

Abstract
The success of mechanism-based drug discovery depends on the definition of the drug target. This definition becomes even more important as we try to link drug response to genetic variation, understand stratified clinical efficacy and safety, rationalize the differences between drugs in the same therapeutic class and predict drug utility in patient subgroups. However, drug targets are often poorly defined in the literature, both for launched drugs and for potential therapeutic agents in discovery and development. Here, we present an updated comprehensive map of molecular targets of approved drugs. We curate a total of 893 human and pathogen-derived biomolecules through which 1,578 US FDA-approved drugs act. These biomolecules include 667 human-genome-derived proteins targeted by drugs for human disease. Analysis of these drug targets indicates the continued dominance of privileged target families across disease areas, but also the growth of novel first-in-class mechanisms, particularly in oncology. We explore the relationships between bioactivity class and clinical success, as well as the presence of orthologues between human and animal models and between pathogen and human genomes. Through the collaboration of three independent teams, we highlight some of the ongoing challenges in accurately defining the targets of molecular therapeutics and present conventions for deconvoluting the complexities of molecular pharmacology and drug efficacy.
James Black famously stated in 2000 that “the best way to discover a new drug is to start with an old one”1. Where available, a deep understanding of the mechanistic action of targeted drugs continues to inform drug discovery, clinical trials and efforts to overcome drug resistance. Thus, maintaining an accurate and up-to-date map of approved drugs and their efficacy targets — that is, the targets through which the drugs exert their therapeutic effect (Box 1) — is an important activity that will guide future drug development and innovation.
Box 1. Definitions used in the article.
Efficacy target
Throughout the paper, we use the term ‘target’ to refer to those proteins or other biomolecules (such as DNA, RNA, heparin and peptides) to which the drug directly binds, and which are responsible for the therapeutic efficacy of the drug. Biomolecules that the drug may also bind to, or be metabolized by, but which are not known to be responsible for its therapeutic effect, are not defined as targets. Although the ChEMBL database assigns identifiers to multi-chain targets such as protein complexes, with annotation of the subunit to which the drug binds (where known), to facilitate the comparison of target annotations between multiple data sets, numbers used here reflect the individual components comprising these targets (in most cases, proteins) with non-ligand-binding subunits of protein complexes excluded (where sufficient binding site annotations are available). DrugCentral uses the same definition but also requires links to bioactivity data.
Drug
The definition of ‘drug’ used here refers to therapeutic ingredients only, and includes all the small molecules and biologics that are currently approved (or have previously been approved) by the US FDA (before June 2015) to enhance human health, and also antimalarial drugs approved elsewhere in the world. The term drug does not include imaging agents, nutritional supplements, sunscreens or vaccines. Furthermore, the numbers reported in the paper refer to parent compounds after the removal of pharmaceutical salts. Identifying an entirely comprehensive set of drugs approved anywhere in the world is a highly challenging task owing to the number of different regulatory agencies involved and the diversity of the information sources required. Moreover, the lack of information is also a challenge; for example, the European Medicines Agency does not have approval data before 1995, and the Japan Pharmaceutical and Medical Devices Agency has published lists of approved drugs in English from 2004 only (see Further information). However, we have additionally identified a set of more than 1,200 drugs approved by other agencies but not currently by the FDA, and we discuss the novel targets within this set separately.
Prodrug
The definition of ‘prodrug’ used here refers to a drug for which the dosed ingredient is an inactive or only mildly efficacious entity, but once in the body it is converted to the active ingredient by either a spontaneous or an enzyme-catalysed reaction. It is estimated that approximately 10% of drugs fall into this category55. There are examples of different prodrugs resulting in the same active ingredient (for example, hydrocortisone is formulated as different prodrugs, including hydrocortisone sodium succinate, hydrocortisone valerate and hydrocortisone probutate) and also examples of prodrugs resulting in multiple active ingredients from a single dosed ingredient (such as azathioprine). For simplicity, we assigned efficacy target information to the inactive prodrug (parent), rather than the active ingredient, which is actually the molecule that interacts with the therapeutic target.
Prescribing information
Prescribing information is a document provided by the company that markets an approved drug, and includes consistently presented and detailed information about the approved drug, including information on clinical pharmacology, such as the mode of action of the drug. This information is provided in the drug label of FDA-approved drugs, which is available as a PDF or mark-up document in the Structured Product Labelling format. In the European Union, the corresponding document is the Summary of Product Characteristics.
ATC code
The Anatomical Therapeutic Chemical Classification System code (ATC code) is attributed to a drug by the WHO Collaborating Centre (WHOCC) for Drug Statistics Methodology. The ATC code classifies drugs according to the following five levels: level 1, the organ or anatomical system on which they act; level 2, the pharmacological action; levels 3 and 4, the chemical, pharmacological and therapeutic subgroups; and level 5, the specific single drug or drug combination. For example, for sildenafil, the ATC code is as follows:
Level 1 (G): genito urinary system and sex hormones
Level 2 (G04): urologicals
Level 3 (G04B): urologicals
Level4 (G04BE): drugs used in erectile dysfunction
Level 5 (G04BE03): sildenafil
Owing to the nature of the ATC classification, a drug can have multiple codes, especially if it acts on multiple anatomical systems; for example, aspirin has five different ATC codes: B01AC06, A01AD05, N02BA01, N02BA51 and N02BA71. A drug can also have multiple codes if it is used as a component of a single product combination therapy.
Arguably, the first attempt to compile a definitive target list dates from 1996, when Drews and Ryser estimated the number of human molecular targets for approved small-molecule drugs2,3. From this article and subsequent analyses4–6, the concept of ‘privileged’ protein families that have had a consistent and successful history of drug discovery began to emerge. In 2006, we published a compendium of drug targets7 and identified that the then available US FDA-approved targeted drugs acted through 324 mechanistic protein targets. Alongside the well-established druggable families, we analysed privileged families and additionally identified a ‘long tail’ of diverse, structurally unrelated protein families with small numbers of members, as well as single proteins.
Several databases now provide data on drug–target interactions, each with different scopes and foci. The first was the Therapeutic Targets Database8. DrugBank9, the most widely used specialist drug information resource, maps drugs to proteins that have been reported to bind to them, and SuperTarget10 is a text-mining-based compilation of direct and indirect drug targets. More recently, Rask-Andersen et al.11 provided an updated view on the status of current drugs and the human targets believed to be responsible for their efficacy in their approved indications. Additionally, Munos12 has highlighted trends in drug classes and target innovation for the past decade, and Agarwal et al.13 have analysed the overlap and uniqueness in the drug targets that are being pursued by industry. Finally, the International Union of Basic and Clinical Pharmacology and British Pharmacological Society (IUPHAR/BPS) Guide to Pharmacology database (see Further information) also compiles information on approved drugs, together with affinity and selectivity data, and assigns primary targets that are supported by experimental evidence14. However, despite the variety of valuable online resources, it is still a challenge to retrieve a consistent and comprehensive view of the targets of approved drugs (covering both small molecules and biologics) with their associated molecular efficacy targets (human and pathogen) organized by therapeutic use. Furthermore, although the concept of a target is a natural one for researchers in the field, there are substantial operational difficulties in consistently mapping this target concept to specific genes and gene products in practice.
Here, we synthesize and build on our previous approaches7 to systematically recompile and comprehensively annotate the current list of FDA-approved drugs (see Box 2 and Box 3 for details of data collection and analysis). We assign to each drug their respective efficacy target or target set from the prescribing information and/or the scientific literature. We emphasize that even with a well-defined concept of efficacy there are challenges in making a clean unambiguous assignment in many cases, especially regarding how to treat protein complexes or drugs that bind to a number of closely related gene products.
Box 2. Data collection and analysis methods.
Efficacy targets assignment and comparison
A list of all US FDA-approved drugs (small molecules and biologics) and antimalarials approved elsewhere in the world was compiled based on the data content of ChEMBL 21, DrugCentral and canSAR (see Further information; information accuracy in DrugCentral was benchmarked against multiple sources including the NCATS Pharmaceutical Collection56 — see table 2 in ref 15). The list was further divided into small molecules and biologics. After the removal of pharmaceutical salts and merging the drug lists, 1,419 unique small-molecule drugs and 250 unique biologic agents were obtained. For each of these drugs, the efficacy target was extracted from the current version of the prescribing information and complemented with the scientific literature in the cases for which either the prescribing information was not available or the mechanism of action was not reported. The following guidelines were used when assigning the efficacy targets:
Identify the target from the “Mechanism of Action” description in the prescribing information. If it is available, assign the therapeutic target (or targets) to the compound.
If the information in the prescribing information is ambiguous, complement this with a literature search for publications related to the mechanism of action of the drug.
For conflicting cases, look at review articles, either about the biology of the disease or the pharmacology of other chemically related drugs, and determine the most plausible mode of action.
For the cases when several subunits or isoforms match the information described in the prescribing information, evaluate which is more likely to be the real therapeutic target by looking at its expression patterns in relevant tissues for the disease.
If the specific subunit (or subunits) or isoform (or isoforms) cannot be identified, assign all of them as targets.
For pathogen targets, if the prescribing information lists several microorganisms against which the drug is effective, pick a representative one rather than assigning all of them as targets.
If the mechanism is still not clear or unknown, and the literature did not provide any conclusive information on the molecular target, do not assign any target to the compound.
All the external data sets were retrieved in June 2015 and included only drugs approved by the US FDA before this date. The mapping of drugs between the data sets was based on the drug names provided by each data set and the parent drug name in ChEMBL 21. To improve the mapping, synonyms associated with each parent drug name were also taken into consideration, as well as active drug names in case of prodrugs. The mapping of targets between the data sets was based purely on UniProt accessions.
Mapping of drugs to the ATC code
For each drug, the respective WHO Anatomical Therapeutic Chemical Classification System code (ATC code) or codes was extracted from ChEMBL 21 or DrugCentral, and assigned to either its respective dosed ingredient (if applicable) or the parent ingredient itself. For the 1,669 drugs, 1,462 could be mapped to current ATC codes and the remaining 207 were labelled “Unclassified”.
Target classification
The protein target classification was made using the existing classification in ChEMBL 21. Both level 1 and 2 were attributed to each of the human and pathogen efficacy targets. For simplicity, all the entries with level 1 labelled as “Cytosolic other”, “Secreted”, “Structural” and “Surface antigen” were all renamed to “Protein other”.
Efficacy targets, orthologues and ATC code mapping
The UniProt IDs of the human protein efficacy targets were mapped to Ensembl Gene IDs and Ensembl Protein IDs through Biomart57. Orthologues of Canis lupus familiaris (dog), Sus scrofa (pig), Rattus norvegicus (rat), Mus musculus (mouse), Danio rerio (zebrafish), Drosophila melanogaster (fruitfly), Caenorhabditis elegans (nematode) and Saccharomyces cerevisiae (yeast) were extracted from Ensembl Compara version 82 57,58. The remaining orthologues among Homo sapiens (human), Plasmodium falciparum and Escherichia coli were extracted from InParanoid version 8.0 59 and mapped via Ensembl Protein IDs. The UniProt IDs of the pathogen protein efficacy targets, composed of a representative set of E. coli proteins, were used to extract the orthologues of H. sapiens, C. l. familiaris, R. norvegicus, M. musculus, D. rerio, D. melanogaster, C. elegans, S. cerevisiae and P. falciparum from InParanoid version 8.0.
Each drug was clustered in an ATC level 4 category, and for each category the orthologues of the protein efficacy targets for those drugs were accounted for. If, within a certain ATC level 4 category, not all protein efficacy targets had orthologues, this would be reflected in the final plot in figure 5 by colouring their presence in a way that it reflects the percentage of efficacy targets with orthologues in a certain species.
Box 3. Additional details on data classification and analysis.
Clinical success
A list of non-approved compounds was compiled based on the data content of ChEMBL 20. Specifically, all of the compounds, the maximum development phase of which differs from phase IV and which have been tested against a human protein, were retrieved from ChEMBL 20. In this context, the protein target was defined as a target whose target type was equal to the following: “single protein”, “protein complex”, “protein complex group” or “protein family”. Compounds were further filtered based on the source of the assay, and only compounds whose activity profile was extracted from the medicinal chemistry literature (assay source equal to 1) were retained for analysis and linked to the protein classification level 2. After the removal of pharmaceutical salts, the total number of non-approved compounds was 382,910. The number of unique compounds associated with a particular protein target class was counted and used to obtain the distribution of tested compounds per family classes. Protein targets whose level 2 classification was null or unclassified were grouped into one single target class named “Other”. Additionally, the following families were also grouped and labelled as “Other” owing to the lower number of tested compounds against members of these families: “Ion channel TRP”, “Ion channel KIR”, “Ion channel SUR”, “Ion channel RYR”, “Ion channel ASIC”, “Ion channel IP3”, “Ion channel K2P”, “Membrane receptor 7tmfz”, “Membrane receptor 7tmtas2r”, “Toll-like membrane receptor”, “Ligase” and “Aminoacyltransferase”.
The same procedure was used to count the distribution of approved drugs per family classes. Briefly, all drugs, the target of which is a human protein, were linked to the protein classification level 2. After the removal of pharmaceutical salts, the total number of drugs was 1,194. The number of unique drugs associated with a particular protein target class was counted and used to obtain the distribution of successful drugs per family class.
Antipsychotics
The list of central nervous system drugs was compiled by combining information from the WHO Anatomical Therapeutic Chemical Classification System code (ATC code) and the United States Pharmacopeial Convention (USP) system60. In summary, a drug with an ATC level 3 code equal to “N05A” and classified as antipsychotic or psychotic by the USP system60 was considered to be an antipsychotic.
pXC50 calculation
Mean potency (pXC50; negative logarithm of XC50) values were calculated as described in Gleeson et al.61, but with minor changes. In summary, bioactivity data for the antipsychotics (shown in Supplementary information S1) were extracted from ChEMBL 20 and filtered to include only XC50 values from assays in which the target was a human protein, and the standardized activity type was flagged as Ki, IC50, EC50, Kd, XC50, AC50 or potency. In the ChEMBL database, standardization of activity data reported in different units to nanomolar units and conversion of logged data, such as pKi and pIC50, to the non-log format has been performed. This standardization enables the maximum comparable bioactivity data to be extracted from the database for use in this analysis. XC50 values not standardized to nanomolar units or reported as “greater than” or “less than” were excluded. In a few cases, the target information in the original publication did not specify the isoform or subunit of the receptor or protein complex. In these cases, the data were kept and analysed independently, not being merged with the data for which the individual proteins were explicitly mentioned. Having extracted the data, XC50 values recorded against the same target and compound were averaged and converted to the pXC50 values.
Image software
All of the figures were produced using a combination of the following programs: R Project for Statistical Computing version 2.15 (see Further information), Circos62 and Inkscape (see Further information).
We also map each drug (and thereby target) to the WHO Anatomical Therapeutic Chemical Classification System code (ATC code; see Further information) as a way of obtaining a standard therapeutic indication for them. The ATC hierarchy consistently classifies drugs according to the organ or system on which they act, and their therapeutic effects, pharmacological actions and chemical class. With this mapping, we explore the footprint of target classes across disease areas and investigate the success of privileged target families given the investment in discovery effort. We also compile a list of drug target orthologues for standard model organisms to develop a foundation for the deeper understanding of species differences, cross-species drug repositioning and applicability of animal model systems.
Complexities in defining efficacy targets
Defining the set of mechanistic drug targets requires unambiguous evidence of the therapeutic action of drugs through clear biomolecular partners. In reality, this association is not always straightforward. Although in many cases it is possible to annotate a widely accepted and unambiguous target, for other drugs there is often disagreement or changes in understanding over time, which is then reflected in differences between primary sources. To address this challenge, we have reassigned efficacy targets afresh from the primary literature and prescribing information and combined annotations made by three independent teams of curators at the European Molecular Biology Laboratory-European Bioinformatics Institute (EMBL–EBI) ChEMBL database, the University of New Mexico DrugCentral database15 and The Institute of Cancer Research canSAR knowledge base16 (see Further information). We defined a simple, consistent set of guidelines to help us assign therapeutic targets (the full set of guidelines is shown in Box 2). Overall, we did not assign targets solely on the basis of reported biochemical and pharmacology data, which are now widely available17. Although there may be evidence for drugs binding with moderate or even high affinity to multiple additional targets, we do not consider these as efficacy targets unless there is evidence for their role in the therapeutic effect of the drug.
For example, antipsychotics are considered to exert their effect largely by acting as antagonists of the dopamine D2 receptor (encoded by DRD2) and sometimes as antagonists or inverse agonists of the 5-hydroxytryptamine (5-HT; also known as serotonin) 2A receptor (encoded by HTR2A)18,19. However, antipsychotics also bind with nanomolar affinity to other 5-HT receptor subtypes, as well as adrenergic, muscarinic and histamine receptors (Supplementary information S1). Despite much speculation and investigation, however, the contribution of these additional targets to the therapeutic effect of antipsychotics has not yet been demonstrated. For example, the therapeutic effect of aripiprazole, a “dopamine–serotonin system stabilizer”20, has been attributed to it acting as a partial agonist of D2 and 5-HT1A receptors and an antagonist of the 5-HT2A receptor21, although it also interacts with other proteins. However, the dopamine stabilizer (-)-OSU6162 appears to occupy a subpopulation of striatal D2/D3 receptors with moderate (micromolar) affinity22, which suggests that a specific interplay between, for example, D2 receptor occupancy and tissue specificity (striate nucleus) may be more therapeutically relevant. Consequently, targets other than D2 and 5-HT2A receptors have not been annotated as efficacy targets of antipsychotic drugs. The fact that a drug has high affinity to an alternative target, or that different drugs from the same class have differentiated target binding profiles, can be important in developing next-generation agents.
Another challenge is how to assign targets to drugs reported to have broad mechanistic effects; examples of such drugs include muscarinic receptor antagonists, voltage-gated potassium channel blockers and broad-spectrum β-lactam antibiotics. For these drugs, one possible solution would be to list the 5 muscarinic receptors for the first case, the 4 α-subunits that may form voltage-gated potassium channels for the second case and, for the third case, to pick all the penicillin-binding proteins (PBPs) from all the bacterial species against which the drug is effective. However, in the case of human targets, a more restricted subset based on selectivity data or expression data could also potentially be chosen. For the pathogen targets, a representative pathogen species could be chosen and only the biomolecules or cellular components of that species could be assigned as drug targets. In making our assignments, we have identified such subsets among the human targets for which sufficient information was available to do so. For targets for which there was inadequate evidence to determine which subunits or family members play a key part, we listed all possible proteins. For example, all anti-muscarinic agents indicated to treat bronchospasms have muscarinic acetylcholine receptor M3 (encoded by CHRM3) assigned as their efficacy target because this muscarinic receptor has the highest expression levels in the airways and is responsible for bronchoconstriction23,24. However, there is evidence to indicate that M1 and M2 receptors cannot be definitively excluded; for example, M1 receptors are responsible for bronchoconstriction in humans25, whereas tissue expression data seem to indicate that M2 receptors might be equally involved24.
For broad-spectrum antibacterials, Escherichia coli was selected as the representative species in ChEMBL. Thus, all broad-spectrum β-lactam antibiotics were linked to the seven PBPs from E. coli, even though it is clear that not all PBPs are targets for all β-lactams in all species26. In DrugCentral, however, susceptible pathogen species were assigned as targets based on antibacterial data reported as minimum inhibitory concentrations against well-defined pathogens. For example, finafloxacin, a fluoroquinolone approved for treating otitis caused by Pseudomonas aeruginosa and/or Staphylococcus aureus, was annotated as targeting both species. With minimum inhibitory concentration and species data available, a microbiologist can compare antibiotic potencies and susceptibilities, which are both important aspects of antibiotic efficacy. This strategy is complementary to the ChEMBL approach of annotating molecular targets; for example, all fluoroquinolones are annotated as E. coli DNA gyrase inhibitors. The DrugCentral approach focuses on the microorganism rather than the bacterial protein target because there is no clear evidence that the antibacterial would be efficacious in other species. Furthermore, bacteria have topical specificity in that some prefer the colon (for example, E. coli), whereas some prefer the nasopharyngeal sinus cavities, lung, kidney or skin. Antibiotics are prescribed differently for different infections. This difference makes target assignment even more complicated because particular antibiotics may be taken up via active transport into a certain tissue (where the infection is), whereas others may not. In addition, infections cause tissues to respond differently. For example, bacterial meningitis makes the blood–brain barrier leaky, enabling the use of antibiotics that otherwise do not cross this barrier but are effective in such infections. Such pathology-related phenomena are even more difficult to account for at a molecular level.
Oncology is a therapeutic area that further illustrates the challenges in defining efficacy targets. The FDA-approved drugs assigned to ATC categories L01 (antineoplastics) and L02 (endocrine therapies) can be broadly divided into three groups. The first group are cytotoxic agents that target human DNA and/or RNA, such as platinum compounds. The second group are cytotoxic agents that act at least partially through protein targets, such as DNA polymerase, DNA topoisomerase and the proteasome. Finally, drugs in the third group are those that are typically considered to be targeted therapeutics, such as kinase inhibitors. However, the assignment of a drug to the third group rather than the second group is complicated by the spectrum of targeting observed. Topoisomerase inhibitors, for example, are selective for their targets but are highly toxic. Conversely, some kinase inhibitors inhibit a wide range of normally functioning kinases and their associated pathways, and adverse reactions to these drugs have been reported in the clinic. A further challenge in the assignment of efficacy targets to cancer drugs is the rapidly growing number of kinase inhibitors (37 approved small-molecule protein kinase inhibitors worldwide as of June 2016). The original clinical hypothesis may be based on the alteration of a specific (EGFR)), but the resultant launched drug may inhibit a broad range of kinase targets, most of which function outside the deregulated pathway — although kinase signalling pathways are largely interconnected in cancer. For example, vandetanib, which was approved for the treatment of metastatic medullary thyroid cancer27, inhibits the kinase product of the oncogene RET, which is mutated in many patients with medullary thyroid cancer28. Vandetanib also inhibits other kinases with interconnecting pathways, such as EGFR and vascular endothelial growth factor receptor (VEGFR) pathways. Indeed, it is common for the prescribing information to list a large number of targets in the section describing the mechanism of action of the drug, and certainty about the importance of a single target can usually only be obtained when the drug is approved in conjunction with a companion diagnostic test (see Further information). However, in contrast to the situation with D2 receptor antagonists described above, the kinases listed as binding a drug in the prescribing information often act on interlinked pathways; thus, we have mechanistic reasons to suspect their involvement in the efficacy of the drug. Therefore, we attempted to include all proteins that are likely to contribute to the observed efficacy of a drug as part of our drug target list, and this list will change as our understanding of drug action improves.
Using these guidelines, the final assignment still requires substantial curation effort. For example, for dronedarone, an anti-arrhythmic drug approved in 2009, the FDA label (see Further information) states that it has anti-arrhythmic properties belonging to all four Vaughan–Williams classes, and that its mechanism of action is “unknown”. This statement implies that at a molecular level, it may have the capacity to modulate sodium channels (class I), β-adrenergic receptors (class II), voltage-gated potassium channels (class III) and L type calcium channels (class IV)23. Recently, blockade of the If current (funny current) via HCN (hyperpolarization-activated cyclic nucleotide-gated) channels — of which there are four subtypes, with HCN4 being the form most highly expressed in the sinoatrial node29 — was identified as the likely mechanism for the bradycardic effect of dronedarone30, rather than modulating L type calcium channels or β-adrenergic receptors. Notably, ivabradine, a recently approved cardiac drug, also blocks If currents via HCN channels29,31. However, the dronedarone study30 did not rule out a role for sodium channels or voltage-gated potassium channels in the overall efficacy of the drug, but focused purely on its bradycardic effect. Other studies have further suggested that the inhibition of inward-rectifier potassium channels (in particular Kir2.132,33) may contribute to the antifibrillatory efficacy of dronedarone. A recent review makes it clear that dronedarone has many anti-arrhythmic effects and has a complex mechanism of action that probably involves many different target classes to a greater or lesser extent34.
This complexity is reflected by the diversity of annotations included in other databases for this drug. For example, Rask-Andersen et al.11 assigned one voltage-gated potassium channel (Kv11.1; encoded by KCNH2) and two adrenergic receptors as targets for dronedarone, whereas the Therapeutic Targets Database lists only Kv1.5 (another voltage-gated potassium channel) as a target. DrugBank lists a total of 18 proteins (adrenergic receptors, sodium and potassium channels and L type calcium channels) for dronedarone, but all flagged with ‘pharmacological action unknown’ because, for their curator, their therapeutic role is uncertain. Finally, the IUPHAR/BPS Guide to Pharmacology database does not include any primary target information (or binding affinity data) for dronedarone, although it does describe its mechanism as involving adrenergic receptors and sodium, potassium and calcium channels35. In January 2016, none of these resources annotated HCN channels as a dronedarone target, even though this specific information was published several years ago and at least one follow up review dedicated to dronedarone agrees that HCN channel blockade may be an important mechanism of action34.
The complex case of dronedarone highlights that our annotations are only a snapshot that represents current knowledge. We will continue to curate and update our assignments in the ChEMBL, DrugCentral and canSAR databases as more experiments are performed and knowledge of drug mechanisms increases. Such complexity is also at the heart of the concept of network pharmacology — the proposal that often several simultaneous distinct points of intervention are required for drug action. It remains to be seen in practice what proportion of drugs absolutely require binding to multiple targets for their efficacy.
Drugs, targets and therapeutic areas
Target annotations were combined from the ChEMBL, DrugCentral and canSAR databases to provide a unified set of drug efficacy targets, provided in Supplementary information S2 (table). Using this approach, we identified 667 unique human protein efficacy targets and 189 pathogen protein efficacy targets (table 1).
Table 1.
Molecular targets of FDA approved drugs
| Targets | Drugs | |||||
|---|---|---|---|---|---|---|
| Drug target Class | Total targets | Small-molecule drug targets | Biologic drug target | Total drugs | Small molecules | Biologics |
| Human Protein | 667 | 549 | 146 | 1194 | 999 | 195 |
| Pathogen Protein | 189 | 184 | 7 | 220 | 215 | 5 |
| Other human biomolecules | 28 | 9 | 22 | 98 | 63 | 35 |
| Other pathogen biomolecules | 9 | 7 | 4 | 79 | 71 | 8 |
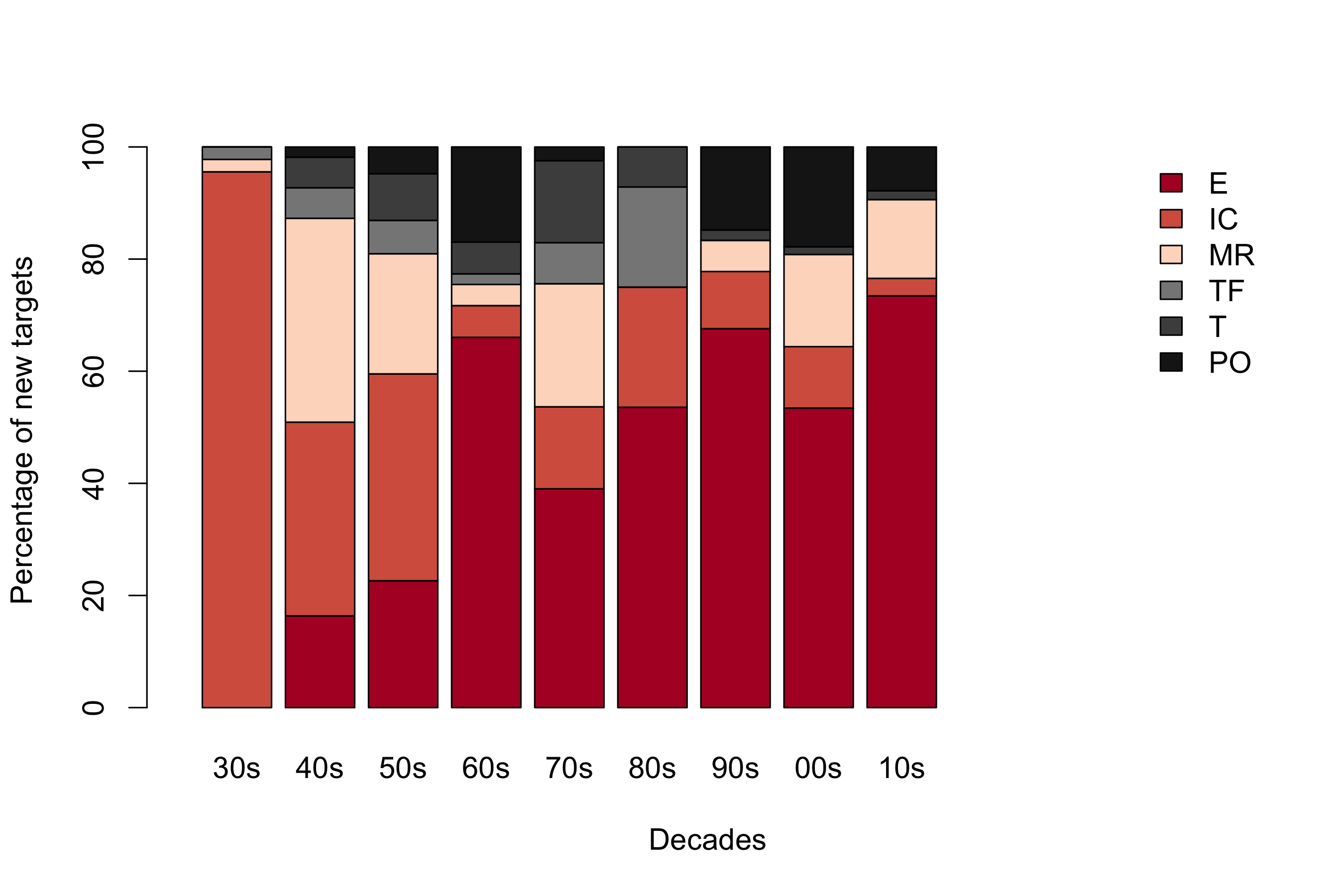
Using the ChEMBL hierarchical target classification system17, we then examined how the human protein targets distribute into homologous families and identified the most enriched ones. Rhodopsin-like G protein-coupled receptors (GPCRs; also known as 7TM1), ion channels, protein kinases and nuclear hormone receptors were considered to be privileged families given that they alone account for 44% of all human protein targets (GPCRs: 12%; ion channels: 19%; kinases: 10%; and nuclear receptors: 3% (figure 1a)). Moreover, owing to the variable number of approved drugs per target, these privileged families are responsible for the therapeutic effect of 70% of small-molecule drugs (GPCRs: 33%; ion channels: 18%; kinases: 3%; and nuclear receptors: 16% (figure 1a)).
Figure 1.

Major protein families as drug targets.
(a) Distribution of human drug targets by gene family. (b) distribution by the fraction of drugs targeting those families; the historical dominance of four families is clear. (c) Clinical success of privileged protein family classes. Distribution of non-approved compounds in ChEMBL 20 (extracted from the medicinal chemistry literature, with bioactivity tested against human protein targets) per family class, and distribution of approved drugs (small molecules and biologics) per human protein family class. 7TM, seven transmembrane family; GPCR, G protein-coupled receptor; LGIC, ligand-gated ion channel; NTPase, nucleoside triphosphatase; VGIC, voltage-gated ion channel.
There is a large difference between the drug and target fractions for protein kinases because of the broad polypharmacology typical of small-molecule kinase inhibitors, whereas the opposite is seen for nuclear receptors. The area of directed protein kinase inhibitors was highlighted in our original 2006 publication as an emerging target class7, and this trend has clearly continued. The remaining human protein efficacy targets are mostly unrelated enzymes. In the case of biologics, secreted or surface antigen proteins are the most important target class. This result is as expected given the highly restricted compartmental distribution of high-molecular-mass drugs within the body.
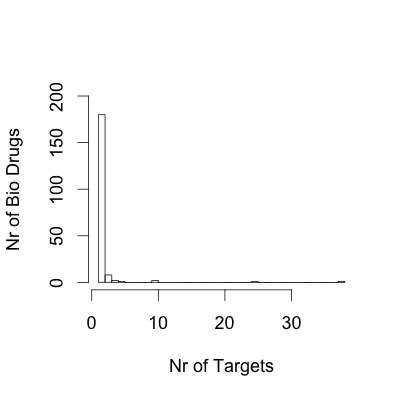
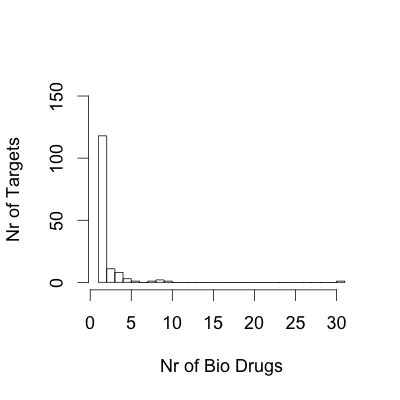
The number of drugs per target and the number of targets per drug are noteworthy in our analysis. By simple averaging, we obtain the figure of two drugs per target. However, this result is an oversimplification of complex pharmacology. Some targets have provided a rich ground for selective drugs, such as the glucocorticoid receptor (which has 61 approved drugs), whereas others fall into the opposite category, such as kinase inhibitors, for which few drugs act on many targets, thus contributing to the overall pharmacological response to those drugs (Supplementary information S3 (figure)). Another key developing trend is monoclonal antibody therapies, which are typically highly specific to a single gene product. This contrasts with small-molecule drugs, for which the interaction with multiple targets (polypharmacology) is more common.
Kinase inhibitors provide some of the best-known examples of polypharmacology because their bioactivity is routinely profiled against many kinases (and other targets) during the drug discovery and development process. This profiling was made possible by the introduction of high-throughput (in vitro) assay technologies. However, for most drugs, which were approved before 1990, this type of target profiling was not systematic; thus, our ability to understand polypharmacology both within and outside target families is a more recent endeavour.
The highly biased distribution in successfully ‘drugged’ protein families is also reflected in the biased distribution of bioactivity data from the ChEMBL database when examining the data at the target class level (figure 1b). ChEMBL is an open-access, large-scale bioactivity database containing manually extracted information from the medicinal chemistry literature together with data from United States Adopted Name (USAN) applications. Consequently, ChEMBL provides an unbiased reflection of small-molecule compounds at the lead optimization phase of drug discovery17. The protein-family-based organization of the data enables detailed examination of attrition during clinical development at a target family level (specifically, potent leads can be identified, but these may then fail in clinical development). Pooling the data by family enables a more robust statistical analysis and reduces the impact of specific targets on the analysis. As shown in figure 1b, it is clear that the discovery-phase investment in rhodopsin-like GPCRs has, at least until now, consistently paid off, because the fraction of approved drugs is slightly higher than the fraction of compounds in ChEMBL targeted to members of this family. The same relative enrichment (or survival) through clinical development is found for nuclear receptors, voltage-gated ion channels (VGICs), various reductases, electrochemical transporters and ligand-gated ion channels (LGICs). Curiously, in the case of nuclear receptors, no new efficacy target belonging to this family has emerged in recent years, although some are currently in trials (Supplementary information S4 (figure)). For protein kinases and proteases, the return in investment has shown the opposite trend. However, interest in protein kinases as drug targets is more recent (data not shown), and many potential kinase-directed drugs are still in active clinical development. For the extensively explored and high-attrition families — for example, the trypsin-like serine proteases — these data support the possibility that, on average, the family has low inherent druggability. Other examples from this simple data-driven analysis point to specific target-based attrition in some cases; for example, more than 40 mitogen-activated protein kinase p38α (also known as MAPK14) inhibitors have entered clinical trials, but have typically only progressed to, or stalled in, phase II trials.
To gain insight into drug innovation patterns by disease area, we linked a target to cognate drugs and then the drugs to their ATC codes. The number of small-molecule and biologic drugs per therapeutic area are shown in Table 2. We then grouped drugs per ATC level 3 code according to their worldwide or FDA approval year. As shown in figure 2, the maturity of the drugs targeting the cardiovascular system (category C) or the dermatological system (category D) is clear. By contrast, figure 2 also illustrates the recent innovation in the oncology and immunology areas (category L), as well as the recent lack of progress and small number of drugs available in the antiparasitic class (category P). A similar analysis at the target family level reveals a higher number of recently approved drugs that modulate kinases compared with the number of recently approved drugs that act through either nuclear receptors or ion channels (figure 3). Specifically, 20 protein kinase inhibitors have been approved by the FDA since 2011, accounting for 28% of all kinase-modulating drugs. This fraction would be even higher if only small molecules were considered in the analysis because biologics such as insulin derivatives (mainly approved before 1990) constitute a substantial portion of the kinase-modulating drugs (although these biologics do not bind to the protein kinase catalytic domain, which is typically used to define family membership).
Table 2.
Therapeutic areas of FDA approved drugs
This list also includes antimalarial drugs approved elsewhere in the world. ATC; WHO Anatomical Therapeutic Chemical Classification System.
| ATC category | Therapeutic area | Number of small molecules | Number of biologics |
|---|---|---|---|
| A | Alimentary tract and metabolism system | 158 | 32 |
| B | Blood and blood forming organs | 33 | 28 |
| C | Cardiovascular system | 200 | 5 |
| D | Dermatologicals | 141 | 5 |
| G | Genito urinary system | 94 | 5 |
| H | Hormonal system | 44 | 31 |
| J | Anti-infectives for systemic use | 194 | 10 |
| L | Antineoplastic and immunomodulating agents | 142 | 67 |
| M | Musculo-skeletal system | 62 | 6 |
| N | Nervous system | 239 | 1 |
| P | Antiparasitic products, insecticides and repellents | 38 | 1 |
| R | Respiratory system | 118 | 4 |
| S | Sensory organs | 143 | 11 |
| V | Various | 30 | 12 |
| U | Unclassified | 156 | 51 |
Figure 2.

Innovation patterns in therapeutic areas. Each node in the inner ring corresponds to a drug represented by its ATC code(s). The inner ring corresponds to the level 1 of the ATC code (see Table 2) scaled to the number of drugs in that category. The outer ring represents the level 3 of the ATC code. Each of the subsequent histograms illustrates the number of drugs (small molecules and biologics) distributed per year of first approval per level 3 of the ATC code. Max histogram scale: 100. The approval year refers to the first known worldwide approval date, if available, otherwise the first FDA approval date.
Figure 3.

Innovation patterns in privileged protein classes. Histogram depicting the number of drugs (small molecules and biologics) that modulate the four privileged families, distributed per year of first approval. On top of each bar, the total number of approved drugs is shown, together with the number and percentage of drugs approved since 2011 in respect to the total drugs modulating these four families. A spreadsheet view of this data is provided in supplementary information S6 (table). 7TM1: G-protein coupled receptor 1 family; Ion channel: Voltage-gated ion channel and Ligand-gated ion channel. Drugs without an ATC code (U – Unclassified) were excluded from this analysis.
Finally, to investigate the relationship between drugs, target classes and therapeutic areas, we again linked a target to cognate drugs and the drugs to their ATC codes, then connected drugs that share efficacy targets belonging to the same target class. In this way, we can analyse target family or functional class promiscuity across diseases or anatomical systems (figure 4). For example, if we look at several of the previously identified privileged target families — membrane receptors belonging to rhodopsin-like GPCRs, VGICs, LGICs and protein kinases — we see that rhodopsin-like GPCRs are targets for small-molecule drugs across almost every ATC class (figure 4a), with the exception of antiparasitic products (category P) and hormonal systems (category H). By contrast, protein kinases, which represent 13% of the protein human efficacy targets assigned to small molecules, only account for 2.4% of the small-molecule drugs, almost all of which are antineoplastic and immunomodulating agents (category L). This category is also represented when linking kinases assigned to biologic drugs, but for this type of drug, kinases seem to have an important role in other anatomical systems too. For biologics overall, only a small fraction of ATC categories are covered (figure 4b). As shown in figure 4a, the patterns created by ion channels are also distinct. Both VGICs and LGICs cover the musculoskeletal system (category M), the nervous system (category N), the alimentary tract and metabolism (category A), the respiratory system (category R), and the cardiovascular system (category C). The VGIC family also covers the dermatological system (category D) and the sensory system (category S). It is interesting to speculate that this clustering reflects a deeply rooted evolutionary relationship of various signalling and control subsystems of the body, and may provide additional guidance and constraints in effective drug repositioning and side-effect liability.
Figure 4.

Promiscuity of privileged protein family classes. Each node in the outer ring corresponds to a drug represented by its ATC code(s). The outer ring corresponds to the level 1 of the ATC code (see Table 2) scaled to the number of drugs in that category. The inner ring represents the level 2 of the ATC code. A node is connected to another when two drugs have an efficacy target that belongs to the same target class. (a) Footprint of privileged family classes modulated by organic small-molecule drugs across disease. (b) Footprint of privileged family classes modulated by biologic drugs across disease. G-protein coupled receptor 1 family (blue); Voltage-gated ion channel (orange); Ligand-gated ion channel (green); Kinase (black). Drugs without an ATC code (U – Unclassified) were excluded from this analysis.
Worldwide drug approvals
Although the analysis presented above is restricted to FDA-approved drugs and antimalarials approved in the rest of the world, we have also collated mechanism-of-action data on an additional set of ~1,200 drugs from WHO International Nonproprietary Names (INN) lists (see Further information) combined with literature searches35,36 to select drugs approved by other regulatory agencies. The vast majority of these drugs are members of the same chemical classes and share the same target (or targets) as an FDA-approved drug. For example, etoricoxib, a selective cyclooxygenase 2 (COX2) inhibitor, is approved in more than 80 countries but has not currently received FDA approval owing to safety concerns, whereas fimasartan, an angiotensin II receptor antagonist, is approved in South Korea only. Inclusion of these drugs identified eight additional, novel drug efficacy targets (table 3).
Table 3.
Drug efficacy targets unique to non FDA approved drugs
Tipiracil is an adjuvant used in the treatment of colorectal cancer to potentiate the action of trifluridine. ATC; WHO Anatomical Therapeutic Chemical Classification System, NA; not applicable
| Target Name | UniProt Accession Number | Example drug | ATC class | Indication |
|---|---|---|---|---|
| Aldose reductase | P15121 | Tolrestat | A10XA | Diabetic complications |
| Melanocyte-stimulating hormone receptor | Q01726 | Afamelanotide | D02BB | Erythropoietic Protoporphyria |
| P2Y purinoceptor 2 | P41231 | Diquafosol | NA | Dry eye |
| Rho-associated protein kinase 1 | Q13464 | Ripasudil | NA | Glaucoma |
| Rho-associated protein kinase 2 | O75116 | Ripasudil | NA | Glaucoma |
| Transthyretin | P02766 | Tafamidis | N07XX | Amyloidosis |
| Troponin C, slow skeletal and cardiac muscles | P63316 | Levosimendan | C01CX | Congestive heart failure |
| Thymidine phosphorylase | P19971 | Tipiracil | NA | Colorectal cancer |
Orthologues in animal models
Selecting the best model organism to study a particular disease or to validate a novel target mechanism involves identifying an induced disease state in a model organism with sufficient similarities to human pathology that a reliable prediction of the effects in humans may be made on the basis of the effects in the model organism. In practice, this is not straightforward. One approach that can be used to select a suitable model organism is to take the core human ‘pharmacolome’ (which we define here as the set of gene products that are modulated by current drugs) to compile a list of orthologues in typical model organisms. These genes can then be mapped back to the respective protein efficacy targets, the efficacy targets to the drugs and the drugs to the therapeutic indication (through the ATC code). Thus, from these data, one can infer which therapeutic areas are potentially best mimicked by which model organism. Figure 5 is a visualization of this information in a single plot (see Supplementary information S5 (figure) for a full-sized version). As in figure 4, the outer ring corresponds to the ATC categories scaled to the number of approved drugs in those categories. The inner ring is composed of ATC level 4 categories, which indicate the chemical, therapeutic and pharmacological subgroup. A series of heatmaps per species is then shown, coloured by how many of the protein efficacy targets are conserved for that ATC level 4 category between model organisms. The dark blue sections in Homo sapiens or E. coli heatmaps indicate that the drug target is a human protein or a bacterial protein. The conservation of efficacy targets is always with respect to the drug target species.
Figure 5.

Protein efficacy targets availability across several model organisms. (a) Each node in the outer ring corresponds to a drug represented by its ATC code(s). The outer ring corresponds to the level 1 of the ATC code (see Table 2), scaled to the number of drugs in that category. The next ring represents the level 4 of the ATC code. Each of the subsequent rings represents a different species, as indicated in the legend, and each section of the ring is coloured according with the presence or absence of orthologues of the efficacy targets of the drugs in that ATC level 4 category. The dark blue sections indicate the species of the protein efficacy targets. (b) An expanded portion of section J of the chart.
Overall, the vertebrates (dog, pig, rat, mouse and zebrafish) all provide comparatively good coverage of the set of human drug targets. In some cases, however, the apparent variation is due to the currently incomplete annotation in genome annotation and/or orthologue assignment for more recently completed genomes. As would be expected, the genomes of Drosophila and Caenorhabditis elegans contain fewer orthologues for human disease targets. The differences reflect anatomical systems that are substantially different or missing compared with humans. However, the degree of conservation varies significantly between the two species and between different therapeutic areas. For example, C. elegans retains many of the targets that are responsible for the efficacy of dermatological and genitourinary drugs, whereas these appear to be absent in Drosophila. When considering even simpler unicellular organisms such as yeast or E. coli, generally only targets reflecting core essential cellular functions, such as DNA, protein and nucleotide synthesis, remain.
When seeking to identify novel anti-infective targets, it is often proposed that absence of the corresponding protein in the host organism (normally humans) is an important prerequisite for success, and such constraints are often applied in bioinformatics filtering of potential targets. However, it can be seen from figure 5 that although several pathogen targets do lack human orthologues, there are a number of proteins that are also present in humans or other mammals. If the ribosome is considered, the number of pathogen targets with human orthologues increases even further. Dihydrofolate reductase (DHFR) inhibitors, for example, are used as antibacterial agents, antineoplastic agents and antiparasitic agents in humans. Antibacterial DHFR-targeted agents, such as trimethoprim, generally achieve sufficient selectivity and therapeutic index over the human systems to avoid mechanism-based toxicity.
Cancer drivers and cancer targets
A substantial proportion of drug discovery efforts in the past decade have involved the rational selection of mechanistic cancer drivers to be targeted37. Moreover, cancer is the area of biggest growth in large-scale systematic efforts to identify disease drivers, powered by major international consortia38–41, and so it is interesting to consider the impact of such efforts on the identification of novel clinically validated targets. The 154 cancer drugs approved by the FDA can be broadly divided into the three groups mentioned above: 26 drugs are cytotoxic agents; 38 drugs are broadly cytotoxic and act at least partially through protein targets, such as proteasome inhibitors; and 85 drugs can be assigned to clear mechanistic protein targets. A further 5 drugs act through unknown or non-protein targets. Systematic efforts to identify cancer drivers based on ‘omics’ data have contributed considerably to the growth in the number of drugs in the third category in recent years. The impact of such approaches is clearly illustrated by the discovery in 2002 of BRAF as the major driver for malignant melanoma, which led to the approval in 2011 of the BRAF inhibitor vemurafenib as the first targeted therapy for melanoma. Subsequently, the MEK inhibitor trametinib, which targets the same signalling pathway as vemurafenib, was approved in 2013. Another example is the discovery of EML4–ALK driver translocation in non-small-cell lung cancer42, leading to the approval of the ALK inhibitor crizotinib in 2011.
The relationship between drug mechanisms and bona fide cancer drivers merits consideration. We have previously analysed the trends in identifying cancer drivers and have shown that multiple studies are converging towards ~600 cancer drivers across different cancers43. We compared the lists of consensus 553 cancer drivers43 to the list of 109 protein targets of the 85 protein-targeted cancer drugs described above (figure 6) and found a small overlap (30 proteins) between the two sets. There are several reasons for this small overlap. Despite the large numbers of patients involved in these studies, they can be biased in their composition and in the statistical methodologies used to select drivers; hence, a gene may fall short of the final statistical prevalence cut-off. Another reason is that many of these drivers are newly discovered cancer-associated genes for which there has been little historical biological investigation; thus, time will tell whether they can yield useful targets for drug discovery. Indeed, our own analysis indicated that at least 10% of cancer drivers are likely to be druggable by small-molecule drugs, but such investigations had not been reported in the medicinal chemistry literature43–45. Finally, and importantly, non-oncogene addiction has been — and will remain — a key aspect of cancer that can be therapeutically targeted46,47. This trend is exemplified by FDA-approved hormone-recognition- and hormone-biosynthesis-targeting agents such as aromatase inhibitors for breast cancer, cytochrome P450 family 17 subfamily A member 1 (CYP17A1) inhibitors for prostate cancer, and poly(ADP-ribose) polymerase (PARP) inhibitors for ovarian cancer. Agents under clinical investigation exploiting synthetic lethality to oncogenes include PARP inhibitors in DNA damage repair-deficient prostate cancer48. Other agents are exploiting non-oncogene addiction; for example, VLX 1570 inhibits proteasome 19s associated protein ubiquitin-specific peptidase 14 (USP14) to exploit 19s addiction in multiple myeloma49. Furthermore, many cancer genes are loss of function drivers; in such cases, the gene has been deleted or disabled through the genomic aberration, and targeting these genes will typically require a synthetic lethality approach. Thus, systematic mapping of disease drivers can indicate future therapeutic strategies both by identifying potential targets and by highlighting key pathways that can be drugged.
Figure 6.

Overlap of cancer drug targets with cancer drivers. We grouped the cancer drugs into the three categories: broadly cytotoxic agents such as platinum complexes and DNA intercalating agents; cytotoxic agents that act through a protein, such as tubulin inhibitors, that do not have biological selectivity; and targeted agents that act through clear protein function-modulating mechanisms such as kinase inhibitors and nuclear hormone receptor antagonists. When we compared the targets of agents in the third group to a consensus reference list on cancer driver genes43 we observe only a small overlap between cancer drivers and current cancer drug targets.
Concluding thoughts and future work
In this article, we have provided an enhanced and updated perspective on the current diversity of approved drugs and their targets, with a focus on the trends and changes over the past 10 years7. Compiling an accurate and agreed list of drug efficacy targets is not a trivial task, and with work from three teams we have made significant progress towards this goal, as well as highlighted some of the practical challenges. These challenges include resolving the non-trivial relationship between a gene and a drug target, assigning the target, and finally establishing a convention to deal with complexes, subunits and splice variants and protein isoforms when counting final effective molecular targets — a major factor in the increase in the number of protein targets to 667 from the 324 identified in our previous study7. All of this is a prerequisite for analysing the diversity of existing drugs and targets in the light of their disease coverage. However, as the literature changes continuously in terms of the knowledge available about the mechanisms of action of drugs, these annotations will need to be updated frequently. Consequently, this data set will be maintained and made publicly accessible. A subset of the merged drug efficacy target data (referred to as Tclin) is currently available on the Illuminating the Druggable Genome website; see Further information).
Interestingly, in the 10 years since the publication of our previous enumeration of drug targets, privileged families such as rhodopsin-like GPCRs, nuclear receptors and VGICs have largely maintained their dominance of the drug target space, underlining the continuous utility of protein families endowed with druggable binding sites. The major changes over the past decade are in the proportion of protein kinase and protease targets; together these previously made up <2% of the total target set, and now represent 6% (protein kinases) and 4% (proteases) of all targets of approved drugs. Reassuringly, the long tail of single exemplar targets from several underrepresented families continues to grow, indicating our ability to innovate in drug discovery.
It is interesting to speculate on the relative contribution of phenotypic versus targeted screens to the discovery of first in class drugs50,51. The simplest view would be that small-molecule drugs for which polypharmacology is required for their action, such as sunitinib, are more likely to have been discovered through phenotypic rather than targeted screens. However, the data may indicate the opposite. Because phenotypic screens are often optimized against mechanistic and pharmacodynamic biomarker modulation, there is pressure towards more specific pharmacology of drugs discovered in this way. By contrast, discovering a small-molecule drug through a target-based screen optimizes the activity of the drug against the desired target, and selectivity against a few identified off-targets, without properly investigating the broader cellular activity of the agent.
As data on tissue expression and causal models mapping molecular to clinical events continue to emerge, the relationship between drug efficacy targets and the tissue localization of disease will progressively be accounted for, because drug action is more likely to be exerted in the tissue of choice. For example, although the anti-Parkinsonian drug ropinirole is more potent at the D3 receptor than the D2 receptor by an order of magnitude, we annotate the D2 receptor as the mechanism of action target because D2 receptors, but not D3 receptors, are expressed in the substantia nigra, the pathologically relevant tissue for anti-Parkinsonian drugs (see Further information). Future efficacy target annotations are anticipated to make extensive use of unambiguous tissue colocalization data for both target and disease.
As our understanding of the causes of complex disease deepens, we find that such diseases involve a combination of environmental factors, genetic and epigenetic dysfunction. Thus, will a reductionist approach to targeted therapy still have a role in the future? Regardless of the initial cause, most human disease is either initiated or mediated by the aberrant action of proteins. Hence, an armoury of mechanistically sophisticated and thoroughly experimentally annotated drugs that target this complexity is required, including incorporation of drug combinations52, network drugs53 and polypharmacology54. These approaches are of particular importance in cancer and infectious disease, for which heterogeneity and evolution under the selective pressure of standard-of care drugs results in the emergence of drug resistance.
Medical care has two goals: to properly diagnose the disease and to select the appropriate therapeutic. As long as (drug-induced) phenotypic alterations are observed under appropriate conditions, it is possible to steer medical care and adjust the therapeutic management for a better outcome. Hence, the key to successful drug discovery and application resides in the seamless integration of pathological mechanisms of disease (that is, molecular and cellular level processes) with diagnoses (clinical embodiments of disease at the organ and/or organism level) and therapeutics (that is, modulating clinical manifestations at the molecular level via therapeutics).
Drug discovery and targeting remains a complex, costly and at times unpredictable process. However, used alongside the new insights into disease and fundamental biology that are emerging, we hope that knowledge about the associations between currently successful drugs, their efficacy targets, phenotypic effects and disease indications that we have reported here can help to contribute to the efficient discovery of a new generation of medicines.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
The work of the authors is supported by the following institutes, organizations and grants: (1) Wellcome Trust Strategic Awards WT086151/Z/08/Z, WT104104/Z/14/Z to J.P.O., A.G., A.H. and A.P.B.; (2) the member states of EMBL to R.S., R.S.D, A.G., A.H., A.P.B.); (3) US National Institutes of Health (NIH) grants 1U54CA189205 01 to O.U., A.G., A.H., A.K., C.G.B., T.I.O. and J.P.O., and NIH grants P30CA118100 and UL1TR001449 to T.I.O.; and (4) B.A.-L. is funded by the Institute of Cancer Research. canSAR is funded by The Cancer Research UK grant to the Cancer Research UK Cancer Therapeutics Unit (grant C309/A11566). The authors thank many of their collaborators for discussions and valuable input in the preparation of this manuscript, in particular the members of the Illuminating the Druggable Genome consortium.
Footnotes
Competing interests
The authors declare no competing interests.
Further Information
canSAR: https://cansar.icr.ac.uk
ChEMBL: https://www.ebi.ac.uk/chembl
Companion diagnostic test: http://www.fda.gov/companiondiagnostics
DRD2 — GTEx entry page: http://www.gtexportal.org/home/gene/DRD2
DRD3 — GTEx entry page: http://www.gtexportal.org/home/gene/DRD3
Dronedarone prescribing information: http://www.accessdata.fda.gov/drugsatfda_docs/label/2013/022425s021lbl.pdf
DrugCentral: http://drugcentral.org
Illuminating the Druggable Genome: https://pharos.nih.gov/idg/index
Inkscape: https://inkscape.org/en
IUPHAR/BPS Guide to Pharmacology: http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?tab=biology&ligandId=7465
NCATS Pharmaceutical Collection: https://tripod.nih.gov/npc/
Pharmaceuticals and Medical Devices Agency list of approved drugs: http://www.pmda.go.jp/english/review-services/reviews/approved-information/drugs/0002.html
R Project for Statistical Computing: http://www.r-project.org
WHO Collaborating Centre for Drug Statistics Methodology — ATC/DDD Index: http://www.whocc.no/atc_ddd_index
WHO INN Drug lists: http://www.who.int/medicines/publications/druginformation/innlists/en
References
- 1.Raju TN. The Nobel chronicles. Lancet. 2000;355:1022. doi: 10.1016/s0140-6736(05)74775-9. [DOI] [PubMed] [Google Scholar]
- 2.Drews J. Genomic sciences and the medicine of tomorrow. Nat Biotechnol. 1996;14:1516–1518. doi: 10.1038/nbt1196-1516. [An early and influential review on the prospects for genomics and drug discovery.] [DOI] [PubMed] [Google Scholar]
- 3.Drews J, Ryser S. The role of innovation in drug development. Nat Biotechnol. 1997;15:1318–1319. doi: 10.1038/nbt1297-1318. [DOI] [PubMed] [Google Scholar]
- 4.Hopkins AL, Groom CR. The druggable genome. Nat Rev Drug Discov. 2002;1:727–30. doi: 10.1038/nrd892. [First attempt to define the future drugabble genome on the basis of successful drug development programs.] [DOI] [PubMed] [Google Scholar]
- 5.Golden JB. Prioritizing the human genome: knowledge management for drug discovery. Curr Opin Drug Discov Devel. 2003;6:310–316. [PubMed] [Google Scholar]
- 6.Imming P, Sinning C, Meyer A. Drugs, their targets and the nature and number of drug targets. Nat Rev Drug Discov. 2006;5:821–835. doi: 10.1038/nrd2132. [DOI] [PubMed] [Google Scholar]
- 7.Overington JP, Al-Lazikani B, Hopkins AL. How many drug targets are there? Nat Rev Drug Discov. 2006;5:993–6. doi: 10.1038/nrd2199. [A ten-year old review on the then-known landscape of drug targets.] [DOI] [PubMed] [Google Scholar]
- 8.Chen X, Ji ZL, Chen YZ. TTD: Therapeutic Target Database. Nucleic Acids Res. 2002;30:412–5. doi: 10.1093/nar/30.1.412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wishart DS, et al. DrugBank: a comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res. 2006;34:D668–72. doi: 10.1093/nar/gkj067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Günther S, et al. SuperTarget and Matador: resources for exploring drug-target relationships. Nucleic Acids Res. 2008;36:D919–22. doi: 10.1093/nar/gkm862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rask-Andersen M, Almén MS, Schiöth HB. Trends in the exploitation of novel drug targets. Nat Rev Drug Discov. 2011;10:579–590. doi: 10.1038/nrd3478. [A more recent review on the landscape of drug targets.] [DOI] [PubMed] [Google Scholar]
- 12.Munos B. A Forensic Analysis of Drug Targets from 2000 through 2012. Clin Pharmacol Ther. 2013;94:407–411. doi: 10.1038/clpt.2013.126. [A recent overview of drug and target approvals for 2000-2012.] [DOI] [PubMed] [Google Scholar]
- 13.Agarwal P, Sanseau P, Cardon LR. Novelty in the target landscape of the pharmaceutical industry. Nat Rev Drug Discov. 2013;12:575–6. doi: 10.1038/nrd4089. [Drug target novelty in industry; this paper addressed the target diversity across large pharma - are all companies pursuing the same targets?] [DOI] [PubMed] [Google Scholar]
- 14.Pawson AJ, et al. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledgebase of drug targets and their ligands. Nucleic Acids Res. 2014;42:D1098–106. doi: 10.1093/nar/gkt1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huttunen KM, Raunio H, Rautio J. Prodrugs—from Serendipity to Rational Design. Pharmacol Rev. 2011;63:750–771. doi: 10.1124/pr.110.003459. [DOI] [PubMed] [Google Scholar]
- 16.Tym JE, et al. canSAR: an updated cancer research and drug discovery knowledgebase. Nucleic Acids Res. 2016;44:D938–43. doi: 10.1093/nar/gkv1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bento AP, et al. The ChEMBL bioactivity database: an update. Nucleic Acids Res. 2014;42:D1083–90. doi: 10.1093/nar/gkt1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Meltzer HY, Roth BL. Lorcaserin and pimavanserin: emerging selectivity of serotonin receptor subtype–targeted drugs. J Clin Invest. 2013;123:4986–4991. doi: 10.1172/JCI70678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Friedman JH. Pimavanserin for the treatment of Parkinson’s disease psychosis. Expert Opin Pharmacother. 2013;14:1969–1975. doi: 10.1517/14656566.2013.819345. [DOI] [PubMed] [Google Scholar]
- 20.Bandelow B, Meier A. Aripiprazole, a ‘Dopamine-Serotonin System Stabilizer’ in the Treatment of Psychosis. German J Psychiatry. 2003;6:9–16. [Google Scholar]
- 21.Mamo D, et al. Differential effects of aripiprazole on D(2), 5-HT(2), and 5-HT(1A) receptor occupancy in patients with schizophrenia: a triple tracer PET study. Am J Psychiatry. 2007;164:1411–1417. doi: 10.1176/appi.ajp.2007.06091479. [DOI] [PubMed] [Google Scholar]
- 22.Tolboom N, et al. The dopamine stabilizer (-)-OSU6162 occupies a subpopulation of striatal dopamine D2/D3 receptors: an [(11)C]raclopride PET study in healthy human subjects. Neuropsychopharmacology. 2015;40:472–479. doi: 10.1038/npp.2014.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rang HP, Dale MM, Ritter JM, Flower RJ, Henderson G. Pharmacology. Elsevier Health Sciences UK; 2012. [A comprehensive and classic book on pharmacology and drug mode of action.] [Google Scholar]
- 24.Koarai A, et al. Expression of muscarinic receptors by human macrophages. Eur Respir J. 2012;39:698–704. doi: 10.1183/09031936.00136710. [DOI] [PubMed] [Google Scholar]
- 25.Lammers JW, Minette P, McCusker M, Barnes PJ. The role of pirenzepine-sensitive (M1) muscarinic receptors in vagally mediated bronchoconstriction in humans. Am Rev Respir Dis. 1989;139:446–449. doi: 10.1164/ajrccm/139.2.446. [DOI] [PubMed] [Google Scholar]
- 26.Krauss J, van der Linden M, Grebe T, Hakenbeck R. Penicillin-binding proteins 2x and 2b as primary PBP targets in Streptococcus pneumoniae. Microb Drug Resist. 1996;2:183–186. doi: 10.1089/mdr.1996.2.183. [DOI] [PubMed] [Google Scholar]
- 27.Wells SA, et al. Vandetanib in Patients with Locally Advanced or Metastatic Medullary Thyroid Cancer: A Randomized, Double-Blind Phase III Trial. J Clin Oncol. 2012;30:134–141. doi: 10.1200/JCO.2011.35.5040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Santoro M, et al. Molecular biology of the MEN2 gene. J Intern Med. 1998;243:505–508. doi: 10.1046/j.1365-2796.1998.00330.x. [DOI] [PubMed] [Google Scholar]
- 29.Thollon C, et al. Use-dependent inhibition of hHCN4 by ivabradine and relationship with reduction in pacemaker activity. Br J Pharmacol. 2007;150:37–46. doi: 10.1038/sj.bjp.0706940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sobrado LF, et al. Dronedarone’s inhibition of If current is the primary mechanism responsible for its bradycardic effect. J Cardiovasc Electrophysiol. 2013;24:914–918. doi: 10.1111/jce.12155. [DOI] [PubMed] [Google Scholar]
- 31.Bucchi A, et al. Identification of the Molecular Site of Ivabradine Binding to HCN4 Channels. PLoS One. 2013;8:e53132. doi: 10.1371/journal.pone.0053132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xynogalos P, et al. Class III antiarrhythmic drug dronedarone inhibits cardiac inwardly rectifying Kir2.1 channels through binding at residue E224. Naunyn Schmiedebergs Arch Pharmacol. 2014;387:1153–1161. doi: 10.1007/s00210-014-1045-6. [DOI] [PubMed] [Google Scholar]
- 33.Gómez R, et al. Structural basis of drugs that increase cardiac inward rectifier Kir2.1 currents. Cardiovasc Res. 2014;104:337–346. doi: 10.1093/cvr/cvu203. [DOI] [PubMed] [Google Scholar]
- 34.Heijman J, Heusch G, Dobrev D. Pleiotropic effects of antiarrhythmic agents: dronedarone in the treatment of atrial fibrillation. Clin Med Insights Cardiol. 2013;7:127–140. doi: 10.4137/CMC.S8445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brayfield A. Martindale: the complete drug reference. Pharmaceutical Press; 2014. [Google Scholar]
- 36.Swiss Pharmaceutical Society. Index Nominum: International Drug Directory. Medpharm Scientific Publishers; 2004. [Google Scholar]
- 37.Allen TM. Ligand-targeted therapeutics in anticancer therapy. Nat Rev Cancer. 2002;2:750–763. doi: 10.1038/nrc903. [DOI] [PubMed] [Google Scholar]
- 38.International Cancer Genome Consortium et al. International network of cancer genome projects. Nature. 2010;464:993–998. doi: 10.1038/nature08987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kandoth C, et al. Mutational landscape and significance across 12 major cancer types. Nature. 2013;502:333–339. doi: 10.1038/nature12634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vogelstein B, et al. Cancer genome landscapes. Science. 2013;339:1546–1558. doi: 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lawrence MS, et al. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature. 2014;505:495–501. doi: 10.1038/nature12912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Soda M, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. 2007;448:561–6. doi: 10.1038/nature05945. [DOI] [PubMed] [Google Scholar]
- 43.Workman P, Al-Lazikani B. Drugging cancer genomes. Nat Rev Drug Discov. 2013;12:889–890. doi: 10.1038/nrd4184. [DOI] [PubMed] [Google Scholar]
- 44.Fletcher JI, Haber M, Henderson MJ, Norris MD. ABC transporters in cancer: more than just drug efflux pumps. Nat Rev Cancer. 2010;10:147–156. doi: 10.1038/nrc2789. [DOI] [PubMed] [Google Scholar]
- 45.Patel MN, Halling-Brown MD, Tym JE, Workman P, Al-Lazikani B. Objective assessment of cancer genes for drug discovery. Nat Rev Drug Discov. 2013;12:35–50. doi: 10.1038/nrd3913. [A review of cancer drug targets, and approaches to prioritise target selection] [DOI] [PubMed] [Google Scholar]
- 46.Luo J, Solimini NL, Elledge SJ. Principles of cancer therapy: oncogene and non-oncogene addiction. Cell. 2009;136:823–837. doi: 10.1016/j.cell.2009.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Weinstein IB, Joe A. Oncogene addiction. Cancer Res. 2008;68:3077–80. doi: 10.1158/0008-5472.CAN-07-3293. discussion 3080. [DOI] [PubMed] [Google Scholar]
- 48.Matteo J, et al. DNA-Repair Defects and Olaparib in Metastatic Prostate Cancer. N Engl J Med. 2015;373:1697–708. doi: 10.1056/NEJMoa1506859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shukla N. Proteasome Addiction Defined in Ewing Sarcoma Is Effectively Targeted by a Novel Class of 19S Proteasome Inhibitors. Cancer Res. 2016;76:4525–34. doi: 10.1158/0008-5472.CAN-16-1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Eder J, Sedrani R, Wiesmann C. The discovery of first-in-class drugs: origins and evolution. Nat Rev Drug Discov. 2014;13:577–587. doi: 10.1038/nrd4336. [DOI] [PubMed] [Google Scholar]
- 51.Swinney DC, Anthony J. How were new medicines discovered? Nat Rev Drug Discov. 2011;10:507–19. doi: 10.1038/nrd3480. [DOI] [PubMed] [Google Scholar]
- 52.Al-Lazikani B, Workman P. Unpicking the combination lock for mutant BRAF and RAS melanomas. Cancer Discov. 2013;3:14–19. doi: 10.1158/2159-8290.CD-12-0520. [DOI] [PubMed] [Google Scholar]
- 53.Workman P, Clarke PA, Al-Lazikani B. Blocking the survival of the nastiest by HSP90 inhibition. Oncotarget. 2016;7:3658–3661. doi: 10.18632/oncotarget.6971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Paolini GV, Shapland RHB, van Hoorn WP, Mason JS, Hopkins AL. Global mapping of pharmacological space. Nat Biotechnol. 2006;24:805–815. doi: 10.1038/nbt1228. [DOI] [PubMed] [Google Scholar]
- 55.Huttunen KM, Raunio H, Rautio J. Prodrugs — from serendipity to rational design. Pharmacol Rev. 2011;63:750–771. doi: 10.1124/pr.110.003459. [DOI] [PubMed] [Google Scholar]
- 56.Huang R, et al. The NCGC pharmaceutical collection: a comprehensive resource of clinically approved drugs enabling repurposing and chemical genomics. Sci Transl Med. 2011;3:80ps16. doi: 10.1126/scitranslmed.3001862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kinsella RJ, et al. Ensembl BioMarts: a hub for data retrieval across taxonomic space. Database (Oxford) 2011;2011 doi: 10.1093/database/bar030. bar030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Flicek P, et al. Ensembl 2012. Nucleic Acids Res. 2012;40:D84–90. doi: 10.1093/nar/gkr991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sonnhammer ELL, Östlund G. InParanoid 8: orthology analysis between 273 proteomes, mostly eukaryotic. Nucleic Acids Res. 2015;43:D234–D239. doi: 10.1093/nar/gku1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.United States Pharmacopeial Convention. USP Dictionary of USAN and International Drug Names 2010. United States Pharmacopeia; 2010. [Google Scholar]
- 61.Gleeson MP, Hersey A, Montanari D, Overington J. Probing the links between in vitro potency, ADMET and physicochemical parameters. Nat Rev Drug Discov. 2011;10:197–208. doi: 10.1038/nrd3367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Krzywinski M, et al. Circos: an information aesthetic for comparative genomics. Genome Res. 2009;19:1639–45. doi: 10.1101/gr.092759.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.