

ABSTRACT
Biliary atresia (BA) is the most common cause of pediatric end-stage liver disease and the etiology is poorly understood. There is no effective therapy for BA partly due to lack of human BA models. Towards developing in vitro human models of BA, disease-specific induced pluripotent stem cells (iPSCs) from 6 BA patients were generated using non-integrating episomal plasmids. In addition, to determine the functional significance of BA-susceptibility genes identified by genome-wide association studies (GWAS) in biliary development, a genome-editing approach was used to create iPSCs with defined mutations in these GWAS BA loci. Using the Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)/Cas9 system, isogenic iPSCs deficient in BA-associated genes (GPC1 and ADD3) were created from healthy iPSCs. Both the BA patient-iPSCs and the knock out (KO) iPSCs were studied for their in vitro biliary differentiation potential. These BA-specific iPSCs demonstrated significantly decreased formation of ductal structures, decreased expression of biliary markers including CK7, EpCAM, SOX9, CK19, AE2, and CFTR and increased fibrosis markers such as alpha smooth muscle actin, Loxl2, and Collagen1 compared to controls. Both the patient- and the KO-iPSCs also showed increased yes-associated protein (YAP, a marker of bile duct proliferation/fibrosis). Collagen and YAP were reduced by treatment with the anti-fibrogenic drug pentoxifylline. In summary, these BA-specific human iPSCs showed deficiency in biliary differentiation along with increased fibrosis, the 2 key disease features of BA. These iPSCs can provide new human BA models for understanding the molecular basis of abnormal biliary development and opportunities to identify drugs that have therapeutic effects on BA.
Keywords: anti-fibrotic drugs, biliary differentiation, neonatal cholestasis, patient-derived stem cells
What Is Known
Although the cholangiopathy and fibrosis of BA are well-documented, the etiology is poorly understood.
GWAS have identified BA-susceptibility genes however the functionality in human is unknown.
YAP has been reported as a marker for biliary proliferation/fibrosis.
What Is New
Patient-iPSCs and BA-gene-knockout-iPSCs showed reduced biliary differentiation with increased fibrosis, two key features of the disease.
These demonstrate the first evidence of modeling BA utilizing human iPSCs and support the feasibility of gene-editing to determine the functionality of GWAS-genes in human biliary differentiation/development.
Increased YAP/collagen in BA-iPSCs were lowered with anti-fibrotic drug treatment.
BA-iPSCs can provide human-relevant models/assays for understanding pathogenesis and identifying therapeutic-drugs.
Biliary atresia (BA) is a serious infantile liver disease of unknown etiology (1–3). Although the inflammatory obliterative cholangiopathy characteristic of BA is well documented, understanding its cause remains a challenge. Animal models with various features of BA have been developed; however, most of them provide incomplete insight to the etiology (3,4). The model that most closely resembles BA was developed by infecting newborn Balb/c mice with Rhesus Rotavirus. The short life-span of the treated pups and the complexity of the experimental procedure, however, limit its application in both mechanistic study and drug development. More importantly, there is a fundamental difference between the murine BA model and the human disease in that mice do not develop severe liver fibrosis (3). A human-specific model is urgently needed to facilitate mechanistic understanding of BA-related genes in human hepatobiliary development.
Human iPSC (induced pluripotent stem cell) technology provides relevant cell sources for disease modeling and drug discovery using patient tissues as we have shown with another liver disease, alpha-1 antitrypsin deficiency (5,6). The human iPSC-based system offers several advantages over conventional cellular and animal models. First, virtually all cellular lineages can be obtained from iPSCs through directed step-wise differentiation due to the pluripotency (7–11). The pluripotency of iPSC permits future studies of genotype-phenotype relationships in multi-cellular or multi-organ systems, which is not feasible with conventional tools. In addition, iPSCs are amenable to genome editing (6,12–17), which allows functional validation of candidate genetic elements in disease development. Combined with their directed differentiation capability, precisely gene edited isogenic iPSC sets will provide novel, human-relevant, and renewable tools for translation of genomic data into knowledge of organogenesis, pathogenesis, and therapeutic possibilities, without using a large number of samples.
The probable cause of BA is multifactorial with genetic predisposition(s), environmental factors and immune dysregulations all been implicated (1,2,18–22). Significant progress has been made in the past few years towards understanding the genetic basis of the disease. GWAS studies have identified glypican 1 (GPC1), adducin 3 (ADD3) and ADP-ribosylation factor 6 (ARF6), genes important in embryonic development and organogenesis, as potential BA susceptibility genes (23–29). Importantly, knock-down of these genes in zebrafish has resulted in developmental biliary defects, a key BA disease feature (25,28). We have previously developed defined and controlled conditions for efficient 2D and 3D bile duct tissue generation from human iPSCs based on our hepatic differentiation methods (5,6,9,30–33) and have recently generated iPSCs from patients with BA (34). To determine if human iPSCs would recapitulate the BA disease phenotypes of biliary differentiation defect and increased fibrosis, the aims of this study were to establish and expand the number of lines of BA patient-specific iPSCs and to use the CRISPR/Cas9-based precise genome editing in healthy iPSC lines to create a panel of lines each having a defined mutation in one of the BA susceptibility loci.
Here we show that in vitro biliary differentiation of BA-specific human iPSC lines, demonstrate decreased formation of ductal structures and decreased expression of phenotypic and functional biliary tissue markers as well as increased fibrosis markers, compared to their respective controls. In addition, these BA-iPSCs also showed increased yes-associated protein (YAP) expression, a marker associated with bile duct proliferation and fibrosis in BA patients (19), along with increased collagen expression, both of which can be lowered with anti-fibrotic drug treatment. These results demonstrate that BA disease features can be recapitulated in vitro using human iPSCs. These disease-specific iPSC lines along with the biliary differentiation technology will provide a new human BA model for understanding molecular basis of abnormal bile duct tissue development, and opportunities to identify agents that have potential therapeutic effects on BA.
METHOD
Cell Culture and Biliary Differentiation of Human Induced Pluripotent Stem Cells
Human iPSC lines were generated from BA patients’ blood samples using our previously established protocol (34) (see Table 1 for clinical characteristics of patients). All human iPSCs were cultured in a feeder free condition on Matrigel (Corning, NY, USA) using mTeSR (STEMCELL Technologies, Vancouver, CA) as we described previously (5,6,8,9,11,30–32,34,35). This study was conducted in accordance with Johns Hopkins Institutional Stem Cell Research Oversight Committee regulations and following a protocol approved by the Johns Hopkins Institutional Review Board. Biliary differentiation was performed by a step-wise protocol described previously (Fig. 1A) (33). Expression of various cholangiocyte markers and ductal structure formation capability after inducing biliary differentiation from the iPSCs were analyzed together with multiple fibrosis markers. Functional differences of these iPSC-derived biliary tissues were also determined using their secretory function assays. For drug testing, the mature stage biliary cells were treated in the biliary differentiation medium (33) containing 0.5 mM pentoxifylline (Sigma, St. Louis, MO, Cat. P1784) for 5 days, while pentoxifylline-free medium was used as a control.
TABLE 1.
Clinical Characteristics of Children With Biliary Atresia and Control Induced Pluripotent Stem Cells
| Age at Enrollment | Total Bilirubin at Diagnosis, mg/dL | Total Bilirubin at Enrollment, mg/dL | Clinical Status | |
| BA patient iPSCs | ||||
| iBA1 | 11y and 4mo | 5.8 | 2.4 | Liver transplantation 265 days old (Figs. 1 and 3) |
| iBA3 | 15mo | 8.7 | 0.9 | Portal hypertension (Figs. 1 and 3) |
| iBA5 | 16y | 11.4 | 0.6 | Liver transplantation 1.6 years old (Figs. 1 and 3) |
| iBA6 | 31days | 6.4 | 6.4 | Enrolled at diagnosis (Figs. 1 and 3) |
| iBA8 | 2y and 5mo | 14.3 | 7.0 | Liver Transplantation 2.5 years old (Figs. 1 and 3) |
| iBA16 | 3y and 4mo | 7.4 | 0.4 | Midline liver, hetereotaxy syndrome with polysplenia; liver transplantation; 1.3 years old (Supplemental Figure 4) |
| Non BA control iPSCs | ||||
| iJS02 | 6y 11mo | n/a | n/a | Underwent endoscopy for gastro-esophageal reflux, anicteric |
| iJS09 | 17y 9mo | n/a | n/a | Underwent endoscopy for gastro-esophageal reflux, anicteric |
| iAAT2 | 3mo | n/a | n/a | Alpha 1 antitrypsin deficiency |
| iAAT3 | 4mo | n/a | n/a | Alpha 1 antitrypsin deficiency |
| iM9 | 1do | n/a | n/a | Healthy |
| iHu71 | 7yo | n/a | n/a | Healthy (used for Figs. 1 and 3, and as a parental control for KO iPSCs in Fig. 2) |
BA = biliary atresia; iPSC = induced pluripotent stem cell.
FIGURE 1.
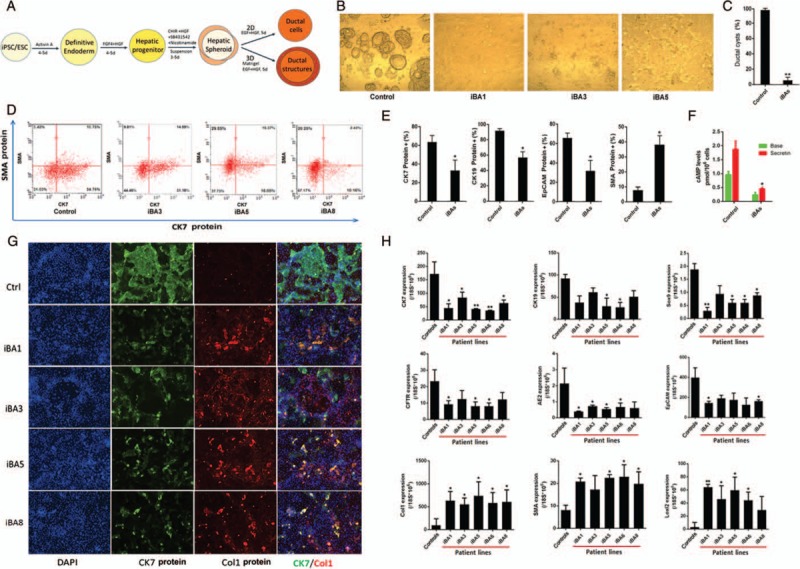
Decreased biliary differentiation and increased fibrosis of biliary atresia (BA) patient iPSCs. A, A schematic diagram of 2D and 3D biliary differentiation procedure. B, C, In a 3D culture, ductal cells from all BA patient induced pluripotent stem cells (iPSCs) formed significantly decreased amount of ductal cysts/tubes at d20 post-differentiation (n = 5, ∗∗P < 0.01). Representative data are shown with iBA1, iBA3, and iBA5. D, E, Flow cytometry (FACS)-based protein analysis after 2D ductal differentiation at d20. Compared to controls, all BA patient iPSCs showed decreased CK7, CK19, and EpCAM (cholangiocyte markers) positive cells and increased alpha smooth muscle actin (SMA) (a fibrosis marker) positive cells. Representative data are shown with iBA3, iBA5, and iBA8. E, FACS based quantification of CK7, CK19, EpCAM protein positive, or SMA protein positive cell populations in biliary differentiation culture. BAs represent data obtained from multiple BA iPSCs (n = 5, ∗P < 0.05). F, Secretin stimulated cholangiocyte functional assays showed increased cAMP in control iPSC-derived cholangiocytes but it was significantly lower in the cholangiocytes derived from patient iPSCs (∗P < 0.05). G, Immunofluorescence based protein analysis of CK7 (green), and another fibrosis marker collagen type 1 (Col1, red). Compared to controls, all BA patient iPSCs expressed lower CK7 protein and higher Col1 protein after biliary differentiation. H, Quantitative real time RT-PCR analysis of biliary and fibrosis markers in multiple BA patient iPSCs after biliary differentiation. Compared to controls, all BA iPSC lines express lower biliary markers and higher fibrosis markers after biliary differentiation. ∗P < 0.05, ∗∗P < 0.01. Controls reflect the combined results were from the combination of all control cell lines listed in Table 1 and iBAs represent the combined results from all BA iPSC lines including iBA1, 3, 5, 6, and 8.
Isogenic Human Induced Pluripotent Stem Cell Lines Deficient in Biliary Atresia-associated Genes Created by Precise Genome Editing
We targeted both GPC1 and ADD3 to create the panel of isogenic iPSCs based on the highly efficient CRISPR/Cas9 method which we have previously used in human iPSCs from another liver disease (6,16). Two sets of isogenic cell lines, derived from 2 different parental iPSC lines (iHu71 and iBC), were used in this study to achieve more robust/unbiased results. In addition, 3 to 6 replicates of each gene-edited iPSCs were examined for biliary differentiation. Representative data are shown using iHu71 parental and isogenic knock out (KO) lines.
Embryoid Body Differentiation
Embryoid Bodies (EBs) were formed using FBS-containing differentiation medium and cultured in suspension for 7 days. The resulting EBs were then plated on gelatin-coated 24-well plates for additional 3 days. The cells were fixed with 4% paraformaldehyde and stained for markers representing the 3 germ layers.
Immunofluorescence and Flow Cytometry
Human iPSCs and iPSC-derived biliary cells grown on matrigel-coated (Corning) plates were fixed with 4% paraformaldehyde (Sigma) for 20 minutes at room temperature, and washed with phosphate-buffered saline (PBS). Primary antibodies against CK7 (1:200, Cell Marque, Cat. 307M-95), Collagen 1 (1:200, Millipore, Burlington, MA, Cat. 234167), Oct4 (1:200, Millipore, Cat. Mab4401), Nanog (1:200, BD Pharmingen, San Jose, CA, Cat. 560109), Tra160 (1:100, Millipore, Cat. Mab4360), and YAP1 (1:100, Sigma, Cat.wh0010413m1) were diluted in PBS with 0.3% BSA and 0.1% Triton X-100. Fixed cells were incubated overnight with appropriate primary antibodies at 4°C for immunochemistry. The next day, cells were washed twice with PBS and incubated with appropriate Alexa Flour 555 or 488 conjugated secondary antibodies (all of the Alexa Fluor Series from Invitrogen, Carlsbad, CA) in PBS at room temperature for 30 to 45 minutes followed by PBS wash. Cells were then counterstained with DAPI before immunofluorescence analysis. Images were taken using the motorized Nikon Ti-E microscope and NIS-Elements software. For SSEA3 (1:50, Biolegend, Cat. 330306), CK7 (1:400, Cell Marque, Cat. 307M-95), EpCAM (1:200, R&D systems, Minneapolis, MN, Cat. AF960), smooth muscle actin (SMA) (1:1000, Sigma, Cat. A5228) and CK19 (1:100, Santa Cruz, Cat. Sc-6278) flow cytometry analysis, cells were digested by Accutase and washed by PBS. 1 × 105 cells were incubated with Alexa 488-SSEA3 or isotype control antibody for 30 min at 4°C. After PBS washing, the cells were analyzed by a Guava EasyCyte Flow Cytometer (Millipore).
RNA Extraction and Real-time Quantitative Real-time Polymerase Chain Reaction
Total RNA was extracted with TRIZOL reagent (Thermo Fisher, Waltham, MA) according to manufacturer's recommendation. Reverse transcription from mRNA to cDNA was performed using High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA). The resulting cDNA was used as template for quantitative polymerase chain reaction (Q-PCR) with StepOnePlus Real-Time PCR System (Applied Biosystems), using TaqMan probes. The final PCR reactions consisted of 20 ng cDNA, 1XTaqMan Fast Advanced Master Mix, and TaqMan gene probe at a 20 μL volume. The TaqMan probes included CK7 (Hs00559840_m1), CK19 (Hs00761767_s1), Sox9 (Hs00165814_m1), CFTR (Hs00357011_m1), AE2 (Hs01586776_m1), EpCAM (Hs00901885_m1), Col1 (Hs00164004_m1), SMA (Hs00426835_g1), Loxl2 (Hs00158757_m1), Yap1 (Hs00902712_g1), and 18S rRNA (Hs03003631_g1) in a total volume of 20 μL.
cAMP Assays
The human iPSC-derived biliary cells were incubated with 100 nM secretin (Sigma, Cat. S7147) in culture media for 20 minutes. The concentration of cAMP was determined using a complete ELISA kit (Abcam, Cat. Ab133051) according to the manufacturer's instruction.
Statistical Analysis
Data is expressed as mean ± standard error of the mean (SEM). For pairwise comparisons, a Student's t test was used. For all tests conducted, P < 0.05 was considered significant.
RESULT
Altered Biliary Differentiation With Increased Fibrogenesis of Biliary Atresia Patient-specific Induced Pluripotent Stem Cell Lines
Multiple BA patient iPSC lines were compared for their biliary differentiation potential with control iPSCs derived from non-BA controls (Fig. 1). These iPSCs were differentiated into biliary epithelial tissues and cells using the step-wise differentiation protocols (Fig. 1A). We did not observe significant differences between BA and non-BA controls in earlier stages including endoderm, hepatic progenitor stages and the very early stage of biliary differentiation (at ∼day 13, Fig S1 left panel, Supplemental Digital Content). We did observe biliary defects in the BA cells as early as d15, when small biliary structures normally start to emerge (Fig S1 middle panel, Supplemental Digital Content). At the end of biliary differentiation (day 20), the BA patient iPSCs showed significantly reduced ductal structure formation in 3D biliary tissue formation assays and did not form detectable levels of ductal cysts/tubes (Fig. 1B, C, and Fig S2, Supplemental Digital Content). This was consistent with 2D biliary differentiation assays which also showed reduced biliary markers such as CK7, CK19, and EpCAM at protein levels and reduced secretory function in the BA patient iPSCs after biliary differentiation (Fig. 1D, E, F, and Fig S3, Supplemental Digital Content). Along with the reduced biliary tissue formation, increased fibrosis markers including alpha SMA and Collagen type 1 in the BA patient iPSCs were detected compared to control lines after biliary differentiation of these iPSCs (Fig. 1D, E, G, and Fig S3). Collagen positive cells are rarely present at any stage of normal biliary differentiation process including the mature stage (Fig. 1G and data not shown). These 2 key disease features, that is, reduced biliary differentiation and increased fibrogenesis, were further confirmed by examining gene expression patterns of more diverse cholangiocyte markers (CK7, CK19, SOX9, CFTR, AE2, and EpCAM) and multiple hepatic fibrosis markers including Collagen type 1, alpha SMA and Loxl2, in diverse iPSC lines derived from 5 different BA patients without anomalies (Fig. 1H). In addition, 1 iPSC line derived from a BA patient with anomalies (midline liver, heterotaxy syndrome with polysplenia), which grows extremely poorly in regular iPSC culture conditions, also showed similar results (ie, reduced biliary marker CK7 and increased fibrosis marker collagen 1 after biliary differentiation, Fig S4, Supplemental Digital Content).
These data together suggest significantly altered biliary differentiation potential of BA patient iPSCs along with increased fibrosis, recapitulating 2 key clinical features of the disease.
Altered Biliary Differentiation of Gene-edited Human Induced Pluripotent Stem Cell With Specific Defects in Biliary Atresia-associated Genes
Using CRISPR/Cas9, we generated human iPSC lines that have defined mutations in the BA susceptibility loci (GPC1 or ADD3) (Fig. 2A–C). These gene KO iPSC lines were capable of growing similarly to the parental iPSC line and maintained their pluripotency (Fig. 2D). In the biliary differentiation assays, these gene-KO iPSC lines formed significantly decreased amount of biliary tissues in both 3D (over 90% decreased, Fig. 2F) and 2D culture conditions (Fig. 2G, H), and generated increased amount of fibrosis marker positive cells compared to their isogenic control iPSCs at protein and RNA levels (Fig. 2G, H). Similar results were also observed in another set of GPC1 and ADD3 KO iPSCs created from a different parental iPSC (reduced biliary marker CK7 and increased fibrosis marker SMA after biliary differentiation, Fig S5, Supplemental Digital Content). The degree of the altered biliary differentiation and increased fibrosis were similar in both ADD3 KO lines and GPC1 KO lines. These data show that human iPSCs edited at BA associated genes (GPC1 and ADD3) can recapitulate the key BA disease features including reduced biliary differentiation and increased fibrosis in vitro.
FIGURE 2.
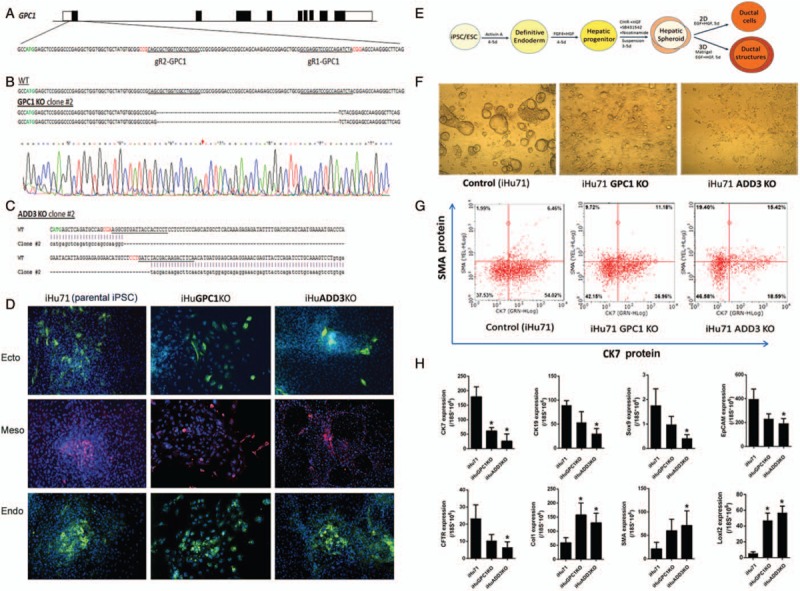
Generation of isogenic human iPSC lines deficient in genes implicated in biliary atresia (BA) development and altered biliary differentiation of these KO iPSCs. A, A diagram of GPC1 locus in chr2 (UTRs in empty boxes) and partial exon 1 sequence are shown (ATG shown in green). Sequences of 2 sgRNAs designed to target this exon with CRISPR/Cas9 are underlined with the PAM sequences in red. B, Sequencing results of a GPC1 knockout (KO) clone is shown in comparison to wildtype un-targeted parental control. Example chromatogram of GPC1−/− clone #2 sequencing is shown with a red arrow indicating the where homozygous deletion occurs. C, A homozygous 109-bp deletion is created in ADD3 exon 2 in ADD3 KO iPSCs using the same targeting strategy. ATG, sgRNA recognition sequences are presented as shown in (A). D, In vitro embryoid body (EB) based spontaneous differentiation of the GPC1−/− and ADD3−/− iPSCs into 3 germ layer tissues. Ectoderm, mesoderm, and endoderm derivatives stained with their respective markers TuJ1, SMA, and AFP. Both KO lines maintained the same pluripotency as their parental iPSC (iHu71) control after gene editing. E, A diagram of 2D and 3D biliary differentiation procedure. F, In a 3D culture, ductal cells from both GPC1 and ADD3 KO iPSC lines formed significantly decreased amount of ductal cysts/tubes. G, Flow cytometry analysis after 2D ductal differentiation. Compared to controls, KO iPSCs showed decreased CK7 (a biliary marker) positive cells and increased SMA (a marker for fibrosis) positive cells. H, Quantitative real-time RT-PCR analysis of biliary and fibrosis markers in KO iPSCs after 2D biliary differentiation. Compared to controls, both KO lines express lower biliary- and higher fibrosis-markers after biliary differentiation. Representative data are shown using iHu71 parental and isogenic KO lines (∗P < 0.05).
Increased Yes-associated Protein Expression in Biliary Atresia Patient Induced Pluripotent Stem Cells and Knock Out Induced Pluripotent Stem Cells, and the Feasibility of Testing Anti-Fibrotic Drugs in These Human Biliary Atresia Induced Pluripotent Stem Cell Assays
Increased YAP1 expression was detected in all the biliary differentiation cultures of BA patient iPSCs and KO iPSCs at varied levels, compared to control iPSCs (Fig. 3A). In addition, the high nuclear YAP expression was detected in the biliary cells from BA patient-specific iPSCs and interestingly it was well co-localized with collagen type 1 protein (Fig. 3B). Collagen postivie cells are rarely present at any stage of normal biliary differentiation including the mature stage (Fig. 1G, Fig. 3B, and data not shown). We further tested the effect of an anti-fibrogenic drug, pentoxiphylline (36–39), in the biliary differentiation culture of high YAP expressing BA patient-specific iPSCs and KO iPSC (Fig. 3B–E). We observed that this anti-fibrotic drug significantly lowered collagen protein levels as well as decreased the nuclear localization of YAP (Fig. 3B). The levels of collagen 1 and YAP1 gene expression were also decreased in the biliary derivatives of both patient iPSCs and GPC1 KO iPSCs (Fig. 3C, D). In contrast, pentoxiphylline had no effect on those 2 markers (Collagen and YAP) in control iPSCs nor did it alter biliary marker CK7 expression in BA iPSCs (Fig. 3C–E), suggesting the effect was specific to fibrosis in BA rather than affecting normal biliary tissues.
FIGURE 3.
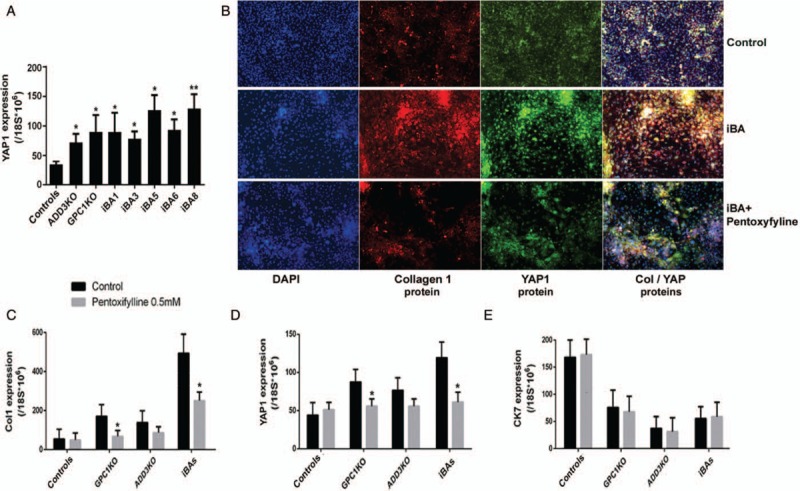
Higher YAP expression in biliary atresia (BA) patient iPSCs and KO iPSCs, and the effect of anti-fibrotic drug treatment. A, YAP mRNA expression in BA and KO iPSCs at mature biliary differentiation stage (d20). Compared to normal controls, BA relevant iPSCs (patient iPSCs and GPC1- or ADD3-KO iPSCs) showed significantly higher YAP expression after biliary differentiation at various levels (∗P < 0.05, ∗∗P < 0.01). B, Immunofluorescent staining of Collagen 1 and YAP1 protein expression in healthy control iPSC (iM9) and BA patient iPSC (iBA8 cells were shown as an example). YAP1 and Collagen 1 were col-localized and YAP was mostly located in the nuclei. An anti-fibrotic drug, Pentoxifylline (0.5 mM 5 day treatment) significantly reduced collagen expression as well as the nuclear localization of YAP protein in the biliary cells derived from the BA patient iPSCs. C–E, Pentoxifylline lowered the levels of gene expression of both collagen 1 and YAP1 in KO lines (iHu71 isogenic GPC1 and ADD3 KO lines) and BA patient lines (iBAs-combined results from all BA iPSCs) while showing no effect on cholangiocyte marker CK7 (∗P < 0.05).
DISCUSSION
Our study demonstrates the feasibility of generating multiple BA-specific iPSC lines to model human BA in vitro. We further showed that these patient-derived iPSCs resulted in defective biliary differentiation, a key feature of BA, in the in vitro biliary differentiation of patient-iPSC lines; we observed dramatically reduced formation of ductal structures and decreased expression of phenotypic and functional biliary tissue markers at both mRNA and protein levels, compared to their respective controls. Interestingly, increased fibrosis associated markers including collagen 1, alpha SMA and Loxl2 were detected in the BA-patient derived iPSCs, suggesting another key disease feature (ie, increased fibrosis) can be recapitulated from these patient iPSCs. Since the BA patient iPSCs are from humans and recapitulate fibrosis, which was not feasible in the animal models, they hold a great promise as a valuable preclinical in vitro human assay for BA research.
GWAS have identified GPC1 and ADD3, genes that play important roles in embryonic development, as BA susceptibility genes (23–29). Subsequently zebrafish studies have suggested the functional relevance of these genes in impaired intrahepatic biliary network formation (25,28,40). To determine the biological consequences of genetic defects in BA susceptibility genes during human biliary differentiation, we also generated human iPSC lines that have defined mutations in these loci. The functional outcomes of these genetic alterations (GPC1 and ADD3) using human iPSC-based biliary differentiation methods were surprisingly similar to those of BA patient derived iPSCs. In addition, these patient-specific and KO iPSCs also showed increased YAP expression along with increased collagen expression, both of which were lowered with treatment of an anti-fibrotic drug. These results demonstrate that BA disease features can be recapitulated in vitro using precisely gene-edited human iPSCs and provide the feasibility of this approach to determine the functional or pathogenetic roles of GWAS identified genes in this well controlled human biological system; since the isogenic parental control provides the controls with the same genetic backgrounds except the gene of interest. The results obtained in this study also cautiously support the previously less appreciated functional roles of genetic factors in BA pathogenesis. However it is very likely that various other factors including environmental factors trigger BA progression or influence severity of the disease. In fact, a majority of BA patient iPSCs have shown more severe fibrosis phenotypes compared to the KO iPSC lines, indicating the possibility of multiple intrinsic or genetic factors playing roles in BA.
The current study reports the first feasibility of using human iPSC technologies for modeling of key disease features of BA and drug testing in vitro, but this is not a detailed pathogenesis study. Based on these human iPSC-based BA models, our future studies will focus on revealing the pathogenesis underlying each of the disease features, and possible mechanisms whereby GPC1/ADD3 mutations could link to the increased YAP or other common pathways in BA.
Our current study which is heavily focused on developing BA disease models using human iPSCs does not reveal whether the increased collagen expressing cells originated from cholangiocytes or elsewhere. It is not likely that these fibrosis marker positive cells have originated from the mesenchymal cells which are rarely present at any stage of normal biliary differentiation including the early and mature stages (Fig. 1G, Fig 3B, and data not shown). However multiple EMT markers were not significantly increased in the differentiated BA-relevant iPSCs at the mature cholangiocyte stage (Fig S6, Supplemental Digital Content). Future studies will be needed to dissect if EMT plays a role in earlier differentiation stages when the biliary defect starts to emerge (Fig S1, Supplemental Digital Content) and identify the cellular origin of BA fibrosis since it could be an important therapeutic target for BA fibrosis prevention/treatment. The phenotypes we observed in the genetically modified iPSCs are similar to those from the BA patients. A limitation of our current study is the absence of direct comparison with primary biliary cells from BA patients since BA patients have no or very limited amount of extrahepatic bile ducts. Another limitation is the lack of complete profiling data comparing the iPSC-cholangiocytes generated from the current differentiation method with human primary cholangiocytes. Their morphology and markers are, however, similar (Fig S7, Supplemental Digital Content). Therefore, future comprehensive genetic analysis of the patient iPSCs could provide useful insight into the disease mechanisms. The disease models based on patient-specific iPSCs and genetically engineered iPSCs can provide an innovative human-specific platform to validate novel disease targets and to uncover new disease mechanisms. Importantly, the patient-derived iPSC lines can provide a highly human relevant preclinical system for developing or discovery of urgently needed new therapies for BA patients. In addition, this approach can be applied to various other cholangiopathies such as primary sclerosing cholangitis that are currently in need of novel tools to develop effective therapies.
Supplementary Material
Footnotes
The study has been supported by Maryland Stem Cell Research Foundation, NIBIB (R01EB023812), NICHD (R03HD091264), Colleen Mitchel BA 5K; Zachary Meehan Memorial Fund for BA Research; Johns Hopkins Pediatric Liver Center, Johns Hopkins Children's Center
The authors report no conflicts of interest.
REFERENCES
- 1.Hartley JL, Davenport M, Kelly DA. Biliary atresia. Lancet 2009; 374:1704–1713. [DOI] [PubMed] [Google Scholar]
- 2.Asai A, Miethke A, Bezerra JA. Pathogenesis of biliary atresia: defining biology to understand clinical phenotypes. Nat Rev Gastroenterol Hepatol 2015; 12:342–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Petersen C, Davenport M. Aetiology of biliary atresia: what is actually known? Orphanet J Rare Dis 2013; 8:128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Petersen C. Biliary atresia: the animal models. Semin Pediatr Surg 2012; 21:185–191. [DOI] [PubMed] [Google Scholar]
- 5.Choi SM, Liu H, Chaudhari P, et al. Reprogramming of EBV-immortalized B-lymphocyte cell lines into induced pluripotent stem cells. Blood 2011; 118:1801–1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Choi SM, Kim Y, Shim JS, et al. Efficient drug screening and gene correction for treating liver disease using patient-specific stem cells. Hepatology 2013; 57:2458–2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chun YS, Chaudhari P, Jang YY. Applications of patient-specific induced pluripotent stem cells; focused on disease modeling, drug screening and therapeutic potentials for liver disease. Int J Biol Sci 2010; 6:796–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ye Z, Zhan H, Mali P, et al. Human-induced pluripotent stem cells from blood cells of healthy donors and patients with acquired blood disorders. Blood 2009; 114:5473–5480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu H, Ye Z, Kim Y, et al. Generation of endoderm-derived human induced pluripotent stem cells from primary hepatocytes. Hepatology 2010; 51:1810–1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jang YY, Ye Z, Cheng L. Molecular imaging and stem cell research. Mol Imaging 2011; 10:111–122. [PubMed] [Google Scholar]
- 11.Sharkis SJ, Jones RJ, Jang YY. Pluripotent stem cell-based cancer therapy: promise and challenges. Sci Transl Med 2012; 4:127ps129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cong L, Ran FA, Cox D, et al. Multiplex Genome Engineering Using CRISPR/Cas Systems. Science 2013; 339:819–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mali P, Yang L, Esvelt KM, et al. RNA-guided human genome engineering via Cas9. Science 2013; 339:823–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Christian M, Cermak T, Doyle EL, et al. Targeting DNA double-strand breaks with TAL effector nucleases. Genetics 2010; 186:757–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bogdanove AJ, Voytas DF. TAL effectors: customizable proteins for DNA targeting. Science 2011; 333:1843–1846. [DOI] [PubMed] [Google Scholar]
- 16.Smith C, Abalde-Atristain L, He C, et al. Efficient and allele-specific genome editing of disease loci in human iPSCs. Mol Ther 2015; 23:570–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jang YY, Cai L, Ye Z. Genome editing systems in novel therapies. Discov Med 2016; 21:57–64. [PubMed] [Google Scholar]
- 18.Clemente MG, Patton JT, Anders RA, et al. Rotavirus infects human biliary epithelial cells and stimulates secretion of cytokines IL-6 and IL-8 via MAPK pathway. Biomed Res Int 2015; 2015:697238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gurda GT, Zhu Q, Bai H, et al. The use of Yes-associated protein expression in the diagnosis of persistent neonatal cholestatic liver disease. Hum Pathol 2014; 45:1057–1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Clemente MG, Patton JT, Yolken R, et al. Prevalence of groups A and C rotavirus antibodies in infants with biliary atresia and cholestatic controls. J Pediatr 2015; 166:79–84. [DOI] [PubMed] [Google Scholar]
- 21.Omenetti A, Bass LM, Anders RA, et al. Hedgehog activity, epithelial-mesenchymal transitions, and biliary dysmorphogenesis in biliary atresia. Hepatology 2011; 53:1246–1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Waisbourd-Zinman O, Koh H, Tsai S, et al. The toxin biliatresone causes mouse extrahepatic cholangiocyte damage and fibrosis through decreased glutathione and SOX17. Hepatology 2016; 64:880–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Garcia-Barceló MM, Yeung MY, Miao XP, et al. Genome-wide association study identifies a susceptibility locus for biliary atresia on 10q24.2. Hum Mol Genet 2010; 19:2917–2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cheng G, Tang CS, Wong EH, et al. Common genetic variants regulating ADD3 gene expression alter biliary atresia risk. J Hepatol 2013; 59:1285–1291. [DOI] [PubMed] [Google Scholar]
- 25.Cui S, Leyva-Vega M, Tsai EA, et al. Evidence from human and zebrafish that GPC1 is a biliary atresia susceptibility gene. Gastroenterology 2013; 144:1107–1115. e1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Miethke AG, Huppert SS. Fishing for biliary atresia susceptibility genes. Gastroenterology 2013; 144:878–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tsai EA, Grochowski CM, Loomes KM, et al. Replication of a GWAS signal in a Caucasian population implicates ADD3 in susceptibility to biliary atresia. Hum Genet 2014; 133:235–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mezina A, Karpen SJ. Genetic contributors and modifiers of biliary atresia. Dig Dis 2015; 33:408–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ningappa M, Min J, Higgs BW, et al. Genome-wide association studies in biliary atresia. Wiley Interdiscip Rev Syst Biol Med 2015; 7:267–273. [DOI] [PubMed] [Google Scholar]
- 30.Liu H, Kim Y, Sharkis S, et al. In vivo liver regeneration potential of human induced pluripotent stem cells from diverse origins. Sci Transl Med 2011; 3: 82ra39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tian L, Deshmukh A, Prasad N, et al. Alcohol increases liver progenitor populations and induces disease phenotypes in human IPSC-derived mature stage hepatic cells. Int J Biol Sci 2016; 12:1052–1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ye Z, Liu CF, Jang YY. Hematopoietic cells as sources for patient-specific iPSCs and disease modeling. Cell Cycle 2011; 10:2840–2844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tian L, Deshmukh A, Ye Z, et al. Efficient and controlled generation of 2D and 3D bile duct tissue from human pluripotent stem cell-derived spheroids. Stem Cell Rev Rep 2016; 12:500–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tian L, Eldridge L, Chaudhari P, et al. Derivation of a disease-specific human induced pluripotent stem cell line from a biliary atresia patient. Stem Cell Res 2017; 24:25–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Choi SM, Kim Y, Liu H, et al. Liver engraftment potential of hepatic cells derived from patient-specific induced pluripotent stem cells. Cell Cycle 2011; 10:2423–2427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Andrade WeC, Tannuri U, da Silva LF, et al. Effects of the administration of pentoxifylline and prednisolone on the evolution of portal fibrogenesis secondary to biliary obstruction-an experimental study in growing animals. J Pediatr Surg 2009; 44:2071–2077. [DOI] [PubMed] [Google Scholar]
- 37.Desmoulière A, Xu G, Costa AM, et al. Effect of pentoxifylline on early proliferation and phenotypic modulation of fibrogenic cells in two rat models of liver fibrosis and on cultured hepatic stellate cells. J Hepatol 1999; 30:621–631. [DOI] [PubMed] [Google Scholar]
- 38.Raetsch C, Jia JD, Boigk G, et al. Pentoxifylline downregulates profibrogenic cytokines and procollagen I expression in rat secondary biliary fibrosis. Gut 2002; 50:241–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Windmeier C, Gressner AM. Pharmacological aspects of pentoxifylline with emphasis on its inhibitory actions on hepatic fibrogenesis. Gen Pharmacol 1997; 29:181–196. [DOI] [PubMed] [Google Scholar]
- 40.Tang V, Cofer ZC, Cui S, et al. Loss of a candidate biliary atresia susceptibility gene, add3a, causes biliary developmental defects in zebrafish. J Pediatr Gastroenterol Nutr 2016; 63:524–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.