

Tissue injury can lead to fibrotic disease and chronic inflammation, for example, in the lungs of patients with idiopathic pulmonary fibrosis. Jürgensen et al. report that fibroblasts regulate immune responses during tissue injury by clearing collectins with pro- or antiinflammatory activity via uPARAP, an endocytic collectin receptor identified in this work.
Abstract
Collectins such as mannose-binding lectin (MBL) and surfactant protein D (SP-D) become temporarily deposited in extravascular compartments after tissue injury and perform immune-stimulatory or inflammation-limiting functions. However, their turnover mechanisms, necessary to prevent excessive tissue damage, are virtually unknown. In this study, we show that fibroblasts in injured tissues undertake the clearance of collectins by using the endocytic collagen receptor uPARAP. In cellular assays, several types of collectins were endocytosed in a highly specific uPARAP-dependent process, not shared by the closely related receptor MR/CD206. When introduced into dermis or bleomycin-injured lungs of mice, collectins MBL and SP-D were endocytosed and routed for lysosomal degradation by uPARAP-positive fibroblasts. Fibroblast-specific expression of uPARAP governed endogenous SP-D levels and overall survival after lung injury. In lung tissue from idiopathic pulmonary fibrosis patients, a strong up-regulation of uPARAP was observed in fibroblasts adjacent to regions with SP-D secretion. This study demonstrates a novel immune-regulatory function of fibroblasts and identifies uPARAP as an endocytic receptor in immunity.
Introduction
Defense collagens, notably including collectins, ficolins, and Class A scavenger receptors, are important components of the innate immune system. These proteins function as pattern recognition receptors and bind to the surface of pathogens as well as to apoptotic cells (Litvack and Palaniyar, 2010; Foo et al., 2015). This event in turn triggers opsonization, agglutination, or complement activation (McCormack and Whitsett, 2002; Cravedi and Heeger, 2014).
Defense collagen expression is enhanced during infection, inflammation, and fibrosis (Forbes and Haczku, 2010). After secretion, soluble defense collagens are found in the circulation, but in many cases, they are also in extravascular compartments. Mannose-binding lectin (MBL), a central blood-borne defense collagen, becomes deposited at injured sites after several types of skin injury, where it acts in the defense against infectious agents as well as in the clearance of apoptotic cells (Lokitz et al., 2005). In the lung, two types of collectins, surfactant proteins D and A (SP-D and SP-A, respectively), are present on mucosal and alveolar surfaces where they play critical roles in innate immune responses (Madan et al., 2001; Wu et al., 2003).
While the increased presence of collectins at sites of tissue injury is well documented, much less is known about their mechanisms of turnover and clearance in tissue repair and homeostasis. In particular, this is the case in the extravascular compartments. Although several cell surface molecules with collectin-binding capacity have been identified, most of these components are engaged directly in processes of opsonization and phagocytosis rather than mediating collectin clearance (Holmskov et al., 1997; Ghiran et al., 2000; Gardai et al., 2003).
All defense collagens share the possession of triple-helical domains with similarity to structural collagens. These collagen-like domains are important for the assembly of single polypeptide units into the biologically active, higher-order oligomers but also take part in various protein–protein interactions (Haczku, 2008). Strikingly, however, it has never been investigated whether these triple-helical domains have an impact on the regulation of collectins or whether they are recognized by collagen receptors with endocytic function.
Two endocytic receptors, urokinase plasminogen activator receptor–associated protein (uPARAP; Endo180; CD280) and mannose receptor (MR; CD206), have been shown to bind and internalize solubilized collagen (Jürgensen et al., 2014). These two receptors, being the products of the MRC2 and the MRC1 genes, respectively, are structurally closely related (Behrendt et al., 2000; Sheikh et al., 2000). Interestingly, however, they have very different expression patterns. uPARAP is expressed on certain mesenchymal cell types after activation, such as the myofibroblasts of tumor stroma (Nielsen et al., 2002; Sulek et al., 2007) and hepatic stellate cells and activated fibroblasts in liver fibrosis (Madsen et al., 2012). In the resting, healthy organism, expression of uPARAP is limited and mostly confined to sites of tissue remodeling (Engelholm et al., 2001; Wagenaar-Miller et al., 2007; Madsen et al., 2013a; Abdelgawad et al., 2014). In contrast, MR is found on leukocyte subpopulations including macrophages and dendritic cells (Martinez-Pomares, 2012; Schuette et al., 2016) as well as in the liver endothelium, where it serves as a clearance receptor for collagen degradation products and various glycoproteins (Lee et al., 2002; Malovic et al., 2007). The expression of MR is particularly enhanced on M2-like macrophages (Madsen et al., 2013b), a cell type considered more closely associated with tissue repair than with inflammation and often associated with cancer invasion (Mantovani et al., 2002). Thus, both receptors are expressed by cells that are abundant at sites of injury, including conditions in which extravascular collectins are also present.
In this study, we demonstrate a novel immunological function of fibroblasts in which these cells act as direct regulators of the innate immune system by using uPARAP to clear collectins from injured tissue.
Results
uPARAP is a unique endocytosis receptor for a wide spectrum of defense collagens
Since the structure of defense collagens includes a triple-helical, collagen-like domain (Haczku, 2008), we hypothesized that this domain could be involved in the clearance of defense collagens through the binding to a specific receptor. To set up a system measuring cellular uptake of these ligands, we first focused on two well-characterized defense collagens, MBL and SP-D, belonging to the group of soluble collectins (Fig. 1 A). Recombinant preparations of both of these proteins were shown to be composed of the disulphide-linked oligomers that constitute the active defense collagen (Fig. S1 A). The proteins were labeled with 125I and shown to retain Ca2+-dependent binding to Escherichia coli, one of their natural biological binding partners (Fig. S1 B). Radioligand uptake experiments were then performed with CHO cells transfected to express established endocytic receptors for collagen (uPARAP and MR; Jürgensen et al., 2014) as well as structurally related receptors within the same protein family (family shown in Fig. 1 B).
Figure 1.

uPARAP-dependent endocytosis of SP-D in vitro. (A) Domain structure of monomeric defense collagens SP-D, SP-A, MBL, ficolins, C1q, collectin-11, collectin-12, SR-AI, and MARCO. The domain structures include a collagen-like domain, which takes part in both homotypic and heterotypic protein interactions. TM, transmembrane domain; CTLD, C-type lectin-like domain; FBG, fibrinogen-like domain; gC1q, globular C1q domain; SRCR, scavenger receptor (SR) cysteine-rich domain. (B) Domain structure of the members of the MR protein family: uPARAP (ENDO180, MRC2, and CD280), MR (MRC1 and CD206), PLA2R (phospholipase A2 receptor), and DEC-205 (CD205). CysRich, cysteine-rich domain; FNII, fibronectin type-II domain. (C–E) Internalization of radiolabeled SP-D (C), MBL (D), and collagen type I (E) in CHO cells transfected to express uPARAP, MR, PLA2R, or DEC-205 or transfected with empty vector (mock). (F) Internalization of radiolabeled SR-AI, MARCO, collectin-12, collectin-11, ficolin-1, and C1q in CHO cells transfected to express uPARAP or mock cells. The following statistical tests were used: one-way ANOVA (C–E) and two-tailed Student’s t test (F). Analysis was performed in triplicate. Data are presented as mean ± SD.
Strikingly, cells transfected with just one of these receptors, uPARAP, displayed a strong endocytic capacity for both collectins (Fig. 1, C and D). This property was indeed caused by the expression of uPARAP since mock-transfected cells had no such capacity. As an independent test of the uPARAP dependence of this process, we treated uPARAP-transfected cells with a function-blocking antibody against uPARAP, mAb 5f4 (Madsen et al., 2011). This treatment led to a strongly reduced ligand uptake (shown for SP-D; Fig. S1 C). None of the other receptors appeared to possess this ability, including MR. This was surprising since both uPARAP and MR were confirmed to mediate collagen endocytosis by the transfected cells used in this study (shown for collagen type I; Fig. 1 E).
Six additional recombinant proteins representing different classes of soluble and membrane-associated defense collagens (Fig. 1 A) were also tested as potential ligands in the same manner. The soluble Collectin-12/CL-P1 and the membrane-associated defense collagens SR-AI and MARCO were all efficiently and specifically internalized by uPARAP-transfected cells, whereas a more moderate but still significant dependence of uPARAP was noted for the cellular uptake of Ficolin-1, Collectin-11/CL-K1, and C1q (Fig. 1 F).
Uptake of MBL by uPARAP-positive dermal fibroblasts in vivo
We next set out to investigate the cellular uptake of collectins in vivo. While collectins other than the SPs are mostly present in the circulation, MBL becomes deposited in the skin after several types of injury, including infection, skin burn, and pemphigus- or systemic lupus erythematosus (SLE)–induced lesions (Møller-Kristensen et al., 2006; Miller et al., 2010; de Messias-Reason et al., 2011; Wallim et al., 2014). To study the uptake of MBL in the skin, we used transgenic mice expressing GFP under the col1a1 promoter (henceforth referred to as col1a1-GFP mice; Krempen et al., 1999). Since this promoter has a high degree of specificity for cells of fibroblastic lineage, it should allow for simple identification of fibroblasts, including cells with a potential expression of uPARAP. This system was first verified by flow cytometry–based analysis of skin cell suspensions using established cell type markers to assess the expression of GFP in defined cell populations (Fig. 2). This experiment confirmed the specific fibroblastic expression of GFP since virtually all of the GFP fluorescence was associated with the CD45−,CD31−,EpCAM− cell population (Figs. 3 A and S2 A). The col1a1-GFP mice were then injected subcutaneously with mixtures of fluorescent MBL and either fluorescent dextran, a general marker for endocytosis (Fig. 3 B; Madsen et al., 2013b), or a fluorescent monoclonal antibody against uPARAP, mAb 2h9 (Fig. 3 C). 24 h after injection, mice were sacrificed, and the dermis was imaged by confocal microscopy.
Figure 2.

Gating strategy for flow cytometry–based analysis of dermal and lung cells. Flow cytometry strategy for analyzing dermal and lung single-cell suspensions for the expression of GFP, the association of cells with fluorescent collectins, or the expression of uPARAP. GFP was expressed under the col1a1 promotor (using col1a1.GFP mice), whereas fluorescent collectins (MBL [dermal cells] and SP-D [lung cells]) and fluorescent anti-uPARAP antibody were taken up by cells in vivo. Single-cell suspensions were stained for CD45, F4/80, CD11b, EpCAM, and CD31. CD45− cells (nonleukocytes) were separated into EpCAM+ epithelial cells, CD31+ endothelial cells, and EpCAM−CD31− cells. Fibroblasts were identified as GFP+ in the EpCAM−CD31− population. CD45+ cells (leukocytes) were divided into F4/80+CD11b+ (macrophages), F4/80−CD11b+ (nonmacrophage, myeloid cells), and CD11b− (nonmyeloid leukocytes).
Figure 3.

Flow cytometry and confocal imaging reveals in vivo uptake of the collectin MBL by uPARAP-positive fibroblasts in dermis. (A) Flow cytometry–based quantification of GFP-positive cells in dermal cell populations acquired from col1a1.GFP transgenic mice. (B and C) Uptake of Alexa Fluor 647–labeled murine MBL-A (white) by dermal fibroblasts in vivo was assessed using confocal imaging 24 h after subcutaneous injection into col1a1.GFP mice. MBL was coinjected with either Alexa Fluor 546–labeled 10 kD dextran (B, red) or Alexa Fluor 555–labeled anti-uPARAP mAb 2h9 (C, red). Fibroblasts (green) with a vesicular uptake of MBL and dextran (B) as well as MBL and the anti-uPARAP antibody (C) were abundant. A nonfibroblast cell population with MBL uptake was also observed, and a very potent uptake of dextran in these cells positively identified them as dermal macrophages (arrows). Hoechst 33342 was used for visualization of cell nuclei (blue). (D and E) Quantification of the number of MBL-associating (D) and uPARAP-positive cells (E) within dermal cell populations assessed using flow cytometry 24 h after subcutaneous injection of Alexa Fluor 647–labeled MBL or Alexa Fluor 647–labeled anti-uPARAP mAb 2h9 into col1a1.GFP mice. Injection of Alexa Fluor 647–labeled irrelevant murine IgG was used as a negative control (E). n = 5 (A), 4 (D), and 6 (E). (F) Internalization of radiolabeled MBL in cultured primary dermal fibroblasts from uPARAP−/− mice and WT (uPARAP+/+) littermates. Internalization was examined in the absence or presence of the uPARAP function-blocking antibody mAb 5f4. One-way ANOVA was used to test for statistical significance. Analysis was performed in triplicate. Data are presented as mean ± SD.
This experiment clearly showed an uptake of MBL by distinct cell types, where it accumulated in a vesicular pattern (Fig. 3, B and C, left), and many of these MBL-positive cells were fibroblasts (GFP positive; green channel). Fibroblasts were also active in the uptake of dextran, known to be routed to the endosomal/lysosomal compartments after endocytosis (Madsen et al., 2013b). After uptake, MBL displayed a vesicular distribution identical to that of dextran (Fig. 3 B, compare white and red channels). Even more strikingly, the MBL-positive fibroblasts were also positive for uPARAP, with the intracellular vesicular patterns of MBL and the anti-uPARAP antibody being identical after uptake (Fig. 3 C).
The uptake of MBL in fibroblasts was confirmed by flow cytometry after MBL injection into live mice in the same manner. When skin cell suspensions were analyzed by this method, an approximately even distribution of MBL in fibroblasts and macrophages was observed (Figs. 3 D and S2 B). Injection of the anti-uPARAP antibody also led to a marked uptake in fibroblasts, with the great majority of GFP+ cells being uPARAP positive (Figs. 3 E and S2 C). Expression of uPARAP in other cell types was very limited in this tissue (Figs. 3 E and S2 C). Although MBL uptake in uPARAP-positive fibroblasts was clearly evident, the experimental model did not allow for a direct evaluation of the impact of uPARAP on the uptake process. We therefore isolated primary fibroblasts from the dermis of newborn WT and littermate-matched uPARAP-deficient mice (Engelholm et al., 2003; Kjøller et al., 2004) and examined their uptake of radiolabeled MBL. These experiments revealed a pronounced uptake of MBL in WT cells, which was strongly reduced in the uPARAP-deficient cells (Fig. 3 F, nonhatched columns) as well as in WT cells pretreated with the function-blocking mAb 5f4 against uPARAP (Fig. 3 F, hatched columns). Thus, uPARAP on primary fibroblasts is active in the endocytosis of MBL.
Uptake of SP-D by uPARAP-positive fibroblasts in the injured lung
We went on to investigate whether lung fibroblasts could perform a similar endocytosis of the lung-associated collectin SP-D from the alveolar space. To do this, we performed intratracheal instillation of fluorescent SP-D into col1a1-GFP mice. 24 h later, mice were sacrificed, and alveoli were inspected by flow cytometry and confocal microscopy. When the cellular composition of lung cell suspensions from these mice was analyzed by flow cytometry using the same markers as employed for dermal cells (Fig. 2), by far the majority of the GFP fluorescence was again associated with fibroblasts (CD45−,CD31−,EpCAM− cells; Figs. 4 A and S3 A). Although a low number of CD45+ were in this case also GFP positive, these cells presented with a fluorescence intensity 10–20-fold lower than that of the fibroblasts. However, when the alveoli from these mice were inspected by confocal microscopy, it turned out that cells with SP-D uptake were mostly GFP negative or displayed a very low GFP signal (i.e., being nonfibroblasts), and in most cases, they appeared to be alveolar macrophages based on localization and morphology (Fig. 4 B).
Figure 4.

Uptake of SP-D in uPARAP-positive fibroblasts in vivo after lung injury. (A and C) Flow cytometry–based quantification of GFP-positive cells and their MFIs in lung cell populations acquired from unchallenged (A) or bleomycin-treated (C) col1a1.GFP mice. (B and D) Uptake of Alexa Fluor 647–labeled SP-D (white) by lung cells was examined using confocal imaging 24 h after intratracheal instillation into the lungs of unchallenged or bleomycin-treated col1a1-GFP mice. SP-D uptake in unchallenged mice (B) was dominated by alveolar macrophages (arrows), but after bleomycin injury (D), fibroblasts (green) with SP-D uptake were abundant. Hoechst 33342 was used for visualization of cell nuclei (blue). (E–G) Flow cytometry–based analysis of ligand uptake by lung cells from unchallenged or bleomycin-treated col1a1-GFP mice. Cellular uptake of fluorescent ligands was recorded 24 h after intratracheal instillation of Alexa Fluor 647–labeled SP-D (E and F), or 24 h after i.v. injection of Alexa Fluor 647–labeled anti-uPARAP mAb2h9 (G). (E) Fraction of GFP+ fibroblasts (left) and other lung cell populations (right) positive for SP-D uptake. Col1a1-GFP mice that had not been injected with fluorescent SP-D were used as negative controls. (F) Level of SP-D associated with SP-D+GFP+ fibroblasts as determined by Alexa Fluor 647 MFI. (G) Fraction of GFP+ fibroblasts (left) and other lung cell populations (right) positive for uptake of fluorescent mAb 2h9. Col1a1-GFP mice injected with Alexa Fluor 647–labeled irrelevant murine IgG was used as negative control. n = 7–9. Two-tailed Student’s t test was used to test significance. (H) Top: Western blotting for uPARAP in lysates prepared from untreated or bleomycin-treated mouse lung cells enriched for fibroblasts (see Materials and methods). Cell lysates from uPARAP-deficient mice (−/−) were included as negative controls. Bottom: Nonreducing SDS-PAGE and Coomassie Brilliant Blue staining of protein lysates as loading controls for the Western blot. (I) Internalization of radiolabeled SP-D in primary dermal fibroblasts from uPARAP−/− mice and WT (uPARAP+/+) littermates. Analysis was performed as in Fig. 3 F.
These observations were in line with the fact that SP-D is confined to the alveolar space in nonpathological conditions. Therefore, to test a possible role of fibroblasts in SP-D endocytosis after lung injury, we subjected col1a1-GFP mice to treatment with bleomycin, inducing a state of acute interstitial lung inflammation, which gradually progresses to fibrosis (Williamson et al., 2015). After 10 d, we then delivered fluorescent SP-D to the lungs by intratracheal instillation and examined the lungs as above. As expected, bleomycin treatment led to a pronounced increase in the number of inflammatory (CD45+) cells (Fig. S3 B) but only modest relative changes in both CD45+ and CD45− subpopulations (Fig. S3, C and D). After treatment, a large fraction of both macrophages (CD45+F4/80+CD11b+) and other myeloid cells (CD45+F4/80−CD11b+) had measurable expression of GFP (Fig. 4 C, left; and Fig. S3 A). However, the GFP intensity of inflammatory cells was again very much lower than that of the fibroblast population (Fig. 4 C, right), with this difference being even larger (30–50-fold) than that observed in the healthy lung. Consequently, this system would still enable a safe identification of fibroblasts based on a strong GFP expression.
Strikingly, confocal analysis of the lungs after injury revealed that GFP-positive fibroblasts with SP-D uptake were now abundant (Fig. 4 D). In these cells, the SP-D fluorescence displayed a vesicular pattern very similar to that observed with fluorescent MBL in uPARAP-positive skin fibroblasts.
The uptake of fluorescent SP-D in different cell types was then determined by flow cytometry of lung cell suspensions from the injected mice (Fig. S4 A; see Fig. 2 for gating system). Fibroblasts (CD45−CD31−EpCAM−GFP+ cells) of unchallenged mice were confirmed to be mostly negative for SP-D uptake (mean value 13% SP-D–positive cells), but in accordance with the confocal imaging, a much larger proportion of fibroblasts (49%) became positive for SP-D after bleomycin-induced injury (Fig. 4 E, left; and Fig. S4 A). Even more importantly, the SP-D fluorescence signal in these SP-D–endocytosing fibroblasts was strongly increased after bleomycin treatment (Fig. 4 F).
In a separate experiment, bleomycin-treated or unchallenged mice were injected with fluorescent anti-uPARAP mAb2h9 into the circulation, followed by flow cytometry analysis of lung cell suspensions as above. Very strikingly, whereas only 14% of fibroblasts in unchallenged mice were positive for endocytosis of the anti-uPARAP antibody, this fraction increased to 48% after bleomycin treatment (Fig. 4 G, left; and Fig. S4 B). A Western blotting of fibroblast-enriched lung cells confirmed a strong up-regulation of uPARAP after bleomycin treatment (Fig. 4 H). Concomitantly, a strong increase in the col1a1 promoter–driven expression of GFP was noted within the CD45−CD31−EpCAM−GFP+ cell population when comparing bleomycin-treated and unchallenged mice (Fig. 4, A and B, right), consistent with fibroblast activation (Kendall and Feghali-Bostwick, 2014).
In addition to fibroblasts, various other cell types also showed increased or decreased association with SP-D after lung injury (Fig. 4 E, right; and Fig. S4 A), but in none of these was uPARAP detected (Fig. 4 G, right). When analyzing the distribution of all SP-D+ cells within the CD45− and CD45+ populations, respectively, fibroblasts were found to make up only 21% of CD45−SP-D+ cells in the unchallenged mice but as much as 61% of CD45−SP-D+ cells after bleomycin treatment (Fig. S4 C, left), supporting an increased role of fibroblasts in SP-D uptake after lung injury. Macrophages were the clearly dominant CD45+SP-D+ cell population in both unchallenged and bleomycin-treated mice (Fig. S4 C, right). Details on the macrophage contribution to SP-D uptake are provided below.
To evaluate a direct role of uPARAP in SP-D endocytosis and turnover, we examined the uptake of this collectin in vitro using primary fibroblasts from WT and uPARAP-deficient mice. As for MBL, a pronounced uptake of radiolabeled SP-D was evident in WT cells, with a strongly reduced uptake being obtained in the uPARAP-deficient cells as well as in WT cells treated with mAb 5f4 (Fig. 4 I).
uPARAP expression regulates endogenous SP-D in the injured lung and affects the sensitivity of mice to lung damage
With fibroblasts evidently involved in clearance of exogenously instilled SP-D after lung injury, we next studied the importance of uPARAP in governing the endogenous SP-D levels and the pathological consequence thereof. To do this, we performed bronchioalveolar lavages (BALs) on uPARAP-deficient and littermate WT mice that were either kept unchallenged or subjected to lung injury as caused by treatment with either bleomycin or lipopolysaccharide (LPS). After determining the lung weight of each specimen (Fig. S5, A–C), BAL fluids were then assayed for SP-D content by ELISA (Fig. 5, A–C). This experiment showed that the uPARAP-mediated uptake of SP-D in injured lungs had a direct impact on the alveolar SP-D concentration levels. Thus, the BAL samples from the injured lungs displayed a significantly elevated SP-D concentration in the uPARAP-deficient mice as compared with WT, with mean values of 134% and 148% for bleomycin and LPS treatment, respectively (Fig. 5, A and B). As expected from the experiment with fluorescence-labeled SP-D above, the SP-D concentrations proved identical in WT and uPARAP-deficient mice in the absence of challenge (Fig. 5 C).
Figure 5.
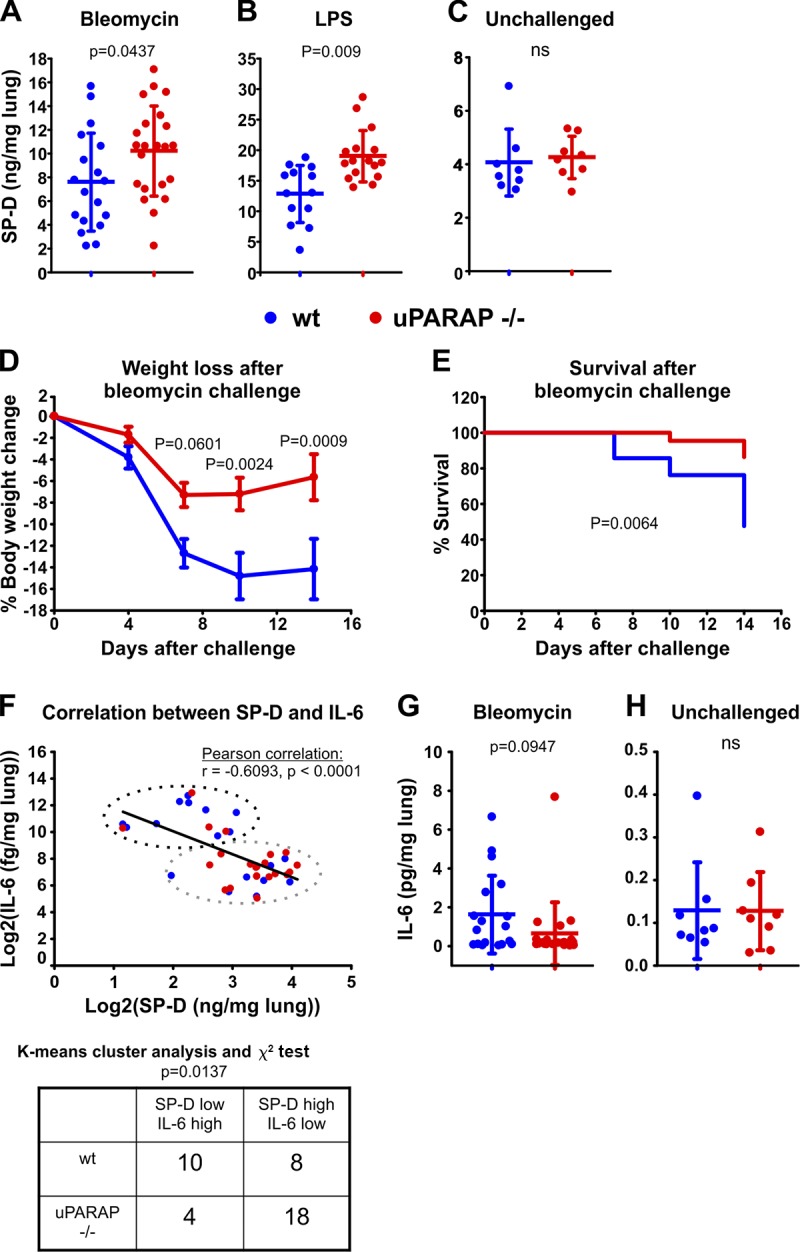
uPARAP regulates endogenous SP-D levels in the injured lung and is decisive for mouse sensitivity toward bleomycin-induced lung injury. (A–C) Endogenous SP-D was determined in BALs collected from bleomycin-treated (A), LPS-treated (B), and unchallenged (C) WT and uPARAP-deficient littermate mice. BALs from bleomycin- and LPS-challenged mice were collected 14 d and 48 h after challenge, respectively. Data are presented as mean ± SD. (D and E) Percent loss of weight (D) and overall survival rate (E) of WT and uPARAP-deficient littermates during a period of 14 d following bleomycin-induced lung injury. n = 22–23. Data are presented as mean ± SEM (D). (F) Correlation between BAL concentrations of SP-D and IL-6 after bleomycin challenge. A combined dataset of log2-transformed SP-D and IL-6 measurements (n = 40) from WT and uPARAP-deficient mice was used to test for correlation (top). An unbiased K means cluster analysis with two clusters was then used to generate an SP-D“low”/IL-6“high” cluster and an SP-D“high”/IL-6“low” cluster, and the number of data points from WT and uPARAP-deficient mice associated with each cluster was determined (top and bottom). (G and H) IL-6 concentrations in BAL from bleomycin-challenged (G) and unchallenged (H) mice. The BAL SP-D and IL-6 concentrations were measured by sandwich ELISA. n = 18–22 (A and G), 13–16 (B), and 8 (C and H). The following statistical tests were used: two-tailed Student’s t test (A–C, G, and H), two-way ANOVA and Sidak’s multiple comparisons test (D), log-rank (Mantel-Cox) test (E), and Pearson correlation and χ2 test (F).
Since SP-D has been shown to have a protective role against bleomycin-induced lung damage (Casey et al., 2005), we also monitored cohorts of uPARAP-deficient and WT mice with respect to body weight and survival during a 14-d period after treatment. Strikingly, the uPARAP-deficient mice proved less sensitive to bleomycin challenge than their WT littermates, evident from a much reduced weight loss (Fig. 5 D) as well as an increased survival rate (Fig. 5 E). As an indicator of inflammation, we measured the BAL content of interleukin 6 (IL-6) in the treated mice. As expected, this experiment demonstrated a negative correlation between the SP-D concentration and the content of the inflammatory cytokine (Fig. 5 F) and also suggested a decreased level of IL-6in uPARAP-deficient mice as compared with WT mice (Fig. 5 G). Although the latter difference did not reach statistical significance, a K means cluster analysis revealed that the difference in the distribution of mouse genotypes with respect to these parameters was indeed statistically different, with WT mice associated with the SP-D–low/IL-6–high cluster and uPARAP knockout mice in the SP-D–high/IL-6–low cluster (Fig. 5 F). No difference in IL-6 levels was found between unchallenged WT and uPARAP-deficient mice (Fig. 5 H).
The macrophage contribution to SP-D uptake is reduced upon lung injury and is independent of MR
Since uPARAP-negative lung macrophages were also found to be active in SP-D uptake (Fig. 4, E and G), we further investigated the contribution from this cell type and subpopulations thereof (Fig. 6 A). These analyses revealed that both alveolar and interstitial macrophages but not monocyte-derived cells took part in the uptake of SP-D (Fig. 6, B and C; and Fig. S4 D). In unchallenged mice, the alveolar subpopulation was the dominant type of macrophage with respect to SP-D uptake, both in terms of number of positive cells and the mean fluorescence intensity (MFI) obtained. This was in accordance with the result of confocal imaging (Fig. 4 B). However, the contribution from these cells declined strongly after bleomycin treatment (Fig. 6, B and C). Under these conditions, the interstitial macrophages became the more abundant SP-D–positive macrophage type (Fig. 6 B). This was consistent with the increased exposure of interstitial tissue, leading to increasing lung fluid contact with both fibroblasts and interstitial macrophages. However, the interstitial macrophages did not show the high levels of uptake as seen for the alveolar macrophages in the untreated mice (Fig. 6 C).
Figure 6.

MR-positive macrophages associate with SP-D in the healthy and injured lung, but MR is not involved in SP-D uptake. (A) Flow cytometry strategy for analyzing lung macrophages for the association with SP-D and their expression of MR. Single-cell suspensions were stained for CD45, F4/80, CD11b, SiglecF, and CD11c. CD45+F4/80+CD11b+ cells were separated into SiglecF+ cells (alveolar macrophages), SiglecF−CD11c+ cells (interstitial macrophages), and SiglecF−CD11c− cells (monocyte-derived macrophages; Misharin et al., 2013). (B) Quantification of the number of macrophages from each subpopulation associating with SP-D in unchallenged lungs and lungs of bleomycin-treated mice. Experimental conditions were as described in Fig. 4 E. (C) Level of SP-D (Alexa Fluor 647 SP-D MFI) associated with SP-D+ subpopulations of macrophages. n = 4. One-way ANOVA was used to test for statistical significance. (D) Fraction of each subpopulation of macrophages positive for MR expression. n = 5. (E and F) Endogenous SP-D was determined in BAL collected from unchallenged (E) and LPS-treated (F) WT mice and MR-deficient littermates by sandwich ELISA. BAL from LPS-challenged mice was collected 48 h after challenge. n = 8 (E) and 7 (F). (G) Internalization of radiolabeled SP-D and collagen type I by alveolar macrophages isolated from lungs of WT mice and MR-deficient littermates. CPM, counts per minute. n = 3. A two-tailed Student’s t test was used for test of significance (E–G).
We also studied whether the macrophage-associated receptor MR was involved in the macrophage-mediated uptake of SP-D in vivo. Even though the collectin uptake studies with transfected cells in vitro (Fig. 1) had pointed to a very low uptake activity of MR for SP-D, the striking activity of MR in internalizing structural collagens still made this receptor an interesting candidate (Madsen et al., 2013b; Jürgensen et al., 2014). We first analyzed whether MR was expressed by the relevant macrophages in the lungs using flow cytometry. This experiment revealed that almost all of the alveolar and a large proportion of the interstitial macrophages but only very few monocyte-derived cells were positive for MR (Figs. 6 D and S4 E). We then compared the BAL SP-D contents and the lung weights of unchallenged and LPS-treated MR-deficient and littermate WT mice (Fig. 6, E and F; and Fig. S5, D and E). However, mice of these genotypes displayed no difference in BAL SP-D concentration irrespective of whether unchallenged mice or mice subjected to lung injury were compared. Finally, since alveolar macrophages were the highest MR-expressing macrophage type, we harvested these cells and assayed for uptake of radiolabeled SP-D. This experiment revealed that the capacity for endocytosis of SP-D was the same in MR-deficient and WT macrophages (Fig. 6 G, left) even though MR was essential for the uptake of collagen into the same macrophage isolates (Fig. 6 G, right).
Expression of SP-D and uPARAP in fibrotic human lungs
In the healthy organism, SP-D is present in the lungs, where it is deposited on alveolar and mucosal surfaces after synthesis in alveolar type II cells and Clara (Club) cells (Madsen et al., 2000). In consistence with this pattern, staining of nonfibrotic human lung specimens with an SP-D–specific antibody displayed a clear localization in distinct alveolar epithelial cells (example in Fig. 7 A, middle). Although some synthesis of uPARAP has been demonstrated previously in the healthy adult human lung at the mRNA level (Wu et al., 1996), we obtained only weak staining when incubating sections adjacent to those above with a uPARAP-specific antibody (Fig. 7 A, right). However, when sections from the lungs of patients with idiopathic pulmonary fibrosis (IPF; Fig. 7 B) or radiotherapy-induced lung fibrosis (Fig. 7 C) were immunostained under the same conditions, a strong expression of both proteins was observed. In these sections, revealing extensive interstitial fibrosis (Fig. 7, B and C, left), SP-D was expressed in the hyperplastic alveolar epithelial cells surrounding the residual alveolar spaces entrapped in the fibrotic tissue (middle) in accordance with results published previously (Nishikiori et al., 2014). While a major part of the epithelial tissue was lost in this condition, the SP-D–positive fraction of the remaining epithelial cells was more than twofold increased relative to the nonfibrotic tissue (Fig. 7 D). In contrast, very little SP-D was detected when examining the bronchiolized region of the same specimen (Fig. 7 E). uPARAP was found to be strongly expressed in fibroblasts inside and around the active fibrosing subepithelial lesions typical of IPF (fibroblast foci; Fig. 7, B and C, right) closely associated with the SP-D–positive epithelial cells. Although the absence of this type of tissue in the nonfibrotic condition precluded a histology-based quantification of uPARAP, it was evident that uPARAP-positive cells were much more abundant in the fibrotic than in nonfibrotic tissue (compare Fig. 7, A–C, right). Thus, in the alveolar region of fibrotic lungs, uPARAP-expressing fibroblasts appeared to be closely associated with cells and regions of SP-D secretion.
Figure 7.

uPARAP is up-regulated in affected regions of human fibrotic lungs and is closely associated with hyperplastic alveolar epithelial cells expressing high levels of SP-D. (A–C) H&E stain (left) and immunostaining for SP-D and uPARAP (middle and right, respectively) in serial sections from surgically resected nonfibrotic alveolar tissue from patients with emphysema (A; nonfibrotic; n = 5), fibrotic tissue from patients with IPF (B; n = 4), and fibrotic tissue from a patient with radiotherapy-induced fibrosis (C). SP-D was detected in single cells with an epithelial appearance along the alveolar walls of nonfibrotic lung tissue (A, middle, arrows) and was extensively expressed in lung tissue from patients with fibrosis (B and C, middle). Here, strong SP-D staining was observed in hyperplastic alveolar epithelial cells (a) in close proximity with active fibrosing subepithelial fibroblast foci (f). These foci included a large number of highly uPARAP-positive fibroblasts (B and C, right). (D) Top: Quantification of the fraction of SP-D–positive epithelial cells in alveolar walls of nonfibrotic tissue and in alveolar compartments entrapped in fibrotic tissue. Bottom: Visiopharm software was used for automated scoring of SP-D–positive and –negative cells in the alveolar wall (see Materials and methods). (E) Immunostaining for SP-D in a bronchiolized area (b) of fibrotic tissue from patient with IPF. Note that cells of the bronchiolized epithelium did not express SP-D. (F) Internalization of radiolabeled SP-D in the human lung fibroblast cell lines MRC-5 and CCL-210 examined in the absence or presence of the uPARAP function-blocking antibody mAb 5f4. A two-tailed Student’s t test was used to test significance (D and F).
To study whether human lung fibroblasts can internalize SP-D in a uPARAP-dependent process, we used two human lung fibroblast cell lines (MRC5 and CCL-210). Just like mouse fibroblasts, these cells displayed a strong uptake of radiolabeled SP-D, which was decreased in the presence of the uPARAP-blocking antibody (Fig. 7 F).
The strong up-regulation of SP-D in hyperplastic epithelial cells of the fibrotic lungs made it important to study whether this was an exclusive property or was a more general phenomenon with surfactant components. Immunohistochemical localization of two such components, SP-A and SP-C, revealed an expression pattern closely similar to that of SP-D. This applied to both the expression in alveolar type II cells of nonfibrotic tissue (Fig. 8 A) and the up-regulation in hyperplastic epithelial cells of the fibrotic lungs (Fig. 8 B). Since all SPs are secreted onto the alveolar surface, this opens the possibility that exposed fibroblasts in fibrotic tissue could facilitate their turnover.
Figure 8.
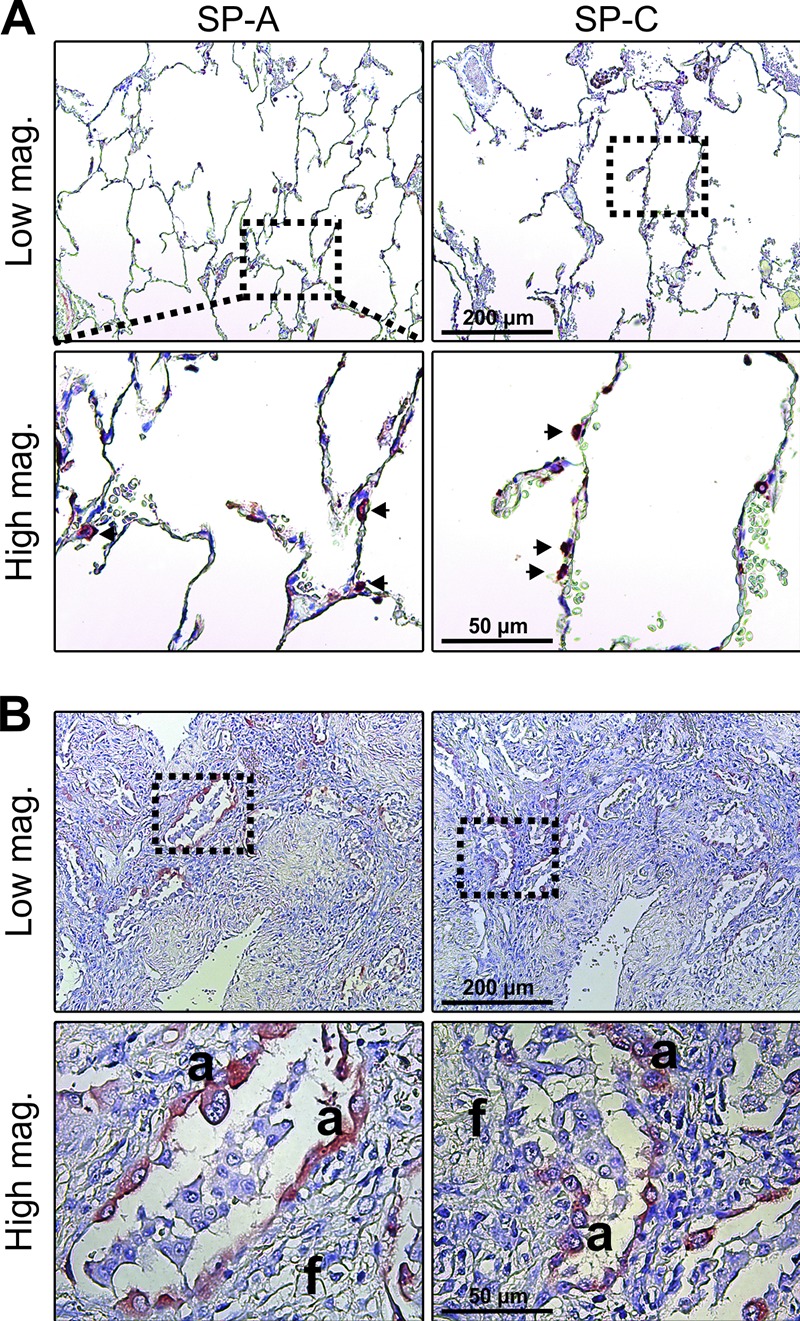
Hyperplastic alveolar epithelial cells express high levels of the collectin SP-A and the surfactant-associated SP-C. (A and B) Immunostaining for SP-A (left) and SP-C (right) in sections of nonfibrotic alveolar tissue (A; arrows highlight positive cells in alveolar wall) and fibrotic tissue in patients with IPF. The number and type of tissue specimens included in the analysis are described in the legend to Fig. 7. Like for SP-D, a strong staining for SP-A and SP-C was observed in hyperplastic alveolar epithelial cells (a) in close proximity with active fibrosing subepithelial fibroblast foci (f).
Discussion
Several types of tissue injury induce the recruitment of collectins to extravascular locations, where they take part in a variety of processes with importance for inflammation and immune regulation. Thus, the role of collectins is not restricted to pathogen pattern recognition but also includes the binding to immunomodulatory receptors (Olde Nordkamp et al., 2014). In some cases, such as the function of SP-D in the injured lung, the up-regulation serves to dampen inflammation (Casey et al., 2005; Aono et al., 2012), whereas in others, increased collectin levels can lead to excessive inflammation and tissue damage (Walsh et al., 2005; van der Pol et al., 2012; Farrar et al., 2016). In all cases, collectin levels need to be regulated in time and space, which makes the controlled clearance of these proteins a task of crucial importance. In this study, we show that fibroblasts are active in the extravascular clearance of SP-D and MBL, two collectins that have diverse functions and completely different expression patterns but share the property of increased secretion into injured tissue. We show that the responsible fibroblasts express the endocytic receptor uPARAP and that this receptor can undertake the endocytosis of collectins for intracellular degradation in vitro. In the case of SP-D, the levels of the endogenous protein could be measured directly in BAL samples after lung injury in mice, enabling the investigation of the functional role of the receptor in vivo. In this situation, the SP-D levels were found to be regulated by the expression of uPARAP in accordance with the receptor being active in SP-D clearance.
This uPARAP-mediated process is particularly intriguing because it constitutes a novel immune-regulatory function of fibroblasts. Activated tissue-specific fibroblasts have been shown to be critical mediators of immune system–related processes in the progression of mouse models of arthritis and intestinal disease (Armaka et al., 2008; Nikitopoulou et al., 2012), but to our knowledge, this study is the first demonstration of a general negative regulatory mechanism for a family of immune-components, in this case collectins, governed directly by fibroblasts (Fig. 9).
Figure 9.
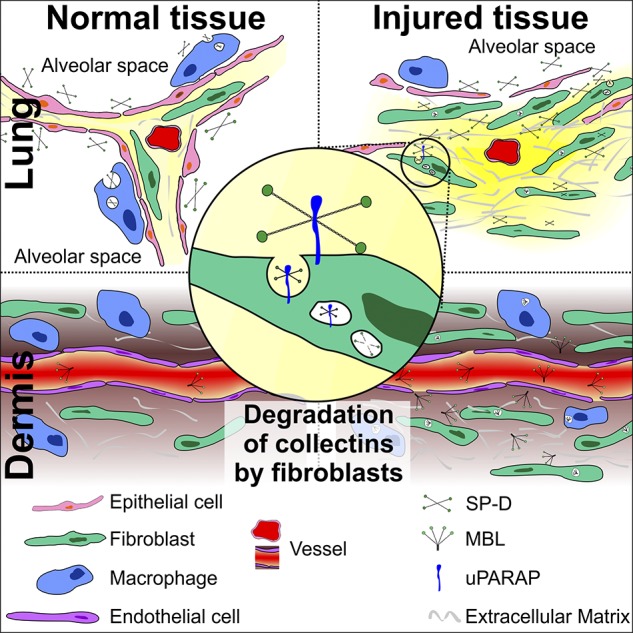
Regulation of extravascular collectins in normal and injured tissue. In normal tissue (left), collectins are largely confined to luminal compartments such as the alveolar space (SP-D), or they are mostly present in the blood (MBL). In the case of SP-D, under normal conditions, alveolar macrophages govern the levels of this collectin through internalization by an unknown mechanism. After injury, disruption of normal tissue architecture leads to breakage of luminal tissue barriers and to extravasation of collectins into the extravascular space (right). Under these conditions, fibroblasts play a crucial role in regulating collectins by undertaking collectin endocytosis alongside tissue macrophages. The endocytosis of collectins by fibroblasts is mediated by the collagen receptor uPARAP (middle).
The novel clearance pathway was found to be tightly linked to tissue injury. When investigating nonfibrotic human lung samples and healthy lungs from mice, we found uPARAP to be expressed at a low level and by a fraction of fibroblasts only. Moreover, under these conditions, we found very limited evidence of contact between fibroblasts and SP-D–secreting epithelial cells, a low association between fibroblasts and exogenously added fluorescent SP-D, and a lack of any effect of uPARAP deficiency on the endogenous SP-D levels in BAL. All of these characteristics changed dramatically after lung injury, where the disruption of normal alveolar tissue architecture and an increased expression of uPARAP appeared to lead to an increased fibroblast–SP-D association (Figs. 4, 5, and 7) and a resulting uPARAP-mediated clearance of SP-D. SP-A, a structurally related SP, was expressed in the same location as SP-D in human fibrotic tissue, opening the possibility that this SP may also be cleared by uPARAP. However, SP-C, being structurally unrelated to SP-A and -D, would be unlikely to interact with this receptor, although it cannot be excluded that fibroblasts are also active in the clearance of this protein.
For SP-D, the importance of this turnover mechanism was demonstrated in two well-characterized lung disease models based on bleomycin or LPS. Importantly, the molecular events that trigger lung injury in these two situations are considered to be completely different (Beutler and Rietschel, 2003; Williamson et al., 2015). Therefore, the fibroblast- and uPARAP-mediated SP-D uptake appears to be a general SP-D clearance pathway in the injured or fibrotic lung, although it should be noted that our bleomycin model focused mostly on the acute inflammation phase; see further below.
The elevated endogenous SP-D levels resulting from uPARAP deficiency were accompanied by pronounced effects on body weight loss and survival in the bleomycin-treated mice (Fig. 5, D and E). This effect of uPARAP deficiency accurately mimics the potent antiinflammatory activity of SP-D demonstrated by genetic manipulation experiments, where SP-D deficiency and overexpression have been shown to induce increased and decreased sensitivity, respectively, in the bleomycin model (Casey et al., 2005). Consequently, the uPARAP-mediated uptake process is likely to contribute to the overall response to bleomycin-induced lung injury by regulating the SP-D concentration, which in turn affects the inflammatory response, including the levels of inflammatory cytokines.
Although the function of uPARAP in internalizing structural collagen may also affect the response to lung fibrosis (Bundesmann et al., 2012), this cannot explain the response to bleomycin treatment observed in our work. First, we observed bleomycin resistance in uPARAP-deficient mice as early as 7–10 d after challenge, which corresponds with the acute inflammation phase of the lung injury model, clearly preceding the fibrotic phase (Williamson et al., 2015). Second, the reduced turnover of structural collagen resulting from loss of uPARAP (Madsen et al., 2012) would exacerbate the fibrotic condition rather than improving mouse resistance to injury. The results of the above-mentioned study (Bundesmann et al., 2012) also included a minor difference in the lung tissue mass between uPARAP-deficient and WT mice after bleomycin treatment. This finding was confirmed in our study, with the lung wet weight being ∼10% lower in deficient mice (Fig. S5) and with the BAL showing a trend toward lower total protein content.
uPARAP has also been found to take part in molecular interactions with the urokinase plasminogen activator (uPA)–uPA receptor (uPAR) complex on certain cell types (Behrendt et al., 1993, 2000). In this connection, it is interesting that uPA has been found to be up-regulated and associated with both fibroblasts and epithelial cells, possibly in a complex with uPAR, in lung fibrosis (Schuliga et al., 2017). However, it is unlikely that this interaction plays a role in the SP-D uptake, or in the collagen interactions, that are governed by uPARAP in this condition. First, the capacity of uPARAP for recognition of collagen-like motifs does not require uPA/uPAR (Behrendt et al., 2000; Madsen et al., 2007). Second, uPAR and uPA are devoid of any collagen recognition capacity in their own right, making them unlikely candidates for a function in the turnover of collagen and collagen-related ligands.
Additional routes of SP-D clearance have been demonstrated in earlier studies by others and are also evident in this work. In the healthy lung, the alveolar macrophage has been well documented to be a general regulator of SPs and lipids (Yoshida et al., 2001; Greenhill and Kotton, 2009). Our data in healthy mice confirm this role of alveolar macrophages also in the specific catabolism of SP-D (Fig. 6, A–C). After LPS-induced lung inflammation, the increased cellular uptake of SP-D has been shown to include a pronounced contribution from neutrophils in rats, whereas the number of uptake-positive macrophages was found to be decreased (Herbein and Wright, 2001), consistent with our observations of a reduced contribution of macrophages to SP-D uptake after injury (Fig. 6). A markedly increased uptake in unidentified (nonleukocyte) cells was noted upon lung injury in the same study, which can now most likely be ascribed to the fibroblast contribution to SP-D turnover. Other investigators have suggested that a direct leakage from the injured lung into the blood could be a potential drainage mechanism for SP-D in IPF (Nishikiori et al., 2014). Although our study does not contradict a contribution from this route, the same underlying tissue damage most likely also leads to the increased SP-D contact with the fibroblast compartment as demonstrated in this study.
Based on the in vivo and in vitro research reported in this study, we conclude that the collectin-clearance capacity of uPARAP is not shared by MR (Figs. 1 and 6). This is particularly surprising because both receptors are endocytic receptors for structural collagens (Jürgensen et al., 2014). Indeed, to our knowledge, they are the only receptors shown so far to have a direct capacity for collagen endocytosis through the clathrin–endosome–lysosome system, and notably, these two receptors are expressed on different cell types, with uPARAP mainly on activated fibroblasts and MR mainly on macrophages. Both receptors use a highly similar FN-II domain–mediated mechanism for collagen uptake (Jürgensen et al., 2014). However, since the organization of functional lectin domains differs between these receptors, with the active domain being CTLD-2 in uPARAP and CTLD-4 in MR (East and Isacke, 2002; Jürgensen et al., 2011), this is one molecular feature which may lie behind the difference in their potential for recognition of collectins.
In this study, we demonstrate an uptake capability of uPARAP not only for MBL and SP-D but also for a broad range of other defense collagens (Fig. 1). Indeed, based on the common possession of collagen-like domains in this group of proteins, uPARAP may have some endocytic capacity for all types of defense collagens, although differences were noted in the efficiency and specificity of uPARAP toward some of the candidates tested. In addition to soluble collectins and ficolins, these potential ligands include membrane proteins such as SR-AI, MARCO, and collectin-12 (Fig. 1 A). While these interaction partners may also be subject to uPARAP-dependent endocytosis after shedding from the cell surface, it is also possible that they interact with uPARAP in their cell-associated form to facilitate fibroblast interactions with their host cells.
Interestingly, with the identification of uPARAP as a novel immune receptor regulating collectins, all four homologous members of the MR protein family now appear to be regulators of immune system–related functions. Thus, MR directly acts as a pattern recognition receptor, a regulator of T cell activity, and has multiple other proposed functions (East and Isacke, 2002; Martinez-Pomares, 2012; Schuette et al., 2016); PLA2R regulates the activity of proinflammatory secretory phospholipase A2 (Tamaru et al., 2013); and DEC-205 delivers antigens for intracellular processing before presentation (Jiang et al., 1995). We believe that the current demonstration of a uPARAP-mediated fibroblast engagement in collectin turnover is critical for the understanding of the complicated pattern of cellular events that are responsible for the regulation of the innate immune system. Due to the broad spectrum of disease-associated targets of collectins and the general importance of their regulation, this may have direct consequences for understanding and treating several pathological conditions.
Materials and methods
Reagents and cells
The following reagents were purchased from commercial sources: bleomycin sulfate and LPS from E. coli (Sigma-Aldrich), Lipofectamine 2000, Alexa Fluor 546–conjugated 10-kD dextran, E. coli bioparticles, Hoechst 33342, Alexa Fluor 647, and Alexa Fluor 555 protein-labeling kits (Life Technologies), iodine-125 (PerkinElmer), E64d (EMD Millipore), proteinase K (Roche), ManBSA (Dextra Laboratories), collagenase type I, DNase type I, and neutral protease (Worthington Biochemical). Monoclonal mouse anti-uPARAP antibodies 2h9 and 5f4 were described previously (Sulek et al., 2007; Madsen et al., 2011). Recombinant mouse MBL-A (used in experiments involving primary fibroblasts and in vivo uptake in the skin), human MBL (used in experiments involving transfected CHO-K1 cells), human SP-D, human ficolin-1, soluble human SR-AI, human CL-P1/collectin-12, human CL-K1/collectin-11 (all produced in mouse myeloma NS0-derived cells), and soluble human MARCO (produced in CHO cells) were purchased from R&D systems. Purified human C1q was purchased from Complement Tech. Antibodies for immunohistochemistry and flow cytometry are described in sections below.
Adherent CHO cells (CHO-K1; CCL-61, RRID: CVCL_U424; ATCC) for use with the Flp-In transfection system were purchased from Life Technologies. Human lung fibroblast cell lines MRC-5 (CCL171) and CCD-19Lu (CCL-210) were acquired from ATCC. Primary cultures of newborn skin fibroblasts and alveolar macrophages are described in the Isolation of primary cells section.
Patient samples
Paraffin-embedded, formalin-fixed, pulmonary alveolar human tissue samples included in this study were surgically resected nonneoplastic and nonfibrotic tissue with emphysema from patients operated for pulmonary tumors (n = 5), fibrotic tissue from patients with IPF (n = 4), and fibrotic tissue from a patient with radiotherapy-induced fibrosis (n = 1). Serial sections from each sample were deparaffinized in xylene, rehydrated in alcohols, and stained with H&E or used for immunostaining for SP-D, SP-A, SP-C, and uPARAP (see below). All H&E-stained and immunostained sections were examined by two lung pathologists (K. Wassilew and E. Santoni-Rugiu) to confirm the diagnoses and to assess morphology and identity of SP-D– and uPARAP-positive cells.
Animal experiments, breeding, and genotyping
Transgenic C57BL/6J mice expressing enhanced GFP under the col1a1 promoter from one allele (Col1a1GFP/0 or col1a1.GFP; Krempen et al., 1999) were bred to WT C57BL/6J mice to generate the experimental mice used in this study. Presence of the Col1a1.GFP transgene was confirmed by observation of green fluorescence in the skin of mice following examination with blue light from a DFP-1 dual fluorescent protein flashlight (Nightsea) and matching filter goggles. Mice homozygous for a targeted mutation in the Mrc1 gene (encoding MR protein; C57BL6/Mrc1tm1Mnz/J; RRID: MGI:3723178; Lee et al., 2002) and WT littermates were generated by interbreeding heterozygous mice (Mrc1+/−). Genotyping of offspring was performed using PCR with the primers 5′-ATGAGGCTTCTCCTGCTTCT-3′ (WT allele forward primer) and 5′-AGCCCTGATCTGTTCCTACG-3′ (common reverse primer) in one reaction and 5′-CAAGATCCGCCACAACATCG-3′ (mutant allele forward primer) and the common reverse primer in a separate reaction (55°C annealing). C57BL/6J mice homozygous for a targeted mutation in the Mrc2 gene (encoding the uPARAP protein; C57BL6/Mrc2tm1Bug; RRID: MGI:2677820; Engelholm et al., 2003) and WT littermates were generated by interbreeding heterozygous mice (Mrc2+/−). Genotyping was performed using PCR with the primers 5′-TCCTACAAATACACGCTGGCGATA-3′ (mutant allele forward primer), 5′-GCAGTTCCCTTTTAAATGCAAATCA-3′ (mutant allele reverse primer), 5′-TCTACACCATCCAGGGAAACTCAC-3′ (WT forward primer), and 5′-TTAAACTGGTAACAGCTGTCAGTC-3′ (WT reverse primer). The annealing temperature for this reaction was 60°C.
Experimental groups of mice consisted of littermates of both sexes with an average age between 13 and 16 wk (age range for all animals: 7–23 wk). The number of males and females were matched to obtain equal distributions for all groups directly compared. The total number of animals included in experiments in this study was 189, distributed according to the following: flow cytometry, 48 mice; lung wet weight recordings and SP-D ELISA readings, 117 mice; confocal imaging of the skin and lungs, 16 mice; and Western blot analysis for uPARAP, 8 mice.
Immunohistochemistry
After deparaffinization and rehydration, antigen retrieval was performed on sections for immunostaining using the following conditions: For SP-D, microwave heat–induced epitope retrieval in citrate buffer, pH 6.0, at 98°C for 15 min; for uPARAP, SP-A, and SP-C, 5 µg/ml proteinase K (Roche) at 37°C for 15 min. Next, endogenous peroxidase was blocked using a solution of 1% H2O2 in water. The sections were then incubated with the following primary antibodies overnight at 4°C: mouse mAb 2h9 against uPARAP/Endo180 (10 µg/ml; Sulek et al., 2007), rabbit anti-human SP-D (1:2,000; Sigma-Aldrich), rabbit anti-human/mouse proSP-C (1:2,000; EMD Millipore), and rabbit anti–human/mouse SP-A (1:500; EMD Millipore). Primary antibodies were diluted in antibody diluent from Dako. For detection, sections were incubated for 45 min with the appropriate detection system for mouse or rabbit antibodies (Envision mouse or rabbit; Dako). NovaRed substrate kit (VWR) was used for development by incubation on sections for 9 min. In between each step, sections were washed twice in TBS-T buffer. To quantify SP-D expression in alveolar compartments of tissue sections from patients suffering from IPF and nonfibrotic tissue sections from patients with emphysema, an unbiased cell classifier was trained using Visiopharm image analysis software (Hoersholm). The cell classifier was fully trained to score SP-D–positive and SP-D–negative cells in defined regions of interest (ROIs) based on NovaRed substrate color recognition. In fibrotic tissue, ROIs were drawn to include one to two cell layers of tissue from alveolar compartments (a minimum of 2,000 cells was counted per patient) with viable epithelial cells visible, entrapped in, or in the vicinity of active fibrosing subepithelial lesions, starting at the interface between the alveolar lumen and cells of the alveolar surfaces and moving toward the underlying interstitial tissue. In nonfibrotic lungs, areas of normal-appearing lung were used as ROI with blood vessels and bronchioles excluded.
Bleomycin- and LPS-induced lung injury
Mice were anesthetized (100 mg/kg ketamine and 10 mg/kg xylazine) and intubated using a small animal intubation kit (Biolite). A single dose of bleomycin or LPS diluted in PBS was then administered to the lungs by intratracheal instillation in a volume of 30–60 µl. For bleomycin, a dose of 0.4 U/kg was used in experiments where uptake of fluorescent SP-D was investigated using confocal imaging or flow cytometry and 0.5 U/kg in experiments where BAL was performed to determine endogenous SP-D levels and where body weight loss and survival were monitored. When studying body weight loss and survival, a weight loss of >20% was used as humane endpoint, and if this criterion was met, the relevant mouse was euthanized and scored as nonsurvivor. LPS was used at a dose of 10 µg/kg in all experiments.
In vivo uptake of fluorescent SP-D, MBL, dextran and mAb 2h9
SP-D, murine MBL-A, anti-uPARAP mAb 2h9, or control pool of mouse IgG (Innovative Research) was fluorescently labeled by using an Alexa Fluor 647 protein labeling or microscale protein labeling kit according to the manufacturer’s instructions (Life Technologies). For in vivo uptake of fluorescent SP-D, Col1a1.GFP transgenic mice (either 10 d after bleomycin treatment or untreated controls) were intubated as stated above. A total of 50 µl PBS containing 3 µg Alexa Fluor 647–labeled SP-D was then administered to the lungs by intratracheal instillation. 24 h later, mice were euthanized in a CO2 chamber, and the lungs were immediately perfused with PBS and resected for flow cytometry or confocal imaging. For in vivo labeling of uPARAP-positive cells in lungs, 20 µg Alexa Fluor 647–labeled mAb 2h9 or control mIgG in a volume of 100 µl sterile PBS was injected into the retroorbital vein of Col1a1.GFP transgenic mice that were either bleomycin treated or controls as above. 24 h later, lungs were excised for flow cytometry. For in vivo uptake of fluorescent MBL in the dermis analyzed using confocal imaging, 10 µg Alexa Fluor 647–labeled MBL with either 25 µg Alexa Fluor 546–labeled, 10 kD dextran, or 10 µg Alexa Fluor 555–labeled anti-uPARAP mAb 2h9 was dissolved in 100 µl PBS and administered to the dermis of Col1a1.GFP transgenic mice by subcutaneous injection. 24 h after the injection, mice were euthanized in a CO2 chamber, and the skin surrounding the injection site was removed for immediate confocal imaging. For in vivo uptake of MBL and labeling of uPARAP-positive cells in the dermis analyzed using flow cytometry, three subcutaneous injections of 60 µl PBS containing 5 µg Alexa Fluor 647–labeled MBL, 4 µg Alexa Fluor 647–labeled mAb 2h9, or 4 µg Alexa Fluor 647–labeled control mIgG, respectively, were performed on each mouse. 24 h after the injections, mice were euthanized in a CO2 chamber, and an ∼1-inch/1-inch piece of the skin surrounding the three injection sites was taken for flow cytometry.
Confocal microscopy
For confocal microscopy on mouse lung tissue, the left lung was resected after perfusion with PBS, rinsed briefly in PBS, and immediately imaged intact. For confocal microscopy of mouse skin, the skin covering the site of injection of MBL and Dextran/mAb 2h9 was resected, rinsed briefly in PBS, and imaged with the dermis facing the objective. For visualization of cell nuclei, 0.6 mg Hoechst 33342 in 150 µl PBS was injected into the retroorbital vein ∼2 h before resection of lung tissue or 4–5 h before resection of skin tissue. Confocal imaging was performed using an IX81 inverted confocal microscope equipped with a FluoView 1000 scanning head (Olympus). All images were acquired using a U Plan S Apochromat 60× 1.2 NA water immersion objective. 405-nm and 488-nm (Olympus), 561-nm (Showa Optronic), and 633-nm (Melles Griot) lasers were used for exciting Hoechst 33342, GFP, Alexa Fluor 546, Alexa Fluor 555, and Alexa Fluor 647, respectively. Image acquisition was performed using FluoView acquisition software (Olympus).
Flow cytometry
Lungs or skin tissue excised for flow cytometry was immediately placed on ice and washed with 1 ml ice-cold DMEM. Next, tissues were cut into small pieces using fine scissors and placed in 1 ml (lungs) or 3 ml (skin) digestion mixture consisting of 2.1 mg/ml collagenase type I, 75 µg/ml deoxyribonuclease type I, and either 0.5 mg/ml (lungs) or 0.125 mg/ml (skin) neutral protease (Worthington Biochemical) dissolved in DMEM supplemented with 5 mM CaCl2. Tissues were then incubated at 4°C with gentle shaking overnight followed by 30–60 min incubation at 37°C. The digestion mixture was then placed in 10 ml ice-cold DMEM, filtered through a 40-µm (lungs) or 70-µm mesh (skin) to remove remaining tissue pieces, and centrifuged at 300 g for 5 min. The resultant cell suspensions were next subjected to red blood cell lysis (lung cells only; 15 min at room temperature; Miltenyi Biotec), subjected to live/dead staining using Zombie Violet or Zombie Aqua (15 min at room temperature; BioLegend), and Fc receptor blocking (15 min on ice; Miltenyi Biotec), all according to the manufacturers’ instructions. Lung cells were next stained for surface markers using three different sets of antibodies. Set 1: Anti-CD45 (clone 30-F11; BV605; BioLegend), anti-CD31 (clone 390; phycoerythrin; BioLegend), anti-EpCAM (clone G8.8; APC-CY7; BioLegend); set 2: anti-CD45 (clone 30-F11; BV605; BioLegend), anti-CD11b (clone M1/70; phycoerythrin; BioLegend), anti-F4/80 (clone BM8; BV785; BioLegend); and set 3: anti-CD45 (clone 30-F11; V450; BioLegend), anti-F4/80 (clone BM8; phycoerythrin; BioLegend), anti-CD11b (clone M1/70; BV785; BioLegend), anti-CD11c (clone N418; APC-eFluor780; eBioscience), anti-SiglecF (clone 1RNM44N; PerCP-eFluor710; eBioscience), and anti-CD206/MR (clone C068C2; BV605; BioLegend). Dermal cells were stained using the following antibodies: anti-CD45 (clone 30-F11; BV605; BioLegend), anti-CD31 (clone 390; PerCP-Cy5.5; BioLegend), anti-EpCAM (clone G8.8; APC-Cy7; BioLegend), anti-F4/80 (clone BM8; phycoerytrhin-Cy7; BioLegend), and anti-CD11b (clone M1/70; phycoerythrin; BioLegend). All antibodies were used at 1:100 dilution except anti-CD206, which was used at 1:50. A staining buffer consisting of PBS with 1% BSA was used for antibody stains and FC block incubation. Antibody staining was run for 1 h on ice. After antibody staining, cells were washed twice in staining buffer and filtered through a 40-µm mesh. Flow analysis for surface markers, GFP expression from Col1a1 promoter, Zombie violet or Zombie aqua live/dead stain, and uptake of Alexa Fluor 647–labeled SP-D, MBL, or anti-uPARAP mAb 2h9 was performed using a Fortessa II instrument (lung cell suspensions; BD) or using an LSR-II instrument (dermal cell suspensions; BD). Positive gates for each marker were defined using fluorescence minus one controls with antibodies, GFP, SP-D/MBL, and live/dead stain in turn excluded from the sample. In experiments with in vivo labeling of uPARAP-positive cells with Alexa Fluor 647–labeled mAb 2h9, uPARAP-positive cell gates were set using samples from mice injected with irrelevant Alexa Fluor 647–labeled murine IgG for comparison (Fig. S4)
Western blotting
Single-cell suspensions from unchallenged or bleomycin-challenged mouse lungs were generated as described in the Flow cytometry section. After lysis of red blood cells, cell suspensions were depleted for leukocytes, endothelial cells, and epithelial cells using a mixture of anti-CD45–, anti-CD31–, and anti-EpCAM–coated magnetic beads, MS columns, and a MACS separator according to the manufacturer’s protocol (Miltenyi Biotec). After depletion, cell suspensions (enriched for CD45−,CD31−,EpCAM− cells including fibroblasts) were washed three times in ice-cold PBS, and cells were lysed in lysis buffer (1% Triton X-100, 50 mM Tris, and 100 mM NaCl, pH 7.4) containing protease inhibitor cocktail III (1:200; Sigma-Aldrich). Protein concentrations in lysates were determined using a BCA kit (Bio-Rad). uPARAP was detected in a total of 3 µg protein from each lysate separated by SDS-PAGE and blotted onto a polyvinylidene difluoride membrane. A 2% BSA blocking solution was applied before detection using 0.5 µg/ml anti-uPARAP mAb 2h9 as primary antibody and rabbit anti-mouse HRP-conjugated secondary antibody (1:3,000; Dako), both diluted in a 0.1% Tween-20 PBS solution. For loading control, 6 µg protein of each lysate was analyzed by SDS-PAGE followed by Coomassie Brilliant Blue staining.
Determination of endogenous SP-D and IL-6 concentrations in BAL fluids
Animals were euthanized with CO2 followed by immediate perfusion of lungs with 3 ml PBS. Next, the trachea was exposed by careful removal of the surrounding tissue to allow for a small incision in the trachea. A 21G needle fitted with a 5-mm piece of rubber tubing was then inserted into the trachea, and the BAL was performed with 1 ml PBS. The harvested BAL fluid was centrifuged at 300 g for 5 min at 4°C to remove any cells present and was stored at −80°C. After harvest of BAL fluids, lungs were excised from the chest cavity, and the wet weight was recorded after removal of excess PBS from the BAL procedure with paper towel. SP-D concentrations in BAL fluids were determined using a mouse SP-D DuoSet ELISA (R&D Systems). BAL fluids were diluted 1:750 (BAL from unchallenged mice) or 1:3,000 (BAL from bleomycin- or LPS-challenged mice) for the ELISA analysis of SP-D content. IL-6 concentrations in BAL fluids were determined using a mouse IL-6 DuoSet ELISA (R&D Systems). BAL samples were diluted 1:2 (BAL from unchallenged and bleomycin-challenged mice) for the ELISA analysis of IL-6 content.
CHO-K1 cell transfection
To generate CHO-K1 clones expressing each of the members of the MR protein family, complete coding sequence cDNAs for murine uPARAP, MR, PLA2R, and DEC-205 were inserted into the pcDNA5/FRT/TO vector as previously described (Jürgensen et al., 2014). CHO-K1 cells were transfected with each construct (or with empty pcDNA5/FRT/TO vector to generate mock-transfected cells) according to Flp-In transfection guidelines (Life Technologies) and using Lipofectamine 2000 as a transfection reagent. Monoclones with stable expression of each construct were subsequently isolated in selection media containing 400 µg/ml hygromycin B (Life Technologies). CHO-K1 monoclones were maintained in DMEM/F12 media supplemented with 10% FCS, 1% penicillin/streptomycin, and 400 µg/ml hygromycin B at 37°C. Endocytic characteristics of cells transfected with each of the four members of the protein family have been published previously, including a demonstration that the respective receptors are expressed in an active form (Jürgensen et al., 2014).
Isolation of primary cells
For the isolation of primary fibroblasts, total skin from newborn male or female uPARAP-deficient mice and WT littermates was placed in a 0.25% Trypsin-EDTA solution and incubated at 4°C overnight. The dermis was then separated from the epidermis and placed in DMEM (high glucose) supplemented with 50% FCS and 1% penicillin/streptomycin in a culture dish at 37°C overnight. The following day, medium was replaced by DMEM containing only 20% serum. After another overnight incubation, serum content was reduced to 10%. The primary fibroblasts recovered from the dermis, now adhering to the culture dish, were maintained in DMEM (high glucose) supplemented with 10% serum and 1% penicillin/streptomycin at 37°C for two to four passages before being assayed for internalization of radiolabeled ligands (see below).
Primary alveolar macrophages were isolated from male MR-deficient mice and WT littermates by performing six sequential BALs with 1 ml PBS. Sequential BAL fluids were pooled and centrifuged at 300 g for 5 min. Pelleted cells were resuspended in RPMI with 10% serum and 1% penicillin/streptomycin and cultured overnight at 37°C. The following day, adherent alveolar macrophages were assayed for internalization of radiolabeled ligands.
Radioligand internalization in cultured cells and assay for binding to E. coli bioparticles
To quantify cellular internalization of receptor ligands, radiolabeling of ligands (collagen type I, SP-D, ficolin-1, human MBL, soluble SR-AI, soluble MARCO, C1q, CL-P1/collectin-12, and CL-K1/collectin-11) with 125I and assay for internalization were performed according to previously established methods (List et al., 2000; Engelholm et al., 2003). In brief, radiolabeled ligands were added to CHO-K1, MRC-5, or CCL-210 cells, primary skin fibroblasts, or primary alveolar macrophages in a concentration of 12.5 ng/ml in serum-free growth medium supplemented with 1.5% BSA and 20 mM Hepes and then incubated for 4–5 h. When using primary cells, the lysosomal protease inhibitor E64d was added to a concentration of 20 µM to increase accumulation of internalized ligand in lysosomes (Kjøller et al., 2004). After the incubation, cells were washed three times in ice-cold PBS and incubated with ice-cold 0.25% trypsin-EDTA and 50 µg/ml proteinase K (Roche) to remove cell surface–associated ligand. Finally, cells were centrifuged, the supernatant was removed, and the internalized fraction of ligand was determined as the 125I activity associated with the cell pellet. In experiments where blocking of uPARAP function through receptor depletion with the anti-uPARAP mAb 5f4 was performed, cells were preincubated overnight with mAb 5f4 (CHO-K1 cells, 2 µg/ml; MRC-5 and CCL-210 cells, 0.5 µg/ml; primary fibroblasts, 20 µg/ml) added to the growth medium. mAb 5f4 was also added during incubation with radiolabeled ligands.
The binding capacity for radiolabeling of SP-D and MBL toward E. coli particles was determined by incubating labeled proteins at a concentration of 0.8 µg/ml with 4.5 × 106 E. coli bioparticles/ml for 60 min at 37°C and gentle shaking in assay buffer (20 mM Tris, 150 mM NaCl, 1.5% BSA, pH 7.4, and 0.1% Tween-20) supplemented with either 5 mM CaCl2 or 10 mM EDTA. Bioparticles were then pelleted by centrifugation at 1,000 g for 5 min and washed three times in assay buffer with centrifugation at 1,000 g for 5 min between the washing steps. After the final wash, bound SP-D, ficolin-1, MBL, or soluble SR-AI was determined as the 125I activity associated with the pellet of E. coli bioparticles.
Statistics
All statistical analyses were performed using GraphPad Prism version 7 for Mac software. For ELISA experiments, a two-tailed Student’s t test was used to test for statistical significance. For flow cytometry experiments and experiments with uptake of radiolabeled ligands in cultured cells, a two-tailed Student’s t test (for single comparisons) or one-way ANOVA (for multiple comparisons) was used. In experiments were the weight loss and survival of mice in response to bleomycin-induced injury was monitored, a two-way ANOVA with Sidak’s multiple comparisons test and log-rank (Mantel-Cox) test were used, respectively. To test for a correlation between SP-D and IL-6 levels in BAL, a Pearson r coefficient was computed using Log2-transformed values for SP-D and IL-6 concentrations. To test for the association of WT and uPARAP-knockout samples with SP-D“low”/IL-6“high” and SP-D“high”/IL-6“low” clusters, a χ2 test was performed. The K means cluster analysis was performed using R software. A P value of <0.05 was considered significant for all statistical tests.
Study approval
All experiments involving mice were performed in an Association for Assessment and Accreditation of Laboratory Animals Care International–accredited vivarium following institutional guidelines and standard operating procedures under animal study proposal 15-766 (National Institute of Dental and Craniofacial Research; National Institutes of Health). Use of all human samples was approved by The National Health Research Ethics Committee in Denmark, and informed consent was obtained from all patients.
Online supplemental material
Fig. S1 shows oligomer composition and biological activity of recombinant collectins and blocking of uPARAP-dependent uptake. Fig. S2 shows GFP expression, MBL uptake, and uPARAP expression in mouse dermal cells. Fig. S3 shows GFP expression and overall distribution of cell populations acquired from lungs of healthy and bleomycin-challenged mice. Fig. S4 shows SP-D uptake and uPARAP expression by distinct cell populations in lungs of healthy or bleomycin-treated mice. Fig. S5 shows wet weight of lungs from unchallenged or bleomycin- and LPS-challenged mice.
Supplementary Material
Acknowledgments
We wish to thank Katharina H. Stegmann, Britt Christoffersen, and Mia Kristine Høg for expert technical assistance.
This work was supported by the Danish Medical Research Council/Danish Council for Independent Research (H.J. Jürgensen, P. Garred, and N. Behrendt), the Danish Cancer Society (H.J. Jürgensen, D.H. Madsen, and N. Behrendt), Region Hovedstadens Forskningsfond (D.H. Madsen and N. Behrendt), the Novo Nordisk Foundation (D.H. Madsen, P. Garred, and N. Behrendt), the European Commission (D.H. Madsen), the National Institute of Dental and Craniofacial Research (National Institutes of Health) Intramural Research Program (T.H. Bugge), the National Institute of Dental and Craniofacial Research Veterinary Resources Core (Z number DE000740-05) and Combined Technical Core (ZIC DE000729-09), the Sven Andersen Research Foundation (P. Garred), the Lundbeck Foundation, and the Danish Cancer Research Foundation.
The authors declare no competing financial interests.
Author contributions: H.J. Jürgensen, L.H. Engelholm, and N. Behrendt formulated the hypotheses and planned the study. H.J. Jürgensen and D.H. Madsen designed the experiments. H.J. Jürgensen, K.S. Nørregaard, and M.M. Sibree conducted the experiments. P. Garred, T.H. Bugge, L.H. Engelholm, and N. Behrendt supervised the experimental work. D. Krustrup, E. Santoni-Rugiu, and K. Wassilew provided human tissue samples, evaluated H&E and immunohistochemistry stainings, and provided pathological expertise. H.J. Jürgensen, L.H. Engelholm, and N. Behrendt prepared the manuscript with contributions from all authors.
References
- Abdelgawad M.E., Søe K., Andersen T.L., Merrild D.M., Christiansen P., Kjærsgaard-Andersen P., and Delaisse J.M.. 2014. Does collagen trigger the recruitment of osteoblasts into vacated bone resorption lacunae during bone remodeling? Bone. 67:181–188. 10.1016/j.bone.2014.07.012 [DOI] [PubMed] [Google Scholar]
- Aono Y., Ledford J.G., Mukherjee S., Ogawa H., Nishioka Y., Sone S., Beers M.F., Noble P.W., and Wright J.R.. 2012. Surfactant protein-D regulates effector cell function and fibrotic lung remodeling in response to bleomycin injury. Am. J. Respir. Crit. Care Med. 185:525–536. 10.1164/rccm.201103-0561OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armaka M., Apostolaki M., Jacques P., Kontoyiannis D.L., Elewaut D., and Kollias G.. 2008. Mesenchymal cell targeting by TNF as a common pathogenic principle in chronic inflammatory joint and intestinal diseases. J. Exp. Med. 205:331–337. 10.1084/jem.20070906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behrendt N., Rønne E., and Danø K.. 1993. A novel, specific pro-urokinase complex on monocyte-like cells, detected by transglutaminase-catalyzed cross-linking. FEBS Lett. 336:394–396. 10.1016/0014-5793(93)80844-K [DOI] [PubMed] [Google Scholar]
- Behrendt N., Jensen O.N., Engelholm L.H., Mørtz E., Mann M., and Danø K.. 2000. A urokinase receptor-associated protein with specific collagen binding properties. J. Biol. Chem. 275:1993–2002. 10.1074/jbc.275.3.1993 [DOI] [PubMed] [Google Scholar]
- Beutler B., and Rietschel E.T.. 2003. Innate immune sensing and its roots: the story of endotoxin. Nat. Rev. Immunol. 3:169–176. 10.1038/nri1004 [DOI] [PubMed] [Google Scholar]
- Bundesmann M.M., Wagner T.E., Chow Y.H., Altemeier W.A., Steinbach T., and Schnapp L.M.. 2012. Role of urokinase plasminogen activator receptor-associated protein in mouse lung. Am. J. Respir. Cell Mol. Biol. 46:233–239. 10.1165/rcmb.2010-0485OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casey J., Kaplan J., Atochina-Vasserman E.N., Gow A.J., Kadire H., Tomer Y., Fisher J.H., Hawgood S., Savani R.C., and Beers M.F.. 2005. Alveolar surfactant protein D content modulates bleomycin-induced lung injury. Am. J. Respir. Crit. Care Med. 172:869–877. 10.1164/rccm.200505-767OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cravedi P., and Heeger P.S.. 2014. Complement as a multifaceted modulator of kidney transplant injury. J. Clin. Invest. 124:2348–2354. 10.1172/JCI72273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Messias-Reason I.J., Nisihara R.M., and Mocelin V.. 2011. Mannan-binding lectin and ficolin deposition in skin lesions of pemphigus. Arch. Dermatol. Res. 303:521–525. 10.1007/s00403-011-1132-1 [DOI] [PubMed] [Google Scholar]
- East L., and Isacke C.M.. 2002. The mannose receptor family. Biochim. Biophys. Acta. 1572:364–386. 10.1016/S0304-4165(02)00319-7 [DOI] [PubMed] [Google Scholar]
- Engelholm L.H., Nielsen B.S., Netzel-Arnett S., Solberg H., Chen X.D., Lopez Garcia J.M., Lopez-Otin C., Young M.F., Birkedal-Hansen H., Danø K., et al. . 2001. The urokinase plasminogen activator receptor-associated protein/endo180 is coexpressed with its interaction partners urokinase plasminogen activator receptor and matrix metalloprotease-13 during osteogenesis. Lab. Invest. 81:1403–1414. 10.1038/labinvest.3780354 [DOI] [PubMed] [Google Scholar]
- Engelholm L.H., List K., Netzel-Arnett S., Cukierman E., Mitola D.J., Aaronson H., Kjøller L., Larsen J.K., Yamada K.M., Strickland D.K., et al. . 2003. uPARAP/Endo180 is essential for cellular uptake of collagen and promotes fibroblast collagen adhesion. J. Cell Biol. 160:1009–1015. 10.1083/jcb.200211091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrar C.A., Tran D., Li K., Wu W., Peng Q., Schwaeble W., Zhou W., and Sacks S.H.. 2016. Collectin-11 detects stress-induced L-fucose pattern to trigger renal epithelial injury. J. Clin. Invest. 126:1911–1925. 10.1172/JCI83000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foo S.S., Reading P.C., Jaillon S., Mantovani A., and Mahalingam S.. 2015. Pentraxins and Collectins: Friend or Foe during Pathogen Invasion? Trends Microbiol. 23:799–811. 10.1016/j.tim.2015.09.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forbes L.R., and Haczku A.. 2010. SP-D and regulation of the pulmonary innate immune system in allergic airway changes. Clin. Exp. Allergy. 40:547–562. 10.1111/j.1365-2222.2010.03483.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardai S.J., Xiao Y.Q., Dickinson M., Nick J.A., Voelker D.R., Greene K.E., and Henson P.M.. 2003. By binding SIRPalpha or calreticulin/CD91, lung collectins act as dual function surveillance molecules to suppress or enhance inflammation. Cell. 115:13–23. 10.1016/S0092-8674(03)00758-X [DOI] [PubMed] [Google Scholar]
- Ghiran I., Barbashov S.F., Klickstein L.B., Tas S.W., Jensenius J.C., and Nicholson-Weller A.. 2000. Complement receptor 1/CD35 is a receptor for mannan-binding lectin. J. Exp. Med. 192:1797–1808. 10.1084/jem.192.12.1797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenhill S.R., and Kotton D.N.. 2009. Pulmonary alveolar proteinosis: a bench-to-bedside story of granulocyte-macrophage colony-stimulating factor dysfunction. Chest. 136:571–577. 10.1378/chest.08-2943 [DOI] [PubMed] [Google Scholar]
- Haczku A. 2008. Protective role of the lung collectins surfactant protein A and surfactant protein D in airway inflammation. J. Allergy Clin. Immunol. 122:861–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbein J.F., and Wright J.R.. 2001. Enhanced clearance of surfactant protein D during LPS-induced acute inflammation in rat lung. Am. J. Physiol. Lung Cell. Mol. Physiol. 281:L268–L277. 10.1152/ajplung.2001.281.1.L268 [DOI] [PubMed] [Google Scholar]
- Holmskov U., Lawson P., Teisner B., Tornoe I., Willis A.C., Morgan C., Koch C., and Reid K.B.. 1997. Isolation and characterization of a new member of the scavenger receptor superfamily, glycoprotein-340 (gp-340), as a lung surfactant protein-D binding molecule. J. Biol. Chem. 272:13743–13749. 10.1074/jbc.272.21.13743 [DOI] [PubMed] [Google Scholar]
- Jiang W., Swiggard W.J., Heufler C., Peng M., Mirza A., Steinman R.M., and Nussenzweig M.C.. 1995. The receptor DEC-205 expressed by dendritic cells and thymic epithelial cells is involved in antigen processing. Nature. 375:151–155. 10.1038/375151a0 [DOI] [PubMed] [Google Scholar]
- Jürgensen H.J., Madsen D.H., Ingvarsen S., Melander M.C., Gårdsvoll H., Patthy L., Engelholm L.H., and Behrendt N.. 2011. A novel functional role of collagen glycosylation: interaction with the endocytic collagen receptor uparap/ENDO180. J. Biol. Chem. 286:32736–32748. 10.1074/jbc.M111.266692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jürgensen H.J., Johansson K., Madsen D.H., Porse A., Melander M.C., Sørensen K.R., Nielsen C., Bugge T.H., Behrendt N., and Engelholm L.H.. 2014. Complex determinants in specific members of the mannose receptor family govern collagen endocytosis. J. Biol. Chem. 289:7935–7947. 10.1074/jbc.M113.512780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kendall R.T., and Feghali-Bostwick C.A.. 2014. Fibroblasts in fibrosis: novel roles and mediators. Front. Pharmacol. 5:123 10.3389/fphar.2014.00123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kjøller L., Engelholm L.H., Høyer-Hansen M., Danø K., Bugge T.H., and Behrendt N.. 2004. uPARAP/endo180 directs lysosomal delivery and degradation of collagen IV. Exp. Cell Res. 293:106–116. 10.1016/j.yexcr.2003.10.008 [DOI] [PubMed] [Google Scholar]
- Krempen K., Grotkopp D., Hall K., Bache A., Gillan A., Rippe R.A., Brenner D.A., and Breindl M.. 1999. Far upstream regulatory elements enhance position-independent and uterus-specific expression of the murine alpha1(I) collagen promoter in transgenic mice. Gene Expr. 8:151–163. [PMC free article] [PubMed] [Google Scholar]
- Lee S.J., Evers S., Roeder D., Parlow A.F., Risteli J., Risteli L., Lee Y.C., Feizi T., Langen H., and Nussenzweig M.C.. 2002. Mannose receptor-mediated regulation of serum glycoprotein homeostasis. Science. 295:1898–1901. 10.1126/science.1069540 [DOI] [PubMed] [Google Scholar]
- List K., Jensen O.N., Bugge T.H., Lund L.R., Ploug M., Danø K., and Behrendt N.. 2000. Plasminogen-independent initiation of the pro-urokinase activation cascade in vivo. Activation of pro-urokinase by glandular kallikrein (mGK-6) in plasminogen-deficient mice. Biochemistry. 39:508–515. 10.1021/bi991701f [DOI] [PubMed] [Google Scholar]
- Litvack M.L., and Palaniyar N.. 2010. Review: Soluble innate immune pattern-recognition proteins for clearing dying cells and cellular components: implications on exacerbating or resolving inflammation. Innate Immun. 16:191–200. 10.1177/1753425910369271 [DOI] [PubMed] [Google Scholar]
- Lokitz M.L., Zhang W., Bashir M., Sullivan K.E., Ang G., Kwon E.J., Lin J.H., and Werth V.P.. 2005. Ultraviolet-B recruits mannose-binding lectin into skin from non-cutaneous sources. J. Invest. Dermatol. 125:166–173. 10.1111/j.0022-202X.2005.23794.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madan T., Kishore U., Singh M., Strong P., Clark H., Hussain E.M., Reid K.B., and Sarma P.U.. 2001. Surfactant proteins A and D protect mice against pulmonary hypersensitivity induced by Aspergillus fumigatus antigens and allergens. J. Clin. Invest. 107:467–475. 10.1172/JCI10124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madsen D.H., Engelholm L.H., Ingvarsen S., Hillig T., Wagenaar-Miller R.A., Kjøller L., Gårdsvoll H., Høyer-Hansen G., Holmbeck K., Bugge T.H., and Behrendt N.. 2007. Extracellular collagenases and the endocytic receptor, urokinase plasminogen activator receptor-associated protein/Endo180, cooperate in fibroblast-mediated collagen degradation. J. Biol. Chem. 282:27037–27045. 10.1074/jbc.M701088200 [DOI] [PubMed] [Google Scholar]
- Madsen D.H., Ingvarsen S., Jürgensen H.J., Melander M.C., Kjøller L., Moyer A., Honoré C., Madsen C.A., Garred P., Burgdorf S., et al. . 2011. The non-phagocytic route of collagen uptake: a distinct degradation pathway. J. Biol. Chem. 286:26996–27010. 10.1074/jbc.M110.208033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madsen D.H., Jürgensen H.J., Ingvarsen S., Melander M.C., Vainer B., Egerod K.L., Hald A., Rønø B., Madsen C.A., Bugge T.H., et al. . 2012. Endocytic collagen degradation: a novel mechanism involved in protection against liver fibrosis. J. Pathol. 227:94–105. 10.1002/path.3981 [DOI] [PubMed] [Google Scholar]
- Madsen D.H., Jürgensen H.J., Ingvarsen S., Melander M.C., Albrechtsen R., Hald A., Holmbeck K., Bugge T.H., Behrendt N., and Engelholm L.H.. 2013a Differential actions of the endocytic collagen receptor uPARAP/Endo180 and the collagenase MMP-2 in bone homeostasis. PLoS One. 8:e71261 10.1371/journal.pone.0071261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madsen D.H., Leonard D., Masedunskas A., Moyer A., Jürgensen H.J., Peters D.E., Amornphimoltham P., Selvaraj A., Yamada S.S., Brenner D.A., et al. . 2013b M2-like macrophages are responsible for collagen degradation through a mannose receptor-mediated pathway. J. Cell Biol. 202:951–966. 10.1083/jcb.201301081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madsen J., Kliem A., Tornoe I., Skjodt K., Koch C., and Holmskov U.. 2000. Localization of lung surfactant protein D on mucosal surfaces in human tissues. J. Immunol. 164:5866–5870. 10.4049/jimmunol.164.11.5866 [DOI] [PubMed] [Google Scholar]
- Malovic I., Sørensen K.K., Elvevold K.H., Nedredal G.I., Paulsen S., Erofeev A.V., Smedsrød B.H., and McCourt P.A.. 2007. The mannose receptor on murine liver sinusoidal endothelial cells is the main denatured collagen clearance receptor. Hepatology. 45:1454–1461. 10.1002/hep.21639 [DOI] [PubMed] [Google Scholar]
- Mantovani A., Sozzani S., Locati M., Allavena P., and Sica A.. 2002. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 23:549–555. 10.1016/S1471-4906(02)02302-5 [DOI] [PubMed] [Google Scholar]
- Martinez-Pomares L. 2012. The mannose receptor. J. Leukoc. Biol. 92:1177–1186. 10.1189/jlb.0512231 [DOI] [PubMed] [Google Scholar]
- McCormack F.X., and Whitsett J.A.. 2002. The pulmonary collectins, SP-A and SP-D, orchestrate innate immunity in the lung. J. Clin. Invest. 109:707–712. 10.1172/JCI0215293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller C., Wilgenbusch S., Michaels M., Chi D.S., Youngberg G., and Krishnaswamy G.. 2010. Molecular defects in the mannose binding lectin pathway in dermatological disease: Case report and literature review. Clin. Mol. Allergy. 8:6 10.1186/1476-7961-8-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misharin A.V., Morales-Nebreda L., Mutlu G.M., Budinger G.R., and Perlman H.. 2013. Flow cytometric analysis of macrophages and dendritic cell subsets in the mouse lung. Am. J. Respir. Cell Mol. Biol. 49:503–510. 10.1165/rcmb.2013-0086MA [DOI] [PMC free article] [PubMed] [Google Scholar]
- Møller-Kristensen M., Ip W.K., Shi L., Gowda L.D., Hamblin M.R., Thiel S., Jensenius J.C., Ezekowitz R.A., and Takahashi K.. 2006. Deficiency of mannose-binding lectin greatly increases susceptibility to postburn infection with Pseudomonas aeruginosa. J. Immunol. 176:1769–1775. 10.4049/jimmunol.176.3.1769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen B., Rank F., Engelholm L.H., Holm A., Danø K., and Behrendt N.. 2002. Urokinase receptor-associated protein (uPARAP) is expressed in connection with malignant as well as benign lesions of the human breast and occurs in specific populations of stromal cells. Int. J. Cancer. 98:656–664. 10.1002/ijc.10227 [DOI] [PubMed] [Google Scholar]
- Nikitopoulou I., Oikonomou N., Karouzakis E., Sevastou I., Nikolaidou-Katsaridou N., Zhao Z., Mersinias V., Armaka M., Xu Y., Masu M., et al. . 2012. Autotaxin expression from synovial fibroblasts is essential for the pathogenesis of modeled arthritis. J. Exp. Med. 209:925–933. 10.1084/jem.20112012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishikiori H., Chiba H., Ariki S., Kuronuma K., Otsuka M., Shiratori M., Ikeda K., Watanabe A., Kuroki Y., and Takahashi H.. 2014. Distinct compartmentalization of SP-A and SP-D in the vasculature and lungs of patients with idiopathic pulmonary fibrosis. BMC Pulm. Med. 14:196 10.1186/1471-2466-14-196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olde Nordkamp M.J., van Eijk M., Urbanus R.T., Bont L., Haagsman H.P., and Meyaard L.. 2014. Leukocyte-associated Ig-like receptor-1 is a novel inhibitory receptor for surfactant protein D. J. Leukoc. Biol. 96:105–111. 10.1189/jlb.3AB0213-092RR [DOI] [PubMed] [Google Scholar]
- Schuette V., Embgenbroich M., Ulas T., Welz M., Schulte-Schrepping J., Draffehn A.M., Quast T., Koch K., Nehring M., König J., et al. . 2016. Mannose receptor induces T-cell tolerance via inhibition of CD45 and up-regulation of CTLA-4. Proc. Natl. Acad. Sci. USA. 113:10649–10654. 10.1073/pnas.1605885113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuliga M., Jaffar J., Harris T., Knight D.A., Westall G., and Stewart A.G.. 2017. The fibrogenic actions of lung fibroblast-derived urokinase: a potential drug target in IPF. Sci. Rep. 7:41770 10.1038/srep41770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheikh H., Yarwood H., Ashworth A., and Isacke C.M.. 2000. Endo180, an endocytic recycling glycoprotein related to the macrophage mannose receptor is expressed on fibroblasts, endothelial cells and macrophages and functions as a lectin receptor. J. Cell Sci. 113:1021–1032. [DOI] [PubMed] [Google Scholar]
- Sulek J., Wagenaar-Miller R.A., Shireman J., Molinolo A., Madsen D.H., Engelholm L.H., Behrendt N., and Bugge T.H.. 2007. Increased expression of the collagen internalization receptor uPARAP/Endo180 in the stroma of head and neck cancer. J. Histochem. Cytochem. 55:347–353. 10.1369/jhc.6A7133.2006 [DOI] [PubMed] [Google Scholar]
- Tamaru S., Mishina H., Watanabe Y., Watanabe K., Fujioka D., Takahashi S., Suzuki K., Nakamura T., Obata J.E., Kawabata K., et al. . 2013. Deficiency of phospholipase A2 receptor exacerbates ovalbumin-induced lung inflammation. J. Immunol. 191:1021–1028. 10.4049/jimmunol.1300738 [DOI] [PubMed] [Google Scholar]
- van der Pol P., Schlagwein N., van Gijlswijk D.J., Berger S.P., Roos A., Bajema I.M., de Boer H.C., de Fijter J.W., Stahl G.L., Daha M.R., and van Kooten C.. 2012. Mannan-binding lectin mediates renal ischemia/reperfusion injury independent of complement activation. Am. J. Transplant. 12:877–887. 10.1111/j.1600-6143.2011.03887.x [DOI] [PubMed] [Google Scholar]
- Wagenaar-Miller R.A., Engelholm L.H., Gavard J., Yamada S.S., Gutkind J.S., Behrendt N., Bugge T.H., and Holmbeck K.. 2007. Complementary roles of intracellular and pericellular collagen degradation pathways in vivo. Mol. Cell. Biol. 27:6309–6322. 10.1128/MCB.00291-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallim L.R., Nisihara R., Skare T., Mocelin V., and Messias-Reason I.J.. 2014. Mannose binding lectin deposition in skin of lupus erythematosus patients: a case series. Hum. Immunol. 75:629–632. 10.1016/j.humimm.2014.04.015 [DOI] [PubMed] [Google Scholar]
- Walsh M.C., Bourcier T., Takahashi K., Shi L., Busche M.N., Rother R.P., Solomon S.D., Ezekowitz R.A., and Stahl G.L.. 2005. Mannose-binding lectin is a regulator of inflammation that accompanies myocardial ischemia and reperfusion injury. J. Immunol. 175:541–546. 10.4049/jimmunol.175.1.541 [DOI] [PubMed] [Google Scholar]
- Williamson J.D., Sadofsky L.R., and Hart S.P.. 2015. The pathogenesis of bleomycin-induced lung injury in animals and its applicability to human idiopathic pulmonary fibrosis. Exp. Lung Res. 41:57–73. 10.3109/01902148.2014.979516 [DOI] [PubMed] [Google Scholar]
- Wu H., Kuzmenko A., Wan S., Schaffer L., Weiss A., Fisher J.H., Kim K.S., and McCormack F.X.. 2003. Surfactant proteins A and D inhibit the growth of Gram-negative bacteria by increasing membrane permeability. J. Clin. Invest. 111:1589–1602. 10.1172/JCI16889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu K., Yuan J., and Lasky L.A.. 1996. Characterization of a novel member of the macrophage mannose receptor type C lectin family. J. Biol. Chem. 271:21323–21330. 10.1074/jbc.271.35.21323 [DOI] [PubMed] [Google Scholar]
- Yoshida M., Ikegami M., Reed J.A., Chroneos Z.C., and Whitsett J.A.. 2001. GM-CSF regulates protein and lipid catabolism by alveolar macrophages. Am. J. Physiol. Lung Cell. Mol. Physiol. 280:L379–L386. 10.1152/ajplung.2001.280.3.L379 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.