanisms of action and the relative strength and weaknesses of therapeutic approach.
Table 1.
Summary of current GBS immunotherapies, including known drugs in clinical development. There is a major dearth in therapeutic options despite significant advances in molecular pathogenesis based on observational human and animal model data.
| Treatment | Status | Molecular Target | Mechanism(s) of Action | Relative strengths/ weaknesses | References |
|---|---|---|---|---|---|
| Plasma Exchange | Marketed | Pathogenic autoantibodies | ? Removal of pathogenic autoantibodies, immune complexes, complement factors, cytokines and other pro-inflammatory humoral mediators. |
Strength: Rapid effect. Weakness: Specialized equipment required, high costs, risk of hemodynamic instability, dilutional coagulopathy, hypocalcemia, thrombosis high. |
95, 102 |
| IVIg | Marketed | Pathogenic autoantibodies | ? Modulate pathogenic autoantibody production, neutralize autoantibodies, inhibition of complement activation and membrane attack complex formation (C5b-9), modulating Fc receptors on macrophages and other effector cells, cytokine, chemokine and adhesion molecule suppression, modulation of T-cell functions, interference with pathogenic antigen recognition. |
Strength: Widespread availability. Specialized equipment not required. Safe in pregnancy. Side effects uncommon. Weakness: Variable efficacy (dependent on brand/ batch), Risk of thrombotic events (e.g. myocardial infarction, deep venous thrombosis), renal failure and anaphylaxis with IgA deficiency, meningismus. |
96 |
| Eculizumab | Phase II Clinical trials | Human complement molecule C5 | inhibition of complement activation and membrane attack complex formation (C5b-9). | Unknown (clinical trials ongoing). | 129, 130 |
Novel perspective: Leukocyte Trafficking at the Blood-Nerve barrier
Although GBS has been recognized as an autoimmune disorder associated with a complex interplay between the systemic immune system and peripheral nerves/nerve roots based on observational data from affected patient nerve biopsies (see Figure 1), cerebrospinal fluid and blood, as well as its representative animal model, experimental autoimmune neuritis (EAN), development of disease-specific molecular therapies is lacking, with a significant failure of translational between animal models and the human disease. Molecules or pathways for therapeutic modulation that have been suggested include T lymphocyte activation with polarization towards CD4+ T helper 1 (Th1) and T helper 17 (Th17) phenotypes, with reduction on T helper 2 (Th2) and possible alterations in CD25+ FoxP3+ regulatory T lymphocytes, polyclonal B-cell maturation and immunoglobulin synthesis within primary and secondary lymphoid organs, complement-mediated lysis, monocyte/macrophage and lymphocyte-mediated demyelination via cytokines, as well as complement- and antibody-dependent cellular cytotoxicity, Schwann cell potentiation of the local innate and adaptive immune response as well as in terminating inflammation by inducing T lymphocyte apoptosis.14,38,55,61,63,64,99,132
Apart from deciphering therapeutic targets, it is imperative to realistically consider the stage in the disease process that patients typically present to the clinician for evaluation and treatment, the specificity of the immune modulatory drug target so as not to disrupt normal physiological or immunological processes in other tissues, whether the drug would modulate GBS pathogenic mechanisms in the systemic circulation or within peripheral nerve/ nerve root endoneurium and if the drug has significant permeability across the restrictive blood-nerve barrier with high endoneurial retention during inflammation. Although it is commonly implied that the blood-nerve barrier is “leaky” in GBS, our recent study that looked at endoneurial microvessel ultrastructure by electron microscopy in untreated AIDP patients demonstrated intact electron dense intercellular tight junctions in regions with infiltrating/ infiltrated leukocytes,133 as shown in Figure 1.
Pathogenic hematogenous leukocyte trafficking has been observed in the most common GBS variant, AIDP and its animal model, EAN, with monocytes being the most common infiltrating leukocyte, followed by T-cells and B-cells,61,62 as shown in Figure 1. We propose pathogenic leukocyte trafficking from the systemic circulation into peripheral nerves/nerve roots across the blood-nerve barrier as a novel plausible site for therapeutic modulation for several reasons: Patients are typically evaluated weeks after disease onset and activation of systemic immune system, and there is an association between hematogenous leukocyte trafficking and disease stage and severity (as seen in EAN)62 and efficacious therapies would work systemically to prevent leukocyte trafficking across the blood-nerve barrier, eliminating the need to consider blood-nerve barrier permeability and endoneurial retention (Figure 2).
Figure 2.
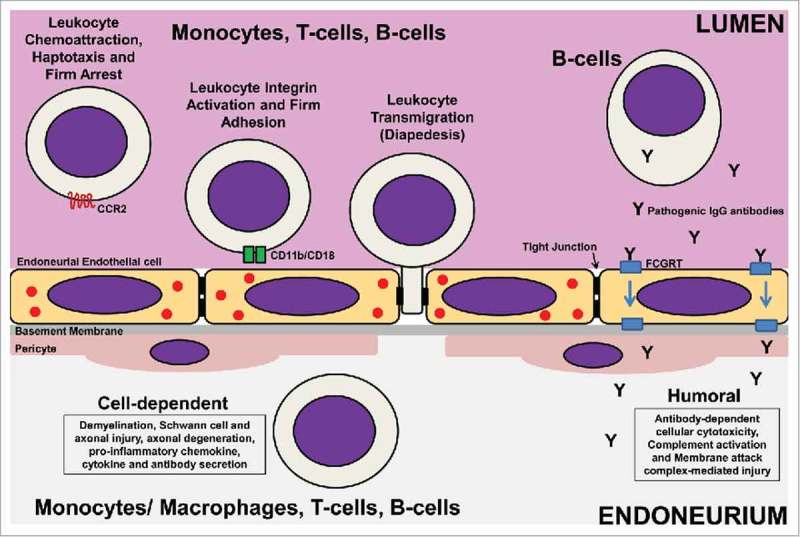
Potential Targets for GBS Immunotherapy Development. Pathogenic leukocyte trafficking across tight junction-forming endoneurial microvessels that form the blood-nerve barrier is pathogenically relevant to AIDP and other demyelinating GBS variants based on human in situ and in vitro data, as well as in vivo data from representative animal models. Taking into account the coordinated process of leukocyte trafficking (multi-step paradigm), leukocyte trafficking antagonists that block pathogenic leukocyte chemoattraction, haptotaxis and firm arrest on activated endoneurial endothelial cells (e.g. Chemokine receptor CCR2 antagonists), firm leukocyte adhesion (e.g. CD11b [αM-integrin] antagonists) or diapedesis (undetermined; with platelet-endothelial cell adhesion molecule-1, CD99, CD99L2 and junctional adhesion molecules-A, -B and –C being potential candidates for antagonism) could result in targeted molecular immunotherapies for GBS. Another unexplored possibility involves modulating pathogenic polyclonal IgG antibody transport from the blood circulation into peripheral nerves and nerve roots across the blood-nerve barrier by Fc gamma receptor and transporter (FCGRT) antagonists. These therapeutic approaches target GBS pathogenesis after systemic immune activation at the critical interphase between the immune system and peripheral nerves/ nerve roots at a time period when patients are symptomatic, with the goal being to limit demyelination and axonal injury/ degeneration. It is envisioned that drugs targeting pathogenic leukocyte trafficking or polyclonal IgG antibody transport can be administered systemically with therapeutic modulation occurring in circulation without need for significant drug blood-nerve barrier permeability to treat GBS. The challenge is elucidating biologically relevant molecules and signaling pathways preferentially activated in GBS at the blood-nerve barrier that are amenable to pharmacologic antagonism to limit potential adverse systemic effects associated with non-specific immune modulation or immunosuppression during the active phase of the disorder.
Leukocyte trafficking into tissues is a coordinated, sequential process (called the multi-step paradigm) which involves leukocyte rolling on activated endothelium mediated by selectins expressed on the endothelium and their counterligands (such as P-selectin glycoprotein ligand-1 and Sialyl Lewis x) expressed on leukocytes, leukocyte chemoattraction, haptotaxis and arrest mediated by specific chemokines expressed on the endothelium bound by glycosaminoglycans binding to chemokine receptors expressed on specific leukocyte subsets, leukocyte integrin activation resulting in firm adhesion to the endothelium via specific cell adhesion molecules such as intercellular adhesion molecule-1 and vascular cell adhesion molecule-1, cytoskeletal modification and migration (diapedesis) across the vascular endothelium via paracellular or transcellular mechanisms that require specific leukocyte-endothelial cell interactions and extravasation across the endothelial basement membrane via secretion of matrix metalloproteinases.134-136
Guided by human observational data,137 we studied a severe murine EAN model and demonstrated an important role for chemokine receptor CCR2 in AIDP pathogenesis by gene knockout and showed efficacy of pharmacologic blockade following disease onset with clinically discernible signs of weakness, associated with reduced peripheral nerve demyelination and inflammation and near complete recovery in motor function.138 Using a novel flow-dependent in vitro human blood-nerve model encompassing cytokine-stimulated primary human endoneurial endothelial cells, we observed the multi-step leukocyte trafficking paradigm via the paracellular route.133,139 We elucidated a crucial role of αM-integrin (also known as CD11b)-intercellular adhesion molecule-1 signaling in untreated GBS patient leukocyte trafficking across the blood-nerve barrier in vitro using function-neutralizing monoclonal antibodies and further validated the pathogenic relevance of this integrin in vivo via monoclonal antibody blockade following disease onset in the severe murine EAN model.133,139 Translational potential to the AIDP variant of GBS was supported by demonstrating clusters of CD11b+ mononuclear leukocytes within the sural nerve endoneurium of nerve biopsies from untreated patients,133 suggesting a pathogenic role and potentially disease-specific molecular therapy for GBS. Figure 2 shows potential targets for novel GBS immunotherapy development, emphasizing specificity of leukocyte trafficking antagonists.
In general, EAN models provide essential tools to study and modulate acute immune-mediated demyelination or axonal injury in a living system with clinical, electrophysiological and histopathological features similar to GBS. This is essential to the development of novel efficacious immunotherapies. However, to increase translational potential, it is imperative to choose molecular targets amenable to small molecular or biologic antagonists modulated after appreciable disease manifestation and design preclinical animal studies with the same degree of scientific rigor applied to clinical trials in GBS patients.
Conclusions
Current immunotherapy for GBS is limited to ameliorating the disease via non-specific systemic pathogenic antibody modulation by PE and IVIg. Despite partial efficacy, it is well established that cohorts of patients show limited or no response, necessitating repeat therapy with the other or the same modality, and in some patients that respond, outcomes are suboptimal with residual disability associated with chronic neuropathic pain and residual motor deficits. Despite significant advances in molecular biology over the last 30 years, numerous publications on GBS pathogenesis, and success in developing disease-specific immune modulatory therapies in rheumatoid arthritis, psoriasis, inflammatory bowel disease, multiple sclerosis and cancers, translational immunotherapies are lacking in GBS and other peripheral nervous system immune-mediated disorders. It is imperative to consider the role of the blood-nerve barrier in GBS pathogenesis and therapeutic development, and we advocate pathogenic leukocyte trafficking as a biologically relevant mechanistic target with translational potential for disease-specific immune modulatory therapy using function-neutralizing antagonists such as humanized monoclonal antibodies that do not require blood-nerve barrier permeability and retention within peripheral nerve/ nerve root endoneurium.
Disclosure of potential conflicts of interest
E.E.U. has a non-exclusive commercial license (held by Baylor Licensing Group) for simian virus-40 large T-antigen immortalized human endoneurial endothelial cells and has received royalties from Springer Science + Business Media for an edited book on laboratory protocols that describes the flow-dependent in vitro blood-nerve barrier leukocyte trafficking assay. S.L. and C.D. have nothing to disclose.
References
- 1.Guillain G, Barre JA, Strohl A. [Radiculoneuritis syndrome with hyperalbuminosis of cerebrospinal fluid without cellular reaction. Notes on clinical features and graphs of tendon reflexes. 1916]. Ann Med Interne (Paris). 1999;150:24–32. [PubMed] [Google Scholar]
- 2.A prospective study on the incidence and prognosis of Guillain-Barre syndrome in Emilia-Romagna region, Italy (1992-1993) Emilia-Romagna Study Group on Clinical and Epidemiological Problems in Neurology. Neurology. 1997; 48:214–21. [PubMed] [Google Scholar]
- 3.Hughes RA, Rees JH. Clinical and epidemiologic features of Guillain-Barre syndrome. J Infect Dis. 1997;176(Suppl 2):S92–8. doi: 10.1086/513793. [DOI] [PubMed] [Google Scholar]
- 4.Alshekhlee A, Hussain Z, Sultan B, Katirji B. Guillain-Barre syndrome: incidence and mortality rates in US hospitals. Neurology. 2008;70:1608–13. doi: 10.1212/01.wnl.0000310983.38724.d4. [DOI] [PubMed] [Google Scholar]
- 5.Sejvar JJ, Baughman AL, Wise M, Morgan OW. Population incidence of Guillain-Barre syndrome: a systematic review and meta-analysis. Neuroepidemiology. 2011;36:123–33. doi: 10.1159/000324710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Van Koningsveld R, Van Doorn PA, Schmitz PI, Ang CW, Van der Meche FG. Mild forms of Guillain-Barre syndrome in an epidemiologic survey in The Netherlands. Neurology. 2000;54:620–5. doi: 10.1212/WNL.54.3.620. [DOI] [PubMed] [Google Scholar]
- 7.Chio A, Cocito D, Leone M, Giordana MT, Mora G, Mutani R, et al. Guillain-Barre syndrome: a prospective, population-based incidence and outcome survey. Neurology. 2003;60:1146–50. doi: 10.1212/01.WNL.0000055091.96905.D0. [DOI] [PubMed] [Google Scholar]
- 8.Bogliun G, Beghi E, Italian GBSRSG. Incidence and clinical features of acute inflammatory polyradiculoneuropathy in Lombardy, Italy, 1996. Acta Neurol Scand. 2004;110:100–6. doi: 10.1111/j.1600-0404.2004.00272.x. [DOI] [PubMed] [Google Scholar]
- 9.Van der Meche FG, Van Doorn PA, Meulstee J, Jennekens FG, Centre GB-cgotDNRS. Diagnostic and classification criteria for the Guillain-Barre syndrome. Eur Neurol. 2001;45:133–9. doi: 10.1159/000052111. [DOI] [PubMed] [Google Scholar]
- 10.Orlikowski D, Prigent H, Sharshar T, Lofaso F, Raphael JC. Respiratory dysfunction in Guillain-Barre Syndrome. Neurocrit Care. 2004;1:415–22. doi: 10.1385/NCC:1:4:415. [DOI] [PubMed] [Google Scholar]
- 11.Ropper AH, Wijdicks EFM, Truax BT. Guillain-Barré syndrome. Philadelphia: F.A. Davis, 1991. [Google Scholar]
- 12.Ruts L, Drenthen J, Jongen JL, Hop WC, Visser GH, Jacobs BC, et al. Pain in Guillain-Barre syndrome: a long-term follow-up study. Neurology. 2010;75:1439–47. doi: 10.1212/WNL.0b013e3181f88345. [DOI] [PubMed] [Google Scholar]
- 13.Lyu RK, Tang LM, Cheng SY, Hsu WC, Chen ST. Guillain-Barre syndrome in Taiwan: a clinical study of 167 patients. J Neurol Neurosurg Psychiatry. 1997;63:494–500. doi: 10.1136/jnnp.63.4.494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.van den Berg B, Walgaard C, Drenthen J, Fokke C, Jacobs BC, van Doorn PA. Guillain-Barre syndrome: pathogenesis, diagnosis, treatment and prognosis. Nat Rev Neurol. 2014;10:469–82. doi: 10.1038/nrneurol.2014.121. [DOI] [PubMed] [Google Scholar]
- 15.Walgaard C, Jacobs BC, van Doorn PA. Emerging drugs for Guillain-Barre syndrome. Expert Opin Emerg Drugs. 2011;16:105–20. doi: 10.1517/14728214.2011.531699. [DOI] [PubMed] [Google Scholar]
- 16.Frenzen PD. Economic cost of Guillain-Barre syndrome in the United States. Neurology. 2008;71:21–7. doi: 10.1212/01.wnl.0000316393.54258.d1. [DOI] [PubMed] [Google Scholar]
- 17.Asbury AK, Arnason BG, Adams RD. The inflammatory lesion in idiopathic polyneuritis. Its role in pathogenesis. Medicine (Baltimore). 1969;48:173–215. doi: 10.1097/00005792-196905000-00001. [DOI] [PubMed] [Google Scholar]
- 18.Prineas JW. Acute idiopathic polyneuritis. An electron microscope study. Lab Invest. 1972;26:133–47. [PubMed] [Google Scholar]
- 19.Feasby TE, Gilbert JJ, Brown WF, Bolton CF, Hahn AF, Koopman WF, et al. An acute axonal form of Guillain-Barre polyneuropathy. Brain. 1986;109(Pt 6):1115–26. doi: 10.1093/brain/109.6.1115. [DOI] [PubMed] [Google Scholar]
- 20.McKhann GM, Cornblath DR, Griffin JW, Ho TW, Li CY, Jiang Z, et al. Acute motor axonal neuropathy: a frequent cause of acute flaccid paralysis in China. Ann Neurol. 1993;33:333–42. doi: 10.1002/ana.410330402. [DOI] [PubMed] [Google Scholar]
- 21.Griffin JW, Li CY, Ho TW, Tian M, Gao CY, Xue P, et al. Pathology of the motor-sensory axonal Guillain-Barre syndrome. Ann Neurol. 1996;39:17–28. doi: 10.1002/ana.410390105. [DOI] [PubMed] [Google Scholar]
- 22.Hadden RD, Cornblath DR, Hughes RA, Zielasek J, Hartung HP, Toyka KV, et al. Electrophysiological classification of Guillain-Barre syndrome: clinical associations and outcome. Plasma Exchange/Sandoglobulin Guillain-Barre Syndrome Trial Group. Ann Neurol. 1998;44:780–8. doi: 10.1002/ana.410440512. [DOI] [PubMed] [Google Scholar]
- 23.Sekiguchi Y, Uncini A, Yuki N, Misawa S, Notturno F, Nasu S, et al. Antiganglioside antibodies are associated with axonal Guillain-Barre syndrome: a Japanese-Italian collaborative study. J Neurol Neurosurg Psychiatry. 2012;83:23–8. doi: 10.1136/jnnp-2011-300309. [DOI] [PubMed] [Google Scholar]
- 24.Ho TW, Mishu B, Li CY, Gao CY, Cornblath DR, Griffin JW, et al. Guillain-Barre syndrome in northern China. Relationship to Campylobacter jejuni infection and anti-glycolipid antibodies. Brain. 1995;118(Pt 3):597–605. [DOI] [PubMed] [Google Scholar]
- 25.Kuwabara S, Yuki N. Axonal Guillain-Barre syndrome: concepts and controversies. Lancet Neurol. 2013;12:1180–8. doi: 10.1016/S1474-4422(13)70215-1. [DOI] [PubMed] [Google Scholar]
- 26.Kim JK, Bae JS, Kim DS, Kusunoki S, Kim JE, Kim JS, et al. Prevalence of anti-ganglioside antibodies and their clinical correlates with guillain-barre syndrome in Korea: a nationwide multicenter study. J Clin Neurol. 2014;10:94–100. doi: 10.3988/jcn.2014.10.2.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hiraga A, Mori M, Ogawara K, Hattori T, Kuwabara S. Differences in patterns of progression in demyelinating and axonal Guillain-Barre syndromes. Neurology. 2003;61:471–4. doi: 10.1212/01.WNL.0000081231.08914.A1. [DOI] [PubMed] [Google Scholar]
- 28.Islam Z, Jacobs BC, van Belkum A, Mohammad QD, Islam MB, Herbrink P, et al. Axonal variant of Guillain-Barre syndrome associated with Campylobacter infection in Bangladesh. Neurology. 2010;74:581–7. doi: 10.1212/WNL.0b013e3181cff735. [DOI] [PubMed] [Google Scholar]
- 29.Fisher M. An unusual variant of acute idiopathic polyneuritis (syndrome of ophthalmoplegia, ataxia and areflexia). N Engl J Med. 1956;255:57–65. doi: 10.1056/NEJM195607122550201. [DOI] [PubMed] [Google Scholar]
- 30.Mori M, Kuwabara S, Fukutake T, Yuki N, Hattori T. Clinical features and prognosis of Miller Fisher syndrome. Neurology. 2001;56:1104–6. doi: 10.1212/WNL.56.8.1104. [DOI] [PubMed] [Google Scholar]
- 31.Ropper AH. Unusual clinical variants and signs in Guillain-Barre syndrome. Arch Neurol. 1986;43:1150–2. doi: 10.1001/archneur.1986.00520110044012. [DOI] [PubMed] [Google Scholar]
- 32.Ropper AH. Further regional variants of acute immune polyneuropathy. Bifacial weakness or sixth nerve paresis with paresthesias, lumbar polyradiculopathy, and ataxia with pharyngeal-cervical-brachial weakness. Arch Neurol. 1994;51:671–5. doi: 10.1001/archneur.1994.00540190051014. [DOI] [PubMed] [Google Scholar]
- 33.Wakerley BR, Yuki N. Pharyngeal-cervical-brachial variant of Guillain-Barre syndrome. J Neurol Neurosurg Psychiatry. 2014;85:339–44. doi: 10.1136/jnnp-2013-305397. [DOI] [PubMed] [Google Scholar]
- 34.Hughes RA, Cornblath DR. Guillain-Barre syndrome. Lancet. 2005;366:1653–66. doi: 10.1016/S0140-6736(05)67665-9. [DOI] [PubMed] [Google Scholar]
- 35.Fokke C, van den Berg B, Drenthen J, Walgaard C, van Doorn PA, Jacobs BC. Diagnosis of Guillain-Barre syndrome and validation of Brighton criteria. Brain. 2014;137:33–43. doi: 10.1093/brain/awt285. [DOI] [PubMed] [Google Scholar]
- 36.Wakerley BR, Uncini A, Yuki N, Group GBSC, Group GBSC. Guillain-Barre and Miller Fisher syndromes–new diagnostic classification. Nat Rev Neurol. 2014;10:537–44. doi: 10.1038/nrneurol.2014.138. [DOI] [PubMed] [Google Scholar]
- 37.Yuki N. Infectious origins of, and molecular mimicry in, Guillain-Barre and Fisher syndromes. Lancet Infect Dis. 2001;1:29–37. doi: 10.1016/S1473-3099(01)00019-6. [DOI] [PubMed] [Google Scholar]
- 38.Yuki N, Hartung HP. Guillain-Barre syndrome. N Engl J Med. 2012;366:2294–304. doi: 10.1056/NEJMra1114525. [DOI] [PubMed] [Google Scholar]
- 39.Yuki N, Taki T, Takahashi M, Saito K, Yoshino H, Tai T, et al. Molecular mimicry between GQ1b ganglioside and lipopolysaccharides of Campylobacter jejuni isolated from patients with Fisher's syndrome. Ann Neurol. 1994;36:791–3. doi: 10.1002/ana.410360517. [DOI] [PubMed] [Google Scholar]
- 40.Jacobs BC, Rothbarth PH, van der Meche FG, Herbrink P, Schmitz PI, de Klerk MA, et al. The spectrum of antecedent infections in Guillain-Barre syndrome: a case-control study. Neurology. 1998;51:1110–5. doi: 10.1212/WNL.51.4.1110. [DOI] [PubMed] [Google Scholar]
- 41.Orlikowski D, Porcher R, Sivadon-Tardy V, Quincampoix JC, Raphael JC, Durand MC, et al. Guillain-Barre syndrome following primary cytomegalovirus infection: a prospective cohort study. Clin Infect Dis. 2011;52:837–44. doi: 10.1093/cid/cir074. [DOI] [PubMed] [Google Scholar]
- 42.McGrogan A, Madle GC, Seaman HE, de Vries CS. The epidemiology of Guillain-Barre syndrome worldwide. A systematic literature review. Neuroepidemiology. 2009;32:150–63. doi: 10.1159/000184748. [DOI] [PubMed] [Google Scholar]
- 43.Jackson BR, Zegarra JA, Lopez-Gatell H, Sejvar J, Arzate F, Waterman S, et al. Binational outbreak of Guillain-Barre syndrome associated with Campylobacter jejuni infection, Mexico and USA, 2011. Epidemiol Infect. 2014;142:1089–99. doi: 10.1017/S0950268813001908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Heikema AP, Islam Z, Horst-Kreft D, Huizinga R, Jacobs BC, Wagenaar JA, et al. Campylobacter jejuni capsular genotypes are related to Guillain-Barre syndrome. Clin Microbiol Infect. 2015;21:852 e1-9. doi: 10.1016/j.cmi.2015.05.031. [DOI] [PubMed] [Google Scholar]
- 45.Meyer Sauteur PM, Huizinga R, Tio-Gillen AP, Drenthen J, Unger WWJ, Jacobs E, et al. Intrathecal antibody responses to GalC in Guillain-Barre syndrome triggered by Mycoplasma pneumoniae. J Neuroimmunol. 2018;314:13–6. doi: 10.1016/j.jneuroim.2017.11.011. [DOI] [PubMed] [Google Scholar]
- 46.Visser LH, van der Meche FG, Meulstee J, Rothbarth PP, Jacobs BC, Schmitz PI, et al. Cytomegalovirus infection and Guillain-Barre syndrome: the clinical, electrophysiologic, and prognostic features. Dutch Guillain-Barre Study Group. Neurology. 1996;47:668–73. [DOI] [PubMed] [Google Scholar]
- 47.Taheraghdam A, Pourkhanjar P, Talebi M, Bonyadi M, Pashapour A, Sharifipour E, et al. Correlations between cytomegalovirus, Epstein-Barr virus, anti-ganglioside antibodies, electrodiagnostic findings and functional status in Guillain-Barre syndrome. Iran J Neurol. 2014;13:7–12. [PMC free article] [PubMed] [Google Scholar]
- 48.Mori M, Kuwabara S, Miyake M, Noda M, Kuroki H, Kanno H, et al. Haemophilus influenzae infection and Guillain-Barre syndrome. Brain. 2000;123(Pt 10):2171–8. doi: 10.1093/brain/123.10.2171. [DOI] [PubMed] [Google Scholar]
- 49.De Wals P Deceuninck G, Toth E, Boulianne N, Brunet D, Boucher RM, et al. Risk of Guillain-Barre syndrome following H1N1 influenza vaccination in Quebec. JAMA. 2012;308:175–81. doi: 10.1001/jama.2012.7342. [DOI] [PubMed] [Google Scholar]
- 50.Brannagan TH, 3rd Zhou Y. HIV-associated Guillain-Barre syndrome. J Neurol Sci. 2003;208:39–42. doi: 10.1016/S0022-510X(02)00418-5. [DOI] [PubMed] [Google Scholar]
- 51.Leis AA, Stokic DS. Neuromuscular manifestations of west nile virus infection. Front Neurol. 2012;3:37. doi: 10.3389/fneur.2012.00037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Anaya JM, Rodriguez Y, Monsalve DM, Vega D, Ojeda E, Gonzalez-Bravo D, et al. A comprehensive analysis and immunobiology of autoimmune neurological syndromes during the Zika virus outbreak in Cucuta, Colombia. J Autoimmun. 2017;77:123–38. doi: 10.1016/j.jaut.2016.12.007. [DOI] [PubMed] [Google Scholar]
- 53.Saiz JC, Vazquez-Calvo A, Blazquez AB, Merino-Ramos T, Escribano-Romero E, Martin-Acebes MA. Zika Virus: the Latest Newcomer. Front Microbiol. 2016;7:496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Prineas JW. Pathology of the Guillain-Barre syndrome. Ann Neurol. 1981;9(Suppl):6–19. doi: 10.1002/ana.410090704. [DOI] [PubMed] [Google Scholar]
- 55.Kieseier BC, Kiefer R, Gold R, Hemmer B, Willison HJ, Hartung HP. Advances in understanding and treatment of immune-mediated disorders of the peripheral nervous system. Muscle Nerve. 2004;30:131–56. doi: 10.1002/mus.20076. [DOI] [PubMed] [Google Scholar]
- 56.Brechenmacher C, Vital C, Deminiere C, Laurentjoye L, Castaing Y, Gbikpi-Benissan G, et al. Guillain-Barre syndrome: an ultrastructural study of peripheral nerve in 65 patients. Clin Neuropathol. 1987;6:19–24. [PubMed] [Google Scholar]
- 57.Csurhes PA, Sullivan AA, Green K, Pender MP, McCombe PA. T cell reactivity to P0, P2, PMP-22, and myelin basic protein in patients with Guillain-Barre syndrome and chronic inflammatory demyelinating polyradiculoneuropathy. J Neurol Neurosurg Psychiatry. 2005;76:1431–9. doi: 10.1136/jnnp.2004.052282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.McCombe PA, Csurhes PA. T cells from patients with Guillain-Barre syndrome produce interferon-gamma in response to stimulation with the ganglioside GM1. J Clin Neurosci 2010;17:537–8. doi: 10.1016/j.jocn.2009.07.096. [DOI] [PubMed] [Google Scholar]
- 59.Chi LJ, Wang HB, Zhang Y, Wang WZ. Abnormality of circulating CD4(+)CD25(+) regulatory T cell in patients with Guillain-Barre syndrome. J Neuroimmunol. 2007;192:206–14. doi: 10.1016/j.jneuroim.2007.09.034. [DOI] [PubMed] [Google Scholar]
- 60.Harness J, McCombe PA. Increased levels of activated T-cells and reduced levels of CD4/CD25+ cells in peripheral blood of Guillain-Barre syndrome patients compared to controls. J Clin Neurosci. 2008;15:1031–5. doi: 10.1016/j.jocn.2007.09.016. [DOI] [PubMed] [Google Scholar]
- 61.Ubogu EE. Inflammatory neuropathies: pathology, molecular markers and targets for specific therapeutic intervention. Acta Neuropathol. 2015;130:445–68. doi: 10.1007/s00401-015-1466-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Xia RH, Yosef N, Ubogu EE. Clinical, electrophysiological and pathologic correlations in a severe murine experimental autoimmune neuritis model of Guillain-Barre syndrome. J Neuroimmunol. 2010;219:54–63. doi: 10.1016/j.jneuroim.2009.11.019. [DOI] [PubMed] [Google Scholar]
- 63.Dalakas MC. Pathogenesis of immune-mediated neuropathies. Biochim Biophys Acta. 2015;1852:658–66. doi: 10.1016/j.bbadis.2014.06.013. [DOI] [PubMed] [Google Scholar]
- 64.Rinaldi S. Update on Guillain-Barre syndrome. J Peripher Nerv Syst. 2013;18:99–112. doi: 10.1111/jns5.12020. [DOI] [PubMed] [Google Scholar]
- 65.Hafer-Macko CE, Sheikh KA, Li CY, Ho TW, Cornblath DR, McKhann GM, et al. Immune attack on the Schwann cell surface in acute inflammatory demyelinating polyneuropathy. Ann Neurol. 1996;39:625–35. doi: 10.1002/ana.410390512. [DOI] [PubMed] [Google Scholar]
- 66.Nyland H, Matre R, Mork S. Immunological characterization of sural nerve biopsies from patients with Guillain-Barre syndrome. Ann Neurol. 1981;9(Suppl):80–6. doi: 10.1002/ana.410090713. [DOI] [PubMed] [Google Scholar]
- 67.Putzu GA, Figarella-Branger D, Bouvier-Labit C, Liprandi A, Bianco N, Pellissier JF. Immunohistochemical localization of cytokines, C5b-9 and ICAM-1 in peripheral nerve of Guillain-Barre syndrome. J Neurol Sci. 2000;174:16–21. doi: 10.1016/S0022-510X(99)00328-7. [DOI] [PubMed] [Google Scholar]
- 68.Zweiman B, Rostami A, Lisak RP, Moskovitz AR, Pleasure DE. Immune reactions to P2 protein in human inflammatory demyelinative neuropathies. Neurology. 1983;33:234–7. doi: 10.1212/WNL.33.2.234. [DOI] [PubMed] [Google Scholar]
- 69.Inglis HR, Csurhes PA, McCombe PA. Antibody responses to peptides of peripheral nerve myelin proteins P0 and P2 in patients with inflammatory demyelinating neuropathy. J Neurol Neurosurg Psychiatry. 2007;78:419–22. doi: 10.1136/jnnp.2006.106617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Makowska A, Pritchard J, Sanvito L, Gregson N, Peakman M, Hayday A, et al. Immune responses to myelin proteins in Guillain-Barre syndrome. J Neurol Neurosurg Psychiatry. 2008;79:664–71. doi: 10.1136/jnnp.2007.123943. [DOI] [PubMed] [Google Scholar]
- 71.Ziganshin RH, Ivanova OM, Lomakin YA, Belogurov AA, Jr., Kovalchuk SI, Azarkin IV, et al. The Pathogenesis of the Demyelinating Form of Guillain-Barre Syndrome (GBS): Proteo-peptidomic and Immunological Profiling of Physiological Fluids. Mol Cell Proteomics. 2016;15:2366–78. doi: 10.1074/mcp.M115.056036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Griffin JW, Li CY, Macko C, Ho TW, Hsieh ST, Xue P, et al. Early nodal changes in the acute motor axonal neuropathy pattern of the Guillain-Barre syndrome. J Neurocytol. 1996;25:33–51. doi: 10.1007/BF02284784. [DOI] [PubMed] [Google Scholar]
- 73.Hafer-Macko C, Hsieh ST, Li CY, Ho TW, Sheikh K, Cornblath DR, et al. Acute motor axonal neuropathy: an antibody-mediated attack on axolemma. Ann Neurol. 1996;40:635–44. doi: 10.1002/ana.410400414. [DOI] [PubMed] [Google Scholar]
- 74.Susuki K, Yuki N, Schafer DP, Hirata K, Zhang G, Funakoshi K, et al. Dysfunction of nodes of Ranvier: a mechanism for anti-ganglioside antibody-mediated neuropathies. Exp Neurol. 2012;233:534–42. doi: 10.1016/j.expneurol.2011.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Willison HJ. The immunobiology of Guillain-Barre syndromes. J Peripher Nerv Syst. 2005;10:94–112. doi: 10.1111/j.1085-9489.2005.0010202.x. [DOI] [PubMed] [Google Scholar]
- 76.Ogawara K, Kuwabara S, Mori M, Hattori T, Koga M, Yuki N. Axonal Guillain-Barre syndrome: relation to anti-ganglioside antibodies and Campylobacter jejuni infection in Japan. Ann Neurol. 2000;48:624–31. doi: 10.1002/1531-8249(200010)48:4%3c624::AID-ANA9%3e3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 77.Sheikh KA, Nachamkin I, Ho TW, Willison HJ, Veitch J, Ung H, et al. Campylobacter jejuni lipopolysaccharides in Guillain-Barre syndrome: molecular mimicry and host susceptibility. Neurology. 1998;51:371–8. doi: 10.1212/WNL.51.2.371. [DOI] [PubMed] [Google Scholar]
- 78.Willison HJ. Ganglioside complexes: new autoantibody targets in Guillain-Barre syndromes. Nat Clin Pract Neurol. 2005;1:2–3. doi: 10.1038/ncpneuro0001. [DOI] [PubMed] [Google Scholar]
- 79.Willison HJ, Goodyear CS. Glycolipid antigens and autoantibodies in autoimmune neuropathies. Trends Immunol. 2013;34:453–9. doi: 10.1016/j.it.2013.05.001. [DOI] [PubMed] [Google Scholar]
- 80.Jacobs BC, Koga M, van Rijs W, Geleijns K, van Doorn PA, Willison HJ, et al. Subclass IgG to motor gangliosides related to infection and clinical course in Guillain-Barre syndrome. J Neuroimmunol. 2008;194:181–90. doi: 10.1016/j.jneuroim.2007.11.017. [DOI] [PubMed] [Google Scholar]
- 81.Jacobs BC, van Doorn PA, Schmitz PI, Tio-Gillen AP, Herbrink P, Visser LH, et al. Campylobacter jejuni infections and anti-GM1 antibodies in Guillain-Barre syndrome. Ann Neurol. 1996;40:181–7. doi: 10.1002/ana.410400209. [DOI] [PubMed] [Google Scholar]
- 82.Phongsisay V, Susuki K, Matsuno K, Yamahashi T, Okamoto S, Funakoshi K, et al. Complement inhibitor prevents disruption of sodium channel clusters in a rabbit model of Guillain-Barre syndrome. J Neuroimmunol. 2008;205:101–4. doi: 10.1016/j.jneuroim.2008.09.016. [DOI] [PubMed] [Google Scholar]
- 83.Ho TW, Li CY, Cornblath DR, Gao CY, Asbury AK, Griffin JW, et al. Patterns of recovery in the Guillain-Barre syndromes. Neurology. 1997;48:695–700. doi: 10.1212/WNL.48.3.695. [DOI] [PubMed] [Google Scholar]
- 84.Kuwabara S, Bostock H, Ogawara K, Sung JY, Kanai K, Mori M, et al. The refractory period of transmission is impaired in axonal Guillain-Barre syndrome. Muscle Nerve. 2003;28:683–9. doi: 10.1002/mus.10488. [DOI] [PubMed] [Google Scholar]
- 85.Feasby TE, Gilbert JJ, Brown WF, Bolton CF, Hahn AF, Koopman WJ, et al. Acute "axonal" Guillain-Barre polyneuropathy. Neurology. 1987;37:357. doi: 10.1212/WNL.37.2.357-b. [DOI] [PubMed] [Google Scholar]
- 86.Kolter T. Ganglioside biochemistry. ISRN Biochem. 2012;2012:506160. doi: 10.5402/2012/506160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Chiba A, Kusunoki S, Obata H, Machinami R, Kanazawa I. Ganglioside composition of the human cranial nerves, with special reference to pathophysiology of Miller Fisher syndrome. Brain Res. 1997;745:32–6. doi: 10.1016/S0006-8993(96)01123-7. [DOI] [PubMed] [Google Scholar]
- 88.Liu JX, Willison HJ, Pedrosa-Domellof F. Immunolocalization of GQ1b and related gangliosides in human extraocular neuromuscular junctions and muscle spindles. Invest Ophthalmol Vis Sci. 2009;50:3226–32. doi: 10.1167/iovs.08-3333. [DOI] [PubMed] [Google Scholar]
- 89.Chiba A, Kusunoki S, Shimizu T, Kanazawa I. Serum IgG antibody to ganglioside GQ1b is a possible marker of Miller Fisher syndrome. Ann Neurol. 1992;31:677–9. doi: 10.1002/ana.410310619. [DOI] [PubMed] [Google Scholar]
- 90.Mori M, Kuwabara S. Fisher syndrome. Curr Treat Options Neurol 2011;13:71–8. doi: 10.1007/s11940-010-0103-8. [DOI] [PubMed] [Google Scholar]
- 91.Jacobs BC, O'Hanlon GM, Bullens RW, Veitch J, Plomp JJ, Willison HJ. Immunoglobulins inhibit pathophysiological effects of anti-GQ1b-positive sera at motor nerve terminals through inhibition of antibody binding. Brain. 2003;126:2220–34. doi: 10.1093/brain/awg235. [DOI] [PubMed] [Google Scholar]
- 92.Overell JR, Willison HJ. Recent developments in Miller Fisher syndrome and related disorders. Curr Opin Neurol. 2005;18:562–6. doi: 10.1097/01.wco.0000173284.25581.2f. [DOI] [PubMed] [Google Scholar]
- 93.Kaida K, Kusunoki S. Antibodies to gangliosides and ganglioside complexes in Guillain-Barre syndrome and Fisher syndrome: mini-review. J Neuroimmunol. 2010;223:5–12. doi: 10.1016/j.jneuroim.2010.02.001. [DOI] [PubMed] [Google Scholar]
- 94.Kleyweg RP, van der Meche FG, Meulstee J. Treatment of Guillain-Barre syndrome with high-dose gammaglobulin. Neurology. 1988;38:1639–41. doi: 10.1212/WNL.38.10.1639. [DOI] [PubMed] [Google Scholar]
- 95.Plasmapheresis and acute Guillain-Barre syndrome The Guillain-Barre syndrome Study Group. Neurology. 1985;35:1096–104. doi: 10.1212/WNL.35.8.1096. [DOI] [PubMed] [Google Scholar]
- 96.van der Meche FG, Schmitz PI. A randomized trial comparing intravenous immune globulin and plasma exchange in Guillain-Barre syndrome. Dutch Guillain-Barre Study Group. N Engl J Med. 1992;326:1123–9. doi: 10.1056/NEJM199204233261705. [DOI] [PubMed] [Google Scholar]
- 97.Plasma exchange in Guillain-Barre syndrome: one-year follow-up French Cooperative Group on Plasma Exchange in Guillain-Barre Syndrome. Ann Neurol. 1992;32:94–7. [DOI] [PubMed] [Google Scholar]
- 98.Randomised trial of plasma exchange, intravenous immunoglobulin, and combined treatments in Guillain-Barre syndrome Plasma Exchange/Sandoglobulin Guillain-Barre Syndrome Trial Group. Lancet. 1997;349:225–30. [PubMed] [Google Scholar]
- 99.Meyer zu Horste G, Hartung HP, Kieseier BC. From bench to bedside–experimental rationale for immune-specific therapies in the inflamed peripheral nerve. Nat Clin Pract Neurol. 2007;3:198–211. doi: 10.1038/ncpneuro0452. [DOI] [PubMed] [Google Scholar]
- 100.Rubinstein MA, Kagan BM, Macgillviray MH, Merliss R, Sacks H. Unusual remission in a case of thrombotic thrombocytopenic purpura syndrome following fresh blood exchange transfusions. Ann Intern Med. 1959;51:1409–19. doi: 10.7326/0003-4819-51-6-1409. [DOI] [PubMed] [Google Scholar]
- 101.Brettle RP, Gross M, Legg NJ, Lockwood M, Pallis C. Treatment of acute polyneuropathy by plasma exchange. Lancet. 1978;2:1100. doi: 10.1016/S0140-6736(78)91837-8. [DOI] [PubMed] [Google Scholar]
- 102.Efficiency of plasma exchange in Guillain-Barre syndrome: role of replacement fluids French Cooperative Group on Plasma Exchange in Guillain-Barre syndrome. Ann Neurol. 1987;22:753–61. [DOI] [PubMed] [Google Scholar]
- 103.Hughes RA, Wijdicks EF, Barohn R, Benson E, Cornblath DR, Hahn AF, et al. Practice parameter: immunotherapy for Guillain-Barre syndrome: report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2003;61:736–40. doi: 10.1212/WNL.61.6.736. [DOI] [PubMed] [Google Scholar]
- 104.Cortese I, Chaudhry V, So YT, Cantor F, Cornblath DR, Rae-Grant A. Evidence-based guideline update: Plasmapheresis in neurologic disorders: report of the Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Neurology. 2011;76:294–300. doi: 10.1212/WNL.0b013e318207b1f6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Donofrio PD. Immunotherapy of idiopathic inflammatory neuropathies. Muscle Nerve. 2003;28:273–92. doi: 10.1002/mus.10402. [DOI] [PubMed] [Google Scholar]
- 106.Appropriate number of plasma exchanges in Guillain-Barre syndrome The French Cooperative Group on Plasma Exchange in Guillain-Barre Syndrome. Ann Neurol. 1997;41:298–306. [DOI] [PubMed] [Google Scholar]
- 107.Raphael JC, Chevret S, Hughes RA, Annane D. Plasma exchange for Guillain-Barre syndrome. Cochrane Database Syst Rev. 2002;2:CD001798. [DOI] [PubMed] [Google Scholar]
- 108.Schroder A, Linker RA, Gold R. Plasmapheresis for neurological disorders. Expert Rev Neurother. 2009;9:1331–9. doi: 10.1586/ern.09.81. [DOI] [PubMed] [Google Scholar]
- 109.Esperou H, Jars-Guincestre MC, Bolgert F, Raphael JC, Durand-Zaleski I. Cost analysis of plasma-exchange therapy for the treatment of Guillain-Barre syndrome. French Cooperative Group on Plasma Exchange in Guillain-Barre Syndrome. Intensive Care Med. 2000;26:1094–100. doi: 10.1007/s001340051323. [DOI] [PubMed] [Google Scholar]
- 110.Osterman PO, Fagius J, Lundemo G, Pihlstedt P, Pirskanen R, Siden A, et al. Beneficial effects of plasma exchange in acute inflammatory polyradiculoneuropathy. Lancet. 1984;2:1296–9. doi: 10.1016/S0140-6736(84)90819-5. [DOI] [PubMed] [Google Scholar]
- 111.Hughes RA, Swan AV, Raphael JC, Annane D, van Koningsveld R, van Doorn PA. Immunotherapy for Guillain-Barre syndrome: a systematic review. Brain. 2007;130:2245–57. doi: 10.1093/brain/awm004. [DOI] [PubMed] [Google Scholar]
- 112.Hughes RA, Swan AV, van Doorn PA. Intravenous immunoglobulin for Guillain-Barre syndrome. Cochrane Database Syst Rev. 2014;9:CD002063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Kuitwaard K, de Gelder J, Tio-Gillen AP, Hop WC, van Gelder T, van Toorenenbergen AW, et al. Pharmacokinetics of intravenous immunoglobulin and outcome in Guillain-Barre syndrome. Ann Neurol. 2009;66:597–603. doi: 10.1002/ana.21737. [DOI] [PubMed] [Google Scholar]
- 114.Buchwald B, Ahangari R, Weishaupt A, Toyka KV. Intravenous immunoglobulins neutralize blocking antibodies in Guillain-Barre syndrome. Ann Neurol. 2002;51:673–80. doi: 10.1002/ana.10205. [DOI] [PubMed] [Google Scholar]
- 115.Zhang G, Lopez PH, Li CY, Mehta NR, Griffin JW, Schnaar RL, et al. Anti-ganglioside antibody-mediated neuronal cytotoxicity and its protection by intravenous immunoglobulin: implications for immune neuropathies. Brain. 2004;127:1085–100. doi: 10.1093/brain/awh127. [DOI] [PubMed] [Google Scholar]
- 116.Dalakas MC. Intravenous immunoglobulin in autoimmune neuromuscular diseases. JAMA. 2004; 291:2367–75. doi: 10.1001/jama.291.19.2367. [DOI] [PubMed] [Google Scholar]
- 117.Dalakas MC. The use of intravenous immunoglobulin in the treatment of autoimmune neuromuscular diseases: evidence-based indications and safety profile. Pharmacol Ther. 2004;102:177–93. doi: 10.1016/j.pharmthera.2004.04.002. [DOI] [PubMed] [Google Scholar]
- 118.Farcas P, Avnun L, Frisher S, Herishanu YO, Wirguin I. Efficacy of repeated intravenous immunoglobulin in severe unresponsive Guillain-Barre syndrome. Lancet. 1997; 350:1747. doi: 10.1016/S0140-6736(97)24050-X. [DOI] [PubMed] [Google Scholar]
- 119.Hughes RA, Swan AV, van Doorn PA. Corticosteroids for Guillain-Barre syndrome. Cochrane Database Syst Rev. 2010;2:CD001446. [DOI] [PubMed] [Google Scholar]
- 120.Hughes RA, Brassington R, Gunn AA, van Doorn PA. Corticosteroids for Guillain-Barre syndrome. Cochrane Database Syst Rev. 2016;10:CD001446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.van Koningsveld R, Schmitz PI, Meche FG, Visser LH, Meulstee J, van Doorn PA, et al. Effect of methylprednisolone when added to standard treatment with intravenous immunoglobulin for Guillain-Barre syndrome: randomised trial. Lancet. 2004;363:192–6. doi: 10.1016/S0140-6736(03)15324-X. [DOI] [PubMed] [Google Scholar]
- 122.Pritchard J, Hughes RA, Hadden RD, Brassington R. Pharmacological treatment other than corticosteroids, intravenous immunoglobulin and plasma exchange for Guillain-Barre syndrome. Cochrane Database Syst Rev. 2016;11:CD008630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Wollinsky KH, Hulser PJ, Brinkmeier H, Aulkemeyer P, Bossenecker W, Huber-Hartmann KH, et al. CSF filtration is an effective treatment of Guillain-Barre syndrome: a randomized clinical trial. Neurology. 2001;57:774–80. doi: 10.1212/WNL.57.5.774. [DOI] [PubMed] [Google Scholar]
- 124.Creange A, Lerat H, Meyrignac C, Degos JD, Gherardi RK, Cesaro P. Treatment of Guillain-Barre syndrome with interferon-beta. Lancet. 1998;352:368–9. doi: 10.1016/S0140-6736(05)60466-7. [DOI] [PubMed] [Google Scholar]
- 125.Pritchard J, Gray IA, Idrissova ZR, Lecky BR, Sutton IJ, Swan AV, et al. A randomized controlled trial of recombinant interferon-beta 1a in Guillain-Barre syndrome. Neurology. 2003;61:1282–4. doi: 10.1212/01.WNL.0000092019.53628.88. [DOI] [PubMed] [Google Scholar]
- 126.Schaller B, Radziwill AJ, Steck AJ. Successful treatment of Guillain-Barre syndrome with combined administration of interferon-beta-1a and intravenous immunoglobulin. Eur Neurol. 2001;46:167–8. doi: 10.1159/000050798. [DOI] [PubMed] [Google Scholar]
- 127.Bensa S, Hadden RD, Hahn A, Hughes RA, Willison HJ. Randomized controlled trial of brain-derived neurotrophic factor in Guillain-Barre syndrome: a pilot study. Eur J Neurol. 2000;7:423–6. doi: 10.1046/j.1468-1331.2000.00096.x. [DOI] [PubMed] [Google Scholar]
- 128.Zhang X, Xia J, Ye H. Effect of Tripterygium polyglycoside on interleukin-6 in patients with Guillain-Barre syndrome. Zhongguo Zhong Xi Yi Jie He Za Zhi. 2000;20:332–4. [PubMed] [Google Scholar]
- 129.Davidson AI, Halstead SK, Goodfellow JA, Chavada G, Mallik A, Overell J, et al. Inhibition of complement in Guillain-Barre syndrome: the ICA-GBS study. J Peripher Nerv Syst. 2017;22:4–12. doi: 10.1111/jns.12194. [DOI] [PubMed] [Google Scholar]
- 130.Yamaguchi N, Misawa S, Sato Y, Nagashima K, Katayama K, Sekiguchi Y, et al. A Prospective, Multicenter, Randomized Phase II Study to Evaluate the Efficacy and Safety of Eculizumab in Patients with Guillain-Barre Syndrome (GBS): Protocol of Japanese Eculizumab Trial for GBS (JET-GBS). JMIR Res Protoc. 2016;5:e210. doi: 10.2196/resprot.6610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Motamed-Gorji N, Matin N, Tabatabaie O, Pavone P, Romano C, Falsaperla R, et al. Biological Drugs in Guillain-Barre Syndrome: An Update. Curr Neuropharmacol. 2017;15:938–50. doi: 10.2174/1570159X14666161213114904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Zhang HL, Zheng XY, Zhu J. Th1/Th2/Th17/Treg cytokines in Guillain-Barre syndrome and experimental autoimmune neuritis. Cytokine Growth Factor Rev. 2013;24:443–53. doi: 10.1016/j.cytogfr.2013.05.005. [DOI] [PubMed] [Google Scholar]
- 133.Dong C, Palladino SP, Helton ES, Ubogu EE. The pathogenic relevance of alphaM-integrin in Guillain-Barre syndrome. Acta Neuropathol. 2016;132:739–52. doi: 10.1007/s00401-016-1599-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Muller WA. Leukocyte-endothelial-cell interactions in leukocyte transmigration and the inflammatory response. Trends Immunol. 2003;24:327–34. doi: 10.1016/S1471-4906(03)00117-0. [DOI] [PubMed] [Google Scholar]
- 135.Man S, Ubogu EE, Ransohoff RM. Inflammatory cell migration into the central nervous system: a few new twists on an old tale. Brain Pathol. 2007;17:243–50. doi: 10.1111/j.1750-3639.2007.00067.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Bianchi E, Molteni R, Pardi R, Dubini G. Microfluidics for in vitro biomimetic shear stress-dependent leukocyte adhesion assays. J Biomech. 2013;46:276–83. doi: 10.1016/j.jbiomech.2012.10.024. [DOI] [PubMed] [Google Scholar]
- 137.Chiang S, Ubogu EE. The role of chemokines in Guillain-Barre syndrome. Muscle Nerve. 2013;48:320–30. doi: 10.1002/mus.23829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Yuan F, Yosef N, Lakshmana Reddy C, Huang A, Chiang SC, Tithi HR, et al. CCR2 gene deletion and pharmacologic blockade ameliorate a severe murine experimental autoimmune neuritis model of Guillain-Barre syndrome. PLoS One. 2014;9:e90463. doi: 10.1371/journal.pone.0090463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Yosef N, Ubogu EE. alpha(M)beta(2)-integrin-intercellular adhesion molecule-1 interactions drive the flow-dependent trafficking of Guillain-Barre syndrome patient derived mononuclear leukocytes at the blood-nerve barrier in vitro. J Cell Physiol. 2012;227:3857–75. doi: 10.1002/jcp.24100. [DOI] [PMC free article] [PubMed] [Google Scholar]