

Abstract
The inhibition of abnormal amyloid β (Aβ) aggregation has been regarded as a good target to control Alzheimer’s disease. The present study adopted 2D-QSAR, HQSAR and 3D QSAR (CoMFA & CoMSIA) modeling approaches to identify the structural and physicochemical requirements for the potential Aβ aggregation inhibition. A structure-based molecular docking technique is utilized to approve the features that are obtained from the ligand-based techniques on 30 curcumin derivatives. The combined outputs were then used to screen the modified 10 compounds. The 2D QSAR model on curcumin derivatives gave statistical values R2 = 0.9086 and SEE = 0.1837. The model was further confirmed by Y-randomization test and Applicability domain analysis by the standardization approach. The HQSAR study (Q2 = 0.615, R2ncv = 0.931, R2pred = 0.956) illustrated the important molecular fingerprints for inhibition. Contour maps of 3D QSAR models, CoMFA (Q2 = 0.687, R2ncv = 0.787, R2pred = 0.731) and CoMSIA (Q2 = 0.743, R2ncv = 0.972, R2pred = 0.713), depict that the models are robust and provide explanation of the important features, like steric, electrostatic and hydrogen bond acceptor, which play important role for interaction with the receptor site cavity. The molecular docking study of the curcumin derivatives elucidates the important interactions between the amino acid residues at the catalytic site of the receptor and the ligands, indicating the structural requirements of the inhibitors. The ligand–receptor interactions of top hits were analyzed to explore the pharmacophore features of Aβ aggregation inhibition. The Aβ aggregation inhibitory activities of novel chemical entities were then obtained through inverse QSAR. The newly designed molecules were further screened through machine learning, prediction of toxicity and nature of metabolism to get the proposed six lead compounds.
Keywords: Alzheimer’s disease, Curcuma longa, 2D-QSAR, 3D-QSAR, Molecular docking
Introduction
Alzheimer’s disease (AD), a devastating neurodegenerative disease, remains epidemic for public health in the twenty-first century (Alzheimer’s Association 2017). AD is characterized by the disintegration of the nervous system, which results in episodic memory problems leading to abnormal behavior and is the leading cause of dementia (Citron 2010). The presence of the extracellular deposits of misfolded and aggregated amyloid-β (Aβ) peptides in the brain is widely considered to be critically concerned in the progression of AD (Hardy and Selkoe 2002; Jack et al. 2010). The sequential enzymatic actions of β-secretase and γ-secretase result in the proteolytic cleavage of amyloid precursor protein (APP) (Selkoe 1997). The formation of Aβ is a two-step process which involves the cleavage of APP by BACE1 to form a β-secretase derived C-terminal fragment of APP, followed by an action of γ-secretase to generate Aβ isoforms ranging from 37 to 42 amino acid residues. Aβ40 is the most abundant isoform, whereas the Aβ, which is mainly associated with AD pathogenesis, is aggregated Aβ42 (Selkoe 1994; Golde et al. 2000). Thus in AD, Aβ monomers form undesirable Aβ aggregates of long insoluble fibrils. They aggregate in the extracellular deposits known as senile plaques. These abnormal changes in Aβ induce abnormal hyperphosphorylation of tau and tangle formation as well as neuronal loss, bringing about cognitive impairment (Haass and Selkoe 2007). The inhibition of abnormal Aβ aggregation is considered as one of the most important etiological agents and is an attractive therapeutic target to control AD (Xiao et al. 2015).
Curcumin is a bioactive phenolic compound present in the rhizome of Curcuma longa L. (Zingiberaceae). Curcumin exhibits various biological and pharmacological activities like anti-inflammatory (Jin et al. 2014), antioxidant (Nishikawa et al. 2013), antimicrobial (Dubey et al. 2008), anti-fungal (Nguyen et al. 2014), and antibacterial activities (Negi et al. 1999). Moreover, various in vivo and in vitro experiments reveal the effects of curcumin on treating or preventing AD pathology (Caesar et al. 2012; Garcia-Alloza et al. 2007; Hamaguchi et al. 2009; Ma et al. 2013). One of the most significant features of curcumin is that it directly inhibits the formation and extension of fibrillar Aβ aggregates and also destabilizes preformed fibrillar Aβ aggregates (Ono et al. 2004). It was reported that chronic dietary curcumin lowered Aβ deposition in Alzheimer Transgenic Mouse (Lim et al. 2001). Some researchers have reported that AD model mice treated with curcumin displayed a reduction in Aβ accumulation in the brain (Begum et al. 2008; Yang et al. 2005a, b). The unique benefit of the curcumin is that it is nontoxic to human even with high dosage (Sharma et al. 2001).
Nowadays, the in vitro assessment of Aβ aggregation inhibitors is still a time consuming and labor intensive task. So we are interested in identifying potential novel leads as Aβ aggregation inhibitors using techniques based on Computer Aided Drug Design (CADD). (Kapetanovic 2008). Molecular modeling in combination with Quantitative Structure–Activity Relationship (QSAR) is used to test the activity of a ligand and the type of interaction into the active site of the protein (Elfiky and Elshemey 2016; Saleh 2015; Aswathy et al. 2017).
The ligand-based molecular modeling techniques used are Pharmacophore mapping and QSAR analysis. The pharmacophore model gives information regarding hydrophobic properties, hydrogen binding properties (acceptor or donor) and aromatic functionality of the compounds in the dataset. The 3D-QSAR studies, which include CoMFA (Cramer et al. 1988) and CoMSIA (Klebe et al. 1994), cover an entire force field around a molecule instead of only spotlighting the pharmacophoric information (Cruciani and Watson 1994). Various properties like electrostatic, steric, hydrophobic and hydrogen-bond donor/acceptor factors were considered in 3D-QSAR for the force field calculations which give the best results when target-recognizing ligands share a unique structural scaffold (Ballante and Ragno 2012; Shibi et al. 2015; Jisha et al. 2017). Molecular docking studies have been extensively employed to identify the exact conformation of ligands in the precise location of the binding cavity of a protein receptor molecule (Lengauer and Rarey 1996). It also predicts the affinity between the ligand and the active site residues of the protein receptor. Therefore, these can be useful to find out the best lead molecule and their further modification by rational drug design approach. On this ground, we chose 30 curcumin derivatives with Aβ aggregation inhibition values reported by Yanagisawa et al. (2015) for the present study.
Materials and methods
Dataset preparation
A desirable set comprising 30 structurally diverse compounds which inhibit abnormal Aβ aggregation has been considered for the study. In this study, the IC50 values of these compounds were converted to pIC50 (− log IC50). The structures of the compounds along with their pIC50 values are specified in Table 1. The compounds were divided into 80% training and 20% test sets. While dividing datasets, a wide range of activity data was confirmed in training as well as test sets. The inhibitory activities on Aβ aggregation (pIC50) have been used as the dependent variables for doing QSAR studies.
Table 1.
Molecular structures and corresponding experimental pIC50 values of the curcumin derivatives


aTest compounds
Calculation of descriptors
Using powerMV software, about 179 descriptors, which include Pharmacophore fingerprints, weighted burden number and eight drug-like properties were computed for the development of 2D-QSAR analysis. Using PAdel-Descriptor, about 15,345 descriptors were also calculated (Yap 2011). In addition, by employing the QuaSAR module of Molecular Operating Environment (MOE), a total of 365 descriptors belonging to three classes: 2D descriptors, which use the atoms and connection information of the molecules, external 3D (x3D), which uses 3D coordinate information with an absolute frame of reference and internal 3D (i3D), which uses 3D coordinate information about each molecule, were calculated. Thus a total pool of 15,889 descriptors was generated for this study.
The pool of 15,889 descriptors was then reduced to 1495 by removing those descriptors that have the same value for 90% of the dataset using “General” descriptor selection algorithm. It is further reduced to 137 by eliminating descriptors highly correlated using “CORCHOP” descriptor selection algorithm. Finally by applying subjective selection using Genetic algorithm method, the pool of descriptors was eventually reduced to six. These six descriptors were used to build the 2D-QSAR model.
2D QSAR study
In the present work, the multiple linear regression (MLR) based QSAR model was developed, to obtain specific information regarding the contribution of different structural and physicochemical characteristics of the compounds towards the inhibitory activity.
Validation of 2D-QSAR model
The leave-one-out (LOO) cross-validation method was used as an internal validation tool to check the predictive ability of the built 2D-QSAR model (Tetko et al. 2001). The internal predictabilities of the model were verified by LOO cross-validated regression coefficient (Q2). Internal validation parameters like standard error of estimate (SEE), the square of correlation coefficient (R2), adjusted R2 (R2A), variance ratio (F) and predicted residual sums of squares standard deviation (PRESS) were used, along with parameters like r2m(LOO) and ∆ r2m(LOO) (Roy et al. 2015a, b). For external validation, the parameter R2pred was used, to verify the predictive ability of the model on the test set. External validation parameters (without scaling) like r2, r20, Concordance Correlation Coefficient (CCC), Q2F1, Q2F2, Q2F3 were also used. Golbraikh and Tropsha (2002) metrics were also found out. Y-randomization technique and Applicability domain analysis were also performed for the MLR model to ascertain the robustness, significance, and reliability of the 2D-QSAR model (Tropsha et al. 2003). The parameter cR2p was taken into consideration as the validation parameters for the Y-based randomization test (Ojha and Roy 2011).
HQSAR
Hologram QSAR (HQSAR) module available at SYBYL-X software v.1.3 was used for the generation of HQSAR models (Sybyl 1.3, Triops Inc, St). A predefined set of rules was used to have a molecule into a molecular fingerprint that encoded the frequency of occurrence of different molecular fragment types. Then the molecular fingerprint was cut into strings at a fixed interval as specified by a hologram length (HL) parameter. All of the generated strings were hashed into a fixed length array. HQSAR does not necessitate 3D alignment for model generation. HQSAR uses different parameters such as atom count, hologram length, and fragment distinction, which are the most important factors involved in the model development. 21 different models were derived using the default fragment size (4–7 atoms) and various combinations of fragment distinctions (A-atoms; C-connections; B-bonds; H-hydrogen; Ch-chirality; DA-donor and acceptors of hydrogen bonds).
3D-QSAR
Molecular modeling and alignment
The partial atomic charges for electrostatic contribution were calculated by the method of Gasteiger–Hückel. Tripos force field was employed for optimization and energy minimization of the compounds with the convergence criterion of 0.01 kcal/mol Å. The most active molecule, compound 8, which has the minimum energy conformation, was selected as a reference molecule (Fig. 1). The remaining compounds were aligned on it by the common substructure alignment.
Fig. 1.

Structure of the template compound (8) and the three regions A, B and C
CoMFA model
The CoMFA model illustrates steric (S) and electrostatic (E) features of the scaffold for showing the selective inhibition of the target. The S and E fields were calculated at each lattice with a grid size of 2 Å. A sp3 hybridized carbon atom was used with + 1 charge serving as a probe atom. Column filtering was set to 1.0 kcal/mol to reduce noise and improve the outcomes of the built model. The cut-off value for steric and electrostatic fields was set to 30 kcal/mol.
CoMSIA model
The CoMSIA model helps to understand the hydrogen bond acceptor (A) and donor (D), the hydrophobic (H) features in addition to the ‘S’ and ‘E’ features. The same lattice box utilized for the creation of the CoMFA model was also employed during the CoMSIA calculation. CoMSIA descriptors were calculated based on a sp3 hybridized carbon as a probe atom with + 1 positive charge, + 1 hydrophobicity, + 1 hydrogen bond donor and + 1 hydrogen bond acceptor at each lattice and grid spacing of 2.0 Å.
Validation of 3D-QSAR models
The partial least squares (PLS) were adopted for the regression analysis to build the 3D-QSAR equations. The optimal number of components and the cross-validation correlation coefficient (Q2) were determined by leave-one-out (LOO) cross-validation procedures. Then, the non-cross-validated analysis was performed to calculate the non-cross-validation correlation coefficient (R2), SEE and F value.
The predictive abilities of the 3D-QSAR models were identified by a test set of 6 compounds, and their pIC50 values were predicted using the following Eq. (1):
| 1 |
where Ypredicted, Yobserved and Ymean are predicted, actual and mean values of the activity, respectively. is the predictive sum of squares (PRESS).
Generation of a pharmacophore model
Using the MOE pharmacophore consensus search module, a pharmacophoric model was generated to highlight the most important key features shown by each group of compounds belonging to the dataset. It was comprised of four different annotation points such as H-bond donor, H-bond acceptor, hydrophobic and aromatic features.
Molecular docking
Primary and secondary structure prediction and validation of the protein molecules
For physico-chemical characterization, theoretical isoelectric point (pI), total number of positive and negative residues, aliphatic index (AI) (Ikai 1980), instability index (II) (Guruprasad et al. 1990), extinction co-efficient (EC) (Gill and Von Hippel 1989) and grand average hydropathy (GRAVY) (Kyte and Doolittle 1982) were computed using the Expasy’s ProtParam server (Gasteiger et al. 2005).
The secondary structural features of the proteins were studied using Self-Optimized Prediction method With Alignment (SOPMA) (Geourjon and Deléage 1995). It give details of Alpha helix, Pi helix, Beta bridge, Extended strand, Beta turn, Bend region, Random coil and Ambiguous states.
Protein–ligand molecular docking analysis
Molecular docking studies representing the correct conformation of the curcumin derivative in protein binding sites was performed using MOE (Chemical Computing Group Inc, Montreal, Quebec, Canada). The 3D structure of the protein complex was downloaded from the Protein Databank (http://www.rcsb.org) with the PDB ID 2BEG (Lührs et al. 2005). This structure was 3D protonated and energy minimized in a MMFF94x force field to a gradient of 0.0001 kcal/mol/Å (Halgren 1996). The active site of the protein molecule was generated using the MOE-Alpha site finder. Then the dummy atoms were created from the obtained alpha spheres. The default settings for all of the parameters including Ligand Placement (Triangle Matcher) and Rescoring (London dG) were found to be suitable for reproduction of the ligand–receptor complexes. After molecular docking, LigX feature of MOE was used to locate the hydrogen bonding interactions between ligand and receptor protein. The top scoring ligand poses from each docking run were used for calculation of binding energy.
Machine learning model
In machine learning approach, predictive models were generated with the help of a set of known active compounds and their extracted properties were used to predict the activity of unknown compounds (Wahi et al. 2015). In the present study, random forest (RF) was employed as a classifier (Shibi et al. 2016). RF is chosen as the classifier, due to its suboptimal performance in cases of strongly unbalanced data (Dong et al. 2015; Hsu et al. 2015; Janitza et al. 2013). The RF algorithm parameters include a forest of ten classification trees with ten attributes randomly selected for splitting at each node, and predictions are based on a majority vote. The WEKA (Waikato University, Hamilton, New Zealand, v3.7), data mining software, was used for the classification. It contains tools for data pre-processing, regression, clustering, association rules, classification, and visualization. The PubChem bioassay dataset AID 647 was selected for model generation which contains 1420 actives and 820 inactives. The machine learning based classification model was generated using training set (80%) and the quality of the generated model was assessed through test set (20%) with tenfold cross validation. Sensitivity, specificity, accuracy and receiver operating characteristic (ROC) were used to understand the performance of the classifier. True positive rate (TPR) or sensitivity is defined as the ratio of true actives, correctly classified as active. True negative rate (TNR) or Specificity is defined as the ratio of true inactives, correctly classified as inactive. The overall effectiveness is assessed by the accuracy.
Prediction of metabolic behavior and ADME properties of the virtual molecules
The metabolic information about compounds in the drug discovery pipeline is critical because of the fact that an extensive first-pass metabolism can result in low bioavailability. Metabolism that occurs too rapidly causes a short therapeutic window requiring a frequent dosing schedule. On the contrary, metabolism that proceeds too slowly can cause an accumulation of the drug molecule in the body which could result in an increase in the risk of toxic effects.
The cytochrome P450s (CYPs) are heme-thiolate enzymes that can metabolize a variety of xenobiotics. There are 57 CYP isoforms in humans, out of which five CYP isoforms, CYP3A4, 2D6, 2C19, 2C9, and 1A2, are accountable for ~ 90% of drug metabolism. Molecular docking method can be utilized to identify the binding conformations of the curcumin derivatives into the site of CYP enzymes (Mannu et al. 2011).
The crystal structures of human CYP3A4 in the unliganded form (1WOE) (Chatake et al. 2005) and bound to substrate (1WOF) (Yang et al. 2005a, b) are downloaded from Protein Data Bank. The protein 1WOE is a wild-type enzyme, except that the N-terminal membrane insertion peptide has been removed to increase solubility for crystallization. There was no substrate or inhibitor bound in the active site of this crystal structure.
In vitro human intestinal absorption (HIA), Caco2 and blood brain barrier (BBB) penetration values were obtained using web based PreADMET program (https://preadmet.bmdrc.kr/).
Results and discussion
2D-QSAR
The Aβ aggregation inhibitory activities of the molecules are mainly dominated by the type of substituent and the substituted positions on curcumin framework. Therefore, 2D QSAR technique can exert its advantages on discovering the dependence of activity on their molecular structures.
A 2D QSAR model was developed through the MLR analysis and the obtained model is depicted in the Eq. (2).
| 2 |
The negative coefficient of the MDEC-44 in the model suggests that a lower value favors the biological activity. MDEC-44 is Molecular distance edge between all quaternary carbons. Similarly, ExtFP728 and GraphFP295 also show negative contribution. WK.eneg is Non-directional WHIM, weighted by Mulliken atomic electronegativities and the descriptor, GraphFP912 show positive contribution to the biological activity.
SEE = 0.1837; R2 = 0.9086; R2A = 0.8763; PRESS = 0.5734; F = 28.162; r2 = 0.8987; r20 = 0.8098; Q2F1 = 0.8211; Q2F2 = 0.8073; Q2F3 = 0.7062; CCC = 0.8656; |r20 − r′20| = 0.2576, [(r2 − r20)/r2] = 0.0989, [(r2 − r′20)/r2] = 0.3856, k = 0.9939, k′ = 1.0032; Average r2m(test) = 0.5063; ∆r2m(test) = 0.2421 indicate a good predictive capability.
Since the R2m(Overall) value is 0.712 and is more than 0.6, indicates the acceptable overall fitting of the developed model. The predicted versus experimental pIC50 values are shown in Fig. 2a. The predicted Aβ aggregation inhibitory activities of the developed model obtained are shown in Table 2.
Fig. 2.
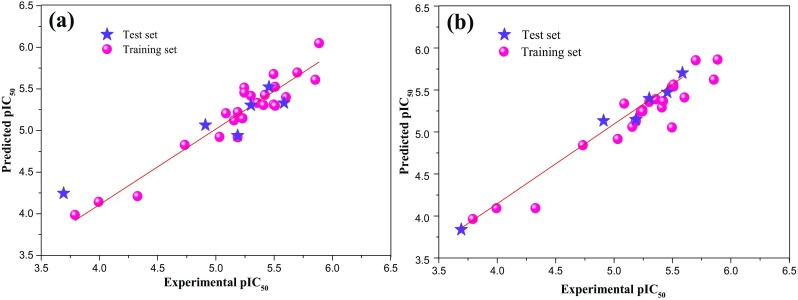
Correlations between actual and predicted pIC50 in the training and test sets for a 2D QSAR and b HQSAR
Table 2.
Experimental and predicted activities (pIC50) of the compounds
| Compounds | Actual pIC50 | Predicted pIC50 | |||
|---|---|---|---|---|---|
| 2D QSAR | HQSAR | CoMFA | CoMSIA | ||
| 1 | 5.699 | 5.698 | 5.854 | 5.384 | 5.395 |
| 2 | 5.495 | 5.312 | 5.054 | 5.412 | 5.445 |
| 3a | 5.585 | 5.331 | 5.703 | 5.407 | 5.414 |
| 4 | 5.357 | 5.332 | 5.390 | 5.343 | 5.304 |
| 5 | 5.086 | 5.208 | 5.339 | 5.345 | 5.372 |
| 6 | 5.244 | 5.516 | 5.246 | 5.314 | 5.283 |
| 7a | 5.456 | 5.523 | 5.472 | 5.365 | 5.328 |
| 8 | 5.886 | 6.050 | 5.862 | 5.417 | 5.462 |
| 9 | 5.420 | 5.428 | 5.369 | 5.347 | 5.324 |
| 10 | 5.187 | 4.919 | 5.135 | 5.304 | 5.296 |
| 11 | 5.409 | 5.306 | 5.293 | 5.345 | 5.322 |
| 12 | 5.229 | 5.148 | 5.241 | 5.368 | 5.386 |
| 13 | 3.993 | 4.144 | 4.092 | 4.172 | 4.158 |
| 14 | 3.791 | 3.987 | 3.964 | 3.426 | 3.431 |
| 15a | 5.301 | 5.305 | 5.398 | 5.378 | 5.419 |
| 16 | 4.327 | 4.212 | 4.093 | 4.786 | 4.747 |
| 17a | 3.693 | 4.246 | 3.838 | 2.987 | 2.996 |
| 18 | 5.495 | 5.679 | 5.541 | 5.375 | 5.359 |
| 19 | 5.509 | 5.296 | 5.541 | 5.405 | 5.455 |
| 20 | 5.032 | 4.924 | 4.916 | 5.284 | 5.268 |
| 21a | 4.910 | 5.065 | 5.134 | 5.266 | 5.307 |
| 22 | 4.733 | 4.828 | 4.843 | 5.139 | 5.083 |
| 23 | 5.301 | 5.417 | 5.359 | 5.353 | 5.359 |
| 24a | 5.187 | 4.939 | 5.147 | 5.338 | 5.382 |
| 25 | 5.155 | 5.122 | 5.062 | 5.304 | 5.297 |
| 26 | 5.244 | 5.456 | 5.262 | 5.310 | 5.307 |
| 27 | 5.602 | 5.401 | 5.411 | 5.395 | 5.406 |
| 28 | 5.854 | 5.610 | 5.624 | 5.453 | 5.467 |
| 29 | 5.509 | 5.526 | 5.564 | 5.424 | 5.437 |
| 30 | 5.187 | 5.222 | 5.126 | 5.337 | 5.382 |
aTest compounds
The robustness of the built 2D-QSAR model was further assessed by applying Y-randomization test. The test is carried out by shuffling the biological activity (pIC50) at 100 random trials for the same number of training set molecules, and the new QSAR models have low R2 values in the range from 0.033 to 0.573 and Q2 values in the range of − 0.537 to − 0.243. But the R2 and Q2 value of the 2D-QSAR model is significantly greater (R2 = 0.908; Q2 = 0.747). Also, for an acceptable QSAR model, the average correlation coefficient (Rr) of randomized models should be less than the correlation coefficient (R) of the non-randomized model. The extent of the difference in the values of the mean squared correlation coefficients of the randomized (R2r) and that of the non-randomized (R2) models is reflected in the value of cR2p parameter. The value of cR2p should be more than 0.5 for passing Y-randomization test.
| 3 |
Since the cR2p value obtained is 0.780, the Y-randomization test was passed for our model.
Applicability domain plays a vital role in checking the reliability of the QSAR models by filtering the chemical structures that cannot be tolerated by the model. We have used applicability domain using standardization approach which uses the molecular descriptors used to build the QSAR model and to predict activity values (Roy et al. 2015a, b). If the training set of the compounds contains properties very dissimilar to the rest of the compounds, then these compounds are considered as X-outliers. In the test set, molecules which are not similar to any of the training set of molecules are considered outside the applicability domain. In our 2D-QSAR model, no compound from the training set or test set was found as X-outlier. As all the compounds of the dataset used to develop 2D-QSAR model fell inside the domain of applicability, our model was not obtained by mere chance.
HQSAR
A total of 21 HQSAR models were constructed by varying the fragment distinction and maintaining the default fragment size (4–7 atoms) with the endeavor of evaluating the influence of the descriptors on the robustness of the models (Table 3). For each constructed model, we also accessed the influence of the hologram length (HL) by building models varying HL as 53, 59, 61, 71, 83, 97, 151 199, 257, 307, 353 and 401 bins. All constructed models showed acceptable LOO validation coefficients (Q2) indicating that the HQSAR method is suitable to generate robust statistical models. According to the highest Q2 value, the best HQSAR model was constructed with A/B/C/Ch. The model had a higher Q2 value and lowered standard error of prediction (SEP) than the two second best models (A/C/DA and A/B/C/H/Ch/DA).
Table 3.
Statistical results of the 21 initial HQSAR models obtained from the variation of the fragment distinction and maintaining the default fragment size (4–7 atoms)
| Fdist | Q2 | SEP | R2ncv | HL | PCs | R2pred |
|---|---|---|---|---|---|---|
| A/B/C | 0.543 | 0.353 | 0.928 | 257 | 5 | 0.423 |
| A/B/H | 0.549 | 0.408 | 0.921 | 61 | 6 | 0.317 |
| A/C/H | 0.446 | 0.428 | 0.876 | 151 | 4 | 0.489 |
| A/B/Ch | 0.439 | 0.455 | 0.948 | 353 | 6 | 0.386 |
| A/C/Ch | 0.550 | 0.349 | 0.942 | 97 | 5 | 0.138 |
| A/B/DA | 0.512 | 0.424 | 0.943 | 83 | 6 | 0.168 |
| A/C/DA | 0.609 | 0.380 | 0.946 | 257 | 6 | 0.423 |
| A/B/C/H | 0.438 | 0.431 | 0.852 | 307 | 4 | 0.861 |
| A/B/C/Ch | 0.615 | 0.306 | 0.931 | 257 | 5 | 0.956 |
| A/C/H/Ch | 0.434 | 0.432 | 0.878 | 151 | 4 | 0.909 |
| A/C/H/DA | 0.565 | 0.400 | 0.954 | 53 | 6 | 0.538 |
| A/B/H/Ch | 0.484 | 0.413 | 0.812 | 59 | 4 | 0.848 |
| A/B/C/DA | 0.577 | 0.384 | 0.917 | 59 | 5 | 0.948 |
| A/H/Ch/DA | 0.542 | 0.400 | 0.897 | 199 | 5 | 0.891 |
| A/B/Ch/DA | 0.502 | 0.417 | 0.934 | 401 | 5 | 0.418 |
| A/B/H/DA | 0.527 | 0.406 | 0.936 | 257 | 5 | 0.898 |
| A/B/C/H/Ch | 0.424 | 0.436 | 0.851 | 307 | 4 | 0.887 |
| A/B/C/Ch/DA | 0.568 | 0.388 | 0.910 | 401 | 5 | 0.948 |
| A/B/H/Ch/DA | 0.522 | 0.408 | 0.916 | 307 | 5 | 0.876 |
| A/B/C/H/DA | 0.578 | 0.373 | 0.912 | 83 | 4 | 0.899 |
| A/B/C/H/Ch/DA | 0.585 | 0.370 | 0.915 | 83 | 4 | 0.904 |
Selected model is given in bold
The model A/B/C/Ch was subjected to external validations employing the test set of compounds. The predicted pIC50 values for all test set of compounds showed a residual error lower than 1 log unit. The predictive potential for the test set (R2m) is equal to 0.910 and the predictive potential for all compounds (R2m overall) is equal to 0.849. The graphical plot of predicted versus actual pIC50 values is represented in Fig. 2b. The predicted Aβ aggregation inhibitory activities of the developed model A/B/C/Ch are shown in Table 2.
Contour maps of the HQSAR analysis (Fig. 3) show the different colors of the atoms or fragments, which determine the overall contribution to the activity profiles of the molecules. The contributions of (i) red colour indicates a bad contribution less than − 0.244 (ii) red–orange color indicates a bad contribution ranging from − 0.244 to − 0.146, (iii) orange color ranges from − 0.146 to − 0.098 (iv) the white color indicates an average contribution ranging from − 0.098 to 0.0303, (v) a yellow color indicates a good contribution of 0.0303 to 0.045, and (vi) the green color signifies the maximum contribution of 0.076 and above. The most significant green contribution is observed in the benzene ring (Fig. 3). Backbone alkyl chains also depict the average contribution as per the most active compound. The contour map of the least active compound 17 is assigned red-orange and yellow color, indicating the most unfavorable fragment contributing to the activity and has a negative impact towards the inhibitory activity.
Fig. 3.

Contribution map generated by HQSAR model
CoMFA analysis
CoMFA study analysis is implemented by the most potent ligand compound 8, used as a template to align all the ligands. The training set is used to build the CoMFA model. The statistical parameters derived from the CoMFA studies are provided in Table 4. The developed CoMFA model has a cross-validated Q2 of 0.687, with five Optimum Number of Components (ONC), a non-cross-validated R2ncv of 0.787, F-value of 181.42 and low SEE of 0.246. Thus the resulted CoMFA model is statistically significant in depicting the novel curcumin derivative’s inhibitory activities.
Table 4.
Statistical results of CoMFA and the best CoMSIA models
| CoMFA | CoMSIA (Model 6) | |
|---|---|---|
| aQ2/ONC | 0.687/5 | 0.743/3 |
| bR2ncv | 0.787 | 0.972 |
| cSEE | 0.246 | 0.094 |
| dR2pred | 0.731 | 0.713 |
| F value | 181.420 | 230.975 |
| Field contribution | ||
| Steric | 55.4 | 19.7 |
| Electrostatic | 44.6 | 52.0 |
| H-bond acceptor | – | 28.3 |
aCross-validated correlation coefficient
bNon cross-validated correlation coefficient
cStandard errors of estimate
dPredicted correlation coefficient for the test set
The predictive ability of the developed model is assessed by a test set of six ligands. The estimated predictive correlation coefficient (R2pred) of the CoMFA model on the test set is 0.731.
The calculated pIC50 values of the compounds for the CoMFA model are listed in Table 2. The correlation between the experimental pIC50 and the calculated ones for the CoMFA model is displayed in Fig. 4. Most of the ligands are located on or nearer to the trend line (R2 = 0.798) specifying that the predictive power of the developed model is good. The analysis of statistical results from CoMFA and graphical output (Fig. 5a) of the correlation analysis reveals that the predicted pIC50 values are in line with the experimental pIC50.
Fig. 4.

Contour maps of CoMFA: a electrostatic and b steric based on compound 8
Fig. 5.
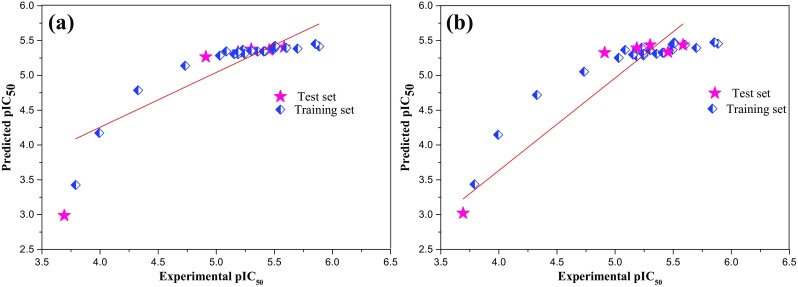
Correlations between actual and predicted pIC50 in the training and test sets for a CoMFA and b CoMSIA models
The higher contributions of electrostatic field (55.4%) than the steric field (44.6%) demonstrate that the electrostatic field is more dominant than the steric field to the inhibitory activity for CoMFA model.
CoMFA contour plots
The information about the steric and electrostatic fields surrounding the molecule can be acquired from the polyhedral contour plots. The contour plots show the regions in 3D space where the variation of the molecular fields is highly related to the corresponding changes in biological activity. The contour plots showing various fields’ contribution of CoMFA model is demonstrated with the template compound 8. Figure 4 represents the steric and electrostatic contour plots of CoMFA respectively. The favorable and unfavorable steric interactions are shown in green and yellow colored polyhedrons (80 and 20% contributions) respectively, while the favorable electropositive and electronegative interactions are given in the blue and red polyhedron (80 and 20% contributions) respectively. 3D color contour plots offer valuable contributions for the change in the design of new ligands.
Steric contour maps (Fig. 4a) indicate that if the bulky groups of the compounds are oriented towards the green region, then it can increase the biological activity. The opposite is true for yellow contours. In template compound 8, which is the most active compound, the contour map shows three large green contours around regions A, B and C, while these regions also show small yellow contours around regions A and C, and large contour around region B, indicating that bulkier substituents are not favorable in these regions. A favorable steric contour around region C can be supported with the good inhibitory potency (pIC50 = 5.854) shown by compound 28, having bulky CH3O–CO–CH2–O group when compared with compound 1 (pIC50 = 5.699). The (CH3)3C–O–CO–CH2–CH2 group attached in the region B of compound 10 is embedded in the yellow region, and the CH3O–CO–CH2–CH2 group in the same region of the compound 4 seems partially embedded in the yellow contour. Thus compound 4 shows highest activity (pIC50 = 5.357) than compound 10. Compounds 20 and 24 with pIC50 value of 5.032 and 5.187, respectively, have poor activity due to the presence of the bulkier groups in the region B oriented in the yellow region. Similarly, for compound 17 (pIC50 = 3.692), compound 22 (pIC50 = 4.733), and compound 10 (pIC50 = 5.187), having bulkier groups at the B region oriented towards the yellow region indicates poor activity for these compounds.
Electrostatic contour maps are shown in Fig. 4b, wherein the blue area specifies the favorable region for electropositive groups and the red area specifies an unfavorable region for electropositive groups. There are 80 and 20% contributions to favorable and unfavorable areas, respectively. Electrostatic contour maps show that the red contour is present over the HOOC–CH2–CH2 group in the region B of the compound 5 with low activity value of pIC50 = 5.086. Compound 23 (pIC50 = 5.301) showed good inhibitory activity due to the blue contour present around the (CH3)2N–CO–CH2–CH2 group at the B region of the compound. Compounds 18 and 7 (pIC50 = 5.495 and 5.456, respectively) showed good inhibitory activity due to the red region present over the electronegative oxygen of the carbonyl group of these compounds. Electronegative oxygen of the carbonyl group and hydroxyl group were oriented towards the blue contour, indicating poor activity of compound 22 (pIC50 = 4.733) and compound 16 (pIC50 = 4.327), respectively. Compounds 20, 24 and 26 (pIC50 = 5.031, 5.187 and 5.244, respectively) contain electronegative oxygen atom oriented towards the blue region, showing lower activity.
CoMSIA analysis
Different combinations of 5 fields (S, E, D, A and H) are combined to generate a total of 16 CoMSIA models. The statistical results of the constructed 16 CoMSIA models are summarized in Table 5. Out of these we selected the model 6 as it has Q2 value greater than 0.5, high F value, high R2ncv when compared to other models.
Table 5.
Statistical parameters of CoMSIA models
| Model | Descriptors | R2LOO(Q2)/ONC | R2ncv/SEEncv | F value | R2m (overall) | R2pred |
|---|---|---|---|---|---|---|
| 1 | S, D and A | 0.661/1 | 0.802/0.238 | 88.968 | 0.7763 | 0.7092 |
| 2 | S, H and A | 0.718/3 | 0.974/0.091 | 245.649 | 0.8182 | 0.5971 |
| 3 | E, D and H | 0.674/1 | 0.807/0.235 | 91.854 | 0.7748 | 0.7219 |
| 4 | E, A and H | 0.683/1 | 0.807/0.234 | 92.100 | 0.7729 | 0.7174 |
| 5 | S, E and H | 0.692/3 | 0.963/0.108 | 173.486 | 0.8248 | 0.6131 |
| 6 | S, E and A | 0.743/3 | 0.972/0.094 | 230.975 | 0.7773 | 0.7127 |
| 7 | S, E and D | 0.673/1 | 0.807/0.235 | 91.907 | 0.7779 | 0.7167 |
| 8 | D, A and H | 0.660/1 | 0.802/0.237 | 89.359 | 0.7735 | 0.7135 |
| 9 | D, A and E | 0.672/1 | 0.811/0.232 | 94.306 | 0.7804 | 0.7210 |
| 10 | S, D and H | 0.664/1 | 0.790/0.245 | 82.890 | 0.7692 | 0.7114 |
| 11 | S, E, D and A | 0.698/1 | 0.820/0.227 | 99.913 | 0.7787 | 0.7194 |
| 12 | S, E, D and H | 0.699/1 | 0.816/0.229 | 97.252 | 0.7856 | 0.7228 |
| 13 | S, E, A and H | 0.726/3 | 0.944/0.132 | 112.754 | 0.8326 | 0.6382 |
| 14 | D, A, H and S | 0.690/1 | 0.812/0.232 | 94.753 | 0.7851 | 0.7119 |
| 15 | D, A, H and E | 0.697/1 | 0.819/0.227 | 99.511 | 0.7867 | 0.7036 |
| 16 | S, E, D, A and H | 0.714/1 | 0.827/0.222 | 105.027 | 0.7913 | 0.7197 |
Selected model is given in bold
The statistical results of the CoMSIA studies are listed in Table 4. The generated CoMSIA model illustrated a Q2 LOO value of 0.743 (> 0.5) by three components. The non-cross validated PLS analysis with the ONC = 3 gave a non-cross-validated R2 (R2ncv = 0.972), a test set R2 (R2pred = 0.713), SEE = 0.094, F value of 230.975, steric = 19.7%, electrostatic contribution = 52.0% and H-bond acceptor contribution = 28.3%. The obtained high R2ncv, Q2 and F values along with the lower SEEncv indicated the satisfactory predictive ability of the derived model. The pIC50 values predicted by the CoMSIA model are listed in Table 2. Figure 5b demonstrates the correlation between experimental and predicted pIC50 values by the CoMSIA model.
CoMSIA contour plots
The steric, electrostatic and H-bond acceptor contour maps derived from the CoMSIA model based on the reference compound 8 are shown in the Fig. 6. As shown in Fig. 6a, a large yellow contour is present near regions B, indicating that the bioactivity of molecules is influenced by the introduction of bulky groups near these regions. The inhibitory activity would be decreased by the introduction of bulky groups in the B region, such as compounds 17 and 16 where the use of bulky groups ((CH3)3C–O–CO–CH2–CH2>CH3O–CO–CH2–CH2) resulted in lower pIC50 values (3.693 < 4.327). This can also be observed by the comparison of molecules 22 (substituted by HO–(CH2–CH2–O)3–CH2–CH2 with pIC50 value of 4.733) and 25 (substituted by HO–CH2–CH2–O–CH2–CH2 with pIC50 value of 5.155). This can also be observed by a comparison of compounds 9 (substituted by CH3–CH2O–CO–CH2–CH2 with pIC50 value of 5.420) and 11 (substituted by (CH3)2CH–O–CO–CH2–CH2 with pIC50 value of 5.409).
Fig. 6.

Contour maps of CoMSIA: a steric; b electrostatic and c H-bond acceptor based on compound 8
Two large green contours are found in the regions A and C, which shows that bulky groups at these regions would lead to increase the inhibitory activity. In compound 28, the green region covers the CH3O–CO–CH2–O group at the region C, which can be supported with the good inhibitory potency (pIC50 = 5.854) shown by compound 28. For instance, the agonist activity of compounds 29 (substituted by HOOC–CH2–O–) and 18 (substituted by –OH) was varied in the order: 18 < 29. This can also be observed by comparing molecules 16 (having CH3O–CH2–O group at regions A and B) and 14 (having CH3O group at regions A and B), where using a bulky group influenced the outcome of pIC50 values (4.327 > 3.790).
Electrostatic contour maps are displayed in Fig. 6b, wherein the blue region designates the favorable region for electropositive groups, and the red region designates an unfavorable region for electropositive groups. There are 80 and 20% contributions to favorable and unfavorable areas, respectively. Electrostatic contour maps show that a medium red contour and a medium blue contour is present over the region B. The compound 5 (pIC50 = 5.086) showed good inhibitory activity due to the red region present over the oxygen of the carboxyl group, when compared with compound 14 (pIC50 = 3.790). Electronegative oxygen of the hydroxyl group was oriented towards the blue contour, indicating poor activity of compound 22 (pIC50 = 4.733). Compounds 7 and 9 (pIC50 = 5.456 and 5.420, respectively) contain an electronegative oxygen atom at the region B oriented towards the red region, showing higher activity. In compound 17 (pIC50 = 3.693) the electronegative oxygen of carbonyl group is oriented towards the blue region, justifying the poor activity of the compound.
H-bond acceptor contour maps are shown in Fig. 6c, wherein the magenta region shows the favorable region for H-bond acceptor groups and the cyan region shows an unfavorable region. Almost 80 and 20% contributions to the favorable and unfavorable region, respectively, are shown. The significantly increased potency of compound 23 (pIC50 = 5.301) may be explained by its atom N as the H-bond acceptor at the region B is oriented towards the magenta contour compared to molecule 14 (pIC50 = 3.790) with a CH3 group at the same position. In fact, compounds with significant activity, for instance compounds 18 (pIC50 = 5.495), 7 (pIC50 = 5.456), 9 (pIC50 = 5.420), 11 (pIC50 = 5.409), 4 (pIC50 = 5.237) and 15 (pIC50 = 5.301), all have such H-bond acceptor groups at the region B, oriented towards the magenta contour map, which are consistent with this contour implication. The compounds 13 (pIC50 = 3.993) and 14 (pIC50 = 3.790) exhibits reduced inhibitory affinity as these compounds do not have the H-bond acceptor.
Pharmacophore analysis
A pharmacophore is an incorporation of steric, electrostatic, H-bond donor, H-bond acceptor and hydrophobic characteristics that were necessary to ensure the optimal molecular interactions with the biological target. Ten structurally diverse curcumin derivatives with high inhibitory activity were used for developing pharmacophore model. Results indicated that pharmacophore model generated have the following requirements: two hydrophobic & aromatic features, two hydrogen bond donor function, and one hydrogen acceptor function as seen in Fig. 7.
Fig. 7.

Pharmacophore models a of all the 30 curcumin derivatives b most active compound 8
Molecular docking
Primary and secondary structure prediction and validation
Isoelectric point (pI) value is the pH at which a protein is stable and compact has no net charge. The computed pI value of 2BEG (5.31) is less than 7 (pI < 7) indicated that these proteins were acidic. The II value provides an assessment of the stability of protein and when the value is smaller than 40, the protein is predicted as stable and when the value is above 40, the protein is predicted to be unstable. The II value for the protein 2BEG is found to be 18.17 suggesting that 2BEG is stable.
The AI is defined as the relative volume of a protein occupied by aliphatic side chains and is a positive factor for the increase of thermal stability of globular proteins. AI for 2BEG is found to be 97.38. The very high AI values of the protein indicate that the protein may be stable for a wide temperature range.
The secondary structures analysis of the protein predicted by SOPMA shows that, the percentages of random coils were more in the protein molecule. The conformational entropy associated with the random coil state contributes to the energetic stabilization of the protein and accounts for much of the energy barrier to protein folding.
Protein–ligand molecular docking analysis
In the present study, the binding mode for 30 curcumin-based molecular hybrids in the active site of the protein structure 2BEG was explored using molecular docking studies. The optimal conformations of these compounds when docked were identified. The active site of the protein 2BEG contains the highly conserved residues. To further elucidate the interaction mechanism, we selected the most potent compound 8 and least potent compound 17, Aβ aggregation inhibitor to perform the deeper docking study and discussion.
The results of the molecular docking studies are summarized in Table 6. These docking results clearly indicate that the most active compounds in the study exhibited significant binding affinities towards the active site of the protein (− 12.250 to − 11.110 kcal/mol), and the energy ranges are comparable to the Aβ aggregation inhibitor.
Table 6.
Molecular docking scores (kcal/mol) of the active molecules in the binding site of the protein 2BEG
| Compound ID | E_score (kcal/mol) |
|---|---|
| 5 | − 11.5212 |
| 6 | − 11.1483 |
| 11 | − 11.9898 |
| 15 | − 11.1105 |
| 16 | − 11.3656 |
| 21 | − 12.2503 |
| 23 | − 11.4561 |
| 25 | − 11.1258 |
| 29 | − 12.2175 |
Molecular design of novel chemical entities
The detailed analysis of 2D-QSAR, HQSAR, 3D-QSAR, Pharmacophore and Molecular docking studies empower us to identify structural requirements for the observed inhibitory activity. According to the information derived from 2D-QSAR, HQSAR, 3D-QSAR, pharmacophore and molecular docking studies, some important facts about the chemical structures requirement was presented to examine the effect of each kind of group as the substituent for regions A, B and C on the inhibitory activity. The HQSAR result shows that the presence of hydroxyl group and oxygen would enhance the inhibitory activity. So it is retained in some of the molecules. 3D-QSAR studies indicate that the presence of bulky groups at regions A and C were considered to enhance the activity. However, the presence of bulky groups at region B would decrease the activity. Molecular docking studies demonstrate that the presence of hydroxyl and carbonyl groups increases the interaction with the active site of the protein moiety. Also, the presence of oxygen atom increases the inhibitory activity of the molecules. Based on these, some novel chemical entities were designed.
For predicting their biological activity, 2D-QSAR model is applied to these new molecules and the corresponding results are listed in Table 7. The descriptors of the newly designed virtual molecules were calculated using PaDEL-Descriptor and MOE softwares. The results show that the eight compounds, M(1), M(2), M(5), M(6), M(7), M(8), M(9) and M(10) show predicted biological activity values higher than before, indicative of their good inhibitory activity. Therefore it is expected that these compounds perhaps should be regarded as the good candidate molecules for experimental synthesis.
Table 7.
Predicted pIC50 values of novel chemical entities using inverse QSAR
| ID | R1 | R2 | R3 | R4 | R5 | Predicted pIC50 |
|---|---|---|---|---|---|---|
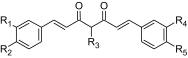
|
||||||
| M(1) | (CH3)3O | HO | (CH3)2CH–CH(NH2)–CO–O | (CH3)3O | HO | 6.5814 |
| M(2) | (CH3)3O | HO | C6H5–CO | (CH3)3O | HO | 6.5314 |
| M(3) | (CH3)3O | (CH3)3O | (CH3)3C–CH2–CH2 | (CH3)3O | (CH3)3O | 5.4683 |
| M(4) | (CH3)3C | (CH3)3C | (CH3)2N(CH3) | (CH3)3C | (CH3)3C | 5.5780 |
| M(5) | (CH3)3O | HO | CH3–CH2–C6H5 | (CH3)3O | HO | 6.5886 |
| M(6) | C6H11O | HO | CH3 | (CH3)3O | C6H11O | 6.8062 |
| M(7) | ((CH3)2CH) 3CO | HO | CH3–CH2 | ((CH3)2CH) 3CO | HO | 6.5836 |
| M(8) | ((CH3)2CH) 3CO | CH3O | CH3–CO–O– | ((CH3)2CH) 3CO | CH3O | 5.9633 |
| M(9) | Cl3O | CH3O | CH3–CO–O– | Cl3O | CH3O | 5.9396 |
| M(10) | Cl3O | CH3O | CH3–CH2 | Cl3O | CH3O | 6.7362 |
Data mining
Data mining techniques can examine the main useful patterns emerging from a set of data. Machine learning (ML) technique can be effective in generating a model out of such data, and the model can be further used to predict the activity any molecule present in a set of selected molecules. This provides good results and accurate information which can be helpful for solving many of the health-related problems. An understanding of the structural features of a set of molecules may thus throw light on the factors that are characteristic of the activity of the molecules.
WEKA (Waikato Environment for Knowledge Analysis) 3.7.3 with ML method was used to screen and to recognize the Aβ aggregation inhibitory activity of the newly designed curcumin derivatives (Frank et al. 2005). The process of classification requires building a classifier (model) which is a mathematical function that assigns class (e.g., active/inactive) labels to instances defined by a set of attributes (e.g., descriptors). The active (n = 1420) and inactive (n = 820) molecules present in the AID 647 dataset downloaded from PubChem were used for the development of classification models. For each molecule, 179 different molecular descriptors were computed using the software called powerMV. The Random Forest (RF) algorithm of WEKA gives high accuracy and time efficiency for predictive data modeling and is regarded as the best classifier (Sajeev et al. 2013; Seal et al. 2012). Therefore in the present study, we tested the activity of the novel chemical entities’ using the classifier based on RF. Using the tenfold cross-validation (CV), the RF classifier was evaluated. A cross-validation is a standard tool in analytics, which helps to develop and fine-tune data mining models. The model was used by taking 80% of the data as the training cum validation set and 20% of data as an independent test set.
Data mining results show that out of the ten novel chemical entities, molecules M(1), M(2), M(3), M(4), M(5), M(7), M(8) and M(9) were active. The model displayed better statistical indices like 63.24% accuracy, 84.8% sensitivity, specificity 30.2%, 63.2% BCR (Balanced Classification Rate). This model achieves a precision of 61.6% and recall of 63.2%. The classifier achieved an F-measure of 0.601 and ROC area of 0.644.
Prediction of site of metabolism and ADME properties
In drug design process, the information of site of metabolism (SOM) is vital to mitigate the toxicity issues and to improve the metabolic behavior of a molecule. For metabolite identification, we have used the Toxtreev2.6.13 software. The methylene group (–CH2–) in between two carbonyl groups is ranked as the best SOM in most of the compounds M(2), M(5), M(7), M(8) and M(9). This SOM involves aliphatic hydroxylation reaction for metabolism. For the compound M(1), the methylene group attached to the –NH2 group is ranked as the best SOM. In compound M(2), the double bonds of the benzene ring adjacent to the oxygen atom are regarded as the SOM with the second rank and the metabolism occurs through aromatic hydroxylation. In compound M(4), the alkyl groups attached to the N atom, are considered as SOM with highest ranks. It undergoes N-dealkylation reaction. This site undergoes O-dealkylation reaction. In compounds M(8) and M(9), the –CH3 of the methoxy group is regarded as the SOM with second position. This site also undergoes O-dealkylation reaction for metabolism.
The ADME properties of the selected six compounds were studied using web based PreADMET program. It gives the details of properties such as HIA rate, BBB penetration and in vitro Caco-2 cell permeability (nm/s). The predicted values of these properties are shown in Table 8.
Table 8.
ADME properties for novel chemical entities
| Compound ID | M(1) | M(2) | M(5) | M(7) | M(9) | M(8) |
|---|---|---|---|---|---|---|
| BBB | 0.1434 | 0.7918 | 4.8833 | 10.4024 | 0.9439 | 0.2978 |
| Caco2 | 21.9193 | 36.4299 | 46.0038 | 51.4046 | 56.2054 | 22.8121 |
| HIA | 94.1330 | 95.7125 | 96.2881 | 96.5479 | 97.7153 | 98.0645 |
The BBB penetration values give us an idea whether a compound can pass across the BBB or not. A compound having BBB value > 2.0 is considered as highly absorbing to CNS (Central Nervous System), that with the value 2.0–0.1 is considered as with middle absorption to CNS and that with the value < 0.1 is to be considered as low absorbing to CNS (Ma et al. 2005; Ajay et al. 1999). The result showed that the compounds M(5) and M(7) have high absorption to CNS and the compounds M(1), M(2), M(8) and M(9) have middle absorption to CNS.
For understanding the intestinal absorption of the compounds under study, several in vitro methods have been used. Caco-2 cell permeability is suggested as a dependable in vitro model for the prediction of oral drug absorption. For a low permeable compound, Caco-2 value should be less than 4; while for a middle permeable one the value should be between 4 and 70; and one with more than 70 is considered as a highly permeable compound (Yamashita et al. 2000). In this study, the result demonstrated that the compounds M(1), M(2), M(5), M(7), M(8) and M(9) have moderate cellular permeability against Caco-2 cells.
The HIA data is the summation of absorption evaluated from the ratio of cumulative excretion and bioavailability. The HIA value for poorly absorbed compounds is between 0 and 20%. For a moderately absorbed compound, the HIA value is 20–70%, and for the well-absorbed compounds, the HIA value ranges from 70 to 100% (Zhao et al. 2001). In this study, we obtained very good HIA values for the compounds M(1), M(2), M(5), M(7), M(8) and M(9).
Molecular docking studies were carried out to propose the binding pose for the identified hit compounds in the binding site of CYP3A4, which would lead to helpful insights for the development of new medications. Here, we present the molecular interactions of Aβ aggregation inhibitors with CYP3A4 using an in silico docking study. CYP3A4 is a complex heme-containing enzyme. It metabolizes more than 50% of the administered drugs. It is the isozyme most implicated in drug-drug interaction profiles (Gibbs and Hosea 2003). The active site of the CYP3A4 enzyme is larger and considerably flexible. The results show that the molecules are not tightly locked into the CYP3A4 active site. They can dissociate and rebind during different stages of the metabolism cycle.
Molecular docking studies of the 10 new virtual active compounds were also performed in the binding pocket of the protein 2BEG. These docking results clearly indicate that the new virtual active compounds in the study exhibited significant binding affinities towards the active site of the protein (− 12.625 to − 10.1492 kcal/mol), and the energy ranges are comparable to the Aβ aggregation inhibitor activity.
Thus compounds M(1), M(2), M(5), M(7), M(8) and M(9) (Fig. 8) are selected as the lead molecules metabolise through aliphatic hydroxylation, aromatic hydroxylation, N-dealkylation, and O-dealkylation reactions.
Fig. 8.
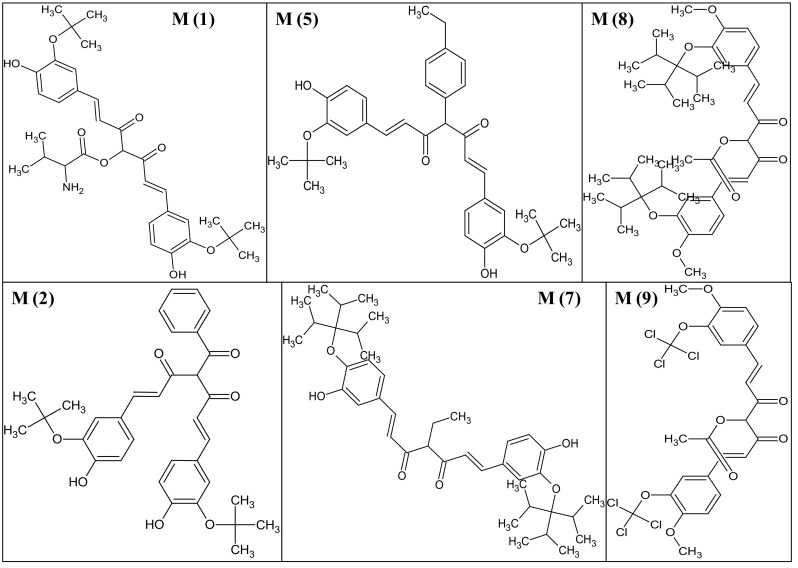
Structures of the lead molecules
Conclusion
In the current study, an effort has been made by extracting the relevant properties of a known Aβ aggregation inhibitory activity set and extrapolating this knowledge to develop predictive cheminformatic models for the classification, identification, and prioritization of new Aβ aggregation inhibitors. For the purpose, a series of 30 curcumin derivatives which are reported as potent Aβ aggregation inhibitors were selected to develop various QSAR and pharmacophore models. The established 2D and 3D-QSAR models showed significant statistical quality and excellent predictive ability. To verify the reliability of the models, the inhibitory activities were evaluated and predicted. Molecular docking analyses of the representative inhibitors were performed to determine the binding modes of the inhibitors at the active site of the protein molecule 2BEG. Based on the QSAR and molecular docking results, some new potent inhibitors were designed, and their activities are then predicted using an inverse QSAR technique. These molecules were further filtered using data mining techniques by Weka, ADME properties were evaluated and the binding interactions understood by molecular docking studies. This resulted in the selection of six lead molecules, M(1), M(2), M(5), M(7), M(8) and M(9). Finally, our in silico results provide compelling evidence for the utility of curcumin derivatives as a preventive medication for neurodegenerative diseases such as Aβ1-42 induced AD. Further investigations are necessary to examine the mechanisms of these molecules and their in vivo effect. Yet, the results of the present study open up the possibility that these derivatives can be developed as promising therapeutics to reach to advanced studies for treatment against Alzheimer’s disease.
Acknowledgements
Aswathy L. is thankful to CSIR, New Delhi for the financial assistance in the form of Senior Research Fellowship. Jisha, R.S. is thankful to the University of Kerala, Thiruvananthapuram for providing financial assistance in the form of University Junior Research Fellowship for this work.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- Ajay, Bemis GW, Murcko MA. Designing libraries with CNS activity. J Med Chem. 1999;42:4942–4951. doi: 10.1021/jm990017w. [DOI] [PubMed] [Google Scholar]
- Alzheimer’s Association 2017 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2017;13:325–373. doi: 10.1016/j.jalz.2017.02.001. [DOI] [Google Scholar]
- Aswathy L, Jisha RS, Masand VH, et al. Computational strategies to explore antimalarial thiazine alkaloid lead compounds based on an Australian marine sponge Plakortis lita. J Biomol Struct Dyn. 2017;35:2407–2429. doi: 10.1080/07391102.2016.1220870. [DOI] [PubMed] [Google Scholar]
- Ballante F, Ragno R. 3-D QSAutogrid/R: an alternative procedure to build 3-D QSAR models. methodologies and applications. J Chem Inf Model. 2012;52:1674–1685. doi: 10.1021/ci300123x. [DOI] [PubMed] [Google Scholar]
- Begum AN, Jones MR, Lim GP, et al. Curcumin structure-function, bioavailability, and efficacy in models of neuroinflammation and Alzheimer’s disease. J Pharmacol Exp Ther. 2008;326:196–208. doi: 10.1124/jpet.108.137455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caesar I, Jonson M, Nilsson KPR, et al. Curcumin promotes A-beta fibrillation and reduces neurotoxicity in transgenic drosophila. PLoS ONE. 2012;7:e31424. doi: 10.1371/journal.pone.0031424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatake T, Tanaka I, Umino H, et al. The hydration structure of a Z-DNA hexameric duplex determined by a neutron diffraction technique. Acta Crystallogr D Biol Crystallogr. 2005;61:1088–1098. doi: 10.1107/S0907444905015581. [DOI] [PubMed] [Google Scholar]
- Citron M. Alzheimer’s disease: strategies for disease modification. Nat Rev Drug Discov. 2010;9:387–398. doi: 10.1038/nrd2896. [DOI] [PubMed] [Google Scholar]
- Cramer RD, Patterson DE, Bunce JD. Comparative molecular field analysis (CoMFA). 1. Effect of shape on binding of steroids to carrier proteins. J Am Chem Soc. 1988;110:5959–5967. doi: 10.1021/ja00226a005. [DOI] [PubMed] [Google Scholar]
- Cruciani G, Watson KA. Comparative molecular field analysis using GRID force-field and GOLPE variable selection methods in a study of inhibitors of glycogen phosphorylase b. J Med Chem. 1994;37:2589–2601. doi: 10.1021/jm00042a012. [DOI] [PubMed] [Google Scholar]
- Dong M, Lu X, Ma Y, et al. An efficient approach for automated mass segmentation and classification in mammograms. J Digit Imaging. 2015;28:613–625. doi: 10.1007/s10278-015-9778-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubey SK, Sharma AK, Narain U, et al. Design, synthesis and characterization of some bioactive conjugates of curcumin with glycine, glutamic acid, valine and demethylenated piperic acid and study of their antimicrobial and antiproliferative properties. Eur J Med Chem. 2008;43:1837–1846. doi: 10.1016/j.ejmech.2007.11.027. [DOI] [PubMed] [Google Scholar]
- Elfiky AA, Elshemey WM. IDX-184 is a superior HCV direct-acting antiviral drug: a QSAR study. Med Chem Res. 2016;25:1005–1008. doi: 10.1007/s00044-016-1533-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank E, Hall M, Holmes G, et al. Weka. In: Maimon O, Rokach L, et al., editors. Data mining and knowledge discovery handbook. Boston, MA: Springer; 2005. pp. 1305–1314. [Google Scholar]
- Garcia-Alloza M, Borrelli LA, Rozkalne A, et al. Curcumin labels amyloid pathology in vivo, disrupts existing plaques, and partially restores distorted neurites in an Alzheimer mouse model: curcumin reverses amyloid pathology in vivo. J Neurochem. 2007;102:1095–1104. doi: 10.1111/j.1471-4159.2007.04613.x. [DOI] [PubMed] [Google Scholar]
- Gasteiger E, Hoogland C, Gattiker A, et al. Protein identification and analysis tools on the ExPASy server. In: Walker JM, et al., editors. The proteomics protocols handbook. Totowa: Humana Press; 2005. pp. 571–607. [Google Scholar]
- Geourjon C, Deléage G. SOPMA: significant improvements in protein secondary structure prediction by consensus prediction from multiple alignments. Comput Appl Biosci. 1995;11:681–684. doi: 10.1093/bioinformatics/11.6.681. [DOI] [PubMed] [Google Scholar]
- Gibbs MA, Hosea NA. Factors affecting the clinical development of cytochrome P450 3A substrates. Clin Pharmacokinet. 2003;42:969–984. doi: 10.2165/00003088-200342110-00003. [DOI] [PubMed] [Google Scholar]
- Gill SC, von Hippel PH. Calculation of protein extinction coefficients from amino acid sequence data. Anal Biochem. 1989;182:319–326. doi: 10.1016/0003-2697(89)90602-7. [DOI] [PubMed] [Google Scholar]
- Golbraikh A, Tropsha A. Beware of q2! J Mol Graph Model. 2002;20:269–276. doi: 10.1016/S1093-3263(01)00123-1. [DOI] [PubMed] [Google Scholar]
- Golde TE, Eckman CB, Younkin SG. Biochemical detection of Abeta isoforms: implications for pathogenesis, diagnosis, and treatment of Alzheimer’s disease. Biochim Biophys Acta. 2000;1502:172–187. doi: 10.1016/S0925-4439(00)00043-0. [DOI] [PubMed] [Google Scholar]
- Guruprasad K, Reddy BV, Pandit MW. Correlation between stability of a protein and its dipeptide composition: a novel approach for predicting in vivo stability of a protein from its primary sequence. Protein Eng. 1990;4:155–161. doi: 10.1093/protein/4.2.155. [DOI] [PubMed] [Google Scholar]
- Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid beta-peptide. Nat Rev Mol Cell Biol. 2007;8:101–112. doi: 10.1038/nrm2101. [DOI] [PubMed] [Google Scholar]
- Halgren TA. Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. J Comput Chem. 1996;17:490–519. doi: 10.1002/(SICI)1096-987X(199604)17:5/6<490::AID-JCC1>3.0.CO;2-P. [DOI] [Google Scholar]
- Hamaguchi T, Ono K, Murase A, Yamada M. Phenolic compounds prevent Alzheimer’s pathology through different effects on the amyloid-β aggregation pathway. Am J Pathol. 2009;175:2557–2565. doi: 10.2353/ajpath.2009.090417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- Hsu J-L, Hung P-C, Lin H-Y, Hsieh C-H. Applying under-sampling techniques and cost-sensitive learning methods on risk assessment of breast cancer. J Med Syst. 2015 doi: 10.1007/s10916-015-0210-x. [DOI] [PubMed] [Google Scholar]
- Ikai A. Thermostability and aliphatic index of globular proteins. J Biochem. 1980;88:1895–1898. [PubMed] [Google Scholar]
- Jack CR, Knopman DS, Jagust WJ, et al. Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurol. 2010;9:119–128. doi: 10.1016/S1474-4422(09)70299-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janitza S, Strobl C, Boulesteix A-L. An AUC-based permutation variable importance measure for random forests. BMC Bioinform. 2013;14:119. doi: 10.1186/1471-2105-14-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin W, Wang J, Zhu T, et al. Anti-inflammatory effects of curcumin in experimental spinal cord injury in rats. Inflamm Res. 2014;63:381–387. doi: 10.1007/s00011-014-0710-z. [DOI] [PubMed] [Google Scholar]
- Jisha RS, Aswathy L, Masand VH, et al. Exploration of 3,6-dihydroimidazo(4,5-d)pyrrolo(2,3-b)pyridin-2(1H)-one derivatives as JAK inhibitors using various in silico techniques. In Silico Pharmacol. 2017 doi: 10.1007/s40203-017-0029-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapetanovic IM. Computer-aided drug discovery and development (CADDD): in silico-chemico-biological approach. Chem Biol Interact. 2008;171:165–176. doi: 10.1016/j.cbi.2006.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klebe G, Abraham U, Mietzner T. Molecular similarity indices in a comparative analysis (CoMSIA) of drug molecules to correlate and predict their biological activity. J Med Chem. 1994;37:4130–4146. doi: 10.1021/jm00050a010. [DOI] [PubMed] [Google Scholar]
- Kyte J, Doolittle RF. A simple method for displaying the hydropathic character of a protein. J Mol Biol. 1982;157:105–132. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- Lengauer T, Rarey M. Computational methods for biomolecular docking. Curr Opin Struct Biol. 1996;6:402–406. doi: 10.1016/S0959-440X(96)80061-3. [DOI] [PubMed] [Google Scholar]
- Lim GP, Chu T, Yang F, et al. The curry spice curcumin reduces oxidative damage and amyloid pathology in an Alzheimer transgenic mouse. J Neurosci. 2001;21:8370–8377. doi: 10.1523/JNEUROSCI.21-21-08370.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lührs T, Ritter C, Adrian M, et al. 3D structure of Alzheimer’s amyloid-beta(1-42) fibrils. Proc Natl Acad Sci USA. 2005;102:17342–17347. doi: 10.1073/pnas.0506723102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma X, Chen C, Yang J. Predictive model of blood–brain barrier penetration of organic compounds. Acta Pharmacol Sin. 2005;26:500–512. doi: 10.1111/j.1745-7254.2005.00068.x. [DOI] [PubMed] [Google Scholar]
- Ma Q-L, Zuo X, Yang F, et al. Curcumin suppresses soluble tau dimers and corrects molecular chaperone, synaptic, and behavioral deficits in aged human tau transgenic mice. J Biol Chem. 2013;288:4056–4065. doi: 10.1074/jbc.M112.393751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannu J, Jenardhanan P, Mathur PP. A computational study of CYP3A4 mediated drug interaction profiles for anti-HIV drugs. J Mol Model. 2011;17:1847–1854. doi: 10.1007/s00894-010-0890-6. [DOI] [PubMed] [Google Scholar]
- Negi PS, Jayaprakasha GK, Jagan Mohan Rao L, et al. Antibacterial activity of turmeric oil: a byproduct from curcumin manufacture. J Agric Food Chem. 1999;47:4297–4300. doi: 10.1021/jf990308d. [DOI] [PubMed] [Google Scholar]
- Nguyen TKC, Dzung TTK, Cuong PV. Assessment of antifungal activity of turmeric essential oil-loaded chitosan nanoparticles. J Chem Bio Phy Sci Sec B. 2014;4:2347–2356. [Google Scholar]
- Nishikawa H, Tsutsumi J, Kitani S. Anti-inflammatory and anti-oxidative effect of curcumin in connective tissue type mast cell. J Funct Foods. 2013;5:763–772. doi: 10.1016/j.jff.2013.01.022. [DOI] [Google Scholar]
- Ojha PK, Roy K. Comparative QSARs for antimalarial endochins: importance of descriptor-thinning and noise reduction prior to feature selection. Chemom Intell Lab Syst. 2011;109:146–161. doi: 10.1016/j.chemolab.2011.08.007. [DOI] [Google Scholar]
- Ono K, Hasegawa K, Naiki H, Yamada M. Curcumin has potent anti-amyloidogenic effects for Alzheimer’s beta-amyloid fibrils in vitro. J Neurosci Res. 2004;75:742–750. doi: 10.1002/jnr.20025. [DOI] [PubMed] [Google Scholar]
- Roy K, Kar S, Ambure P. On a simple approach for determining applicability domain of QSAR models. Chemom Intell Lab Syst. 2015;145:22–29. doi: 10.1016/j.chemolab.2015.04.013. [DOI] [Google Scholar]
- Roy K, Kar S, Ambure P. On a simple approach for determining applicability domain of QSAR models. Chemom Intell Lab Syst. 2015;145:22–29. doi: 10.1016/j.chemolab.2015.04.013. [DOI] [Google Scholar]
- Sajeev R, Athira RS, Nufail M, et al. Computational predictive models for organic semiconductors. J Comput Electron. 2013;12:790–795. doi: 10.1007/s10825-013-0486-3. [DOI] [Google Scholar]
- Saleh NA. The QSAR and docking calculations of fullerene derivatives as HIV-1 protease inhibitors. Spectrochim Acta Part A Mol Biomol Spectrosc. 2015;136:1523–1529. doi: 10.1016/j.saa.2014.10.045. [DOI] [PubMed] [Google Scholar]
- Seal A, Passi A, Jaleel UA, et al. In-silico predictive mutagenicity model generation using supervised learning approaches. J Cheminform. 2012;4:10. doi: 10.1186/1758-2946-4-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe DJ. Cell biology of the amyloid beta-protein precursor and the mechanism of Alzheimer’s disease. Annu Rev Cell Biol. 1994;10:373–403. doi: 10.1146/annurev.cb.10.110194.002105. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer’s disease: genotypes, phenotypes, and treatments. Science. 1997;275:630–631. doi: 10.1126/science.275.5300.630. [DOI] [PubMed] [Google Scholar]
- Sharma RA, McLelland HR, Hill KA, et al. Pharmacodynamic and pharmacokinetic study of oral Curcuma extract in patients with colorectal cancer. Clin Cancer Res. 2001;7:1894–1900. [PubMed] [Google Scholar]
- Shibi IG, Aswathy L, Jisha RS, et al. Molecular docking and QSAR analyses for understanding the antimalarial activity of some 7-substituted-4-aminoquinoline derivatives. Eur J Pharm Sci. 2015;77:9–23. doi: 10.1016/j.ejps.2015.05.025. [DOI] [PubMed] [Google Scholar]
- Shibi IG, Aswathy L, Jisha RS, et al. Virtual screening techniques to probe the antimalarial activity of some traditionally used phytochemicals. Comb Chem High Throughput Screen. 2016;19:572–591. doi: 10.2174/1386207319666160420141200. [DOI] [PubMed] [Google Scholar]
- Tetko IV, Tanchuk VY, Villa AE. Prediction of n-octanol/water partition coefficients from PHYSPROP database using artificial neural networks and E-state indices. J Chem Inf Comput Sci. 2001;41:1407–1421. doi: 10.1021/ci010368v. [DOI] [PubMed] [Google Scholar]
- Tropsha A, Gramatica P, Gombar V. The importance of being earnest: validation is the absolute essential for successful application and interpretation of QSPR models. QSAR Comb Sci. 2003;22:69–77. doi: 10.1002/qsar.200390007. [DOI] [Google Scholar]
- Wahi D, Jamal S, Goyal S, et al. Cheminformatics models based on machine learning approaches for design of USP1/UAF1 abrogators as anticancer agents. Syst Synth Biol. 2015;9:33–43. doi: 10.1007/s11693-015-9162-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Y, Ma B, McElheny D, et al. Aβ(1–42) fibril structure illuminates self-recognition and replication of amyloid in Alzheimer’s disease. Nat Struct Mol Biol. 2015;22:499–505. doi: 10.1038/nsmb.2991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita S, Furubayashi T, Kataoka M, et al. Optimized conditions for prediction of intestinal drug permeability using Caco-2 cells. Eur J Pharm Sci. 2000;10:195–204. doi: 10.1016/S0928-0987(00)00076-2. [DOI] [PubMed] [Google Scholar]
- Yanagisawa D, Taguchi H, Morikawa S, et al. Novel curcumin derivatives as potent inhibitors of amyloid β aggregation. Biochem Biophys Rep. 2015;4:357–368. doi: 10.1016/j.bbrep.2015.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang F, Lim GP, Begum AN, et al. Curcumin inhibits formation of amyloid β oligomers and fibrils, binds plaques, and reduces amyloid in vivo. J Biol Chem. 2005;280:5892–5901. doi: 10.1074/jbc.M404751200. [DOI] [PubMed] [Google Scholar]
- Yang H, Xie W, Xue X, et al. Design of wide-spectrum inhibitors targeting coronavirus main proteases. PLoS Biol. 2005;3:e324. doi: 10.1371/journal.pbio.0030324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yap CW. PaDEL-descriptor: an open source software to calculate molecular descriptors and fingerprints. J Comput Chem. 2011;32:1466–1474. doi: 10.1002/jcc.21707. [DOI] [PubMed] [Google Scholar]
- Zhao YH, Le J, Abraham MH, et al. Evaluation of human intestinal absorption data and subsequent derivation of a quantitative structure-activity relationship (QSAR) with the Abraham descriptors. J Pharm Sci. 2001;90:749–784. doi: 10.1002/jps.1031. [DOI] [PubMed] [Google Scholar]